

Abstract
Introduction
This investigator-initiated study explores the safety, maximum tolerated dose (MTD), clinical response, and pharmacokinetics (PK) of hydroxychloroquine (HCQ) with and without erlotinib in patients with advanced non-small cell lung cancer (NSCLC).
Methods
Patients with prior clinical benefit from an epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor were randomized to HCQ or HCQ plus erlotinib in a 3+3 dose escalation schema.
Results
Twenty-seven patients were treated, 8 with HCQ (arm A) and 19 with HCQ plus erlotinib (arm B). EGFR mutations were detected in 74% of patients and 85% had received ≥2 prior therapies. Arm A had no dose-limiting toxicities (DLTs), but the MTD was not reached as this arm closed early to increase overall study accrual. In arm B, 1 patient each experienced grade 3 rash, nail changes, skin changes, nausea, dehydration, and neutropenia, 1 had grade 4 anemia, and 1 developed fatal pneumonitis, all considered unrelated to HCQ. There were no DLTs, therefore the highest tested dose for HCQ with erlotinib 150mg was 1000mg daily. One patient had a partial response (PR) to erlotinib/HCQ, for an overall response rate of 5% (95% CI, 1–25). This patient had an EGFR mutation and remained on therapy for 20 months. Administration of HCQ did not alter the PK of erlotinib.
Conclusions
HCQ with or without erlotinib was safe and well-tolerated. The recommended phase 2 dose of HCQ was 1000mg when given in combination with erlotinib 150mg.
Introduction
Non-small cell lung cancer (NSCLC) is the most common cause of cancer mortality in the United States1 and chemotherapy is only modestly effective. A subset of NSCLCs harbor a mutation in the epidermal growth factor receptor gene (EGFR) that imparts an oncogene-addicted biology, conferring a distinct sensitivity to EGFR tyrosine kinase inhibitors (TKIs)2–5. Acquired resistance arises after approximately one year, so developing strategies to delay and treat resistance is of utmost clinical importance. 6,7,8
Hydroxychloroquine (HCQ) and chloroquine (CQ) are immunomodulating agents widely prescribed for their anti-malarial and anti-rheumatic effects. Additionally HCQ and CQ have been implicated as possible adjuncts to anti-cancer treatment. This has been explored most extensively in a prospective study of patients with glioblastoma multiforme in which adding CQ to standard treatment (surgery, radiation therapy, and chemotherapy) improved survival compared with standard treatment alone6.
We recently described a subpopulation of cells present within several “oncogene-addicted” tumor-derived cell lines that display a reversible drug-tolerant phenotype7 that can be spontaneously acquired and relinquished and allows them to survive within an otherwise drug-sensitive cancer. The drug-tolerant phenotype is associated with an altered chromatin structure, rendering them sensitive to histone deacetylase (HDAC) inhibitors7. Since HDAC inhibitors had been associated with poor efficacy and significant toxicity in clinical studies available at the time we conceived of this project8, we hypothesized that CQ and HCQ, which appear to similarly alter chromatin structure and are clinically well-tolerated9, could potentially be used to disrupt this reversible drug-tolerant state. Pre-clinical studies exploring this hypothesis are presented herein, and overall demonstrated that HCQ might have efficacy in EGFR-mutant NSCLC, either alone or in combination with erlotinib. Building from these results, we conducted a phase I trial investigating the safety and tolerability of HCQ with or without erlotinib in advanced NSCLC, with the long-term objective of studying the combination in EGFR-mutant NSCLC.
Materials and Methods
Laboratory Studies
We used the PC9 NSCLC model (exon 19 EGFR mutation) to test the ability of CQ and HCQ to selectively promote cell death in the drug-tolerant subpopulation in vitro. We generated PC9-derived clones with reversible drug-tolerance to gefitinib or erlotinib by continuous propagation in pharmacologically-relevant TKI concentrations, as previously described 7. The resultant clones exhibit distinct morphology, have ~400-fold reduced EGFR TKI sensitivity compared to parental lines, and can be propagated indefinitely in TKI. Parental and TKI-tolerant clones were treated with variable concentrations of CQ and HCQ and cell viability was assayed 7.
