

Abstract
Copy number variation (CNV) is an important determinant of human diversity and plays important roles in susceptibility to disease. Most studies of CNV carried out to date have made use of chromosome microarray and have had a lower size limit for detection of about 30 kilobases (kb). With the emergence of whole-exome sequencing studies, we asked whether such data could be used to reliably call rare exonic CNV in the size range of 1–30 kilobases (kb), making use of the eXome Hidden Markov Model (XHMM) program. By using both transmission information and validation by molecular methods, we confirmed that small CNV encompassing as few as three exons can be reliably called from whole-exome data. We applied this approach to an autism case-control sample (n = 811, mean per-target read depth = 161) and observed a significant increase in the burden of rare (MAF ≤1%) 1–30 kb CNV, 1–30 kb deletions, and 1–10 kb deletions in ASD. CNV in the 1–30 kb range frequently hit just a single gene, and we were therefore able to carry out enrichment and pathway analyses, where we observed enrichment for disruption of genes in cytoskeletal and autophagy pathways in ASD. In summary, our results showed that XHMM provided an effective means to assess small exonic CNV from whole-exome data, indicated that rare 1–30 kb exonic deletions could contribute to risk in up to 7% of individuals with ASD, and implicated a candidate pathway in developmental delay syndromes.
Introduction
Copy number variation (CNV) has been identified as a major determinant of genetic diversity and disease and has been implicated in many neuropsychiatric disorders including developmental delay syndromes.1–7 Most studies examining CNV in human disease have used chromosome microarray and have an effective lower resolution of ca. 30 kilobases (kb); however, there is reason to believe that CNV in the 1–30 kb range are important in both human diversity and disease because CNV in this range have been detected in whole-genome studies.8
There is evidence for a role for CNV in autism spectrum disorder (ASD [MIM 209850]).2,6,7 ASD is characterized by impairments in reciprocal social interaction and communication and by the presence of restricted interests and/or repetitive and stereotyped behaviors.9–11 ASD affects ∼1% of the population and is highly heritable, and estimates from multiple studies have indicated that there might be 1,000 genes or loci that contribute to ASD.2,12–15 Rare, deleterious genetic variation at all scales can contribute to ASD, from de novo, recessive, or X-linked single nucleotide variation (SNV) to aneuploidy.16 Rare CNV, both inherited and de novo, has been repeatedly implicated in ASD, but for practical reasons the primary focus has been on CNV larger than 30 kb.2,7,17,18
Whole-exome sequencing (WES) has emerged as a cost-effective and efficient means to identify rare genic SNV contributing to risk for multiple disorders including ASD.12–15 With the widespread use of WES, methods have been developed to call CNV from WES data. XHMM (exome-hidden Markov model) is a recent approach that uses principal-component analysis to normalize exome read depth and a hidden Markov model (HMM) to identify exonic CNV from WES data.19 In the first report of this method, XHMM calls were validated with two approaches. First, transmission of CNV from parent to child was used to show that median per-family transmission rates were at about 50% for all CNV with estimated size >100 kb, with similar results for all CNV <100 kb. Second, XHMM calls of CNV >100 kb in length were compared with calls from chromosome microarray run on the same samples. These results provided strong support for XHMM as a method for calling larger exonic CNV and indicated that smaller CNV (encompassing as few as three exons) could also be reliably called by XHMM. Although validation of smaller CNV calling by XHMM was not carried out to the same extent as for larger CNV, smaller exonic CNV were quite common, with 82% of CNV identified being <100 kb and the median size of CNV being 18 kb in length. This indicates that effective identification of exonic CNV in the 1–30 kb range could be an important means to understand genetic risk in human disease.
In the current study, we further evaluated XHMM for the reliable identification of rare exonic CNV from WES, focusing exclusively on CNV with an estimated size of 1–30 kb in length. We used transmission data to set the calling quality threshold and employed molecular validation in an independent data set to confirm that XHMM can reliably call CNV in this size range. We applied XHMM to an ancestry-matched sample of ASD cases and controls and observed an increase in small CNV in ASD cases. The CNV in cases disrupted genes coding for proteins in the actin cytoskeleton network and genes involved in autophagy, highlighting potential new pathways in ASD risk. Our results indicated that XHMM is useful for identifying small exonic CNV from whole-exome data and implicated a previously unidentified pathway in ASD.
Material and Methods
Processing and Validation Pipeline Overview
Our processing pipeline (see Figure S1 available online) started with WES. We then called CNV with XHMM, filtered the CNV, and obtained burden statistics with PLINK. We subsequently performed computational and molecular validation. We describe these stages in detail below.
CNV Calling with XHMM
The study was approved by the appropriate institutional review board of all participating institutions, and written informed consent from all subjects was obtained. We analyzed two data sets. The first data set was a set of 261 trios that we used solely for assessing transmission of parental CNV. Our second data set, sequenced on the same platform and at the same site as the trio samples, consisted of 432 ASD cases of European ancestry and 379 ancestry-matched controls, totaling 811 individuals.20 Exonic DNA was captured with Agilent SureSelect Human All Exon v.2. Mean per-target depth of coverage across all targets was 161, with 90% of targets sequenced to an average of 17× or greater. Further details on sample collection, ancestry, control matching, and lab procedures can be found in Lim et al.20 After whole-exome sequencing, the data were mapped to the hg19 human reference genome with BWA21 and processed with Picard to mark duplicate reads, realign around indels, and recalibrate quality scores.
We calculated read depth per target in the WES by using GATK for each of the 189,979 targets in our exome capture. To call CNV, we ran XHMM,19 which infers CNV from read depth calculated from WES, by using the steps summarized in the online tutorial. Additional GATK arguments were as follows: -dt BY_SAMPLE -dcov 5000 --minBaseQuality 0 --minMappingQuality 20 --start 1 --stop 5000 --nBins 200 --includeRefNSites.
As described in detail previously,19 XHMM consists of two main steps. In the first step, systematic noise is removed by transforming the data into PCA space and removing the highest-variance dimensions. Many of these dimensions show high correlation with quantities such as GC content, mean sample read depth, mean target read depth, platform, and batch, which are not related to CNV; other dimensions do not correlate clearly with these systematic effects but are also not indicative of CNV. In the second step, average read depths at each target are converted to Z scores based on the distribution across samples. These Z scores are used as input to a hidden Markov model that calculates, for each sample, the most likely series of states (diploid, deletion, duplication) across all targets, which is finally output as a set of regions of deletion or duplication (composed of contiguous targets) for each sample. As a result, XHMM is not dependent on a minimum-read depth threshold, but only on sufficient dynamic range in read depth signal to reflect changes in copy number (see the Fromer et al.19 for a full discussion of XHMM’s handling of read depth).
