

Abstract
In eucaryotes, many secreted proteins and peptides are proteolytically excised from larger precursor proteins by a specific class of serine proteases, the proprotein/prohormone convertases (PCs). This cleavage is essential for substrate activation, making the PCs very interesting pharmacological targets in cancer and infectious disease research. Correspondingly, their structure, function and inhibition are intensely studied – studies that require the respective target proteins in large amounts and at high purity. Here we describe the development of a novel purification protocol of furin, the best-studied member of the PC family. We combined the heterologous expression of furin from CHO cells with a novel purification scheme employing an affinity step that efficiently extracts only active furin from the conditioned medium by using furin-specific inhibitor moieties as bait. Several potential affinity tags were synthesized and their binding to furin characterized. The best compound, Biotin-(Adoa)2-Arg-Pro-Arg-4-Amba coupled to streptavidin-Sepharose beads, was used in a three-step chromatographic protocol and routinely resulted in a high yield of a homogeneous furin preparation with a specific activity of ~60 units/mg protein. This purification and the general strategy can easily be adapted to the efficient purification of other PC family members.
Keywords: affinity chromatography, furin, proprotein convertase, protease inhibitor, protein purification
Introduction
Many secreted proteins and peptides are initially synthesized as larger inactive precursor proteins (or proproteins) and are subsequently converted to their mature, active forms by specific proteolytic cleavages within the secretory pathway (Seidah and Chretien, 1997; Steiner, 1998). The respective processing proteases belong to the family of proprotein/ prohormone convertases (PCs) and share an overall subtilisin-like fold in their catalytic domains. In mammals, they can be grouped into the classical PCs that cleave mostly after dibasic residues (PC1/3, PC2, furin, PC4, PC5/6, PACE4 and PC7) and are homologous to the yeast endoproteinase Kex2/ kexin, as well as two PCs that cleave after nonbasic sites [subtilisin-kexin-isoenzyme-1 (SKI-1)/site one protease (S1P) and proprotein convertase subtilisin/kexin type nine (PCSK9)/neural apoptosis regulated convertase one (NARC-1)] (Seidah et al., 2008). These enzymes proteolytically activate various precursors including those of peptide hormones (such as insulin), extracellular proteases, growth and differentiation factors (implicated in neurodegenerative diseases, tumor growth and metastasis) and bacterial toxins as well as viral coat proteins. The fact that the PC family members are involved in such a large number of physiological and pathological processes (Hallenberger et al., 1992; Nakayama, 1997; Seidah and Prat, 2002; Bassi et al., 2005; Scamuffa et al., 2006) renders them very interesting drug targets (von Eggelkraut-Gottanka and Beck-Sickinger, 2004; Basak, 2005; Fugere and Day, 2005; Bontemps et al., 2007; Seidah and Prat, 2007).
Furin, the best-characterized family member which is often regarded as the PC prototype, cleaves its target proteins with high specificity following the R-X-K/R-R motif. It represents a ubiquitously expressed, type I membrane protein consisting of multiple domains; it itself is initially translated as a proprotein (Thomas, 2002). Furin becomes activated by a pH-dependent, two-step proteolytic processing event, during which the autoinhibitory prodomain is cleaved off, followed by destabilization and dissociation of the prodomain and structural reorganization of the newly created N-terminus (Henrich et al., 2005; Than et al., 2005; Feliciangeli et al., 2006). The mature enzyme consists of the closely associated catalytic and P-domains consisting of 336 and 128 amino acids, respectively, followed by 222 additional residues comprising a cysteine-rich domain, a single transmembrane helix, a short cytoplasmic domain and intervening linker regions. The first two domains, which intimately contact each other (Henrich et al., 2003), are sufficient to effect catalytic activity. Constructs lacking the P-domain are inactive, probably due to insufficient folding and stability (Hatsuzawa et al., 1992; Creemers et al., 1993).
The crystal structure of furin in complex with the covalently bound decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone inhibitor (Henrich et al., 2003), the only experimental structure of a mammalian, classical PC (but see, e.g., Holyoak et al., 2003, for the structure of the homologous yeast enzyme) show how this enzyme is able to recognize its substrates with high specificity and permitted the development of specific peptidic inhibitors targeted towards furin (reviewed in Rockwell and Thorner, 2004). The other classical members of the PC family are highly homologous (up to ~70% identity in the catalytic domain) and based on homology modeling show very similar 3D structures (Henrich et al., 2005). Correspondingly, one can either develop inhibitors that block the entire PC family (and given the overlapping function of the different family members, this approach might be beneficial for certain pharmacological applications) or specifically target one or only a small sub-fraction of the PCs. The latter strategy most probably will require the use of non-peptidic backbones and must address surface residues at the edge or outside of the catalytic cleft to gain the required specificity (Henrich et al., 2005). Several furin inhibitors belonging to different molecular classes (Hallenberger et al., 1992; Angliker, 1995; Cameron et al., 2000; Kacprzak et al., 2004; Basak, 2005; Komiyama, 2005; Jiao et al., 2006; Komiyama et al., 2009; Becker et al., 2010; Sielaff et al., 2011) have been developed thus far. However, it has been proven difficult to predict and to model the binding mode, especially of the non-peptidic inhibitors (Sielaff et al., 2011).
To investigate the inhibitory mode of known inhibitors, and to develop novel inhibitors, the target enzyme must be available in pure form. Especially for the crystallographic study of protein-inhibitor complexes, milligram amounts of highly pure and homogeneous protein preparations are needed. Soluble human and mouse furin have been expressed in Chinese hamster ovary (CHO) cells in large amounts and purified using a classical chromatographic approach consisting of ion exchange and size exclusion chromatography (Cameron et al., 2000; Kacprzak et al., 2004). The resulting protein preparations have been successfully used for crystallization and structure determination (Henrich et al., 2003). Over the past years, however, it has become apparent that active furin, representing an empty active site cleft, and protein preparations that are inhibited by non-covalent and weak binding inhibitors are difficult to crystallize (Sielaff et al., 2011; and M.E. Than, unpublished data). It is generally known that bound substrate-analogs and/or inhibitors can reduce the inherent flexibility of enzymes, rendering them more amenable to crystallization. In addition, the crystallization tendency of proteins is negatively influenced by flexible surface features (such as flexible loops of the polypeptide chain or the attachment of sugar chains) and impurities. To address the latter issue, we have developed a novel purification scheme for mouse furin, employing heterologous expression from CHO cells followed by an affinity-based chromatographic purification scheme, which results in significantly increased purity and yield.