Clinical trial design and patients
To translate the laboratory findings to the clinic, we initiated a single-center, open-label, dose-escalation phase I trial of HCQ with or without erlotinib in advanced NSCLC patients who previously responded to an EGFR TKI. The primary outcome was safety and to determine the maximum tolerated dose (MTD) of HCQ. Secondary objectives included response and pharmacokinetics (PK). The study was approved and monitored by our institutional review board and all subjects provided written informed consent. Funding for this investigator-initiated study was provided by Genentech and the Dana-Farber/Harvard Cancer Center SPORE in Lung Cancer, NCI P50 CA090578 (J.S.), and the trial was listed on clinicaltrials.gov prior to patient enrollment (NCT #NCT01026844).
Eligible patients had advanced NSCLC (stage IIIB/pleural effusion or IV by AJCC 6th edition 10)with an Eastern Cooperative Oncology Group performance status of 0–211. Prior clinical benefit from EGFR TKI, defined as ≥12 weeks of erlotinib or gefitinib was required. There was no restriction on prior therapies and central nervous system (CNS) disease was allowed if definitively treated and clinically stable. All patients had measurable disease by RECIST 1.012, adequate end organ function, and an eye exam excluding retinal disease given the potential for HCQ-induced retinopathy13.
The study was initially designed as a two-armed randomized phase I trial in which each arm independently underwent HCQ dose escalation starting at 400mg daily and increasing by 200mg increments to a maximum planned dose of 1000mg. Patients were randomly assigned to HCQ monotherapy (arm A) or HCQ plus erlotinib at 150 mg daily (arm B). Arm B patients received a 1 week erlotinib run-in prior to HCQ initiation for PK purposes. Treatment was divided into continuous 28-day cycles and proceeded until disease progression or unacceptable toxicity. All subjects in a given dose cohort were observed for one complete cycle before accrual to the next dose level began.
Adverse events (AEs) were assessed using the National Cancer Institute Common Toxicity Criteria (CTC) version 3.014. Dose limiting toxicities (DLTs) were defined as ≥CTC grade 2 retinopathy or keratitis, or ≥CTC grade 3 hematologic, skin, CNS, neuropathic, cardiac, respiratory, gastrointestinal, or renal AEs in the first cycle considered at least possibly related to HCQ. If a DLT was observed, an additional 3 patients were enrolled at that dose level. The MTD for HCQ in each arm would be defined as one dose level below that at which ≥ 2 of 6 patients experienced a DLT, or if no DLTs were observed, the highest tested dose. Upon reaching the MTD or highest tested dose, an expansion cohort of up to 12 patients was planned in each arm.
In October 2008, after enrollment of 18 patients (8 on arm A and 10 on arm B), the study was amended to limit enrollment to arm B only (HCQ plus erlotinib) given increasing pre-clinical evidence supporting a role for combination therapy (but not HCQ monotherapy) and to help increase overall patient accrual.
Trial assessments and pharmacokinetic studies
Patients were assessed every 4 weeks through cycle 6, and every 8 weeks thereafter. Dilated eye exams occurred at baseline and every 3 months; radiographic imaging for efficacy was performed every 8 weeks and determination of best response to therapy was made using RECIST 1.012.
PK samples were obtained weekly during cycle 1 and monthly thereafter from all patients to monitor the steady-state minimum concentration (Cminss) of HCQ. For patients in arm B, sampling over two 24-hour intervals on day 7 of erlotinib monotherapy and again on day 7 of combination erlotinib and HCQ was performed to define the plasma concentration-time profile of erlotinib. HCQ Cminss was determined by liquid chromatography/mass spectrometry. Erlotinib PK was assessed by liquid chromatography-tandem mass spectrometry, as previously reported with minor modifications15,16. For additional details see Supplemental Digital Content 1.
Statistical considerations
All patients were included in all analyses. Safety data was analyzed using summary statistics. Response rate was calculated separately for each arm. Progression-free survival (PFS) was the time from enrollment until progression or death and overall survival (OS), from enrollment until death. Survival analyses were performed using the Kaplan-Meier method.
The Cminss of HCQ was calculated for each patient as the geometric mean of all acceptable determinations. Individual erlotinib plasma concentration-time curves were analyzed by standard noncompartmental methods using WinNonlin Professional version 5.0 software (Pharsight Corp., Cary, NC). Area under the plasma concentration-time curve(AUC24) for erlotinib was estimated using the log-linear trapezoidal algorithm. Apparent oral clearance (CL/F) was calculated as the dose divided by AUC24. The t-test (two-sided) was used to compare PK parameters after logarithmic transformation.