XHMM accepts threshold arguments to remove outlier samples and targets based on properties of the read depth distribution. The values we used are shown in Table S1. Values were chosen to include as many samples and targets as possible, excluding only the extreme outliers, with the intention of finding as many instances of potential CNV as possible and then applying more stringent filters to the results. For the ASD data set, these parameter settings removed 25 individuals from the data set, and removed 16,528 targets from the set of targets considered in step two of the XHMM algorithm. With the values given, XHMM called 4,608 CNV in 770 individuals.
The genotyping stage of XHMM (scoring CNV called in one or more samples across all samples) uses values from a parameter file, and we used the default values (Table S2). CNV and samples were then filtered on six attributes: XHMM quality score (SQ) ≥65, exons spanned ≥3, estimated CNV length ≥1 kb, minor allele frequency (MAF) <1%, per sample CNV count >0 CNV and ≤55 CNV, and per sample total CNV ≤18 Mb. The SQ threshold was set on the basis of a transmission analysis of CNV called by XHMM on the set of 261 trios, which, like the ASD case-control data set, were sequenced at the Broad Institute with identical approaches. These analyses showed a stable median transmission rate per trio of 50% for CNV in 1–30 kb at SQ thresholds from 55–85 (Figure 1). Given this, as well as the prior work of Fromer et al. 19 (which used a threshold of 60), and similar studies in additional data sets (M.F., unpublished data), we set a conservative threshold of 65; note that evidence for excess small CNV in ASD was robust to various thresholds, as described below. All filtering was performed with PLINK.22 After applying the above filters, we retained 1,386 CNV (803 case, 583 control) in 559 samples (299 cases, 260 controls) in the ASD data set. This set of CNV constitutes our set of high-confidence CNV calls and is the base set on which all later stratification was performed. These CNV calls are publicly available as dbVar nstd86.
Figure 1.
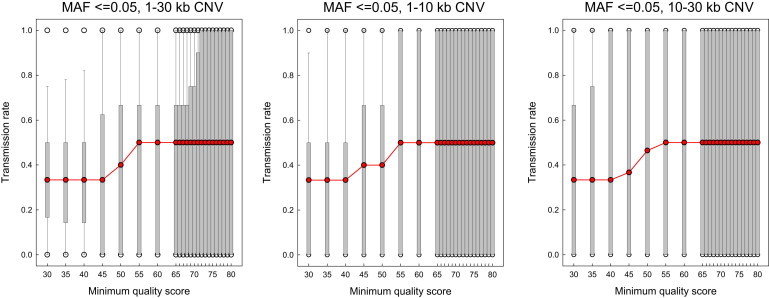
Transmission Analysis
Transmission rate of CNV from parents to child is shown as a function of XHMM quality score threshold (SQ) for CNV of size 1–30 kb (left), 1–10 kb (center), and 10–30 kb (right). At each SQ threshold, transmission was calculated and aggregated per family. The red line indicates the median per-family transmission rate at each threshold, with gray bars indicating the interquartile range at each threshold. A minor allele frequency (MAF) filter of 5% was applied to the entire set of CNV before size stratification and transmission analysis.
Because a goal was to probe the lower range (≤30 kb) of CNV in ASD and to also do some comparisons across ranges of these small CNV, we stratified our high-confidence set by size (1–10 kb and 10–30 kb). This yielded subsets by type and by size for further analysis. Size and type stratification was performed with PLINK.
Burden Analysis
For the ASD sample, we used burden analysis as implemented in PLINK22 to evaluate sets for increased burden in cases. PLINK burden analysis was performed by permutation, generating p values for a one-sided comparison of case versus controls. Our analyses were confined to the three PLINK tests that have been used previously in ASD:2,7 RATE, which reflects the number of CNV per sample; PROP, reflecting the proportion of samples with one or more CNV; and GRATE, reflecting the average number of genes spanned by CNV per sample.
We computed burden analysis by using the --cnv-indiv-perm and --mperm 10000 options, with the --cnv-count option and a list of genes with corresponding hg19 coordinates to enable per-gene tests. The hg19 gene coordinate list was produced by downloading the RefSeq genes track from the UCSC Table Browser23 and then removing most duplicate entries by (1) collapsing multiple entries with identical gene names and transcription start and end positions and (2) merging entries with identical gene names and overlapping transcription start and end positions. This produced a list of 24,527 genes (23,642 unique). Per-segment enrichment was calculated with the --mperm 50000 option; per-gene enrichment was calculated with the --cnv-intersect, --cnv-test-region, and --mperm 10000 options.
Computational Validation
We performed several types of computational validation of our results in ASD to confirm XHMM quality score cutoffs, assess the stability of our findings, and reduce the effect of potential covariates such as mean sample read depth.
We chose to further validate our SQ cutoff and investigate our choice of MAF by assessing the sensitivity of our findings to SQ and MAF cutoffs across a range of values. For each combination of SQ and MAF value, we performed PLINK burden analysis on the 1–30 kb deletion set and plotted the results of the three tests (Figure S2). If our results were due to a particular combination of cutoff values, we would expect to see a sharp change from one cutoff value to another. Instead, values are reasonably stable, particularly in the SQ range 55–70 for MAF from 0.5% to 2.0%.
It was important to ensure that our finding of burden in small CNV in ASD was not an artifact of differences in mean read depth caused by different sequencing platform and batch. In detailed exploratory analyses, we saw some borderline evidence that mean sample read depth differed between cases and controls. In order to minimize the influence of mean read depth, we created three subsets of samples, each of similar read depth, based on the distribution of read depth by batch. These sets were then used as “clusters” by PLINK to specify that permutation should only be performed within clusters while calculating empirical p values. This was done by adding the --within argument when assessing burden with --cnv-indiv-perm and the other arguments specified in PLINK. The results (Figure S3) showed the same enrichment pattern as burden tests performed without within-cluster permutation (Figure 2). Hence, variation in read depth captured in the clusters that we defined based on sequencing batch did not seem to have appreciable effect on the burden in 1–10 or 1–30 kb deletions.
Figure 2.
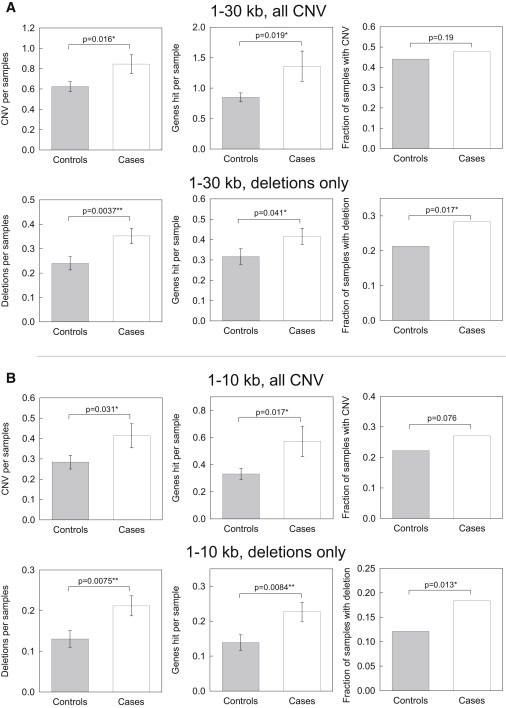
Enrichment of Small Deletions in Autism
(A) Case/control comparisons for 1–30 kb CNV (deletions and duplications) in the top row, and 1–30 kb deletions in the bottom row.
