

Abstract
The early diagnostic value of glucose hypometabolism and atrophy as potential neuroimaging biomarkers of mild cognitive impairment (MCI) and Alzheimer's disease (AD) have been extensively explored using [18F]fluorodeoxyglucose positron emission tomography (FDG-PET) and structural magnetic resonance imaging (MRI). The vast majority of previous imaging studies neglected the effects of single factors, such as age, symptom severity or time to conversion in MCI thus limiting generalisability of results across studies. Here, we investigated the impact of these factors on metabolic and structural differences. FDG-PET and MRI data from AD patients (n = 80), MCI converters (n = 65) and MCI non-converters (n = 64) were compared to data of healthy subjects (n = 79). All patient groups were split into subgroups by age, time to conversion (for MCI), or symptom severity and compared to the control group. AD patients showed a strongly age-dependent pattern, with younger patients showing significantly more extensive reductions in gray matter volume and glucose utilisation. In the MCI converter group, the amount of glucose utilisation reduction was linked to the time to conversion but not to atrophy. Our findings indicate that FDG-PET might be more closely linked to future cognitive decline whilst MRI being more closely related to the current cognitive state reflects potentially irreversible damage.
Keywords: Ageing, Alzheimer's disease, [18F]fluorodeoxyglucose positron emission tomography (FDG-PET), Magnetic resonance imaging (MRI), Mild cognitive impairment
Highlights
-
•
Age related differences in Alzheimer’s disease are examined using FDG-PET and MRI.
-
•
Effects of disease progression and symptom severity on FDG-PET and MRI are evaluated.
-
•
AD patients show an age-dependent atrophy and glucose hypometabolism pattern.
-
•
In MCI, disease progression was linked to FDG-PET changes but not to atrophy.
1. Introduction
Neuroimaging research in the field of dementia focuses on the impact of behavioral, neuropsychological, genetic and demographic factors on brain atrophy associated with transition from healthy aging through mild cognitive impairment (MCI) and further to Alzheimer's disease (AD) (Buckner, 2004; Ewers et al., 2011; Fonteijn et al., 2012; Förster et al., 2012; Gao et al., 1998; Gomar et al., 2011; Good et al., 2001; Jedynak et al., 2012; Johnson et al., 2009; Tisserand et al., 2004). We face a steadily increasing number of structural magnetic resonance imaging (sMRI, (Davatzikos et al., 2008b; Kloppel et al., 2008)) and [18F]fluorodeoxyglucose positron emission tomography (FDG-PET, (Habeck et al., 2008; Haense et al., 2009; Sadeghi et al., 2008)) studies showing high sensitivity to the disease process when used separately (Alexander et al., 2002; Anchisi et al., 2005; Jack et al., 2010; Jagust et al., 2007; Kinkingnehun et al., 2008; Kloppel et al., 2008; Schroeter et al., 2009) and a gain in diagnostic accuracy when used together in a multimodal approach (Dukart et al., 2011a; Fan et al., 2008).
A viable way of understanding pathophysiology and further improving diagnostic accuracy in MCI and AD is the integration of previous knowledge about characteristic features of progression from a healthy state to AD. However, the straightforward interpretation of such an integrative approach could be limited by either systematic differences in demographic and cognitive abilities within patients and healthy controls or the use of particular image data processing algorithms (Crivello et al., 2002; Dukart et al., 2010; Jones et al., 2005). One such demographic factor that has been shown to have a strong differential impact on disease progression is age. Previous studies have indicated that early-onset (age < 65 years) and late-onset (age ≥ 65 years) AD patients show a differential pathology in MRI and FDG-PET (Canu et al., 2012; Frisoni et al., 2007; Ishii et al., 2005; Kaiser et al., 2012; Karas et al., 2007; Möller et al., 2013; Sakamoto et al., 2002). Nonetheless, the interpretation of these differences is still controversial as the differences might be explained by a differential age specific baseline resulting from comparison of early- and late-onset AD patients to different age-matched control group or to each other, by differential underlying pathology or by an interaction of age and disease related brain network changes. Also the link between FDG-PET and MRI and the current (as indicated by symptom severity) and future cognitive state (as indicated by future cognitive decline) is a matter of debate. Both imaging modalities have been reported to correlate with both current symptom severity and the future cognitive decline (Chételat et al., 2005; Drzezga et al., 2003; Edison et al., 2007; Fox et al., 1999; Vemuri et al., 2009a, b). These findings on the other side are not fully compatible with the currently widely accepted model of progression of both imaging markers in AD suggesting a differential time-line of progression with FDG-PET showing first a rapid decline and atrophy developing rather later in the progression from healthy aging to MCI and further to AD (Jack et al., 2010). An accurate estimation of the unique impact of these specific factors and their interaction on the progression of MCI or AD is crucial to integrate findings across different studies. Longitudinal studies offer a potential solution; although they are logistically difficult to carry out and lack complete control for all possible confounds within a cohort (Davatzikos et al., 2008a; Drzezga et al., 2003; Johnson et al., 2009).
The aim of this study is to disentangle the effect of age, symptom severity as measured by Mini Mental State Examination (MMSE, (Folstein et al., 1975)) and time to conversion (TTC) on changes in two prominent imaging biomarkers of AD—glucose metabolism and brain atrophy pattern. Therefore, we systematically investigate the effect of the factors age, symptom severity and TTC (in MCI patients) on brain pathology observed in both imaging modalities using the same group of control subjects whilst accounting for the effect of healthy aging using a recently proposed approach (Dukart et al., 2011b). The use of the same group of patients for investigation of the effects of different variables of interest is expected to reduce potential confounding effects in other uncontrolled factors. By additionally accounting for the variance explained by healthy aging we target the question on how much AD related pathology is required at different ages to induce the same level of cognitive impairment.
Given the fact that age interacts with AD-specific atrophy patterns (Franke et al., 2010; Good et al., 2001), we expected that younger MCI converters (cMCI) and younger AD patients would show a more extensive pattern of atrophy and glucose hypometabolism than older patients with similar symptom severity. According to a recent model of MCI progression characterised by rapid reduction in glucose metabolism followed by brain atrophy in much later stages of disease progression (Jack et al., 2010), we hypothesized that cMCI would initially show a more extensive decrease in glucose utilisation compared to the amount of atrophy associated with closeness to conversion. Keeping in mind that both FDG-PET and sMRI correlate with cognitive decline (Fox et al., 1999; Herholz et al., 2011), we hypothesized that by accounting for partial volume effects (PVE) in FDG-PET, changes in symptom severity would be more strongly linked to gray matter (GM) volume reductions than to glucose hypometabolism during progression from MCI to AD.
2. Methods
2.1. Subjects
To evaluate the effect of age, symptom severity and TTC on differences in FDG-PET and sMRI, we used multicenter sMRI and FDG-PET data of AD patients (n = 80), cMCI (n = 65), MCI non-converters (ncMCI, n = 64) and healthy control subjects (n = 79) (Table 1). The mean follow-up ± standard deviation for ncMCI was 26.8 ± 9.1 month, for cMCI 28.1 ± 8.0 month, for AD 19.8 ± 7.7 month and for the control group 30.5 ± 8.3 month. All data were extracted from the Alzheimer's disease Neuroimaging Initiative (ADNI) database (www.adni-info.org). Data were selected for availability of FDG-PET (50% of the ADNI cohort). Additionally, selection criteria were, if possible, matching for age and gender. Exclusion criteria for the current study were reported quality problems in imaging acquisition (e.g. moderate to severe motion artifacts). The cMCI group consisted of patients who converted to AD within 36 months of follow-up. Diagnosis of AD patients was based on NINCDS/ARDRA criteria (McKhann et al., 1984). Only stable MCI according to all available follow-up information were included in the ncMCI group. Similarly, group definition for all other groups comprised all available follow-up information, e.g. only subjects without reported conversion to MCI or AD in the available follow-up were included in the control group. Exclusion criteria for the ADNI data were the presence of any significant neurological disease other than AD, history of head trauma followed by persistent neurological deficits or structural brain abnormalities, psychotic features, agitation or behavioral problems within the previous three months or history of alcohol or substance abuse. A detailed description of the ADNI inclusion and exclusion criteria can be found under the following url: http://www.adni-info.org/pdfs/adni_protocol_9_19_08.pdf. Additionally, a list of subjects included in our study is provided in Supplement 1. For each subject, only data from the first study time-point were used for further analysis. The study was conducted according to the Declaration of Helsinki. Written informed consent was obtained from all participants before protocol-specific procedures were performed.
Table 1.