Results
Laboratory Studies
PC9-derived clones with reversible tolerance to gefitinib (gefitinib resistant, GR) or erlotinib (erlotinib resistant, ER) were made (Figure 1A). CQ at 10μM did not affect viability or growth of the parental PC9 cells, but was associated with dramatic reduction in cell viability in both GR and ER, with 70–98% reduced cell survival after 72 hours of CQ treatment (Figure 1B). HCQ at 10μM led to similar findings (Figure 1C). To examine whether HCQ could affect the initial emergence of the drug-tolerant clones, co-treatment of parental PC9 cells with erlotinib and HCQ was performed. We observed an approximate 90% reduction in the number of TKI-tolerant clones detected at 33 days, under conditions where HCQ monotherapy did not detectably affect growth or survival (Figure 1D).
Figure 1. Disruption of drug resistance by chloroquine or hydroxychloroquine.

(A) Photomicrographs illustrating the morphology of untreated PC9 NSCLC cells and three representative clones that had been selected for resistance either to gefitinib (GR3) or erlotinib (ER3 and ER4) by continuous culture in 1 micromolar (μM) drug for several weeks. (B). Viability of parental PC9 cells and several gefitinib-resistant (GR) or erlotinib-resistant (ER) PC9-derived clones following 72 hr treatment with 10 μM chloroquine (CQ), as measured using SYTO60 nuclear staining. (C) Comparison of the effects of CQ and hydroxychloroquine (HCQ) on the viability of representative tested resistant clones using the assay described in B. (D). Upper panels: Representative plates of cells stained with SYTO60 following drug treatment in the presence or absence of HCQ for the indicated number of days. Included are PC9 cells treated with erlotinib (ERL) or cisplatin (CISPLAT), the HER2-amplified breast cancer cell line SKBR3, treated with the HER2 kinase inhibitor lapatinib (LAPAT), and the BRAF mutant melanoma cell line M14 treated with the RAF kinase inhibitor AZ628. Lower panels: Graphs depicting the quantitative analysis of drug-resistant colonies detected, as averaged from three independent plates, with error bars representing the standard deviation.
We evaluated three additional treatment settings to address the potentially broader ability of HCQ to disrupt drug-tolerant clone formation. Co-administration of HCQ substantially suppressed formation of drug-tolerant clones in PC9 cells treated with cisplatin, HER2-amplified SKBR3 breast cancer cells treatedwith the HER2 TKI lapatinib, and BRAF-mutant melanoma cells treated with the BRAF TKI AZ628 (Figure 1D).
Patients
Twenty-seven patients were enrolled between August 2007 and May 2010; 8 to arm A, 12 to the dose-escalation portion of arm B, and 7 to the arm B MTD expansion. There were 14 women and 13 men with a median age of 66 years (range 27–76, Table 1). All tumors were adenocarcinoma and 20 (74%) were EGFR mutation-positive (5 patients were EGFR wild-type, 2 had unknown EGFR status). Patients were heavily pretreated with 10 (37%) having 2 prior treatments and 13 (48%) having ≥3.
Table 1.