(B) Case/control comparisons for 1–10 kb CNV (deletions and duplications) in the top row, and 1–10 kb deletions in the bottom row. Error bars represent SEM.
We chose a set of 66 1–30 kb deletions for molecular validation by using real-time quantitative PCR (qPCR). All but 3 of the 66 examples of CNV were chosen from the set of singleton CNV in the 1–30 kb range: Singleton CNV were a subset of the base 1% MAF set that were chosen because events observed only once are more likely to be false-positive events although at the same time, if real, are more likely to be contributing to risk. Samples with CNV were chosen to span a range of CNV length (1–30 kb), number of exons (3–19), and XHMM quality score (65–99). Universal Probe Library (Roche) probe and primers sets were designed for all target regions with ProbeFinder software (Roche), and qPCR was performed on an ABI 7500 Real Time PCR instrument. Each sample was analyzed in duplicate in a 10 μl volume (100 nM UPL probe, 200 nM of each primer, 1 × KAPA PROBE FAST Universal qPCR Master Mix with ROX from KAPA Biosystem, and 25 ng of genomic DNA). Results were analyzed with the qBase+ software package (Biogazelle) with normalization of all assays to two control genes (COBL [MIM 610317] and SNCA [MIM 163890]). Each sample was further compared to DNA from a control subject without any known CNV. Finally, for each primer set, data were normalized to give a value of 1 for the control subject, and the relative levels of DNA for the case DNA were determined at each probe. For the purposes of this study, we consider a CNV to be validated with a CNV-directed probe showing 75% or less of control levels.
We also attempted to further validate seven CNV and determine precise start and end points by using PCR followed by Sanger sequencing. In two of the seven cases, we could not localize the deletion breakpoints. This is not surprising given the nature of CNV called from WES: breakpoints will be located in intronic or intragenic regions upstream and downstream of the called CNV extent and might be located as far away as the next upstream or downstream target evaluated by XHMM. In addition, XHMM only makes use of reliably called exons, which is dependent on the existence of appropriate target capture probes, read depth, and additional parameters. Breakpoint localization is therefore difficult given this large possible range for breakpoint location, combined with the limitation of amplicon size to 6 kb for optimal speed and accuracy. For the other five deletions, genomic fragments harboring expected deletions were amplified with Phusion Hot Start High-Fidelity polymerase. Sanger sequencing of PCR-amplified fragments was carried out at Genewiz (South Plainfield). For MRPS15 (MIM 611979), amplification was carried out with primers 5′-accacagaggatgagagttgg-3′ and 5′-aggggtgggctatttaagga-3′; for RPGRIP1 (MIM 605446), with primers 5′-agacgggaggccttgtctat-3′ and 5′-tttggctctgggaaatt-3′; for DNAH5 (MIM 603335), with primers 5′-tcatggactactctaacaaaactggt-3′ and 5′-ggaaaacattatggcagtctgaa-3; for ATP8B3 (MIM 605866), 5′-ggtggcacgcaactgtaat-3′ and 5′-gcctgcttgacggtttctta-3′; and for SULT1A2 (MIM 601292), 5′-gagcagcccctcctgtct-3′ and 5′-ctgggaaagtcgcctcact-3′.
Pathway and Network Analyses
We derived case and control gene lists from genes overlapped by 1–30 kb deletions. By using the filtering described above, including the 1% MAF filter, this produced case and control lists of 142 and 96 genes, respectively, with 21 genes appearing in both lists. We also derived gene lists from singleton CNV (by using the same filtering steps as before, but replacing the 1% MAF filter with a filter to retain only singleton CNV). This produced case and control lists of 86 and 60 genes respectively, with two genes in both lists (nonoverlapping CNV can still hit the same gene).
The case and control gene lists with MAF ≤1% were analyzed with DAPPLE24 to build separate networks for each list and compare connectivity within each list of encoded proteins. The number of genes submitted, gene symbols recognized, genes in the source protein-protein interaction (PPI) network InWeb,25 and genes sharing a common interactor (CI) in InWeb were 142, 123, 75, and 59 for case genes and 96, 83, 55, and 38 for control genes. In terms of percents, 42% of case genes and 40% of control genes were among the genes with CIs in InWeb. (A CI is an intermediate gene with direct connections to two genes in the input gene list; input genes that are in InWeb but that do not share common interactors cannot be connected to any other input gene via only one intermediate gene.)
In addition to the DAPPLE network, a larger PPI network, created from HPRD,26 MINT,27 BioGRID,28 IntAct,29 and KEGG,30 was also made use of, allowing for a more detailed look at intermediate proteins that connect the case genes and 115 gene products previously identified from genes showing de novo loss-of-function (LoF) mutations in ASD12–15. It is estimated that about half of these genes will be true ASD genes.14 The case (MAF ≤ 1%) and de novo LoF genes were seeded into the PPI network previously described.31 An ASD network was then generated including direct interactions between seed genes and interaction through adjacent intermediates that connect two seed genes. Each intermediate node was given a p value from a proportion test, which indicates the specificity of the intermediate protein to the seed genes compared with all other interacting partners in the background network. The network clustering was implemented with the organic clustering method in yEd.
Finally, we used the gene-set enrichment tool Enrichr32 to separately analyze case and control gene lists for overlap with pathway gene-set libraries (specifically, with the Enrichr database PPI Hub Proteins, which uses PPI from Genes2Networks33) and gene-set libraries created from Gene Ontology.34 We considered a pathway or ontology term enriched for a gene list if the Benjamini-Hochberg corrected p value was significant at p < 0.01 and if there were more than two overlapping genes.
Results
Reliable Calling of Rare Small CNV from Whole-Exome Sequencing Data
We called rare, small (1–30 kb) exonic CNV in two data sets for which DNA had been subjected to whole-exome sequencing (WES). Depth of read coverage was calculated with the genome analysis toolkit (GATK35), and the average per-exon coverage was used to call CNV with the eXome Hidden Markov Model (XHMM19) program. CNV called by XHMM were then filtered with PLINK22 to retain only CNV meeting quality, length, and MAF thresholds, as well as per-sample CNV number and length thresholds. The CNV quality threshold was determined on a separate set of exomes obtained from trios, where we observed a median 50% transmission per trio of CNV from parents to the child (indicative of random Mendelian transmission and hence reliable CNV calling) in both the smaller (1–10 kb) and larger (10–30 kb) CNV bins. A conservative quality score of 65 was used in further analyses, similar to the value of 60 used previously.19 Molecular validation (see below) in an independent sample provided further evidence that XHMM was reliable for calling small CNV from WES data.