Subject group characteristic.
| Controls | AD | MCI con | MCI noncon | ANOVA (df,F,P) | |
|---|---|---|---|---|---|
| Number | 79 | 80 | 65 | 64 | – |
| Male/Female | 41/38 | 40/40 | 41/24 | 38/26 | – |
| Age (years) | 75.8 ± 4.9 | 75.7 ± 7.0 | 75.1 ± 6.9 | 75.6 ± 6.8 | 3,0.1,.938 |
| Age range (min–max) | 62–87 | 55–88 | 58–88 | 60–87 | – |
| MMSE (score) | 28.7 ± 1.6 | 23.6 ± 2.2 | 26.8 ± 1.8 | 27.5 ± 1.7 | 3,109.9,<.001 |
Mean ± standard deviation. AD Alzheimer’s disease, ANOVA analysis of variance, con converters, MCI mild cognitive impairment, MMSE Mini Mental State Examination, noncon non-converters.
To evaluate the effect of age, symptom severity and TTC on differences in FDG-PET and sMRI, we used multicenter sMRI and FDG-PET data of AD patients (n = 80), cMCI (n = 65), MCI non-converters (ncMCI, n = 64) and healthy control subjects (n = 79) (Table 1). The mean follow-up ± standard deviation for ncMCI was 26.8 ± 9.1 month, for cMCI 28.1 ± 8.0 month, for AD 19.8 ± 7.7 month and for the control group 30.5 ± 8.3 month. All data were extracted from the Alzheimer's disease Neuroimaging Initiative (ADNI) database (www.adni-info.org). Data were selected for availability of FDG-PET (50% of the ADNI cohort). Additionally, selection criteria were, if possible, matching for age and gender. Exclusion criteria for the current study were reported quality problems in imaging acquisition (e.g. moderate to severe motion artifacts). The cMCI group consisted of patients who converted to AD within 36 months of follow-up. Diagnosis of AD patients was based on NINCDS/ARDRA criteria (McKhann et al., 1984). Only stable MCI according to all available follow-up information were included in the ncMCI group. Similarly, group definition for all other groups comprised all available follow-up information, e.g. only subjects without reported conversion to MCI or AD in the available follow-up were included in the control group. Exclusion criteria for the ADNI data were the presence of any significant neurological disease other than AD, history of head trauma followed by persistent neurological deficits or structural brain abnormalities, psychotic features, agitation or behavioral problems within the previous three months or history of alcohol or substance abuse. A detailed description of the ADNI inclusion and exclusion criteria can be found under the following url: http://www.adni-info.org/pdfs/adni_protocol_9_19_08.pdf. Additionally, a list of subjects included in our study is provided in Supplement 1. For each subject, only data from the first study time-point were used for further analysis. The study was conducted according to the Declaration of Helsinki. Written informed consent was obtained from all participants before protocol-specific procedures were performed.
2.2. MRI data
The MRI dataset included standard T1-weighted images obtained with different 1.5 T scanner types using a 3D MP-RAGE sequence varying in TR and TE with an in-plane resolution of 1.25 × 1.25 mm and 1.2 mm slice thickness. All images were preprocessed as described on the ADNI website (http://www.loni.ucla.edu/ADNI/Data/ADNI_Data.shtml) including distortion and B1 non-uniformity corrections.
2.3. FDG-PET data
FDG-PET scanning at different PET-scanner types was performed using one of the following three protocols: 1) dynamic: a 30 min six frame acquisition (6 five-minute frames), with scanning from 30 to 60 min post FDG injection; 2) static: a single-frame, 30 min acquisition with scanning 30–60 min post injection; and 3) quantitative: a 60 min dynamic protocol consisting of 33 frames, with scanning beginning at injection and continuing for 60 min. Images also differed in resolution, orientation, voxel and image dimensions and count statistics. The mean images of realigned frames from 30 to 60 min post injection were used for further analysis.
2.4. MRI pre-processing
All image-processing steps were carried out using the SPM5 software package (Wellcome Trust Centre for Neuroimaging: http://www.fil.ion.ucl.ac.uk/spm/) implemented in Matlab 7.7 (MathWorks Inc., Sherborn, MA). For common pre-processing sMRI and FDG-PET data we applied the same preprocessing procedure as described in detail in Dukart et al. (Dukart et al., 2011a). In this procedure, structural MRI data were first interpolated to an isotropic resolution of 1 × 1 × 1 mm3, bias corrected for inhomogeneity artifacts and segmented into grey matter, white matter and cerebrospinal fluid (only the grey matter segments were used for further analyses). The segmented grey matter images were spatially normalized to a study specific template created from all subjects using a diffeomorphic registration algorithm—DARTEL (Ashburner, 2007). In this step, the images were modulated using Jacobian determinants to preserve the total amount of signal from each region. The obtained grey matter volume images were smoothed with a Gaussian kernel of 12 mm full width at half maximum and masked using a grey matter mask created by thresholding the first and the last DARTEL template by 0.2 to avoid big edge effects.
2.5. FDG-PET pre-processing
FDG-PET data were interpolated to an isotropic resolution of 1 × 1 × 1 mm3 and co-registered to the corresponding sMRI scan of each subject using the co-registration function in SPM5. Further pre-processing included a convolution based PVE correction (Muller-Gartner et al., 1992; Quarantelli et al., 2004) of FDG-PET using sMRI data and subsequent masking of non-GM voxels. Spatial normalisation to a study specific template was performed using flow-fields obtained from sMRI data (Ashburner, 2007). As for sMRI a smoothing kernel of 12 mm full width at half maximum was applied to all FDG-PET data. The obtained FDG-PET and grey matter volume images had a very similar effective smoothness as indicated by subsequent SPM smoothness estimation during statistical analyses. FDG-PET data were intensity normalized to the cerebellar mean glucose uptake and masked using a grey matter mask to avoid big edge effects. The cerebellar region was chosen for intensity normalisation of FDG-PET as it has been shown to be a region of choice for intensity normalisation which is unaffected in healthy ageing and early stages of AD when correcting for PVE caused by atrophy (Dukart et al., 2010; Kushner et al., 1987).
2.6. Correction for age effects
To remove the effect of healthy aging on glucose utilisation and regional grey matter volumes we first computed voxel-wise linear regressions only in control subjects and separately for both imaging modalities. In a second step the beta coefficients obtained from the voxel-wise linear regressions are applied to remove the variance explained by healthy ageing in patients' and control subjects' imaging data (Dukart et al., 2011b).
2.7. Statistical analysis of imaging data
All statistical analyses of imaging data were carried out within the SPM5 framework. To investigate the effect of age on differences observed in FDG-PET and MRI in MCI and AD, the entire groups of AD, cMCI or ncMCI subjects were split based on age, symptom severity (MMSE score) or TTC (only for MCI converters) into 3 equally sized subgroups (Tables 2, 3, 4) matching for the remaining one (or two for cMCI) variables. For example for the subdivision of the AD group by age the 80 AD subjects were sorted in an ascending order by age and then divided into three equally sized biggest possible subgroups (for AD 26 subjects per group: 26 ∗ 3 = 78) and leaving the remaining subjects for potential matching. Analyses of variance and if significant subsequent post-hoc t-tests were then computed to evaluate the subgroups for potential differences in variables of no-interest for the particular comparison (e.g. for age: MMSE (for cMCI also for TTC) and vice versa). If one of the tests indicated significant differences between the subgroups (e.g. significantly lower MMSE scores in younger AD), subjects with the extreme values in the corresponding group (in this case low MMSE scores) were replaced by some of the subjects left for matching and showing similar age values but higher MMSE scores. The statistical tests were then recomputed. If a test again revealed significant differences the subgroup size was reduced by removing from each subgroup one subject showing “extreme” relative values as compared to the mean of the subgroup in the significant variable of no interest. This procedure of replacing subjects or removing one subject from each subgroup was then repeated until matching in variables of no interest was achieved for each group and each subgroup splitting. FDG-PET and voxel-based morphometry (VBM) data of each subgroup were compared separately with the entire group of control subjects. Additionally, subgroups were matched for gender. There were no significant differences in the distribution of the FDG-PET protocols between the obtained subgroups.
Table 2.
Subgroup characteristics for AD patients.
| Age evaluation | ||||
|---|---|---|---|---|
| Young AD | Mean AD | Old AD | ANOVA (df,F,p) | |
| Number | 25 | 25 | 25 | – |
| Male/Female | 12/13 | 12/13 | 14/11 | – |
| Age (years) | 69.2 ± 3.7 | 75.6 ± 1.3 | 82.2 ± 3.0 | 2,131.6,<.001 |
| Age range (min–max) | 59–73 | 73–78 | 78–87 | – |
| MMSE (score) | 23.8 ± 2.1 | 23.8 ± 2.1 | 23.5 ± 2.3 | 2,0.2,.833 |
| Symptom severity evaluation | ||||
| High MMSE |
Mean MMSE |
Low MMSE |
ANOVA (df,F,p) |
|
| Number | 25 | 25 | 25 | – |
| Male/Female | 11/14 | 14/11 | 12/13 | – |
| Age (years) | 75.4 ± 6.2 | 75.5 ± 5.8 | 76.5 ± 7.0 | 2,0.2,0.791 |
| MMSE (score) | 26.0 ± 0.7 | 23.9 ± 0.9 | 21.0 ± 0.7 | 2,257.9,<.001 |
Mean ± standard deviation. AD Alzheimer’s disease, ANOVA analysis of variance, MCI mild cognitive impairment, MMSE Mini Mental State Examination.