Patient Characteristics
| Variable | HCQ alone (N=8) | HCQ + Erlotinib (N=19) |
|---|---|---|
| Age in years, median (range) | 61 (48–76) | 65 (27–73) |
| Sex, n (%) | ||
| Female | 5 (63) | 9 (47) |
| Male | 3 (38) | 10 (53) |
| Race, n (%) | ||
| White | 7 (88) | 16 (84) |
| Asian | 1 (13) | 2 (11) |
| Other | - | 1 (5) |
| Baseline PS, n (%) | ||
| 0 | 3 (38) | 9 (47) |
| 1 | 5 (63) | 8 (42) |
| 2 | - | 2 (11) |
| Smoking status, n (%) | ||
| Never | 5 (63) | 10 (53) |
| Former | 1 (13) | 9 (47) |
| Current | 2 (25) | - |
| Histology, n (%) | ||
| Adenocarcinoma | 8 (100) | 19 (100) |
| EGFR mutation status, n (%) | ||
| Mutant: Exon 19 del | 4 (50) | 9 (47) |
| Mutant: L858R | 1 (13) | 2 (11) |
| Mutant: other | 1 (13) | 3 (16) |
| Wild-type | 2 (25) | 3 (16) |
| Unknown | - | 2 (11) |
| Number of prior regimens, n (%) | ||
| 1 | 1 (13) | 3 (16) |
| 2 | 4 (50) | 6 (32) |
| ≥3 | 3 (38) | 10 (53) |
| Last prior therapy, n (%) | ||
| EGFR TKI | 6 (75) | 10 (53) |
| Other | 3 (38) | 9 (47) |
Safety and Toxicity
Treatment was generally well-tolerated, with most AEs categorized as grades 1 and 2 (Table 2). The most commonly observed treatment-related AEs were rash (37%), nausea (33%), diarrhea (33%), and fatigue (30%). Vomiting, dyspepsia, anorexia, and dry skin occurred in < 20% of patients. Grade 3 or greater AEs were uncommon and were all considered unrelated to HCQ. One patient each experienced grade 3 rash, nail changes, skin changes, nausea, dehydration, and neutropenia; 1 developed grade 4 anemia; and 1 had fatal pneumonitis about 1 month into erlotinib plus HCQ 400 mg daily (note this patient had previously received 10 months of gefitinib and 2 months of dacomitinib without pneumonitis). Grade 3 or greater toxicity did not appear to be dose-dependent (4 at HCQ 400mg, 1 at HCQ 600mg and 3 at HCQ 1000mg, all with concurrent erlotinib). Overall, the majority of treatment-related AEs were observed in arm B.
Table 2.
Treatment-related toxicities that occurred in at least 10% of subjects or were serious
| Event | HCQ Alone, n=8 | HCQ + Erlotinib, n =19 | Total, N=27 | |||||
|---|---|---|---|---|---|---|---|---|
| Gr 1 | Gr 2 | Gr 1 | Gr 2 | Gr 3 | Gr 4 | Gr 5 | All Grades | |
| n (%) | n (%) | n (%) | ||||||
| Rash | - | - | 8 (42) | 1 (5) | 1 (5) | - | - | 10 (37) |
| Diarrhea | - | - | 5 (26) | 4 (21) | - | - | - | 9 (33) |
| Nausea | 2 (25) | 1 (13) | 2 (11) | 3 (16) | 1 (5) | - | - | 9 (33) |
| Fatigue | 1 (13) | - | 4 (21) | 3 (16) | - | - | - | 8 (30) |
| Vomiting | 3 (38) | - | 2 (11) | - | - | - | - | 5 (19) |
| Dyspepsia | - | - | 4 (21) | 1 (5) | - | - | - | 5 (19) |
| Anorexia | - | - | 3 (16) | 2 (11) | - | - | - | 5 (19) |
| Dry skin | - | - | 3 (16) | 2 (11) | - | - | - | 5 (19) |
| Nail changes | - | - | 1 (5) | 1 (5) | 1 (5) | - | - | 3 (11) |
| Skin, other | - | - | - | 2 (11) | 1 (5) | - | - | 3 (11) |
| Ocular, other | - | - | 2 (11) | 1 (5) | - | - | - | 3 (11) |
| Anemia | - | 1 (13) | - | 1 (5) | - | 1 (5) | - | 3 (11) |
| Neutropenia | - | - | - | - | 1 (5) | - | - | 1 (4) |
| Pneumonitis | - | - | - | - | - | - | 1 (5) | 1 (4) |
| Dehydration | - | - | - | - | 1 (5) | - | - | 1 (4) |
No DLTs were observed in either arm. Therefore, the MTD for HCQ when given in combination with erlotinib 150mg daily is 1000mg, which was the highest dose tested. The MTD for HCQ alone could not be determined since enrollment to that arm was closed early.