Excess of Small Deletions in ASD
We called CNV in a sample consisting of 811 subjects, including 432 individuals with ASD and 379 controls, all matched for European ancestry by using available genome-wide SNP data.20 After filtering, we retained 559 samples (299 cases, 260 controls) with 1,386 CNV events (803 in cases and 583 in controls). When we compared rare 1–30 kb exonic CNV in ASD cases as compared to the ancestry matched controls, we observed enrichment in cases for 1–30 kb and for the subset of 1–10 kb CNV, as measured by CNV per sample and the correlated metric of genes hit by CNV per sample (Figure 2). Permutation p values correcting for mean sample read depth, sex, sequencing batch, and sequencing platform confirmed these significant findings (Figure S3).
The increased numbers of CNV in individuals with ASD appeared to be primarily driven by enrichment for deletions (Figure 2) because deletions, whether considering the entire size range (1–30 kb) or just the bin of smallest (1–10 kb) CNV, were enriched in individuals with ASD when examining CNV per subject, genes hit by CNV per subject, or fraction of subjects with CNV. The findings of increased small deletions in ASD were observed across an SQ range from 55 from 70 and a MAF range from 0.5% to 2.0% (Figure S2). In fact, even singleton deletions showed some enrichment for deletions in ASD even in this significantly smaller subsample (Figure S4). In contrast to the robust findings with deletions, we observed only nonsignificant enrichment of duplications in cases versus controls (Figure S5).
Subjects with ASD also demonstrated a greater likelihood of having multiple small events (Figure S6). For example, 22 subjects with ASD (5.65%) had two or three small exonic deletions in the 1–30 kb range, whereas only eight controls (2.4%) did. Similarly, nine subjects with ASD (2.3%) had two or three small exonic deletions in the 1–10 kb range, whereas only two controls (0.6%) did.
To further confirm the accuracy of XHMM for deletions in the ≤30 kb range, we chose a set of 66 1–30 kb deletions for molecular validation by using PCR. All but three of the 66 CNV were chosen from singleton CNV in the 1–30 kb range, a subset of CNV that should be most likely to be false positive. In addition, we chose the CNV to span a range of CNV length (1–30 kb), number of exons (3–19), and XHMM quality score (65–99). For qPCR, for each predicted deletion, we chose two target segments predicted to lie within the deletion extent. Validation rates averaged 85% (Figure 3; Table 1), with no evidence for better or worse performance of XHMM in any of three size bins. For five CNV that encompassed just 3 exons, we were able to amplify the disrupted allele by PCR and sequence the product. We observed agreement of XHMM calls with the deletions identified by Sanger sequencing (Figure 4).
Figure 3.
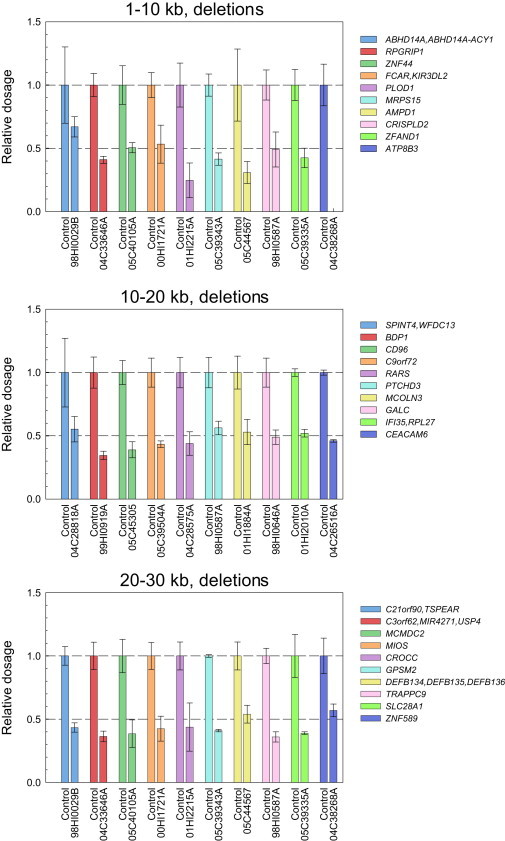
Molecular Validation of XHMM Calls
Each panel shows ten examples of qPCR-validated deletions in differing size ranges, as indicated in the panel. Each deletion is represented by a pair of bars representing the dosage of DNA relative to control as assayed by using qPCR for a probe placed within the called deletion. The left bar shows the result, normalized to 1.0, for the control probe; the right bar shows the normalized dosage for the probe in the sample with the called deletion. Error bars represent SEM. Note that the rightmost sample in the upper panel (04C38268A, gene ATP8B3) is likely a homozygous deletion.
Table 1.
Validation Rate of Deletions as a Function of CNV Size
| Size Range (kb) | Tested | # Validated | % Validated |
|---|---|---|---|
| 1–10 | 31 | 25 | 80.7 |
| 10–20 | 22 | 20 | 90.9 |
| 20–30 | 13 | 11 | 84.6 |
| Total | 66 | 56 | 84.9 |
Example CNV (n = 66) were chosen from among all CNV to include CNV in different size bins and quality scores. Sixty-three of the chosen CNV were singleton. qPCR probes were designed within the predicted deleted region. A CNV was considered validated if one internal probe showed a >25% decrease in dosage as compared to a control sample without the CNV. Examples are shown in Figure 3.
Figure 4.