Results
Design and synthesis of the affinity tag
To develop a novel, affinity-based purification method for furin showing characteristics superior to existing methodologies (Cameron et al., 2000), we employed furin-specific inhibitors as affinity tags that we immobilized by their biotin moieties on streptavidin-Sepharose for column-chromatographic use. We have previously described a series of substrate analog furin inhibitors containing decarboxylated arginine mimetics as the P1 residue. The most potent inhibitor of this series, Phenylacetyl-Arg-Val-Arg-4-amidinobenzylamide [1, Phac-Arg-Val-Arg-4-Amba (Becker et al., 2010)], inhibits furin with a Ki-value of 0.81 nM (Table 1). Based on this inhibitor structure, we designed various biotinylated analogs for use as ligands in affinity purification of furin. In general, such a ligand should have an intermediate affinity for its binding partner (Urh et al., 2009), to enable both sufficient specific binding to allow impurity washout and also permit elution of the target protein under gentle conditions. Based on the X-ray structure of mouse furin inhibited by decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone (Henrich et al., 2003), it is clear that the decanoyl moiety is directed towards the solvent; therefore, we assumed that it would be possible to attach a biotin moiety via an appropriate spacer at the N-terminus of the inhibitor. An initial analog was designed based on reference inhibitor 2 lacking the P4 arginine residue, which is important in dictating furin affinity, and therefore has a 3000-fold reduced Ki-value compared with 1. After coupling the N-terminal biotin moiety via a flexible 8-(amino)-3,6-dioxa-octanoyl (Adoa) spacer, compound 3 was obtained, which has a Ki-value in the lower μM range similar to compound 2.
Table 1.
Analytical data and inhibition constants of synthesized furin inhibitors.
| No. | Structure | HPLC (min) | MS calc./found (M + H) + | Ki (nM)b |
|---|---|---|---|---|
| 1 | Phac-Arg-Val-Arg-4-Amba | 24.40 | 678.4/679.4 | 0.81 |
| 2 | Acetyl-Val-Arg-4-Amba | 18.58 | 446.3/447.3 | 2390 |
| 3 | Biotin-Adoa-Val-Arg-4-Amba | 24.62 | 775.4/776.5 | 4200 |
| 4 | Phac-Arg-Pro-Arg-4-Amba | 22.77 | 676.4/339.2a | 36.7 |
| 5 | Biotin-Adoa-Arg-Pro-Arg-4-Amba | 21.36 | 929.5/930.6 | 38.3 |
| 6 | Biotin-(Adoa)2-Arg-Pro-Arg-4-Amba | 22.82 | 1074.6/1075.5 | 57.4 |
| 7 | Biotin-(Adoa)3-Arg-Pro-Arg-4-Amba | 24.07 | 1219.7/611.1a | 15.1 |
| 8 | Phac-Arg-Thr-Arg-4-Amba | 21.40 | 680.4/341.3a | 6.0 |
| 9 | Biotin-(Adoa)2-Arg-Thr-Arg-4-Amba | 22.53 | 1078.6/540.5a | 9.0 |
(M+2H)2+/2.
Ki measurements were performed as described previously (Becker et al., 2010; Sielaff et al., 2011).
As the next step, we designed a slightly more potent inhibitor by incorporation of a P4 Arg and modification of the P3 residue. The replacement of the P3 Val residue by proline leads to inhibitor 4 that has an approximately 50-fold lower inhibitory potency compared with 1 (Becker et al., 2011), most probably due to the elimination of an important hydrogen bond between the P3 amino acid and the carbonyl oxygen of the furin residue Gly255. Its biotinylated analog 5 shows, as expected, an affinity for furin very similar to the original inhibitor compound 4. To test for the influence on the affinity purification of the length of the linker moiety between the two headgroups, we designed compound 6 (Ki-value of 57.4 nM; Figure 1), which harbors two Adoa spacers corresponding to an 18-atom long backbone. It exhibits a similar inhibitory potency as found for 4 or 5, and was the first derivative suitable for affinity purification (see below). We designed two additional compounds for this study: compound 7 harbors three Adoa spacers (corresponding to 27 backbone atoms) designed to test if a further increase in linker length is beneficial. With a Ki-value of 15.1 nM, compound 7 showed an inhibitory constant in a similar lower nM range as compounds 4, 5 and 6 (Ki: 36.7, 38.3 and 57.4 nM, respectively). In a second attempt, the P3 proline of 4 was substituted with threonine to generate compound 8, which we predicted would result in tighter binding by enabling the formation of an antiparallel β-sheet with Gly255 and by reducing the conformational strain imposed by proline on the peptide backbone. Indeed, this exchange reduced the inhibitory constant ~6-fold, a value basically retained in its analog compound 9, which contains two Adoa spacers and the biotin moiety.
Figure 1.
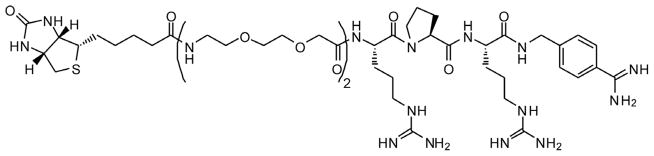
Chemical structure of Biotin-(Adoa)2-Arg-Pro-Arg-4-Amba (inhibitor 6) for final use in the affinity-based purification.