Efficacy
Twenty-three patients (7 on arm A, 16 on arm B) underwent radiographic evaluation (Figure 2). One patient electively withdrew from the study in cycle 1 and 3 patients progressed clinically prior to restaging. All patients on arm A had progressive disease as their best response to therapy (RR 0%; 95% CI, 0–34). Within arm B, 1 patient out of 19 had a partial response (PR) and 4 had confirmed stable disease (SD), for an overall response rate of 5% (95% CI, 1–25) and a disease control rate (PR + SD) of 26% (95% CI, 12–49). The patient who had a PR received HCQ 600mg plus erlotinib and remained on therapy for 20 months. Her tumor had an exon 19 deletion EGFR mutation and there was an interval of 2 years since prior EGFR TKI. All 4 patients who attained SD remained on therapy for ≥16 weeks, including one EGFR-mutant, 2 EGFR wild-type, and 1 EGFR unknown. All 4 had ≥3 prior regimens; only one had received erlotinib as the immediate prior regimen, and she achieved SD for 16 weeks with HCQ 400mg plus erlotinib.
Figure 2. Individual response to therapy.
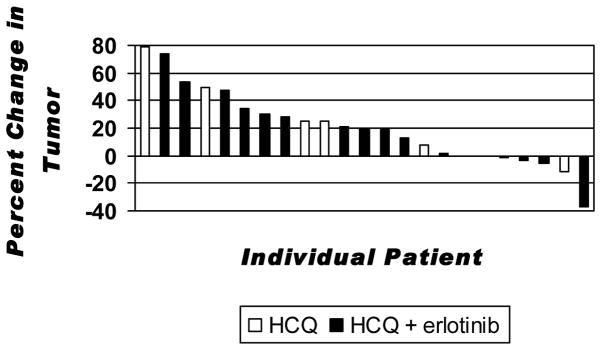
Bars indicate the individual patient best response to treatment expressed as a percent change of tumor burden compared to baseline for HCQ alone (white) and HCQ plus erlotinib (black).
Overall median follow-up time was 9.1 months. The median PFS was 1.8 months (95% CI 0.7, 1.8) in arm A and 2.0 months (95% CI 1.5, 3.8) in arm B. Median OS was 9.0 months (95% CI 1.4, 38.5) in arm A and 10.6 months (95% CI 3.6, 14.1) in arm B.
Pharmacokinetics
Data was available to estimate the Cminss of HCQ in whole blood and plasma for 8 patients in arm A and 13 patients in arm B. As shown in Figure 3, the average Cminss of HCQ in whole blood and plasma increased progressively as the daily HCQ dose was escalated from 400 to 800 mg/day in both study arms. Accordingly, dose-normalized Cminss values in patients treated with 400–800 mg of HCQ were used to assess the effect of erlotinib on the steady-state PK of HCQ. The mean dose-normalized Cminss of HCQ was lower when given with erlotinib than as monotherapy, in both whole blood (5.93 ± 2.51 μM/g, n = 9 for combination and 9.40 ± 5.85 μM/g, n = 8 for monotherapy, p=0.12) and plasma (1.00 ± 0.40 μM/g, n = 9 for combination and 1.64 ± 0.61 μM/g, n = 8 for monotherapy, p=0.015). The blood-to-plasma ratio for the Cminss of HCQ was similar (P=0.33) in the combination (6.14 ± 1.54) and in HCQ monotherapy (5.42 ± 1.55)..
Figure 3. Pharmacokinetic Assays.

Cminss is measured in whole blood (A) and plasma (B). Data points represent Cminss values for individual patients who received HCQ alone (open markers) or in combination with erlotinib (closed markers). The solid bars depict mean values of the Cminss for the group of patients evaluated at each dose level.
Steady-state erlotinib plasma concentration-time profiles were obtained in 10 patients during two 24-hour dosing intervals – once after the 7-day erlotinib run-in and once a week into combination therapy with HCQ 400–800 mg/day, Table 3. There were no significant differences between any measured parameters, suggesting that concurrent administration of HCQ does not affect the plasma PK of erlotinib in this patient population.
Table 3.
Comparison of erlotinib steady-state pharmacokinetic parameters when given alone and in combination with daily hydroxychloroquine.
| Parameter | Concurrent HCQ
|
Difference (%) | P-value a | |
|---|---|---|---|---|
| − | + | |||
| C0 (μM) | 2.52 ± 1.92 | 2.13 ± 1.51 | −15.2 | 0.17 |
| Cmax (μM) | 5.48 ± 2.09 | 4.71 ± 4.95 | −14.0 | 0.13 |
| AUC24 (μM · h) | 88.7 ± 43.9 | 76.2 ± 29.9 | −14.1 | 0.19 |
| CL/F (L/h) | 4.3 ± 2.1 | 5.0 ± 2.0 | 16.5 | 0.190 |
Paired two-sample t-test for means of log transformed data.