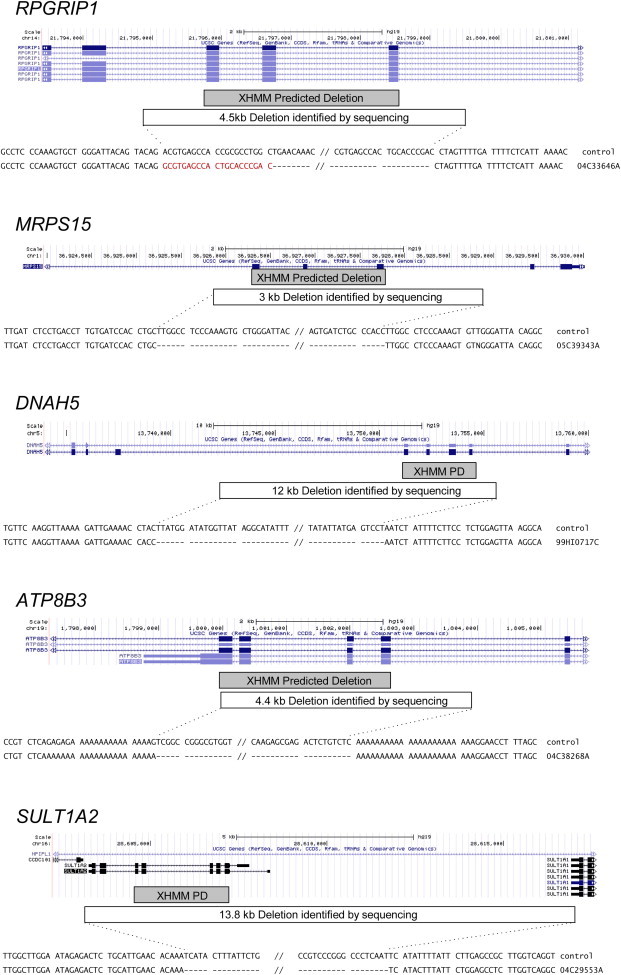
Breakpoint Determination via Sanger Sequencing
Each panel shows the results from Sanger sequencing of one of five validated deletions. In each panel, exons and transcripts are shown on the top, followed by the extent of the called and validated deletions, and finally the sequence surrounding deletion start and end points. Regions of surrounding sequence that differ from control are shown in red.
For completeness, we also examined larger CNV size bins of 30–100 kb and 100+ kb. There was no significant increased burden in deletions or duplications in CNV in these size bins.
Small CNV Implicates Dysregulation of Autophagy in ASD
Given the small size of CNV studied here, most (68%) hit only one gene, which, in contrast to larger CNV, makes them easily suited for gene discovery. We carried out three analyses of the genes disrupted by rare small CNV. First, we constructed separate networks from genes hit by case and control CNV by using DAPPLE.24 The case gene network (Figure 5, left) showed significant enrichment for direct connections between input genes (p = 0.006) and mean number of direct connections per input gene (p = 0.002), while the control gene network (Figure 5, right) was not enriched for either (p = 0.106, p = 0.855, respectively). This indicates that the genes disrupted by small CNV in cases tend to cluster into pathways.
Figure 5.
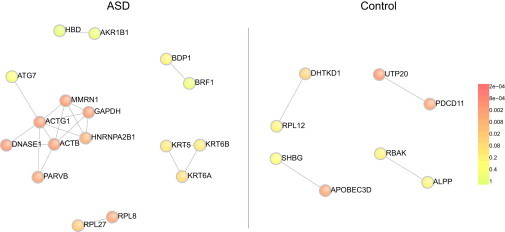
DAPPLE Network Derived from Genes Hit by 1–30 kb Deletions
The subnetwork of direct connections is shown for genes disrupted in ASD cases (left) or controls (right). For case genes, there were 21 connections between 17 genes and the network was significantly enriched for direct connections between input genes (p = 0.006) and mean number of direct connections per input gene (p = 0.002). For control genes, there were four connections between eight genes and the network showed no enrichment for direct connections between input genes (p = 0.106) or mean number of direct connections per input gene (p = 0.855).
We next used Enrichr32 to look at enrichment with prior gene sets, focusing on gene ontology (GO) categories. The genes disrupted by deletions in cases were enriched for the GO Molecular Function (MF) categories of structural molecule activity (GO:0005198; Padj = 4.32E-04) and structural constituent of cytoskeleton (GO:0005200; Padj = 4.32E-04). The disrupted genes overlapped for these two enriched categories (ACTG1 [MIM 102560], MYH4 [MIM 160742], VILL, MYOM2 [MIM 603509], RPL27 [MIM 607526], RPL8 [MIM 604177], KRT6A [MIM 148041], KRT6B [MIM 148042], KRT5 [MIM 148040], KRT3 [MIM 148043], and ACTB [MIM 102630] for GO:0005198, and VILL, ACTG1, KRT6A, KRT6B, KRT5, and ACTB for GO:0005200). For genes disrupted by deletions in controls, there were no similarly significant enrichments, with the two most significant findings observed in chloride channel activity (GO:0005254 with genes CLCNKA [MIM 602024], CLCNKB [MIM 602023], and BEST1 [MIM 607854]) and the overlapping anion transmembrane transporter activity category (GO:0008509, with genes CLCNKA, CLCNKB, BEST1, and SLCO1B3 [MIM 605495]) (Padj = 0.027 for both).
We also used Enrichr to determine whether there could be enrichment for networks associated with specific PPI Hub Proteins33 (Table 2; Figure 6). We observed very significant enrichment with five related PPI Hub Proteins. These five proteins are mammalian orthologs to the yeast autophagy gene Atg8 and include GABARAPL2 (MIM 607452; Padj = 4.88E-10), GABARAPL1 (MIM 607420; Padj = 1.19E-8), MAP1LC3A (MIM 601242; Padj = 3.98E-5), GABARAP (MIM 605125; Padj = 3.98E-5), and MAP1LC3B (MIM 609604; Padj = 3.31E-3). Two actin related PPI Hub Proteins, ACTB and ACTG1, also showed significant finding in case genes with p values of 8.87E-05 and 0.00510, respectively. There were no significant findings in the control gene lists (Padj > 0.5).
Table 2.
Top PPI Hub Protein Enrichments for ASD CNV
| PPI Hub Protein | Overlap | Adjusted p Value | Genes |
|---|---|---|---|
| GABARAPL2 | 17/539 | 4.88E-10 | RPS18, RPL27, ATG7, HNRNPA2B1, RPL8, HBG1, CALCOCO2, PLOD1, EPRS, TRAP1, KRT6C, KRT6A, KRT6B, KRT5, KRT3, GAPDH, ACTB |
| GABARAPL1 | 15/499 | 1.19E-08 | RPS18, RPL27, ATG7, HNRNPA2B1, RPL8, CALCOCO2, PLOD1, TRAP1, KRT6C, KRT6A, KRT6B, KRT5, KRT3, GAPDH, ACTB |
| MAP3K14 | 7/166 | 8.35E-05 | ACTG1, RPS18, RPL8, EPRS, GAPDH, ACTB, RPL27 |
| MAP1LC3A | 10/383 | 3.98E-05 | PLOD1, EPRS, TRAP1, ATG7, HNRNPA2B1, RPL8, CALCOCO2, KRT5, GAPDH, ACTB |
| GABARAP | 11/479 | 3.98E-05 | RPS18, RPL27, ATG7, HNRNPA2B1, RPL8, PLOD1, TRAP1, KRT6C, KRT5, GAPDH, ACTB |
| ACTB | 9/339 | 8.87E-05 | ACTG1, TANC1, MYH4, RPS18, PARVB, HNRNPA2B1, DNASE1, GAPDH, ACTB |
| PRKCE | 6/193 | 0.00180 | ACTG1, RPS18, HNRNPA2B1, GAPDH, ACTB, AKR1B1 |
| ACTG1 | 6/246 | 0.00510 | ACTG1, MYH4, PARVB, DNASE1, GAPDH, ACTB |
| MAP1LC3B | 7/322 | 0.00331 | TRAP1, ATG7, HNRNPA2B1, RPL8, KRT5, GAPDH, ACTB |
For each PPI Hub Protein, the number of genes disrupted by CNV is shown as a fraction of the total number of gene products known to associate with the hub protein. Adjusted p values and gene names are shown as well. All enrichment with Padj < 0.05 are shown for case CNV. There was no equivalent enrichment with control genes (all p > 0.5).