Table 3.
Subgroup characteristics for MCI converters.
| Age evaluation | ||||
|---|---|---|---|---|
| Young MCI | Mean MCI | Old MCI | ANOVA (df,F,p) | |
| Number | 21 | 21 | 21 | – |
| Male/Female | 11/10 | 14/7 | 14/7 | – |
| Age (years) | 67.9 ± 4.1 | 75.6 ± 1.8 | 82.0 ± 2.9 | 2,109.2,<.001 |
| Age range (min–max) | 58–72 | 73–78 | 79–88 | – |
| MMSE (score) | 27.3 ± 1.9 | 26.9 ± 1.6 | 26.1 ± 1.7 | 2,2.8,.068 |
| TTC (months) | 21.1 ± 7.7 | 19.4 ± 9.8 | 18.0 ± 7.8 | 2,0.7,.493 |
| Symptom severity evaluation | ||||
| High MMSE |
Mean MMSE |
Low MMSE |
ANOVA (df,F,p) |
|
| Number | 21 | 21 | 21 | – |
| Male/Female | 14/7 | 15/6 | 11/10 | – |
| Age (years) | 72.6 ± 6.8 | 76.3 ± 5.0 | 76.5 ± 7.5 | 2,2.4,.1 |
| MMSE (score) | 28.8 ± 0.7 | 26.9 ± 0.6 | 24.8 ± 0.6 | 2,218.6,<.001 |
| TTC (months) | 21.4 ± 8.6 | 20.3 ± 8.2 | 16.9 ± 8.4 | 2,1.7,.193 |
| Time to conversion evaluation | ||||
| 24 months |
18 months |
12 months |
ANOVA (df,F,p) |
|
| Number | 15 | 15 | 15 | – |
| Male/Female | 10/5 | 9/6 | 8/7 | – |
| Age (years) | 76.3 ± 5.1 | 76.8 ± 7.7 | 76.8 ± 5.9 | 2,0.0,.973 |
| MMSE (score) | 27.1 ± 1.7 | 26.7 ± 1.8 | 26.5 ± 1.8 | 2,0.4,.701 |
Mean ± standard deviation. ANOVA analysis of variance, MCI mild cognitive impairment, MMSE Mini Mental State Examination.
Table 4.
Subgroup characteristics for MCI non-converters.
| Age evaluation | ||||
|---|---|---|---|---|
| Young MCI | Mean MCI | Old MCI | ANOVA (df,F,p) | |
| Number | 21 | 21 | 21 | – |
| Male/Female | 11/10 | 13/8 | 13/8 | – |
| Age (years) | 68.0 ± 4.6 | 75.7 ± 1.8 | 82.6 ± 2.4 | 2,111.2,. < .001 |
| Age range (min–max) | 60–74 | 74–79 | 79–87 | – |
| MMSE (score) | 27.7 ± 1.4 | 28.1 ± 1.5 | 26.9 ± 1.8 | 2,3.1,.051 |
| Symptom severity evaluation | ||||
| High MMSE |
Mean MMSE |
Low MMSE |
ANOVA (df,F,p) |
|
| Number | 21 | 21 | 21 | – |
| Male/Female | 13/8 | 12/9 | 12/9 | – |
| Age (years) | 75.1 ± 6.2 | 75.4 ± 6.8 | 75.7 ± 7.3 | 2,0.0,.957 |
| MMSE (score) | 29.2 ± 0.5 | 27.7 ± 0.5 | 25.6 ± 1.8 | 2,105.4,<.001 |
Mean ± standard deviation. ANOVA analysis of variance, MCI mild cognitive impairment, MMSE Mini Mental State Examination.
All subgroup comparisons of AD, cMCI and ncMCI subjects with healthy controls were performed using a full factorial analysis of variance (ANOVA) design controlling for gender and total intracranial volume (for VBM). Total intracranial volume was calculated by counting all voxels in a sum image of grey matter, white matter and cerebrospinal fluid probability maps of each subject exceeding an absolute threshold of 0.2. This volume measurement was then recomputed into litres. In this, the three clinical subgroups (depending on split for age, symptom severity or TTC [only for cMCI]) and the control group were entered as independent factors. T-contrasts were used to evaluate differences between subgroups and control subjects. In contrast to classical parametric analyses, subgroup comparisons also provide the possibility to detect more complex relationships between age, symptom severity and TTC and differences observed in both imaging modalities. Such a complex relationship would be for example an increase in glucose hypometabolism only in middle-aged but not in younger or older AD patients. This kind of u-shaped relationships would not be detected by a regression analysis. A significance threshold of p < 0.001 uncorrected at voxel-level and p < 0.05 FWE-corrected for multiple comparisons at cluster level accounting for non-stationary smoothness in imaging data (as suggested for voxel-based morphometry data, Worsley et al., 1999; Hayasaka et al., 2004) was used for all imaging contrasts. This approach has been explicitly suggested for VBM analyses. It is based on the random field theory and is determining for any chosen voxel-based statistical threshold a corresponding cluster extent threshold which each cluster has to exceed taking into account i) the voxel-wise statistical threshold, ii) the smoothness of the data and iii) the total amount of comparisons.
As in our study we further aimed to investigate differences in the extent of atrophy and glucose hypometabolism related to age, symptom severity and TTC in the subgroups, the extents of detected differences within each modality between different conditions and between the modalities for each condition were compared to each other using two proportions z-tests. Thereby the total amount of detected significant voxels for each comparison was expressed as a proportion relative to the total amount of voxels within the statistical mask. Further, as the difference in proportions of significant voxels is highly dependent on the chosen significance threshold (e.g. at a high threshold the same difference in cluster extent is less likely than at a lower threshold) and on data smoothness, the sample size was calculated as the total amount of tested voxels scaled by a factor calculated as the number of expected clusters times number of expected voxels per cluster. This information about number and extent of expected clusters is provided within the SPM software in the statistical evaluation of each contrast and is based on estimates of data smoothness and the chosen statistical threshold. For these comparisons a statistical threshold of p < .05 Bonferroni corrected for the total amount of comparisons within each group was chosen.
2.8. Parametric analyses
To additionally evaluate parametric differences related to age, symptom severity and TTC in FDG-PET and sMRI we performed three regression analyses for each of the modalities using either only AD, cMCI or ncMCI subjects. Age, gender and MMSE were included in all regression analyses with additional inclusion of TTC for the cMCI group. For all sMRI regression analyses total intracranial volume was included in the design matrix. Correlations of imaging data with age and MMSE were evaluated for all clinical groups. In addition, correlations with TTC were calculated for cMCI.
A significance threshold of p < 0.001 uncorrected at voxel-level and p < 0.05 FWE-corrected for non-stationarity of smoothness at cluster level was used (Hayasaka et al., 2004).
2.9. Statistical analysis of behavioral data
We performed ANOVAs for group comparisons for age and MMSE in the entire dataset and for all subgroups (Tables 1, 2, 3). cMCI subgroups were additionally compared to each other for TTC. If an ANOVA revealed a significant between-group effect, a Bonferroni t-test was calculated with a significance threshold of p < .05 (corrected for multiple comparisons, two-tailed). Group differences regarding sex were evaluated using a chi-square test for independent samples. The statistical analyses were performed with the commercial software package SPSS 17.0 (http://www.spss.com/statistics/). Additionally, we calculated correlations between all variables of interest for each clinical group separately with a significance threshold of p < .05 (Bonferroni corrected for multiple comparisons, where deemed appropriate).
3. Results
3.1. Imaging results
3.1.1. Age
In young AD patients relative GM volume loss affected almost the whole cortex, sparing only sensorimotor and occipital regions (Fig. 1). In subcortical areas, both thalami were also atrophic. In middle-aged AD patients GM atrophy extended bilaterally to hippocampus, thalamus, cerebellum, cingulate, precuneate, parietal, temporal, occipital, frontal and insular cortices and to right putamen. In older AD patients we observed atrophy in bilateral hippocampi, thalami, temporal, temporo-parietal, sensorimotor and right occipital regions. In young AD patients glucose hypometabolism was observed in bilateral temporo-parietal, precuneus, posterior cingulate, lateral frontal, medial frontal and posterior thalamic regions. Glucose hypometabolism in middle-aged AD patients was restricted to bilateral temporo-parietal, right frontal, bilateral posterior cingulate regions and precuneus and in older patients to bilateral precuneus and to the right parietal cortex.