Abbreviations: C0, drug concentration in plasma measured immediately before dosing; Cmax, maximum concentration of drug in plasma; AUC24, area under the plasma concentration-time curve during the 24 h interval between two doses; CL/F apparent oral clearance.
Discussion
We studied PC9 models of acquired resistance to EGFR inhibitors and identified two experimental conditions in which HCQ had cytotoxic activity in the context of drug tolerance in EGFR mutant NSCLC. Cells with acquired resistance were effectively killed by HCQ at concentrations which had not affected the parental TKI-sensitive cells. In addition, we observed that initial treatment with HCQ plus EGFR TKI could prevent the development of drug-tolerant clones. We therefore designed and performed a phase I clinical study of HCQ with or without erlotinib to assess safety, as a foundation for a future study to evaluate the suppression of acquired TKI-resistance. We found that HCQ was safe and well-tolerated in NSCLC patients previously treated with EGFR TKIs. The MTD of HCQ when given with 150mg of erlotinib daily was the highest tested dose, 1000 mg daily. Treatment-related side effects were mild (predominantly grades 1–2) and were consistent with the known toxicity profile of erlotinib, including rash, fatigue, diarrhea, and nausea17. There were few grade 3–5 AEs and no DLTs.
We did not observe robust clinical activity in this heavily pre-treated cohort, with the notable exception of an EGFR-mutant patient with a durable response to erlotinib plus HCQ. The etiology of her initial response may have been “retreatment effect”, which is a well-documented phenomenon in a subset of EGFR-mutants18, 19. However, the 20-month duration of her response was longer than we have anecdotally experienced with erlotinib retreatment.
To our knowledge, this is the first clinical report assessing HCQ in combination with erlotinib, though there have been several clinical trials examining CQ or HCQ in cancer patients6, 20. One relevant study was a small prospective randomized trial of glioblastoma multiforme patients in which CQ was administered with standard surgery, radiation, and temozolomide chemotherapy. Those treated with CQ had a significantly longer OS than controls, with an increase in the median survival from 11 to 24 months6. Another single-arm study of patients with EGFR-mutated, TKI-naïve NSCLC treated with HCQ plus gefitinib is ongoing. Interim analysis of 13 patients on this study yielded an 11.5 month median PFS, suggesting that the combination might delay acquired resistance compared to historic gefitinib controls (Chin TM, submitted for publication). Other ongoing studies are examining the effect of HCQ plus chemotherapy in breast, colorectal, pancreas and NSCLC21.
There are several mechanisms by which HCQ could potentiate the effects of erlotinib, specifically in TKI-resistant tumors. This manuscript describes preclinical evidence that HCQ may disrupt the drug-tolerant state that occurs after lethal drug exposure, possibly through effects on chromatin structure similar to those induced by HDAC inhibitors7. Interestingly, HCQ also appears to be effective at delaying the development of the drug-tolerant state in NSCLC cells treated with erlotinib or cisplatin, breast cancer cells treated with a HER2 TKI, and melanoma cells treated with a BRAF TKI, suggesting that HCQ may be a beneficial adjunct to therapy in a broad range of clinical applications.
CQ and HCQ have also been postulated to directly inhibit autophagy. Under conditions of cellular stress such as TKI-mediated inhibition of critical signaling pathways, autophagy is activated to clear damaged cellular debris and allow for cell survival22. Preclinical studies have shown that blocking autophagy along with a TKI appears to enhance cell death23. In pancreatic cancer cell lines and mouse xenograft models with constitutive autophagic activation, the addition of CQ resulted in autophagy inhibition with decreased proliferation of cells and marked tumor regression24. Additionally, the use of CQ in a mouse xenograft model of pancreatic cancer caused marked tumor regression and prolonged survival with evidence of autophagy inhibition and elevated DNA damage24.