Figure 6.
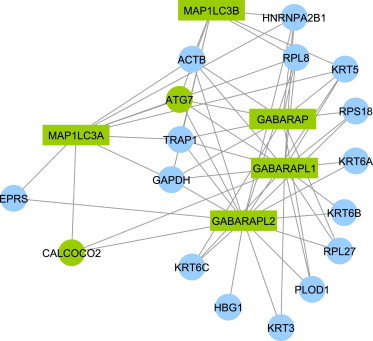
Interaction of Genes Hit by 1–30 kb Deletions in Cases with Autophagy Genes
We made use of a carefully curated PPI network13 to explore the autophagy pathway and its relation with genes hit by small deletions in ASD subjects. Circular nodes represent genes hit by CNV in ASD subjects. Green nodes participate in the autophagy pathway according to the NCBI Gene Database and green rectangular nodes are categorized as hub proteins in the pathway.32
We also examined singleton CNV for enrichment in these PPI Hub Proteins. There were two Hub Proteins that showed significant findings with case singleton CNV: GABARAPL2 (Padj = 0.02, with genes RPL27, ATG7 [MIM 608760], RPL8, HBG1 [MIM 142200], CALCOCO2 [MIM 604587], and EPRS [MIM 138295]) and MAP3K14 (MIM 604655; Padj = 0.02, with genes ACTG1, RPL8, EPRS, and RPL27). There were no significant findings with control singleton CNV. These results provide further support for both autophagy and actin dynamics as potential ASD pathways.
Finally, to determine whether the current findings from small CNV show relationship to recent whole-exome findings of de novo loss-of-function (LoF) mutations,12–15,26 we examined the relationship of the CNV with these LoF mutations by using the larger PPI network we assembled (Figure 7). We observed many modules that included genes identified both in the current study and in the WES studies on de novo LoF genes in ASD. Of the genes hit by CNV in Figure 7, 65% were the only gene hit by that CNV. Interestingly, GABARAPL2 is a central node in a module (circled) that includes CNV findings as identified here, as well as de novo LoF findings from the prior WES studies in ASD trios.
Figure 7.
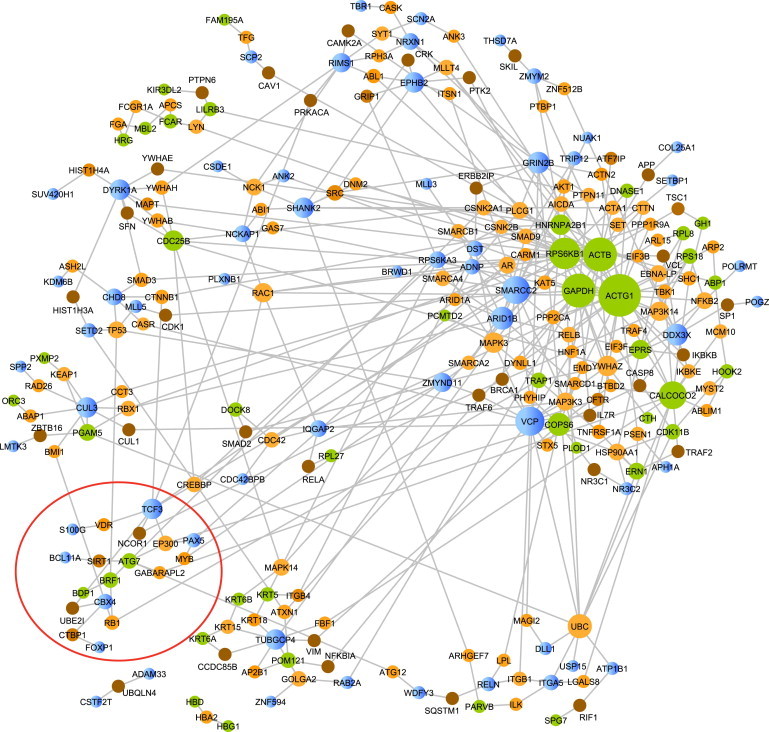
Networks of ASD Genes Disrupted by Small CNV or De Novo Loss-of-Function Mutations
We made use of a carefully curated PPI network31 to explore the relationship of genes disrupted by small CNV (this study) or de novo loss-of-function mutations12–15. All nodes were sized based on connectivity degree. Green nodes denote small CNV case genes, blue nodes represent LoF genes, and the orange- to brown-color nodes are intermediate proteins where the shade is based on p value computed by using a proportion test, with darker color indicating smaller p value.
Discussion
There is now abundant evidence for a major role of CNV in human disease.1–7 To date, such studies most commonly focused on CNV that were >30 kb because of the presumed limits of reliable calling of CNV from chromosome microarray studies. With the widespread adoption of WES in genetic studies, we asked whether exonic CNV smaller than 30 kb could be reliably called. We made use of XHMM, which had been developed and validated for a broad range of CNV, and assessed how reliably it performed for smaller CNV, focusing on rare CNV.
XHMM takes per-target read depth data, uses principle component analysis to remove systematic batch effects unrelated to underlying changes in copy number, and then uses a hidden Markov model to remove noise and identify regions of deletion or duplication. As previously shown,19 XHMM converges to the expected rate of 50% transmission of CNV from parents to children as the quality cutoff is raised. We reproduced this result on an independent data set for CNV in the 1–30 kb range. This indicates that XHMM exhibits high specificity and sensitivity at sufficiently high threshold, because inaccurate or missed calls would manifest as Mendelian errors. We have added evidence of specificity by demonstrating a validation rate of 85% for 1–30 kb deletions by using independent molecular methods. XHMM also exhibits high sensitivity for calls of three or more exons when compared to Birdsuite36 calls on Affymetrix arrays for the same samples.19
We identified breakpoints for five small deletions called by XHMM. In four cases, there were stretches of homologous sequences of 25 bp to 1.8 kb flanking the deletion, which could have mediated nonallelic homologous recombination.