Preparation of the affinity matrix
We initially tested binding of the biotinylated compounds 3, 5, 6, 7 and 9 to the streptavidin-Sepharose column and selected the best-suited biotinylated inhibitor for affinity chromatography. All compounds bound tightly to the streptavidin-Sepharose beads, exceeding the minimum stoichiometries given by the manufacturer. This was analyzed either by SDS-PAGE-based qualitative binding tests employing streptavidin-Sepharose beads or by quantitative binding assays based on the UV-absorption of the 4-amidinobenzylamide moiety at the P1 position of the inhibitors using streptavidin-Sepharose columns, or both (data not shown). Binding of furin to the different affinity beads was estimated from the relative band intensity in Coomassie-stained SDS-PAGE analyses, comparing the furin concentrations in the applied solution, the non-bound protein (supernatant) and the protein eluting from the beads in buffers containing 150, 250, 500 and 1000 mM NaCl as well as the protein fraction that remained bound to the beads after the high-salt wash (pellet fraction). The data are summarized in Table 2 and typical gel images are shown in Figure 2 for the negative control (i.e., binding of furin to unmodified streptavidin-Sepharose beads) and for the binding test of furin to compound 6 immobilized on the beads. In addition to the clear binding of furin to the affinity beads loaded with inhibitor 6, we observed a dilution of furin upon mixing with the bead suspension. In addition, a slight reduction in signal intensity was observed with each wash/ elution step due to some loss of the affinity beads upon repeated washing. This was evident from the apparently lower concentration of furin in the supernatant in comparison to the applied protein solution for all samples.
Table 2.
Binding of furin to affinity tags immobilized on streptavidin-Sepharose.
| No. | Protein applied | Supernatant | Elution with (mM) NaCl
|
Pellet | |||
|---|---|---|---|---|---|---|---|
| 150 | 250 | 500 | 1000 | ||||
| 0a | +++ b | ++ | − | − | − | − | − |
| 3 | +++ | ++ | − | − | − | − | − |
| 5 | +++ | ++ | − | − | − | − | − |
| 6 | +++ | + | + | + | ++ | ++ | − |
| 7 | +++ | + | − | + | ++ | ++ | − |
| 9 | +++ | + | − | + | ++ | ++ | − |
Negative control, i.e., plain streptavidin-Sepharose beads.
The band intensity of the SDS-PAGE analysis observed in three independent experiments is given from ‘−’(no band) to ‘+++’ (strong band).
Figure 2. Qualitative binding assay of furin to different affinity tags: SDS-PAGE of inhibitor 6 and negative control.

(A) Negative control. Strong furin bands are visible for the applied material (A) and the supernatant (S) showing protein that does not bind to the beads. No protein can be observed in the fractions of the elution steps with 150–1000 mM NaCl, indicated by 150, 250, 500 and 1000, and in the pellet (P). (B) Qualitative binding assay of inhibitor 6. Furin is seen in all fractions except for the pellet; the strongest bands are visible for the applied material (A) and the elution steps at 500 mM and 1 M NaCl. ‘M’ indicates the molecular weight marker; the respective weights are given on the left-hand side in kDa. Each lane corresponds to 40 μl of the respective fractions.
Using unmodified streptavidin-Sepharose beads (negative control), no protein could be detected in any of the elution steps nor in the final analysis of the beads by Coomassie-stained SDS-PAGE. Basically, all furin applied to the beads is recovered in the supernatant and the subsequent wash steps; the observed reduction in concentration is well explained by the dilution effect. Combined with the results presented above, this shows that the binding of mouse furin is primarily caused by the specific interaction with the inhibitor coupled to the column material. The first two biotinylated compounds 3 and 5 show very similar behavior. Although both compounds bind well to streptavidin-Sepharose, the resulting affinity beads are not able to significantly retain furin. The stronger binding inhibitor 5 showed very weak retention of furin in an additional column-based experiment using completely desalted buffers. However, furin eluted at only 20 mM NaCl, preventing any practical use of compound 5 in the purification of furin (data not shown). We reasoned that the weak binding of compound 5 to furin is a non-specific effect most probably due to electrostatic interaction between this positively charged compound and the negatively charged active site cleft of furin. However, the linker between the two headgroups of compounds 3 and 5 seems to be too short to permit strong, concomitant binding of the affinity ligand to both furin and the matrix-bound streptavidin.
Compound 6 immobilized at the streptavidin-Sepharose, distinguished from compound 5 by one additional Adoa spacer residue only, shows significant binding to furin. The concentration of furin in the supernatant fraction is decreased and elution of furin from the beads is observed over a large range of salt concentrations, with peak furin elution at the 500 mM NaCl step. Application of this affinity matrix in a quantitative, column chromatography-based purification scheme (see below) showed that its binding to furin falls exactly in the correct affinity range to provide both sufficiently tight binding for impurity washout and also to enable gentle release of the purified target protein from the matrix. The elution of furin from the compound 6 affinity matrix at intermediate salt concentrations shows that the interaction between the affinity tag and furin is dominated by electrostatic interactions. Given the many hydrogen bond interactions observed between the covalently bound decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone inhibitor and furin in the complex crystal structure (Henrich et al., 2003), we next asked whether compound 6 is possibly still conformationally restrained by the (Adoa)2 spacer or by the proline residue at position P3, and hence hampered in the formation of all possible contacts with the protein surface. Correspondingly, we tested compounds 7 and 9, which showed, similar to compound 6, a significant decrease in the furin concentration of the supernatant, illustrating tight binding of the protein to the affinity matrix. In contrast to compound 6, both compounds 7 and 9 release furin only at more elevated NaCl concentrations from the affinity matrix, and no furin is found in the 150-mM NaCl elution. These data show that releasing remaining conformational strain in compound 6 results in tighter binding of the resulting affinity tags. However, this tighter binding of both compounds to the target protein and the respective elution only at high salt concentrations might render the elution step less efficient in comparison to compound 6. All further experiments were therefore performed with a streptavidin-Sepharose column carrying immobilized compound 6.