Fig. 1.
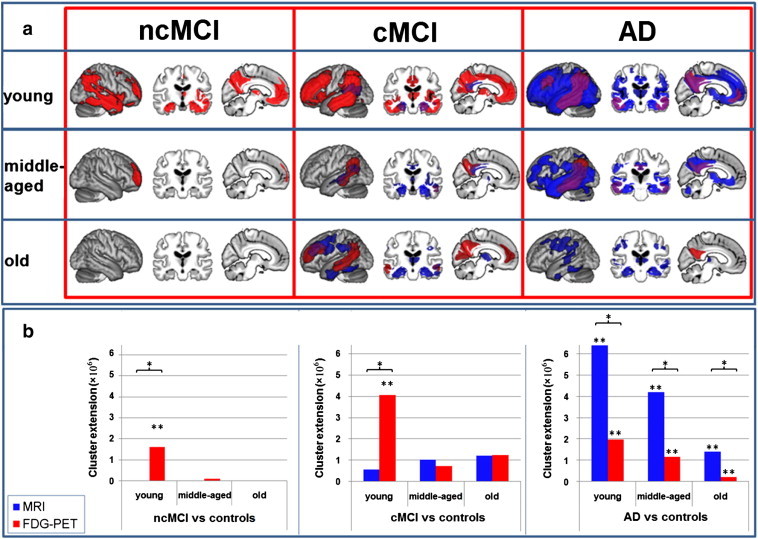
MRI and FDG-PET results for the comparison of three groups of AD patients, MCI converters (cMCI) and non-converters (ncMCI) split by age compared to the same group of control subjects. a) MRI (blue: atrophy) and FDG-PET (red: hypometabolism) results plotted onto an averaged brain. Overlapping results are displayed in violet. Results for differently aged subgroups are separated by rows. Results for different clinical groups are separated by columns. Cortical hypometabolism in ncMCI patients was restricted to the right hemisphere. In contrast, in both other groups the results were much more symmetric however with more extensive changes in the left hemisphere. b) Number of significant voxels (p = 0.001 uncorrected at voxel level and p = 0.05 FWE corrected at cluster level) detected in each comparison in FDG-PET (red) and MRI (blue). AD Alzheimer’s disease, FDG-PET [18F]fluorodeoxyglucose positron emission tomography, FWE family-wise error, MCI Mild Cognitive Impairment, MRI structural magnetic resonance imaging, * indicates a significant difference between modalities within the condition, ** indicates a significant difference to all other conditions within the modality. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Young cMCI showed atrophy in bilateral hippocampal, precuneate, posterior cingulate, left parietal and right temporal cortices (Fig. 1). Middle-aged cMCI showed a very similar but more widespread pattern of atrophy additionally involving the right insula and both anterior thalami. In older cMCI, the most widespread atrophy was found that involved bilateral parietal, temporal and frontal cortices, the thalamus and the hippocampus. In contrast, glucose utilisation was most strongly reduced in young MCI converters, involving medial and lateral frontal, temporal, parietal and insular cortices, the cingulum, basal ganglia, thalamus and hippocampus. Both middle-aged and old cMCI showed glucose hypometabolism in precuneate and temporo-parietal regions. Additionally, older cMCI had reduced glucose metabolism in medial and lateral frontal cortical regions.
There was no significant atrophy in any of the ncMCI groups. Glucose utilisation was reduced in younger ncMCI in precuneate, temporal, parietal, lateral and medial frontal, hippocampal and thalamic regions. Middle-aged ncMCI showed decreased glucose metabolism in the right polar frontal cortex. No hypometabolism was observed in older ncMCI.
3.1.2. Symptom severity
In mild AD, atrophy was mainly restricted to precuneus, posterior cingulate, hippocampal, anterior thalamic, insular, lateral frontal, temporal and parietal regions. In contrast, in the moderately impaired AD group, atrophy was more extensive in parietal and temporal cortices and additionally involved orbito-frontal regions. In the more severe AD group, atrophy involved almost all cortical and subcortical structures, sparing only some motor regions and most parts of the cerebellum. AD patients with mild symptom severity showed decreased glucose utilisation in precuneus, bilateral temporal and right parietal regions. In moderately and severely impaired AD patients, we detected impaired glucose utilisation in a bilateral network consisting of lateral frontal, temporo-parietal and precuneate regions.
In both mild and moderate cMCI, atrophy was restricted to bilateral hippocampal and right temporal regions. cMCI with lowest MMSE scores revealed a more wide-spread pattern of atrophy covering lateral and orbital frontal, parietal, temporal, insular and occipital cortices in the patient group. Further, in this group, atrophy was significant in precuneate, hippocampal and anterior thalamic regions. In all three groups of cMCI glucose hypometabolism extended to temporo-parietal, lateral and medial frontal regions and precuneus. Additionally, cMCI with moderate symptom severity showed reduced glucose metabolism in both thalami and right caudate nuclei. Further, in cMCI with comparably mild symptom severity, we observed reduced glucose utilisation in orbito-frontal and insular cortices.
In ncMCI, only the group with the lowest MMSE showed significant atrophy in the right hippocampus whilst glucose hypometabolism was restricted to right precuneus and parietal regions (Fig. 2).
Fig. 2.
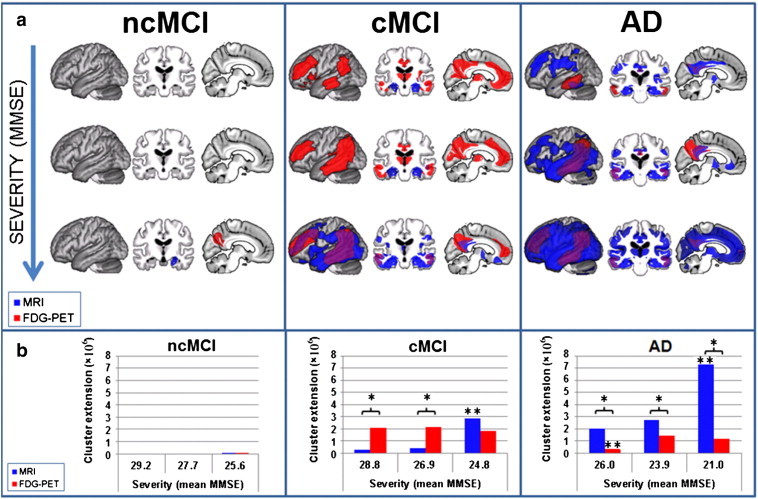
MRI and FDG-PET results for the comparison of three groups of AD patients, MCI converters (cMCI) and non-converters (ncMCI) split by symptom severity compared to the same group of control subjects. a) MRI (blue: atrophy) and FDG-PET (red: hypometabolism) results plotted onto an averaged brain. Overlapping results are displayed in violet. Results for differently affected subgroups are separated by rows. Results for different clinical groups are separated by columns. b) Number of significant voxels (p = 0.001 uncorrected at voxel level and p = 0.05 FWE corrected at cluster level) detected in each comparison in FDG-PET (red) and MRI (blue). AD Alzheimer's disease, FDG-PET [18F]fluorodeoxyglucose positron emission tomography, FWE family-wise error, MCI Mild Cognitive Impairment, MRI structural magnetic resonance imaging, * indicates a significant difference between modalities within the condition, ** indicates a significant difference to all other conditions within the modality. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.1.3. Time to conversion (TTC)
In the cMCI group converting to AD 24 months after initial presentation, GM atrophy was observed in hippocampal, anterior thalamic and right temporal regions (Fig. 3). In cMCI with a TTC of 18 months, GM atrophy was only detected in bilateral hippocampus. In the cMCI group converting to AD in 12 months, atrophy was restricted to bilateral hippocampus and to right temporo-parietal and temporal cortices. In cMCI with a TTC of 24 months glucose utilisation reductions were restricted to right temporo-parietal, right temporal and bilateral precuneate regions. In the cMCI group converting to AD 18 months glucose hypometabolism was found in temporal, cingulate, lateral and medial frontal and parietal regions. Glucose utilisation reduction was even more prominent in the group converting to AD in 12 months, involving precuneate, parietal, temporal, lateral and medial frontal, cingulate and insular regions.
Fig. 3.