We observed an average of 0.49 small rare duplications and 0.35 small rare deletions in affected individuals, with 0.38 small rare duplications and 0.24 small rare deletions in controls. Such CNV is genic and is hence potentially associated with functional changes in many cases. In addition, such CNV often disrupts just one or two genes, such that the disruption that contributes to a given phenotype can be more readily interpreted.
There is extensive evidence for rare CNV in risk for ASD,2,6,7 particularly for CNV >30 kb. Consistent with previous studies looking at larger CNV, we observed an increased number of CNV in ASD cases as compared to ancestry-matched controls. This was associated with an increased number of genes disrupted by small rare CNV in ASD. Looking at deletions and duplications separately, rare, small, genic deletions were specifically associated with ASD. The difference in proportion of individuals with one or more 1–30 kb deletions (28% versus 21%, p = 0.017) indicated that potentially disease-associated 1–30 kb exonic deletions could be present in up to 7% of individuals with ASD.
While previous studies showed burden in larger CNV (>100 kb or >500 kb), we did not reproduce those results here. Given the low false-positive and false-negative rates for XHMM discussed above, this likely reflects an issue of sample size rather than caller reliability. This is not unexpected given the premise that large CNV have larger effect sizes, but are far less frequent due to strong selective pressure. Previous studies showing burden in larger CNV2,7 used samples of several thousand individuals.
Previous studies have also shown that de novo CNV show increased association with disease. Because this study was carried out on case-control data, we could do no large-scale assessment of de novo CNV on those data. When we reanalyzed the independent trio data set by using the standard XHMM rules for determining transmission19 and considering only CNV for which transmission could be reliably determined, we observed that 6% of 1–30 kb deletions and 5% of 1–30 kb duplications were de novo according to PLINK/SEQ. In addition, for a small proportion of the cases in our study, we had parental DNA available and assessed de novo status by attempting to validate case (child) CNV in the parents by using qPCR as described earlier. Out of 27 CNV in trios where we had complete DNA, 2 out of the 27 validated small exonic deletions were de novo (7.4%). While these numbers are small, the proportion that are de novo is consistent with the in silico analyses.
The genes disrupted by deletions in ASD cases showed significant connectivity and were enriched for the GO Molecular Function categories of structural molecule activity (GO:0005198; Padj = 4.32E-04) and structural constituent of cytoskeleton (GO:0005200; Padj = 4.32E-04). The actin cytoskeleton plays a critical role in synaptic development and plasticity.37 Dysregulation of this process in ASD is implicated by mutations in genes that directly or indirectly modulate the synaptic cytoskeleton (e.g., SHANK3 [MIM 606230]38) and by alterations in gene expression of actin-associated genes in postmortem ASD brain samples.39 Our finding of disruption of networks involving the actin cytoskeleton by small CNV in ASD is consistent with these prior findings.
Looking at PPI Hub Proteins to understand the pathways that might be implicated by genes disrupted by deletions in cases, we observed very significant enrichment with five related PPI Hub Proteins (GABARAP, GABARAPL1, GABARAPL2, MAP1LC3A, and MAP1LC3B), which are all mammalian orthologs to the yeast autophagy gene Atg8. Autophagy is a process responsible for the lysosomal turnover of organelles and proteins.
The sample size for this study was too small to have complete confidence in the pathways implicated by the ASD-associated CNV; however, the findings are consistent with some prior publications. In a very recent study of individuals with a deletion at 16q24.2, 14 individuals with ASD and/or intellectual disability (ID) showed deletions that overlap the coding region of MAP1LC3B.40 In addition, a recent study of CNV in ASD identified a CNV disrupting GABARAPL1 in an individual with ASD.41 A boy presenting with moderate ID, intractable epilepsy, and dysmorphic features had a 2.3 Mb microdeletion of 17p13.2p13.1, which involved 17 genes, including GABARAP.42 Knockdown of the zebrafish gabarap gene resulted in a small head and micrognathism, indicating a role for GABARAP in the phenotype of the individual. Overlapping deletions disrupting GABARAP were observed in additional individuals with ID.43 Finally, overexpression of the Fragile X (MIM 300624)-associated gene FMR1 (MIM 309550) in mice appears to regulate levels of GABARAPL2.44 These results, when taken together with our findings, provide evidence for a role for the pathways involving the related proteins GABARAP, GABARAPL1, GABARAPL2, MAP1LC3A, and MAP1LC3B in some forms of ASD.
Alterations in autophagy have been implicated in neurodegenerative disorders, including Huntington disease (MIM 143100), Alzheimer disease (MIM 104300), Parkinson disease (MIM 168600), and Lewy body disease (MIM 127750);45 however, there are emerging data showing important roles for autophagy in synaptic development.46 Consistent with an important role in development, it has recently been shown that GABARAPL1 (the only member of the group studied) is robustly expressed throughout brain and neuronal development.47
The protein products of ATG7 and CALCOCO2, both autophagy genes and two of the genes disrupted by CNV in ASD subjects, bind PPI hub proteins involved in autophagy as do many of the genes disrupted by CNV in ASD (Figure 5). More broadly, the PI3K/AKT/mTOR pathway regulates autophagy, and many genes in this pathway have been implicated in ASD and ID, including PTEN (MIM 601728), TSC1 (MIM 605284), and TSC2 (MIM 191092).16 Although still speculative at this point, it might be that one impact of dysregulation of the PI3K/AKT/mTOR pathway is a deleterious change in neural development due to defects in autophagy.
In summary, we found that XHMM19 can reliably call rare (MAF ≤ 1%) and small (1–30 kb, and three or more exons) exonic CNV from WES data. This provides an important tool in dissecting the genomic architecture of disease. In addition, we found an enrichment of rare small deletions in subjects with ASD (p = 0.0037). Deletions (1–30 kb) were found in 28% of cases but only 21% of controls (p = 0.017) indicating that small CNV could contribute to risk in as much as 7% of individuals with ASD. Because small CNV hit few genes (68% of the 208 small deletions hit only one gene) we were able to easily perform network analysis on the genes hit. We found significant enrichment of five related PPI hub proteins that are mammalian orthologs to the yeast autophagy gene Atg8: GABARAPL2, GABARAPL1, MAP1LC3A, GABARAP, and MAP1LC3B. These findings indicate that small CNV contribute to ASD risk and that disruption of autophagy may be an important pathway in ASD.
Acknowledgments
This work was supported by the National Institute of Mental Health, National Institutes of Health (grants MH089025, MH097849, and MH100233 to J.D.B.) and the Seaver Foundation. C.S.P. is a Seaver Fellow and A.P.G. is a Seaver Fellow.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
PLINK CNV Handling, http://pngu.mgh.harvard.edu/∼purcell/plink/cnv.shtml
PLINK/SEQ, http://atgu.mgh.harvard.edu/plinkseq
UCSC Table Browser, http://genome.ucsc.edu/cgi-bin/hgTables
XHMM Tutorial, http://atgu.mgh.harvard.edu/xhmm/tutorial.shtml
Accession Numbers
The accession number for the copy number variants in the dbVar database is nstd86.