Furin purification using immobilized Biotin-(Adoa)2-Arg-Pro-Arg-4-Amba (6)
The dihydrofolate reductase-coupled amplification method to overexpress truncated furin secreted at high concentration into the conditioned medium (Cameron et al., 2000) was successfully employed to produce mg amounts of active furin. An initial concentration step significantly reduced the sample volume. Hence, this step rendered storage and handling of the expression medium much easier and accelerated the entire purification. As shown in Table 3, a highly efficient, three-step chromatographic purification scheme was developed. The initial ion-exchange chromatographic step (Figure 3A) proved valuable for removal of several low-molecular weight constituents in the conditioned medium, such as phenol red, and already resulted in an approximate two-fold increase in specific activity by separating late-eluting proteins while retaining most of the furin activity. All fractions showing significant furin activity were pooled to maximize yield at this step. The second purification step employed compound 6 coupled to streptavidin-Sepharose by its biotin moiety, chosen based on its superior binding properties for affinity chromatography among the compounds tested in this study. The chromatogram (Figure 3B) and the SDS-PAGE of this purification step (Figure 4) show that most proteins did not bind to the column at all; only active furin was specifically retained at the affinity matrix, as evidenced by the barely detectable levels of furin activity in the flow-through (total activity: F1, 0.7 U; F2, 1.2 U; F3, 1.8 U) in comparison with the eluted furin (total activity of pooled fractions 1–10: 130±3 U, Table 3). Upon linearly increasing the ionic strength of the elution buffer, the peak fraction of the highly pure and active furin eluted at ~400 mM NaCl. As expected from an efficient affinity chromatography, the relative activity (green bars in Figure 3B) correlates well with the furin concentration (band intensity in Figure 4). This purification step increased the specific activity almost five-fold in a single step, while retaining ~75% of the activity applied to the affinity column. A final gel filtration step (Figure 3C) was used to polish the purified material by removing possible aggregates and sodium chloride, further increasing its specific activity slightly. This column run also shows the homogeneity of the furin preparation. Owing to the specific isolation of active furin in the previous step, not much gain in purity can be expected here. Correspondingly, gel filtration might not be needed for most applications. As even smallest amounts of aggregates can be crucial for the crystallization of proteins, it should be employed for such use. All together, this three-step purification procedure is highly efficient resulting in a final specific activity of almost 60 units per milligram protein and an overall yield of ~50% of the initial furin activity. The efficiency of the purification procedure can further be visualized by the SDS-PAGE analysis of conditioned medium applied to the first column and of the pooled fractions from each purification step (Figure 5).
Table 3.
Purification of recombinant furin by affinity chromatography.
| Purification step | Total activity unitsa | Total protein mg | Specific activity units/mga | Yield % | Purification factor |
|---|---|---|---|---|---|
| Mediumb | 193 ±5 | 42 ±1 | 4.6 ±0.2 | 100 | 1 |
| DEAE | 176 ±4 | 16 ±0.5 | 11 ±0.4 | 91 | 2.4 |
| Affinity | 130 ±3 | 2.6 ±0.1 | 50 ±3 | 67 | 11 |
| Gel filtration | 97 ±9 | 1.7 ±0.1 | 57 ±7 | 50 | 12 |
1 unit = 1 μmol of AMC/h.
Approximately 40 ml 10-fold concentrated conditioned medium was used for the purification.
Figure 3. Chromatographic purification of mouse furin.
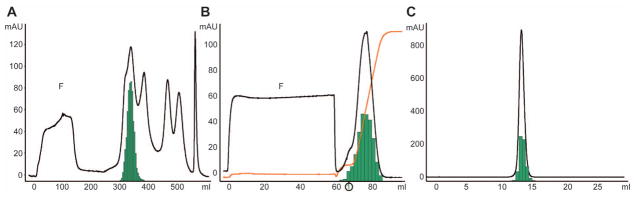
The absorbance at 280 nm (black line, left ordinate) and the relative furin activity of selected fractions (green bars) is shown as function of total volume of the chromatography and the sample application step (F) is indicated in panels (A) and (B). (A) DEAE-chromatography; (B) affinity chromatography on Biotin-(Adoa)2-Arg-Pro-Arg-4-Amba coupled to streptavidin-Sepharose showing in addition the applied salt gradient in orange; and (C): size exclusion chromatography. In panel (B), the first fraction of the protein elution applied to the SDS-PAGE (Figure 4) is indicated with (1).
Figure 4. SDS-PAGE of affinity chromatography.
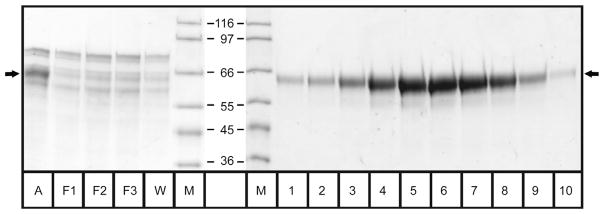
Selected fractions of the affinity chromatography step were analyzed by SDS-PAGE. ‘A’ indicates the applied material, ‘F1’–‘F3’ the fractions of the flow-through, ‘W’ the washing step, ‘M’ the molecular mass markers (the respective weights are given in kDa) and lanes 1–10 show the peak elution fractions. The arrows on both sides mark the furin band.
Figure 5. Purification overview.
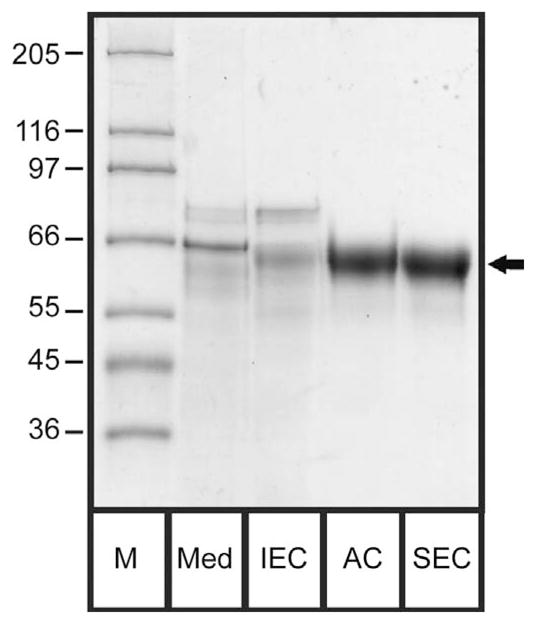
The conditioned medium and pool fraction of the different purification steps were analyzed by SDS-PAGE. The lanes from left to right show the molecular weight marker (M; the respective weights are given on the left-hand side in kDa), the conditioned medium (Med), the ion exchange chromatography pool (IEC), the affinity chromatography pool (AC) and the size exclusion chromatography pool (SEC), containing 2.5 μg total protein each. The arrow marks the furin band.