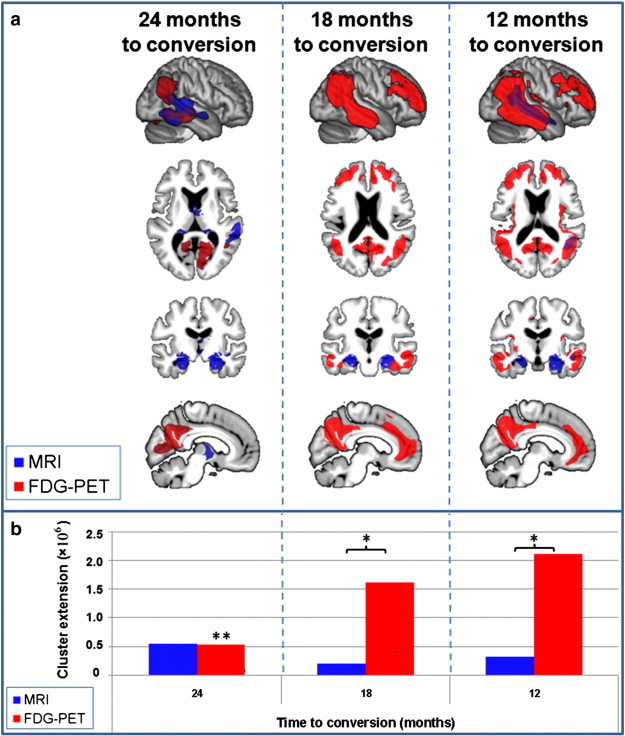
MRI and FDG-PET results for the comparison of three subgroups of MCI converters split by time to conversion compared to the same group of control subjects. a) MRI (blue: atrophy) and FDG-PET (red: hypometabolism) results plotted onto an averaged brain. Overlapping results are displayed in violet. Results for different subgroups are separated by columns. b) Number of significant voxels (p = 0.001 uncorrected at voxel level and p = 0.05 FWE corrected at cluster level) detected in each comparison in FDG-PET (red) and MRI (blue). AD Alzheimer's disease, FDG-PET [18F]fluorodeoxyglucose positron emission tomography, FWE family-wise error, MCI Mild Cognitive Impairment, MRI structural magnetic resonance imaging, * indicates a significant difference between modalities within the condition, ** indicates a significant difference to all other conditions within the modality. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Parametric results
In the AD group, we observed significant positive correlations of age with GM volume in bilateral occipital, parietal, temporal, frontal, posterior cingulate and cerebellar regions (Fig. 4). Given that we removed the voxel-wise effect of healthy aging on glucose utilisation and regional grey matter volumes in all patients based on estimates from healthy controls (Dukart et al., 2011b), positive correlations with age in AD indicate that less disease-related GM atrophy or glucose hypometabolism is required in higher age compared to lower age to induce the same symptom severity. Further significant correlations were observed in right insula and right dorsal nucleus caudatus. MMSE correlated with GM volume in bilateral parietal, temporal, frontal regions, posterior hippocampal and thalamic regions. In all clinical groups no further significant sMRI correlations were observed with any of the variables.
Fig. 4.
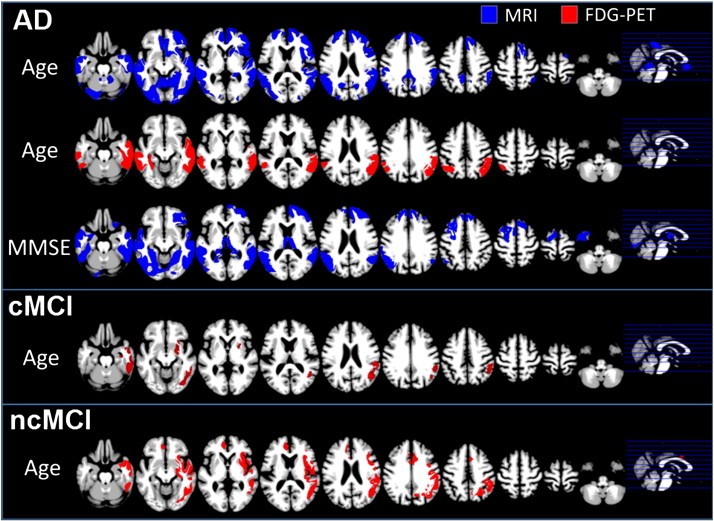
Significant age and MMSE related positive correlations observed in MRI (blue) and FDG-PET (red) in AD patients, MCI converters (cMCI) and MCI non-converters (ncMCI). AD Alzheimer's disease, FDG-PET [18F]fluorodeoxyglucose positron emission tomography, MCI Mild Cognitive Impairment, MRI structural magnetic resonance imaging. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In AD significant positive correlations between age and glucose metabolism were observed in bilateral parietal and temporal cortices. In cMCI significant positive right-sided correlations with age were observed in parietal and temporal regions and in putamen. In ncMCI we observed significant positive correlations of age with glucose metabolism in right putamen, right parietal, temporal, premotor and insular regions and in left anterior cingulate cortex.
3.3. Behavioral results
In comparison of all groups of AD patients, cMCI, ncMCI and control subjects, there was no significant difference of age or sex [χ2(3) = 3.2;p = .348]. MMSE differed significantly between control subjects and patients [Controls vs. MCI converters: t(142) = 6.5;p < .001; Controls vs. ncMCI: t(103) = 4.2;p = .001; Controls vs. AD: t(141) = 16.6;p < .001]. There was a significant difference in MMSE between MCI and AD patients [cMCI vs. AD: t(143) = 9.6;p < .001; ncMCI vs. AD: t(142) = 11.8;p < .001]. The difference between cMCI and ncMCI was not significant [t(127) = 2.3;p = .209].
The subgroups of AD patients, cMCI and ncMCI, used for evaluation of age effects on FDG-PET and sMRI did not differ in symptom severity (Tables 1, 2, 3) or in relation to sex. Of course, the between-group differences in age for the three subgroup comparisons were all highly significant (p < .001). For AD, cMCI and ncMCI subgroups used for symptom severity evaluation, age (Tables 2, 3, 4) and sex did not differ between subgroups. As expected, MMSE scores differed significantly (p < .001) for these comparisons for AD, cMCI and ncMCI subgroups. Subgroups of cMCI for evaluation of TTC-related differences in FDG-PET and sMRI did not differ in age, sex or symptom severity.
Correlational analyses in the AD group revealed only very weak, insignificant correlations between age and symptom severity as measured by MMSE [r = − .09;p = .444]. In the cMCI group, the variables age, symptom severity and TTC were also only weakly but insignificantly correlated [age and MMSE: r = − .24;p = .156; age and TTC: r = − .11;p = 1.0; MMSE and TTC: r = − .10;p = 1.0]. Similarly, there was no significant correlation between age and MMSE in ncMCI [r = − .21;p = .095].
4. Discussion
In this study we demonstrate and dissociate differential patterns of structural and metabolic brain differences related to age, symptom severity and disease progression in AD, cMCI and ncMCI subjects. An important aspect of our study is the focus on detection of the link between the spatial extent of AD related imaging pathology and age, symptom severity and TTC. This focus on spatial extents of pathology contrasts our study from previous research targeting rather the correlational link between these variables and the grade of glucose hypometabolism or atrophy at each particular voxel/region. Both types of relationships are rather independent. For example a reduction of neural function in a particular region under a particular threshold might have an impact on cognitive performance. However, any further increase in AD pathology in the very same region might not have any additional effect on cognition and would be therefore not detected by a correlational analysis. In this case only additional impairment of other brain regions would lead to further cognitive decline. By comparing all patient groups to the same control group and adjusting for healthy aging we further for the first time account for the potential confounding effect of the use of different control groups that was neglected in previous research.
The cMCI and ncMCI differ in the amount of glucose hypometabolism and brain atrophy explained by age and symptom severity with cMCI resembling AD patients. We confirm previous findings of a correlation between TTC and glucose metabolism as well as an inverse relationship between age increase and amount of atrophy and hypometabolism associated with a particular symptom severity in AD (Matsunari et al., 2007; Salmon et al., 2000). Given that we account for differences associated with healthy aging, our findings of differences in the amount of glucose hypometabolism and atrophy in AD subgroups subdivided by age can be interpreted as pathological age-specific add-ons that relate to a degree of functional impairment.
Our study results in three important findings. Firstly, the differential age-related glucose hypometabolism and atrophy patterns between clinical groups have implications for the use of neuroimaging biomarkers for improving diagnostic accuracy and prediction of conversion to AD. While FDG-PET is the dominant biomarker in young cMCI and atrophy is mainly restricted to the hippocampus, the pattern observed in old cMCI is of prominent atrophy associated with equally marked glucose hypometabolism. This observation is consistent with a relative diagnostic superiority of FDG-PET compared to sMRI in younger patients with mild AD symptoms (Kawachi et al., 2006; Matsunari et al., 2007). The age-related differential pattern of abnormality discovered with FDG-PET and sMRI supports previous findings of better diagnostic performance with FDG-PET only in early-onset AD patients, but equivalent performance for the two in elderly patients (Matsunari et al., 2007).
The second finding concerns the relationship between symptom severity as measured by classical screening and the progression of changes in FDG-PET and sMRI. We demonstrate for cMCI and AD patients a strong link between symptom severity and the amount of atrophy, which is not evident in ncMCI. Interestingly, no such pattern was seen in glucose metabolism with any of the patient cohorts. This result indicates that changes measured by sMRI are more strongly related to the current symptom severity in AD patients.