A final mechanism by which CQ and HCQ may enhance erlotinib-mediated cell killing is through lysosomal pathway disruption. Cell line and human tumor models show that the CQ-mediated impairment of maturation from endosome to lysosome results in decreased intralysosomal degradation of phosphorylated EGFR, which in turn causes over-expression of activated EGFR. This appears to make TKI-resistant cells more dependent on the EGFR signaling pathway, thereby increasing their sensitivity to EGFR TKI (Chin TM, submitted for publication).
Pharmacokinetic (PK) analyses from our study showed that administration of HCQ at doses of 400–800 mg/day for 7 days did not result in significant changes in the mean value of any PK variable for erlotinib. The mean trough blood and plasma concentrations of HCQ in arm A patients were in good agreement with prior studies25, 26. Concurrent erlotinib resulted in a decrease in trough concentration of HCQ, consistent with enhanced first-pass HCQ metabolism from erlotinib-mediated CYP3A4 induction27–29.
The study must be interpreted within the context of its limitations. This was a phase I study designed to evaluate safety. The low response to treatment in the study cohort may have been influenced by potentially ineffective doses of HCQ in the early cohorts, or by a heavily pre-treated patient population. Furthermore, the laboratory work leading to the phase I trial concept focused on EGFR-mutant models while the inclusion criteria for the study dictated only prior receipt of ≥12 weeks of TKI, thus enriching for EGFR mutants but not excluding wild-type patients. Mandating EGFR mutations may ultimately prove a better strategy for studying the effect of HCQ, and we have initiated a randomized phase II study in TKI-naive EGFR-mutants (NCT00977470). Patients are assigned to erlotinib alone or erlotinib plus HCQ, under the hypothesis that the HCQ arm will have delayed emergence of drug-resistance and prolonged PFS.
In conclusion, laboratory studies of PC9-derived EGFR TKI-tolerant cells suggested sensitivity to HCQ, and that co-treatment with HCQ and EGFR TKI could delay the emergence of the drug-tolerant state. We have found in this phase I study that the combination of HCQ 1000mg and erlotonib 150mg daily is safe and well-tolerated, with a suggestion of clinical benefit in a subset of patients with advanced NSCLC who had previously benefitted from an EGFR TKI. A phase II trial assessing the added benefit of HCQ with erlotinib in EGFR-mutant NSCLC is currently underway.
Supplementary Material
Supplemental Digital Content 1: Detailed pharmacokinetic methods. Goldberg_SDC.doc.
Acknowledgments
Financial support: This work was supported by Genentech and the Dana-Farber/Harvard Cancer Center SPORE in Lung Cancer, NCI P50 CA090578 (J.S.).
Footnotes
Conflicts of Interest and Source of Funding: Joel Neal received research funding from Genentech. Panos Fidias is on the advisory board for Genentech. Alice Shaw has consulted for Pfizer, Ariad, Chugai, Novartis, Millenium, and Daiichi. Thomas Lynch is a consultant for Merck, Supergen, and Boehringer-Ingelheim, is on the Board of Directors for Infinity, and is a joint holder for a patent for EGFR mutation testing. Sreenath Sharma received a grant from the LUNGevity Foundation & Goldman Philanthropic Partnerships, consulted for Concert Pharmaceuticals, and is employed by Norvartis. Jeffrey Settleman is employed by Genentech. Lecia Sequist received a grant from Genentech and is on the advisory boards for Clovis Oncology, Celgene, GSK, Daiichi Sankyo, and Merrimack. For the remaining authors none were declared.