Discussion
Obtaining milligram amounts of highly pure and homogeneous protein is important for many analytic applications, especially for the development of novel inhibitors of medically interesting target proteins by rational, structure-based methods. Such studies typically rely on the protein crystallographic study of enzyme-inhibitor complexes. Based on an established method to efficiently overexpress the proprotein convertase furin from CHO cell conditioned medium and to purify it primarily based on ion-exchange chromatography (Cameron et al., 2000; Kacprzak et al., 2004), we have developed a novel purification scheme using affinity chromatography as a central purification step. The affinity purification step relies on efficient binding of the protein to a competitive, furin-specific inhibitor.
The development of the affinity matrix utilized our knowledge of the binding mode, chemical synthesis and affinity of several peptidic inhibitors of furin (Henrich et al., 2003; Becker et al., 2010, 2011). We reasoned that we could immobilize inhibitors by attaching them via a biotin moiety at their N-termini to a streptavidin-Sepharose matrix, while being able to fine-tune their affinity towards furin both by modifying the amino acid sequence binding into the active site cleft of the enzyme and by varying the spacer length connecting the biotin moiety and the inhibitory headgroup. As expected, the attachment of the flexible Adoa spacer and of the biotin moiety did not significantly reduce the affinity of the ligands towards furin, as evidenced by the similar Ki-values of the unmodified inhibitors and their biotinylated counterparts. Also, all compounds carrying the biotin moiety did bind well to the matrix, a prerequisite for performing affinity purification.
As the next step, we varied both the length of the Adoa spacer and the basic affinity of the inhibitory portion of the compounds, to select the compound best suited for affinity purification. Interestingly and unexpectedly, the compounds 3 and 5 carrying only one Adoa group (corresponding to 9 backbone atoms) between their biotin moieties and the inhibitory headgroups did not retain furin on the corresponding affinity beads, although the soluble compounds (i.e., not bound to the streptavidin-Sepharose matrix) did bind well to furin as evidenced by their Ki-values. Consulting the respective structural data (Weber et al., 1989; Henrich et al., 2003), we concluded that both the biotin moiety attached to the N-terminus of the inhibitor molecules, which binds to streptavidin, as well as the peptide segment at the C-terminus, which binds to furin, must interact with rather deep pockets. Correspondingly, one Adoa spacer is probably not sufficiently long to permit concomitant binding of both headgroups to their respective interaction partners. Indeed, the incorporation of two Adoa residues in between the two headgroups (corresponding to 18 backbone atoms) resulted in compound 6, which shows the desired binding properties to both the matrix-coupled streptavidin and to furin. Both a further increase in spacer length (compound 7) and the formation of an additional hydrogen bond to furin (compound 9) resulted in tighter binding of the respective affinity beads to furin. This result shows that even the (Adoa)2 spacer in compound 6 prohibits optimal binding of the affinity resin to furin. Hence, it prevents the formation of all possible interactions between the C-terminal peptide region and the deep active site cleft of furin (Henrich et al., 2003). Further increasing the affinity of the inhibitory segment towards furin (compound 9) also increases binding, as expected. The interaction with the negatively charged active site cleft of furin must be predominantly electrostatic for all binding compounds, as furin can be washed off of the beads at elevated salt concentrations, even for the tighter binding compounds 7 and 9. Based on the rather tight binding of the latter two affinity tags to furin and the resulting potential difficulties in eluting furin from the respective affinity columns, we concluded that compound 6 was the best-suited inhibitor for application in the affinity-based chromatographic purification of furin.
Using compound 6 as affinity tag, we developed a novel three-step furin purification scheme employing (i) an initial ion-exchange chromatographic step to collect most of the active furin present in the expression medium while removing non-protein and several protein impurities; (ii) the highly efficient affinity purification step that retains only active furin at high specificity and removes all other proteins, including those of similar size and electrostatic surface properties; and (iii) a final polishing step by gel filtration chromatography. This procedure proved to be superior in both yield and purification capacity compared to existing methodologies (Cameron et al., 2000), resulting in a highly homogeneous furin preparation with an apparent molecular weight of ~60 kDa in SDS-PAGE of unprecedented specific activity. In addition to providing extremely pure protein, this purification strategy is fast and delivers high yield. It should hence be the preferred future method for the preparation of large quantities of highly active furin for use in protein crystallization and for inhibitor studies. Although experimentally tested only for the mouse enzyme, we expect that this purification procedure employing compound 6 as an affinity tag will be equally efficient for the human enzyme, which is expected to have an identical active site cleft. In addition, Sepharose-bound compound 6 or possibly minor variations thereof should also permit the affinity-based purification of other basic PCs, as they all share a highly homologous structure of the active site cleft (Henrich et al., 2005). It remains to be evaluated if this purification procedure, which relies on exposure of active sites, can also quantitatively separate the multiple ionic forms of PC1/3 observed during ion exchange separation (Hoshino et al., 2011), thereby contributing to a better understanding of these various species on a molecular level.
In addition, the purification of SKI-1, one of the two PCs cleaving after nonbasic sites, could also greatly benefit from this general procedure. The expression of active SKI-1 in an insect cell culture system has been established (Bodvard et al., 2007). The affinity purification scheme developed here is based on binding only catalytically competent protease and hence represents a purification principle that is orthogonal to the current His-tag purification strategy. It should result in a significant improvement of the existing purification scheme for SKI-1, either by replacing or by complementing it. A potential inhibitor for this purpose might base on the sequences RRLL or RSLK successfully employed in substrates of SKI-1 (Bodvard et al., 2007). We expect the need for an experimental optimization of the exact inhibitory sequence and spacer length possibly similar to the work described here.