A third, more surprising result was obtained when MCI converters were split by TTC. We expected that symptom severity and TTC would be strongly linked, as neuropsychological measurements are used to determine AD stage. However, the measurements are only weakly correlated, which suggests that different mechanisms produce current (as measured by MMSE) and future cognitive states (as measured by TTC). Thus, a comparison of TTC duration subgroups with healthy control subjects showed a TTC-dependant amount of glucose hypometabolism, but no relationship with the amount of atrophy. The link between FDG-PET measurements and subsequent cognitive decline is consistent with previous reports showing the superiority of FDG-PET compared to neuropsychological measurements for prediction of subsequent cognitive decline and conversion to AD in MCI patients (Arnaiz et al., 2001; Chételat et al., 2005; Hinrichs et al., 2011).
The differential results for TTC and symptom severity-related differences observed with FDG-PET and sMRI cannot be explained by different pathologies or other factors because the cMCI subgroups came from splitting the same group of subjects.
The current view of the progression of FDG-PET and sMRI changes in AD is that functional abnormalities occur earlier than those with sMRI (Jack et al., 2010). Our results suggest a modification of the current model of biomarker changes related to progression from MCI to AD (Jack et al., 2010). Imaging abnormalities appear at an early stage in sMRI as well as FDG-PET; however, structural changes are restricted mainly to the hippocampus. In contrast, but consistent with the suggested model, glucose hypometabolism progresses rapidly in this early stage of AD. The model suggested by Jack et al. further implies that the magnitude of biomarker changes is equal in both FDG-PET and structural MRI data (Jack et al., 2010). The assumption behind this idea is that structural changes follow functional changes. In contrast, our data suggest that FDG-PET changes reach their peak already at the MCI stage (Figs. 2 and 3) and do not further increase but rather decrease (e.g. because the preserved tissue is maintaining its functional activity level) at later disease stages when adjusting for atrophy effects using PVE correction. In contrast, atrophy further increases at later AD stages in our study population and even extends to regions not showing any significant hypometabolism. For this reason we expect that if one would not use PVE correction, one would observe further reductions in glucose hypometabolism even in AD in agreement with Jack et al (2010).
Although the results of our study are relevant to improving understanding of progression from MCI to AD, some limitations need consideration to enable valid interpretation. Firstly, we used the same clinical groups to examine different variables of interest, with splitting into different subgroups depending on expression of the variables of interest. Therefore, the results may be confounded by between-group differences in uncontrolled factors such as functional asymmetries, intelligence quotient and other more specific cognitive domains associated with Alzheimer's disease. Such differences in uncontrolled factors might have contributed to the observed differences in the amount of atrophy and hypometabolism and shall be overcome in future large-scale longitudinal studies. Secondly, although subgroup comparisons for confounding variables were not significant in ANOVAs, they could have contributed to the differential results. We feel this is unlikely to have introduced any systematic bias, as the between-group differences were small and restricted to only one of the subgroups.
When removing the variance explained by healthy ageing in the imaging data we make the explicit assumption of linearity of age related differences in GM volume and glucose metabolism. This issue has been a major focus of several large scale volumetric and PET studies performed in the past two decades (Gonoi et al., 2009; Good et al., 2001; Loessner et al., 1995; Smith et al., 2007). All of these studies provided strong evidence that linear as compared to more complex models are sufficient to describe global and local atrophy and glucose metabolism reductions observed in the course of normal ageing (from the age of 20 to 100 years. Our assumption is therefore in line with previous research performed on that topic. Nonetheless, imprecisions of the applied voxel-wise healthy ageing models e.g. due to wrong linearity assumptions are a general possible confounding effect for studying age-specific differences in diseased populations. In this framework it is also important to note that age-related findings we report in our study only reflect cross-sectional differences in sMRI and FDG-PET between differently aged AD/MCI patient groups. They should therefore not be confused with within subject longitudinal changes related to AD progression.
A further limitation of our study is that a proportion of control subjects used in our study might have a preclinical AD pathology. The inclusion of such subjects could have lowered the effects observed in subgroup comparisons and therefore lead to an underestimation of the amount of AD pathology. Similarly, one cannot exclude that also some of the ncMCI would have converted to AD (or reverted from MCI to normal range) if followed for a longer time period. As indicated in previous studies on MCI with a follow-up of 10 years more than 80% of MCI are expected to convert to AD in the long term (Visser et al., 2006). Current ADNI group assignments should be therefore considered carefully and the results need to be validated using cohorts with a longer follow-up than currently available from the ADNI. However, this limitation is common to all imaging studies including not pathologically proven AD patients and control subjects. Furthermore, as we compare all subgroups exactly to the same control group, the potential inclusion of control subjects with preclinical AD would affect all comparisons in the same way without introducing a bias towards a specific subgroup. Also the finding of only a very weak link between TTC and symptom severity in our cohort requires careful interpretation. TTC is only a crude and general estimate of cognitive function. It is therefore likely that some more specific cognitive tests might be more closely related to disease progression as indicated by TTC. It is important to note that all differential results reported in our study are detected by comparing all patient subgroups to the same control group. We therefore provide only indirect evidence for between-subgroup differences. In contrast, the direct subgroup comparisons were not significant. These findings therefore need to be validated in future studies by direct comparisons of patient subgroups using larger data sets than currently available from ADNI and preferably in longitudinal study designs.
In our study we observed a slight decrease in sensitivity of FDG-PET at later as compared to earlier AD stages. We interpret this effect as an interaction of pathophysiology with the PVE correction performed in our study. However, this effect might be also caused by methodical artifacts, e.g. due to intensity normalisation (cerebellar metabolism is known to decline in AD) or due to increasing inaccuracy of PVE correction caused by increasing cortical atrophy and associated problems with accurate MRI segmentation.
Our results have potential implications for the selection of biomarkers, MCI staging and for treatment evaluation. While FDG-PET appears to be a more sensitive biomarker of disease progression at the MCI stage, structural differences as measured by sMRI seem to be more strongly linked to the current global cognitive state, potentially reflecting irreversible damage. Therefore, we suggest that for treatment evaluation it may be crucial to use both structural and functional biomarkers.
The following is the supplementary data related to this article.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nicl.2013.07.005.
Disclosure/Conflict of interest
Dr. Juergen Dukart is supported by LIFE—Leipzig Research Center for 635 Civilization Diseases at the University of Leipzig. LIFE is funded by means of the European Union, by the European Regional Development Fund (ERFD) and by means of the Free State of Saxony within the frame- work of the excellence initiative. He is also supported by the Swiss National Science Foundation (NCCR Synapsy).
Dr. Karsten Mueller reports no disclosures.
Dr. Arno Villringer reports no disclosures.
Dr. Ferath Kherif is supported by the Velux Stiftung.
Dr. Bogdan Draganski is supported by the Swiss National Science Foundation (project grant Nr 320030_135679, NCCR Synapsy and SPUM 33CM30_140332/1) Foundation Parkinson Switzerland, Foundation Synapsis, Novartis Foundation for medical–biological research and Deutsche Forschungsgemeinschaft (Kfo 247).
Dr. Richard Frackowiak reports no disclosures.
Dr. Matthias L. Schroeter is supported by the German Federal Ministry of Education and Research (BMBF; German FTLD consortium) and by the LIFE—Leipzig Research Center for Civilization Diseases at the University of Leipzig.
Acknowledgments
Juergen Dukart, Arno Villringer and Matthias L. Schroeter are supported by LIFE—Leipzig Research Center for Civilization Diseases at the University of Leipzig. LIFE is funded by means of the European Union, by the European Regional Development Fund (ERFD) and by means of the Free State of Saxony within the framework of the excellence initiative. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc., F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., as well as non-profit partners the Alzheimer's Association and Alzheimer's Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organisation is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129, K01 AG030514, and the Dana Foundation.