References
- 1.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Månér S., Massa H., Walker M., Chi M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 4.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.-C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 6.Cook E.H., Jr., Scherer S.W. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 7.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The 1000 Genomes Project Consortium. Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Durand C.M., Betancur C., Boeckers T.M., Bockmann J., Chaste P., Fauchereau F., Nygren G., Rastam M., Gillberg I.C., Anckarsäter H. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elsabbagh M., Divan G., Koh Y.-J., Kim Y.S., Kauchali S., Marcín C., Montiel-Nava C., Patel V., Paula C.S., Wang C. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012;5:160–179. doi: 10.1002/aur.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J., Nyholt D.R., Magnussen P., Parano E., Pavone P., Geschwind D., Lord C., Iversen P., Hoh J., Ott J., Gilliam T.C., Autism Genetic Resource Exchange Consortium A genomewide screen for autism susceptibility loci. Am. J. Hum. Genet. 2001;69:327–340. doi: 10.1086/321980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 17.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fromer M., Moran J.L., Chambert K., Banks E., Bergen S.E., Ruderfer D.M., Handsaker R.E., McCarroll S.A., O’Donovan M.C., Owen M.J. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 2012;91:597–607. doi: 10.1016/j.ajhg.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim E.T., Raychaudhuri S., Sanders S.J., Stevens C., Sabo A., MacArthur D.G., Neale B.M., Kirby A., Ruderfer D.M., Fromer M., NHLBI Exome Sequencing Project Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77:235–242. doi: 10.1016/j.neuron.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32(Database issue):D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossin E.J., Lage K., Raychaudhuri S., Xavier R.J., Tatar D., Benita Y., Cotsapas C., Daly M.J., International Inflammatory Bowel Disease Genetics Constortium Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lage K., Karlberg E.O., Størling Z.M., Olason P.I., Pedersen A.G., Rigina O., Hinsby A.M., Tümer Z., Pociot F., Tommerup N. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 2007;25:309–316. doi: 10.1038/nbt1295. [DOI] [PubMed] [Google Scholar]
- 26.Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009;37(Database issue):D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ceol A., Chatr Aryamontri A., Licata L., Peluso D., Briganti L., Perfetto L., Castagnoli L., Cesareni G. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010;38(Database issue):D532–D539. doi: 10.1093/nar/gkp983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stark C., Breitkreutz B.J., Chatr-Aryamontri A., Boucher L., Oughtred R., Livstone M.S., Nixon J., Van Auken K., Wang X., Shi X. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39(Database issue):D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aranda B., Achuthan P., Alam-Faruque Y., Armean I., Bridge A., Derow C., Feuermann M., Ghanbarian A.T., Kerrien S., Khadake J. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38(Database issue):D525–D531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aoki K.F., Kanehisa M. Using the KEGG database resource. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis [et al] 2005;Chapter 1 doi: 10.1002/0471250953.bi0112s11. Unit 1 12. [DOI] [PubMed] [Google Scholar]
- 31.O’Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger S.I., Posner J.M., Ma’ayan A. Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinformatics. 2007;8:372. doi: 10.1186/1471-2105-8-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Korn J.M., Kuruvilla F.G., McCarroll S.A., Wysoker A., Nemesh J., Cawley S., Hubbell E., Veitch J., Collins P.J., Darvishi K. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melom J.E., Littleton J.T. Synapse development in health and disease. Curr. Opin. Genet. Dev. 2011;21:256–261. doi: 10.1016/j.gde.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Betancur C., Buxbaum J.D. SHANK3 haploinsufficiency: a “common” but underdiagnosed highly penetrant monogenic cause of autism spectrum disorders. Molecular Autism. 2013;4:17. doi: 10.1186/2040-2392-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voineagu I., Wang X., Johnston P., Lowe J.K., Tian Y., Horvath S., Mill J., Cantor R.M., Blencowe B.J., Geschwind D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Handrigan G.R., Chitayat D., Lionel A.C., Pinsk M., Vaags A.K., Marshall C.R., Dyack S., Escobar L.F., Fernandez B.A., Stegman J.C. Deletions in 16q24.2 are associated with autism spectrum disorder, intellectual disability and congenital renal malformation. J. Med. Genet. 2013;50:163–173. doi: 10.1136/jmedgenet-2012-101288. [DOI] [PubMed] [Google Scholar]
- 41.Griswold A.J., Ma D., Cukier H.N., Nations L.D., Schmidt M.A., Chung R.H., Jaworski J.M., Salyakina D., Konidari I., Whitehead P.L. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum. Mol. Genet. 2012;21:3513–3523. doi: 10.1093/hmg/dds164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Komoike Y., Shimojima K., Liang J.S., Fujii H., Maegaki Y., Osawa M., Fujii S., Higashinakagawa T., Yamamoto T. A functional analysis of GABARAP on 17p13.1 by knockdown zebrafish. J. Hum. Genet. 2010;55:155–162. doi: 10.1038/jhg.2010.1. [DOI] [PubMed] [Google Scholar]
- 43.Krepischi-Santos A.C., Rajan D., Temple I.K., Shrubb V., Crolla J.A., Huang S., Beal S., Otto P.A., Carter N.P., Vianna-Morgante A.M., Rosenberg C. Constitutional haploinsufficiency of tumor suppressor genes in mentally retarded patients with microdeletions in 17p13.1. Cytogenet. Genome Res. 2009;125:1–7. doi: 10.1159/000218743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernández J.J., Martínez R., Andújar E., Pérez-Alegre M., Costa A., Bonilla-Henao V., Sobrino F., Pintado C.O., Pintado E. Gene expression profiles in the cerebellum of transgenic mice over expressing the human FMR1 gene with CGG repeats in the normal range. Genet. Mol. Res. 2012;11:467–483. doi: 10.4238/2012.March.1.4. [DOI] [PubMed] [Google Scholar]
- 45.Choi A.M., Ryter S.W., Levine B. Autophagy in human health and disease. N. Engl. J. Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 46.Shen W., Ganetzky B. Nibbling away at synaptic development. Autophagy. 2010;6:168–169. doi: 10.4161/auto.6.1.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le Grand J.N., Bon K., Fraichard A., Zhang J., Jouvenot M., Risold P.Y., Boyer-Guittaut M., Delage-Mourroux R. Specific distribution of the autophagic protein GABARAPL1/GEC1 in the developing and adult mouse brain and identification of neuronal populations expressing GABARAPL1/GEC1. PLoS ONE. 2013;8:e63133. doi: 10.1371/journal.pone.0063133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.