Materials and methods
Recombinant furin expression in CHO cells
Five liters of conditioned OptiMem medium (Gibco, Gaithersburg, MD, USA; containing 0.1 mg/ml bovine aprotinin) were obtained by daily collection of 100 ml from each of 10 confluent roller bottles of mouse furin-expressing CHO cells (Cameron et al., 2000). The conditioned medium was concentrated 10-fold using a Vivaflow 200 tangential flow apparatus (PES, 30 kDa cut-off value; Sartorius Stedim Biotech GmbH, Goettingen, Germany) at 4°C and was stored frozen prior to use for affinity chromatography.
Chemical synthesis of affinity tags
All inhibitors were synthesized by a combination of solid phase and solution synthesis as described previously (Becker et al., 2010). Briefly, Fmoc-Arg(Pbf)-OH [1 equivalent based on resin loading in the presence of four equivalents of diisopropylethylamine (DIPEA) in dry dichloromethane (DCM), 2 h at room temperature, followed by washing with DCM/MeOH/DIPEA (17/2/1, v/v/v, 3×1 min) and several times with DCM, dimethylformamide (DMF), DCM] was loaded on 2-chlorotritylchloride resin. The following standard amino acids were coupled by manual solid phase peptide synthesis in polypropylene syringes (Intavis, Köln, Germany) using a Fmoc protocol with a four-fold excess of Fmoc-amino acid, hydroxybenzotriazole (HOBt) and o-benzotriazolyl-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), respectively, and eight equivalents of DIPEA in DMF. Fmoc-Adoa-OH (Bachem, Weil am Rhein, Germany) or Fmoc-(Adoa)2-OH (Iris Biotech GmbH, Marktredwitz, Germany) were coupled with only a 2.5-fold excess. The N-terminal biotinylation was performed with a five-fold excess of biotin, HOBt and HBTU, respectively, in the presence of 10 equivalents of DIPEA in dimethyl sulfoxide (DMSO)/DMF (1:1) overnight. The side chain-protected peptide was cleaved from resin by treatment with 1% trifluoroacetic acid (TFA) in DCM (2×30 min) at room temperature. After evaporation of the solvent the peptides were coupled to 4-amidinobenzylamine×2 HCl (Becker et al., 2010) using (benzotriazolyl)-N-oxy-pyrrolidinium phosphonium hexafluorophosphate/DIPEA in the presence of 6-Cl-HOBT (3 h in DMF), followed by evaporation of the solvent and side chain deprotection with TFA/triisopropylsilane/H2O (95/2.5/2.5, v/v/v, stirring for 4 h at room temperature). All products were purified by preparative HPLC (purity >95% based on detection at 220 nm), lyophilized from 80% tert-BuOH in water, and finally obtained as TFA salts.
Analytical HPLC experiments were performed on a Shimadzu LC-10A system (column: Nucleodur C18, 5 μm, 100 Å, 4.6 ×250 mm; Machery-Nagel, Düren, Germany) with a linear gradient of acetonitrile (1–60% in 59 min, detection at 220 nm) containing 0.1% TFA at a flow rate of 1 ml/min. The final inhibitors were purified to more than 95% purity (detection at 220 nm) by preparative HPLC (pumps: Varian PrepStar Model 218 gradient system, detector: ProStar Model 320, fraction collector; Varian Model 701; Varian, Darmstadt, Germany) using a C8 column (Nucleodur, 5 μm, 100 Å, 32×250 mm; Macherey-Nagel, Düren, Germany) and a linear gradient of acetonitrile containing 0.1% TFA at a flow rate of 20 ml/ min. All inhibitors were finally obtained as TFA salts after lyophilization. The molecular masses of the synthesized compounds were determined using a QTrap 2000 ESI spectrometer (Applied Biosystems, Darmstadt, Germany).
Reagents for synthesis: Fmoc-amino acids, coupling reagents and reagents for synthesis were obtained from Orpegen, Bachem, Iris, Fluka, Aldrich or Acros.
Binding tests of the affinity beads to furin
To prepare the affinity beads, 50 μl of streptavidin-Sepharose beads (GE Healthcare, Uppsala, Sweden) were washed three times with 0.9 ml cold buffer A [20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2 mM CaCl2, pH 7.4] supplemented with 150 mM NaCl and incubated with 0.75 ml 0.2 mM biotinylated inhibitor (Table 2) in buffer A supplemented with 150 mM NaCl for 30 min at 4°C. A negative control was prepared using 0.75 ml of the same buffer without inhibitor. The supernatant including unbound inhibitor was carefully removed and three washing steps of 0.9 ml each were performed to ensure that no unbound inhibitor remained in the tube.
The affinity beads were incubated in 100 μl buffer A supplemented with 150 mM NaCl with 15 μg of pure furin for 30 min at 4°C. The supernatant including unbound furin was transferred into a fresh tube and prepared for SDS-PAGE analysis. The affinity beads were washed three times with buffer A to ensure the complete removal of unbound furin. Afterwards, four elution steps (5 min incubation time each) were performed with increasing amounts of NaCl (basically buffer A, supplemented with 150 mM, 250 mM, 500 mM or 1 M NaCl) and each time the supernatant was prepared for SDS-PAGE analysis. Following the last elution step, the beads were washed two more times with buffer containing 1 M NaCl to remove the last furin traces that eluted at high salt and the resulting bead pellet was again prepared for SDS-PAGE analysis. Every incubation of the bead suspension was finished by a centrifugation step (0.5 min at 1000 g) to sediment the beads prior to removal of the supernatant. All experiments were performed in triplicate.