Furthermore, Matthias L. Schroeter is supported by the German Federal Ministry of Education and Research (BMBF; German FTLD consortium).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- Alexander G.E., Chen K., Pietrini P., Rapoport S.I., Reiman E.M. Longitudinal PET evaluation of cerebral metabolic decline in dementia: a potential outcome measure in Alzheimer's disease treatment studies. The American Journal of Psychiatry. 2002;159:738–745. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- Anchisi D., Borroni B., Franceschi M., Kerrouche N., Kalbe E., Beuthien-Beumann B., Cappa S., Lenz O., Ludecke S., Marcone A., Mielke R., Ortelli P., Padovani A., Pelati O., Pupi A., Scarpini E., Weisenbach S., Herholz K., Salmon E., Holthoff V., Sorbi S., Fazio F., Perani D. Heterogeneity of brain glucose metabolism in mild cognitive impairment and clinical progression to Alzheimer disease. Archives of Neurology. 2005;62:1728–1733. doi: 10.1001/archneur.62.11.1728. [DOI] [PubMed] [Google Scholar]
- Arnaiz E., Jelic V., Almkvist O., Wahlund L., Winblad B., Valind S., Nordberg A. Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment. NeuroReport. 2001;12:851–855. doi: 10.1097/00001756-200103260-00045. [DOI] [PubMed] [Google Scholar]
- Ashburner J. A fast diffeomorphic image registration algorithm. NeuroImage. 2007;38:95–113. doi: 10.1016/j.neuroimage.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Buckner R.L. Memory and executive function in aging and AD: multiple factors that cause decline and reserve factors that compensate. Neuron. 2004;44:195–208. doi: 10.1016/j.neuron.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Canu E., Frisoni G.B., Agosta F., Pievani M., Bonetti M., Filippi M. Early and late onset Alzheimer's disease patients have distinct patterns of white matter damage. Neurobiology of Aging. 2012;33:1023–1033. doi: 10.1016/j.neurobiolaging.2010.09.021. [DOI] [PubMed] [Google Scholar]
- Chételat G., Eustache F., Viader F., De La Sayette V., Pélerin A., Mézenge F., Hannequin D., Dupuy B., Baron J.C., Desgranges B. FDG-PET measurement is more accurate than neuropsychological assessments to predict global cognitive deterioration in patients with mild cognitive impairment. Neurocase. 2005;11:14–25. doi: 10.1080/13554790490896938. [DOI] [PubMed] [Google Scholar]
- Crivello F., Schormann T., Tzourio-Mazoyer N., Roland P.E., Zilles K., Mazoyer B.M. Comparison of spatial normalization procedures and their impact on functional maps. Human Brain Mapping. 2002;16:228–250. doi: 10.1002/hbm.10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davatzikos C., Fan Y., Wu X., Shen D., Resnick S.M. Detection of prodromal Alzheimer's disease via pattern classification of magnetic resonance imaging. Neurobiology of Aging. 2008;29:514–523. doi: 10.1016/j.neurobiolaging.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davatzikos C., Resnick S.M., Wu X., Parmpi P., Clark C.M. Individual patient diagnosis of AD and FTD via high-dimensional pattern classification of MRI. NeuroImage. 2008;41:1220–1227. doi: 10.1016/j.neuroimage.2008.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drzezga A., Lautenschlager N., Siebner H., Riemenschneider M., Willoch F., Minoshima S., Schwaiger M., Kurz A. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer's disease: a PET follow-up study. European Journal of Nuclear Medicine and Molecular Imaging. 2003;30:1104–1113. doi: 10.1007/s00259-003-1194-1. [DOI] [PubMed] [Google Scholar]
- Dukart J., Mueller K., Horstmann A., Vogt B., Frisch S., Barthel H., Becker G., Moller H.E., Villringer A., Sabri O., Schroeter M.L. Differential effects of global and cerebellar normalization on detection and differentiation of dementia in FDG-PET studies. NeuroImage. 2010;49:1490–1495. doi: 10.1016/j.neuroimage.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Dukart J., Mueller K., Horstmann A., Barthel H., Moller H.E., Villringer A., Sabri O., Schroeter M.L. Combined evaluation of FDG-PET and MRI improves detection and differentiation of dementia. PLoS One. 2011;6 doi: 10.1371/journal.pone.0018111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukart J., Schroeter M.L., Mueller K., Initiative T.A.s.D.N. Age correction in dementia—matching to a healthy brain. PLoS One. 2011;6:e22193. doi: 10.1371/journal.pone.0022193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison P., Archer H.A., Hinz R., Hammers A., Pavese N., Tai Y.F., Hotton G., Cutler D., Fox N., Kennedy A., Rossor M., Brooks D.J. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Ewers M., Frisoni G.B., Teipel S.J., Grinberg L.T., Amaro E., Heinsen H., Thompson P.M., Hampel H. Staging Alzheimer's disease progression with multimodality neuroimaging. Progress in Neurobiology. 2011;95:535–546. doi: 10.1016/j.pneurobio.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y., Resnick S.M., Wu X., Davatzikos C. Structural and functional biomarkers of prodromal Alzheimer's disease: a high-dimensional pattern classification study. NeuroImage. 2008;41:277–285. doi: 10.1016/j.neuroimage.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein M.F., Folstein S.E., McHugh P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Fonteijn H.M., Modat M., Clarkson M.J., Barnes J., Lehmann M., Hobbs N.Z., Scahill R.I., Tabrizi S.J., Ourselin S., Fox N.C. An event-based model for disease progression and its application in familial Alzheimer's disease and Huntington's disease. NeuroImage. 2012;60:1880–1889. doi: 10.1016/j.neuroimage.2012.01.062. [DOI] [PubMed] [Google Scholar]
- Förster S., Grimmer T., Miederer I., Henriksen G., Yousefi B.H., Graner P., Wester H.J., Förstl H., Kurz A., Dickerson B.C., Bartenstein P., Drzezga A. Regional expansion of hypometabolism in Alzheimer's disease follows amyloid deposition with temporal delay. Biological Psychiatry. 2012;71:792–797. doi: 10.1016/j.biopsych.2011.04.023. [DOI] [PubMed] [Google Scholar]
- Fox N.C., Scahill R.I., Crum W.R., Rossor M.N. Correlation between rates of brain atrophy and cognitive decline in AD. Neurology. 1999;52:1687–1689. doi: 10.1212/wnl.52.8.1687. [DOI] [PubMed] [Google Scholar]
- Franke K., Ziegler G., Kloppel S., Gaser C. Estimating the age of healthy subjects from T1-weighted MRI scans using kernel methods: exploring the influence of various parameters. NeuroImage. 2010;50:883–892. doi: 10.1016/j.neuroimage.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Frisoni G.B., Pievani M., Testa C., Sabattoli F., Bresciani L., Bonetti M., Beltramello A., Hayashi K.M., Toga A.W., Thompson P.M. The topography of grey matter involvement in early and late onset Alzheimer's disease. Brain. 2007;130:720–730. doi: 10.1093/brain/awl377. [DOI] [PubMed] [Google Scholar]
- Gao S., Hendrie H., Hall K. The relationships between age, sex, and the incidence of dementia and Alzheimer disease—a meta-analysis. Archives of General Psychiatry. 1998:809–815. doi: 10.1001/archpsyc.55.9.809. [DOI] [PubMed] [Google Scholar]
- Gomar J.J., Bobes-Bascaran M.T., Conejero-Goldberg C., Davies P., Goldberg T.E., Initiative A.s.D.N. Utility of combinations of biomarkers, cognitive markers, and risk factors to predict conversion from mild cognitive impairment to Alzheimer disease in patients in the Alzheimer's disease neuroimaging initiative. Archives of General Psychiatry. 2011;68:961–969. doi: 10.1001/archgenpsychiatry.2011.96. [DOI] [PubMed] [Google Scholar]
- Gonoi W., Abe O., Yamasue H., Yamada H., Masutani Y., Takao H., Kasai K., Aoki S., Ohtomo K. Age-related changes in regional brain volume evaluated by atlas-based method. Neuroradiology. 2009;10:865–873. doi: 10.1007/s00234-009-0641-5. [DOI] [PubMed] [Google Scholar]
- Good C.D., Johnsrude I.S., Ashburner J., Henson R.N., Friston K.J., Frackowiak R.S. A voxel-based morphometric study of ageing in 465 normal adult human brains. NeuroImage. 2001;14:21–36. doi: 10.1006/nimg.2001.0786. [DOI] [PubMed] [Google Scholar]
- Habeck C., Foster N.L., Perneczky R., Kurz A., Alexopoulos P., Koeppe R.A., Drzezga A., Stern Y. Multivariate and univariate neuroimaging biomarkers of Alzheimer's disease. NeuroImage. 2008;40:1503–1515. doi: 10.1016/j.neuroimage.2008.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haense C., Herholz K., Jagust W.J., Heiss W.D. Performance of FDG PET for detection of Alzheimer's disease in two independent multicentre samples (NEST-DD and ADNI) Dementia and Geriatric Cognitive Disorders. 2009;28:259–266. doi: 10.1159/000241879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayasaka S., Phan K.L., Liberzon I., Worsley K.J., Nichols T.E. Nonstationary cluster-size inference with random field and permutation methods. NeuroImage. 2004;22:676–687. doi: 10.1016/j.neuroimage.2004.01.041. [DOI] [PubMed] [Google Scholar]