References
- 1.Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep-Oct;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004 May 20;350(21):2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009 Sep 3;361(10):947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 4.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010 Jun 24;362(25):2380–8. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 5.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010 Feb;11(2):121–8. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 6.Sotelo J, Briceno E, Lopez-Gonzalez MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006 Mar 7;144(5):337–43. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]
- 7.Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010 Apr 2;141(1):69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramalingam SS, Maitland ML, Frankel P, et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol. 2010 Jan 1;28(1):56–62. doi: 10.1200/JCO.2009.24.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furst DE, Lindsley H, Baethge B, et al. Dose-loading with hydroxychloroquine improves the rate of response in early, active rheumatoid arthritis: A randomized, double-blind six-week trial with eighteen-week extension. Arthritis Rheum. 1999 Feb;42(2):357–65. doi: 10.1002/1529-0131(199902)42:2<357::AID-ANR19>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 10.AJCC cancer staging manual. 6. New York, NY: Springer-Verlad New York, Inc; 2002. [Google Scholar]
- 11.Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the eastern cooperative oncology group. Am J Clin Oncol. 1982 Dec;5(6):649–55. [PubMed] [Google Scholar]
- 12.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. european organization for research and treatment of cancer, national cancer institute of the united states, national cancer institute of canada. J Natl Cancer Inst. 2000 Feb 2;92(3):205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 13.Rynes RI, Bernstein HN. Ophthalmologic safety profile of antimalarial drugs. Lupus. 1993 Feb;2(Suppl 1):S17–9. [PubMed] [Google Scholar]
- 14.Trotti A, Colevas AD, Setser A, et al. CTCAE v3.0: Development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003 Jul;13(3):176–81. doi: 10.1016/S1053-4296(03)00031-6. [DOI] [PubMed] [Google Scholar]
- 15.Zhao M, He P, Rudek MA, et al. Specific method for determination of OSI-774 and its metabolite OSI-420 in human plasma by using liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2003 Aug 15;793(2):413–20. doi: 10.1016/s1570-0232(03)00356-8. [DOI] [PubMed] [Google Scholar]
- 16.Shah VP, Midha KK, Findlay JW, et al. Bioanalytical method validation--a revisit with a decade of progress. Pharm Res. 2000 Dec;17(12):1551–7. doi: 10.1023/a:1007669411738. [DOI] [PubMed] [Google Scholar]
- 17.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005 Jul 14;353(2):123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 18.Kurata T, Tamura K, Kaneda H, et al. Effect of re-treatment with gefitinib (‘iressa’, ZD1839) after acquisition of resistance. Ann Oncol. 2004 Jan;15(1):173–4. doi: 10.1093/annonc/mdh006. [DOI] [PubMed] [Google Scholar]
- 19.Yano S, Nakataki E, Ohtsuka S, et al. Retreatment of lung adenocarcinoma patients with gefitinib who had experienced favorable results from their initial treatment with this selective epidermal growth factor receptor inhibitor: A report of three cases. Oncol Res. 2005;15(2):107–11. [PubMed] [Google Scholar]
- 20.Briceno E, Calderon A, Sotelo J. Institutional experience with chloroquine as an adjuvant to the therapy for glioblastoma multiforme. Surg Neurol. 2007 Apr;67(4):388–91. doi: 10.1016/j.surneu.2006.08.080. [DOI] [PubMed] [Google Scholar]
- 21.www.clinicaltrials.gov [Internet].
- 22.Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011 Feb 15;17(4):654–66. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Degtyarev M, De Maziere A, Orr C, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008 Oct 6;183(1):101–16. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011 Apr 1;25(7):717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munster T, Gibbs JP, Shen D, et al. Hydroxychloroquine concentration-response relationships in patients with rheumatoid arthritis. Arthritis Rheum. 2002 Jun;46(6):1460–9. doi: 10.1002/art.10307. [DOI] [PubMed] [Google Scholar]
- 26.Khoury H, Trinkaus K, Zhang MJ, et al. Hydroxychloroquine for the prevention of acute graft-versus-host disease after unrelated donor transplantation. Biol Blood Marrow Transplant. 2003 Nov;9(11):714–21. doi: 10.1016/j.bbmt.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Zhao M, He P, et al. Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes. Clin Cancer Res. 2007 Jun 15;13(12):3731–7. doi: 10.1158/1078-0432.CCR-07-0088. [DOI] [PubMed] [Google Scholar]
- 28.Harmsen S, Meijerman I, Beijnen JH, et al. Nuclear receptor mediated induction of cytochrome P450 3A4 by anticancer drugs: A key role for the pregnane X receptor. Cancer Chemother Pharmacol. 2009 Jun;64(1):35–43. doi: 10.1007/s00280-008-0842-3. [DOI] [PubMed] [Google Scholar]
- 29.Dong PP, Fang ZZ, Zhang YY, et al. Substrate-dependent modulation of the catalytic activity of CYP3A by erlotinib. Acta Pharmacol Sin. 2011 Mar;32(3):399–407. doi: 10.1038/aps.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content 1: Detailed pharmacokinetic methods. Goldberg_SDC.doc.