Affinity column preparation
For this purpose, 15 ml 0.2 mM Biotin-(Adoa)2-Arg-Pro-Arg-4-amidinobenzylamide (4-Amba) in buffer A supplemented with 150 mM NaCl was applied to a 1-ml streptavidin-Sepharose column (GE Healthcare) with a flow rate of 0.1 ml/min. The column was washed with buffer A including up to 1 M NaCl and stored in 20% ethanol.
Affinity based purification of furin
Ten-times concentrated conditioned medium was thawed, diluted 1:3 with buffer A2 (buffer A complemented with 0.4 mM DDM) and pumped on a 50-ml diethylaminoethyl (DEAE) anion exchange column (GE Healthcare) at a flow rate of 1 ml/min. Following a wash with 100 ml of buffer A2, a linear gradient of 0–250 mM NaCl (over 4.5 column volumes; 4 ml fractions) was used to elute furin and the column was washed with 100 ml of each 50% buffer B2 and 100% buffer B2 (20 mM HEPES, 500 mM NaCl, 2 mM CaCl2, 0.4 mM DDM, pH 7.4). Fractions containing significant activity were pooled and applied to the 1-ml affinity chromatography column (see above) at a flow rate of 0.5 ml/min. Following a 10-ml wash with buffer A supplemented with 150 mM NaCl, a linear gradient (15 column volumes) up to 700 mM NaCl was used for the elution of furin. Fractions containing activity were pooled, concentrated by ultrafiltration (Amicon Ultra 30 K centrifugation devices; Millipore, Carrigtwohill, Ireland) and applied to a Superdex 200 10/300 size exclusion chromatography column (GE Healthcare) equilibrated in 10 mM HEPES, 150 mM NaCl, 2 mM CaCl2, pH 7.4 at a flow rate of 0.5 ml/min. Fractions of 0.5 ml were collected and screened for activity. All purification steps were carried out at 4°C.
Enzyme assays
Fractions eluting from the different columns were screened in a kinetic assay for relative furin activity in 96-well plates at 37°C in a fluorometer (POLARstar Galaxy, BMG Labtech, Offenburg, Germany) using 380 nm as the excitation and 460 nm as the emission wavelength. The activity assay was basically performed as described previously (Cameron et al., 2000) in 100 mM HEPES, 5 mM CaCl2, 0.1% Brij 35, pH 7.0 using 200 μM Pyr-Arg-Thr-Lys-Arg-7-amino-4-methylcoumarin (AMC) (Bachem, Bubendorf, Switzerland) as substrate. In addition, the activity of each pool was determined in a static assay, giving absolute activity data: 100 μl reactions were incubated at 37°C for 5 min and the reaction was stopped by the addition of 500 μl 5 mM ethylenediaminetetraacetic acid (EDTA). The quantitative furin activity was determined using a FP-6500 spectrofluorometer (Jasco, Gross-Umstadt, Germany) and AMC (Fluka, Buchs, Switzerland) was used as a standard. The amount of protein was determined according to the Bradford method. The mean values and the standard deviations are given from three independent determinations of the protein and AMC concentrations of a representative purification. To also estimate the absolute activity in the flow-through, the respective fractions (F1–F3) were measured once.
Acknowledgments
This research was supported by a National Institutes of Health grant DA05084 to I.L. and a Deutsche Forschungsgemeinschaft grant TH862/1-4 to M.E.T.
References
- Angliker H. Synthesis of tight binding inhibitors and their action on the proprotein-processing enzyme furin. J Med Chem. 1995;38:4014–4018. doi: 10.1021/jm00020a016. [DOI] [PubMed] [Google Scholar]
- Basak A. Inhibitors of proprotein convertases. J Mol Med. 2005;83:844–855. doi: 10.1007/s00109-005-0710-0. [DOI] [PubMed] [Google Scholar]
- Bassi DE, Fu J, Lopez de Cicco R, Klein-Szanto AJ. Proprotein convertases: “master switches” in the regulation of tumor growth and progression. Mol Carcinog. 2005;44:151–161. doi: 10.1002/mc.20134. [DOI] [PubMed] [Google Scholar]
- Becker GL, Hardes K, Steinmetzer T. New substrate analogue furin inhibitors derived from 4-amidinobenzylamide. Bioorg Med Chem Lett. 2011;21:4695–4697. doi: 10.1016/j.bmcl.2011.06.091. [DOI] [PubMed] [Google Scholar]
- Becker GL, Sielaff F, Than ME, Lindberg I, Routhier S, Day R, Lu Y, Garten W, Steinmetzer T. Potent inhibitors of furin and furin-like proprotein convertases containing decarboxylated P1 arginine mimetics. J Med Chem. 2010;53:1067–1075. doi: 10.1021/jm9012455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodvard K, Mohlin J, Knecht W. Recombinant expression, purification, and kinetic and inhibitor characterisation of human site-1-protease. Protein Expr Purif. 2007;51:308–319. doi: 10.1016/j.pep.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Bontemps Y, Scamuffa N, Calvo F, Khatib AM. Potential opportunity in the development of new therapeutic agents based on endogenous and exogenous inhibitors of the proprotein convertases. Med Res Rev. 2007;27:631–648. doi: 10.1002/med.20072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron A, Appel J, Houghten RA, Lindberg I. Polyarginines are potent furin inhibitors. J Biol Chem. 2000;275:36741–36749. doi: 10.1074/jbc.M003848200. [DOI] [PubMed] [Google Scholar]
- Creemers JW, Siezen RJ, Roebroek AJ, Ayoubi TA, Huylebroeck D, Van de Ven WJ. Modulation of furin-mediated proprotein processing activity by site-directed mutagenesis. J Biol Chem. 1993;268:21826–21834. [PubMed] [Google Scholar]