- Herholz K., Westwood S., Haense C., Dunn G. Evaluation of a calibrated 18F-FDG PET score as a biomarker for progression in Alzheimer disease and mild cognitive impairment. Journal of Nuclear Medicine. 2011;52:1218–1226. doi: 10.2967/jnumed.111.090902. [DOI] [PubMed] [Google Scholar]
- Hinrichs C., Singh V., Xu G., Johnson S.C., Initiative A.D.N. Predictive markers for AD in a multi-modality framework: an analysis of MCI progression in the ADNI population. NeuroImage. 2011;55:574–589. doi: 10.1016/j.neuroimage.2010.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K., Kawachi T., Sasaki H., Kono A.K., Fukuda T., Kojima Y., Mori E. Voxel-based morphometric comparison between early- and late-onset mild Alzheimer's disease and assessment of diagnostic performance of z score images. AJNR. American Journal of Neuroradiology. 2005;26:333–340. [PMC free article] [PubMed] [Google Scholar]
- Jack C.R., Knopman D.S., Jagust W.J., Shaw L.M., Aisen P.S., Weiner M.W., Petersen R.C., Trojanowski J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurology. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W., Reed B., Mungas D., Ellis W., Decarli C. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology. 2007;69:871–877. doi: 10.1212/01.wnl.0000269790.05105.16. [DOI] [PubMed] [Google Scholar]
- Jedynak B.M., Lang A., Liu B., Katz E., Zhang Y., Wyman B.T., Raunig D., Jedynak C.P., Caffo B., Prince J.L. A computational neurodegenerative disease progression score: method and results with the Alzheimer's disease neuroimaging initiative cohort. NeuroImage. 2012;63:1478–1486. doi: 10.1016/j.neuroimage.2012.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.K., Storandt M., Morris J.C., Galvin J.E. Longitudinal study of the transition from healthy aging to Alzheimer disease. Archives of Neurology. 2009;66:1254–1259. doi: 10.1001/archneurol.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.K., Symms M.R., Cercignani M., Howard R.J. The effect of filter size on VBM analyses of DT-MRI data. NeuroImage. 2005;26:546–554. doi: 10.1016/j.neuroimage.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Kaiser N.C., Melrose R.J., Liu C., Sultzer D.L., Jimenez E., Su M., Monserratt L., Mendez M.F. Neuropsychological and neuroimaging markers in early versus late-onset Alzheimer's disease. American Journal of Alzheimer's Disease and Other Dementias. 2012;27:520–529. doi: 10.1177/1533317512459798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas G., Scheltens P., Rombouts S., van Schijndel R., Klein M., Jones B., van der Flier W., Vrenken H., Barkhof F. Precuneus atrophy in early-onset Alzheimer’s disease: a morphometric structural MRI study. Neuroradiology. 2007;49:967–976. doi: 10.1007/s00234-007-0269-2. [DOI] [PubMed] [Google Scholar]
- Kawachi T., Ishii K., Sakamoto S., Sasaki M., Mori T., Yamashita F., Matsuda H., Mori E. Comparison of the diagnostic performance of FDG-PET and VBM-MRI in very mild Alzheimer's disease. European Journal of Nuclear Medicine and Molecular Imaging. 2006;33:801–809. doi: 10.1007/s00259-005-0050-x. [DOI] [PubMed] [Google Scholar]
- Kinkingnehun S., Sarazin M., Lehericy S., Guichart-Gomez E., Hergueta T., Dubois B. VBM anticipates the rate of progression of Alzheimer disease: a 3-year longitudinal study. Neurology. 2008;70:2201–2211. doi: 10.1212/01.wnl.0000303960.01039.43. [DOI] [PubMed] [Google Scholar]
- Kloppel S., Stonnington C.M., Chu C., Draganski B., Scahill R.I., Rohrer J.D., Fox N.C., Jack C.R., Jr., Ashburner J., Frackowiak R.S. Automatic classification of MR scans in Alzheimer's disease. Brain. 2008;131:681–689. doi: 10.1093/brain/awm319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner M., Tobin M., Alavi A., Chawluk J., Rosen M., Fazekas F., Alavi J., Reivich M. Cerebellar glucose consumption in normal and pathologic states using fluorine-FDG and PET. Journal of Nuclear Medicine. 1987;28:1667–1670. [PubMed] [Google Scholar]
- Loessner A., Alavi A., Lewandrowski K.U., Mozley D., Souder E., Gur R.E. Regional cerebral function determined by FDG-PET in healthy volunteers: normal patterns and changes with age. Journal of Nuclear Medicine. 1995;36:1141–1149. [PubMed] [Google Scholar]
- Matsunari I., Samuraki M., Chen W.P., Yanase D., Takeda N., Ono K., Yoshita M., Matsuda H., Yamada M., Kinuya S. Comparison of 18F-FDG PET and optimized voxel-based morphometry for detection of Alzheimer's disease: aging effect on diagnostic performance. Journal of Nuclear Medicine. 2007;48:1961–1970. doi: 10.2967/jnumed.107.042820. [DOI] [PubMed] [Google Scholar]
- McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E.M. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Möller C., Vrenken H., Jiskoot L., Versteeg A., Barkhof F., Scheltens P., van der Flier W.M. Different patterns of gray matter atrophy in early-and late-onset Alzheimer’s disease. Neurobiology of Aging. 2013;34:2014–2022. doi: 10.1016/j.neurobiolaging.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Muller-Gartner H.W., Links J.M., Prince J.L., Bryan R.N., McVeigh E., Leal J.P., Davatzikos C., Frost J.J. Measurement of radiotracer concentration in brain gray matter using positron emission tomography: MRI-based correction for partial volume effects. Journal of Cerebral Blood Flow and Metabolism. 1992;12:571–583. doi: 10.1038/jcbfm.1992.81. [DOI] [PubMed] [Google Scholar]
- Quarantelli M., Berkouk K., Prinster A., Landeau B., Svarer C., Balkay L., Alfano B., Brunetti A., Baron J.C., Salvatore M. Integrated software for the analysis of brain PET/SPECT studies with partial-volume-effect correction. Journal of Nuclear Medicine. 2004;45:192–201. [PubMed] [Google Scholar]
- Sadeghi N., Foster N.L., Wang A.Y., Minoshima S., Lieberman A.P., Tasdizen T. vol. 1–4. 2008. Automatic classification of Alzheimer's disease vs. frontotemporal dementia: a spatial decision tree approach with FDG-PET; pp. 408–411. (2008 Ieee International Symposium on Biomedical Imaging: From Nano to Macro). [Google Scholar]
- Sakamoto S., Ishii K., Sasaki M., Hosaka K., Mori T., Matsui M., Hirono N., Mori E. Differences in cerebral metabolic impairment between early and late onset types of Alzheimer's disease. Journal of Neurological Sciences. 2002;200:27–32. doi: 10.1016/s0022-510x(02)00114-4. [DOI] [PubMed] [Google Scholar]
- Salmon E., Collette F., Degueldre C., Lemaire C., Franck G. Voxel-based analysis of confounding effects of age and dementia severity on cerebral metabolism in Alzheimer's disease. Human Brain Mapping. 2000;10:39–48. doi: 10.1002/(SICI)1097-0193(200005)10:1<39::AID-HBM50>3.0.CO;2-B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter M.L., Stein T., Maslowski N., Neumann J. Neural correlates of Alzheimer's disease and mild cognitive impairment: a systematic and quantitative meta-analysis involving 1351 patients. NeuroImage. 2009;47:1196–1206. doi: 10.1016/j.neuroimage.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.D., Chebrolu H., Wekstein D.R., Schmitt F.A., Markesbery W.R. Age and gender effects on human brain anatomy: a voxel-based morphometric study in healthy elderly. Neurobiology of Aging. 2007;28:1075–1087. doi: 10.1016/j.neurobiolaging.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Tisserand D.J., van Boxtel M.P., Pruessner J.C., Hofman P., Evans A.C., Jolles J. A voxel-based morphometric study to determine individual differences in gray matter density associated with age and cognitive change over time. Cerebral Cortex. 2004;14:966–973. doi: 10.1093/cercor/bhh057. [DOI] [PubMed] [Google Scholar]
- Vemuri P., Wiste H., Weigand S., Shaw L., Trojanowski J., Weiner M., Knopman D., Petersen R., Jack C. MRI and CSF biomarkers in normal, MCI, and AD subjects diagnostic discrimination and cognitive correlations. Neurology. 2009;73:287–293. doi: 10.1212/WNL.0b013e3181af79e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P., Wiste H., Weigand S., Shaw L., Trojanowski J., Weiner M., Knopman D., Petersen R., Jack C. MRI and CSF biomarkers in normal, MCI, and AD subjects predicting future clinical change. Neurology. 2009;73:294–301. doi: 10.1212/WNL.0b013e3181af79fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser P.J., Kester A., Jolles J., Verhey F. Ten-year risk of dementia in subjects with mild cognitive impairment. Neurology. 2006;67:1201–1207. doi: 10.1212/01.wnl.0000238517.59286.c5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.