- Feliciangeli SF, Thomas L, Scott GK, Subbian E, Hung CH, Molloy SS, Jean F, Shinde U, Thomas G. Identification of a pH sensor in the furin propeptide that regulates enzyme activation. J Biol Chem. 2006;281:16108–16116. doi: 10.1074/jbc.M600760200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugere M, Day R. Cutting back on pro-protein convertases: the latest approaches to pharmacological inhibition. Trends Pharmacol Sci. 2005;26:294–301. doi: 10.1016/j.tips.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature. 1992;360:358–361. doi: 10.1038/360358a0. [DOI] [PubMed] [Google Scholar]
- Hatsuzawa K, Murakami K, Nakayama K. Molecular and enzymatic properties of furin, a Kex2-like endoprotease involved in precursor cleavage at Arg-X-Lys/Arg-Arg sites. J Biochem (Tokyo) 1992;111:296–301. doi: 10.1093/oxfordjournals.jbchem.a123753. [DOI] [PubMed] [Google Scholar]
- Henrich S, Cameron A, Bourenkov GP, Kiefersauer R, Huber R, Lindberg I, Bode W, Than ME. The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat Struct Biol. 2003;10:520–526. doi: 10.1038/nsb941. [DOI] [PubMed] [Google Scholar]
- Henrich S, Lindberg I, Bode W, Than ME. Proprotein convertase models based on the crystal structures of furin and kexin: explanation of their specificity. J Mol Biol. 2005;345:211–227. doi: 10.1016/j.jmb.2004.10.050. [DOI] [PubMed] [Google Scholar]
- Holyoak T, Wilson MA, Fenn TD, Kettner CA, Petsko GA, Fuller RS, Ringe D. 2.4 A resolution crystal structure of the prototypical hormone-processing protease Kex2 in complex with an Ala-Lys-Arg boronic acid inhibitor. Biochemistry. 2003;42:6709–6718. doi: 10.1021/bi034434t. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Kowalska D, Jean F, Lazure C, Lindberg I. Modulation of PC1/3 activity by self-interaction and substrate binding. Endocrinology. 2011;152:1402–1411. doi: 10.1210/en.2010-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao GS, Cregar L, Wang J, Millis SZ, Tang C, O’Malley S, Johnson AT, Sareth S, Larson J, Thomas G. Synthetic small molecule furin inhibitors derived from 2,5-dideoxystreptamine. Proc Natl Acad Sci USA. 2006;103:19707–19712. doi: 10.1073/pnas.0606555104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacprzak MM, Peinado JR, Than ME, Appel J, Henrich S, Lipkind G, Houghten RA, Bode W, Lindberg I. Inhibition of furin by polyarginine-containing peptides: nanomolar inhibition by nona-D-arginine. J Biol Chem. 2004;279:36788–36794. doi: 10.1074/jbc.M400484200. [DOI] [PubMed] [Google Scholar]
- Komiyama T. Interaction of EGLIN C variants with the extended subsites of the precursor processing proteases. Protein Pept Lett. 2005;12:415–420. doi: 10.2174/0929866054395220. [DOI] [PubMed] [Google Scholar]
- Komiyama T, Coppola JM, Larsen MJ, van Dort ME, Ross BD, Day R, Rehemtulla A, Fuller RS. Inhibition of furin/proprotein convertase-catalyzed surface and intracellular processing by small molecules. J Biol Chem. 2009;284:15729–15738. doi: 10.1074/jbc.M901540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J. 1997;327:625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell NC, Thorner JW. The kindest cuts of all: crystal structures of Kex2 and furin reveal secrets of precursor processing. Trends Biochem Sci. 2004;29:80–87. doi: 10.1016/j.tibs.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Scamuffa N, Calvo F, Chretien M, Seidah NG, Khatib AM. Proprotein convertases: lessons from knockouts. FASEB J. 2006;20:1954–1963. doi: 10.1096/fj.05-5491rev. [DOI] [PubMed] [Google Scholar]
- Seidah NG, Chretien M. Eukaryotic protein processing: endoproteolysis of precursor proteins. Curr Opin Biotechnol. 1997;8:602–607. doi: 10.1016/s0958-1669(97)80036-5. [DOI] [PubMed] [Google Scholar]
- Seidah NG, Prat A. Precursor convertases in the secretory pathway, cytosol and extracellular milieu. Essays Biochem. 2002;38:79–94. doi: 10.1042/bse0380079. [DOI] [PubMed] [Google Scholar]
- Seidah NG, Prat A. The proprotein convertases are potential targets in the treatment of dyslipidemia. J Mol Med. 2007;85:685–696. doi: 10.1007/s00109-007-0172-7. [DOI] [PubMed] [Google Scholar]
- Seidah NG, Mayer G, Zaid A, Rousselet E, Nassoury N, Poirier S, Essalmani R, Prat A. The activation and physiological functions of the proprotein convertases. Int J Biochem Cell Biol. 2008;40:1111–1125. doi: 10.1016/j.biocel.2008.01.030. [DOI] [PubMed] [Google Scholar]
- Sielaff F, Than ME, Bevec D, Lindberg I, Steinmetzer T. New furin inhibitors based on weakly basic amidinohydrazones. Bioorg Med Chem Lett. 2011;21:836–840. doi: 10.1016/j.bmcl.2010.11.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2:31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- Than ME, Henrich S, Bourenkov GP, Bartunik HD, Huber R, Bode W. The endoproteinase furin contains two essential Ca2+ ions stabilizing its N-terminus and the unique S1 specificity pocket. Acta Crystallogr D Biol Crystallogr. 2005;61:505–512. doi: 10.1107/S0907444905002556. [DOI] [PubMed] [Google Scholar]
- Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urh M, Simpson D, Zhao K. Affinity chromatography: general methods. Methods Enzymol. 2009;463:417–438. doi: 10.1016/S0076-6879(09)63026-3. [DOI] [PubMed] [Google Scholar]
- von Eggelkraut-Gottanka R, Beck-Sickinger AG. Biosynthesis of peptide hormones derived from precursor sequences. Curr Med Chem. 2004;11:2651–2665. doi: 10.2174/0929867043364405. [DOI] [PubMed] [Google Scholar]
- Weber PC, Ohlendorf DH, Wendoloski JJ, Salemme FR. Structural origins of high-affinity biotin binding to streptavidin. Science. 1989;243:85–88. doi: 10.1126/science.2911722. [DOI] [PubMed] [Google Scholar]