

Abstract
A low-error 16S rRNA amplicon sequencing method (LEA-Seq) plus whole genome sequencing of >500 cultured isolates were used to characterize bacterial strain composition in the fecal microbiota of 37 USA adults sampled for up to five years. Microbiota stability follows a power law function which, when extrapolated, suggests that most strains in an individual are residents for decades. Shared strains were recovered from family members, but not from unrelated individuals. Sampling individuals for up to 32 weeks while consuming a monotonous liquid diet indicated that changes in weight are more predictive of changes in strain composition than sampling interval. This combination of stability and responsiveness to physiologic change confirms the potential of the gut microbiota as a diagnostic tool and therapeutic target.
Our growing understanding of the human gut microbiota as an indicator of and contributor to human health suggests that it will play important roles in the diagnosis, treatment, and ultimately prevention of human disease. These applications require an understanding of the dynamics and stability of the microbiota over the lifespan of an individual. Amplicon sequencing of the bacterial 16S rRNA gene from fecal microbial communities (microbiota) has revealed that each individual harbors a unique collection of species (1–3). Estimates of the number of species present in an individual’s microbiota have varied greatly; from ~100 with culture-based techniques (4) to ~160 with culture-independent deep shotgun sequencing of fecal community DNA (5) to several fold higher based on 16S rRNA amplicon sequencing even after in silico attempts to remove chimeric molecules formed during PCR and errors introduced during sequencing. These artifacts complicate tracking of individual bacterial taxa across time by inflating the set of strains in each sample with false positives. Shotgun sequencing of the community’s microbiome is another approach for defining diversity (6), but it is difficult to associate gene sequences with their genome of origin. With these limitations in mind, we have developed a method for amplicon sequencing to assay the bacterial composition of the gut microbiota of individuals at high depth with high precision over time. When combined with high throughput methods for culturing and sequencing the genomes of anaerobic bacteria, these results reveal that the majority of the bacterial strains in an individual’s microbiota persist for years, and suggest that our gut colonizers have the potential to shape many aspects of our biological features for most if not the entirety of our lives.
A Method for Low Error Amplicon Sequencing (LEA-Seq) of Bacterial 16S rRNA genes
A 16S rRNA sequencing method for assaying the stability of an individual’s microbiota over time would ideally retain high precision at high sequencing depth ( ). Low precision data complicate comparison of sequences between samples, as it becomes difficult to differentiate species (typically defined as isolates that share ≥97% sequence identity in their 16S rRNA genes), and strains (isolates of a given species with more minor variations in their 16S rRNA gene sequences) from sequencing errors. Standard amplicon sequencing is limited in its precision by the overall error rate of the sequencing method. Low sequencing depth prevents determining if a strain has dropped out of a given individual’s microbiota or has fallen below the limits of detection at the sampling depth employed.
In many applications it would be advantageous to exchange sequence depth for improved sequence quality. Despite several optimizations we developed to increase the precision of standard amplicon sequencing at shallow depths, we found that sequencing a sample beyond 10,000 reads did not substantially increase the lower detection limit possible at high precision (Supplemental Results). Exchanging sequence quantity for sequence quality is inherent to shotgun genome sequencing where redundant sequencing of genomes at 10- to 50-fold coverage enables a far lower error rate than is attainable from single-reads alone. In general, to redundantly sequence DNA fragments it is necessary to create a finite DNA pool that is smaller than the amount of sequencing available (i.e., create a bottleneck) and to have a method of labeling the molecules in the pool (7–9). To adapt these techniques to redundantly sequence PCR amplicons, the initial template DNA could be diluted to create a bottleneck. However, this dilution would likely need to be empirically determined for every input sample (e.g., using qPCR), and one would still need to label each template molecule. As an alternative, we developed a method that we named Low Error Amplicon Sequencing (LEA-Seq).
As outlined in Fig. 1A, LEA-Seq is based on redundant sequencing of a set of linear PCR template extensions of 16S rRNA genes to trade sequence quantity for quality. In this method, we create the bottleneck with a linear PCR extension of the template DNA with a dilute, barcoded, oligonucleotide primer solution. Each oligonucleotide is labeled with a random barcode positioned 5′ to the universal 16S rRNA primer sequence (Fig. 1A, fig. S1). We then amplify the labeled, bottlenecked linear PCR pool with exponential PCR using primers that specifically amplify only the linear PCR molecules. During the exponential PCR, an index primer is added to the amplicons with a third primer to allow pooling of multiple samples in the same sequencing run (Fig. 1A). This exponential PCR pool is then sequenced at sufficient depth to redundantly sequence (~20x coverage) the bottlenecked linear amplicons. The resulting sequences are separated by sample using the index sequence, and the amplicon sequences within each sample are separated by the unique barcode; the multiple reads for each barcode allow the generation of an error-corrected consensus sequence for the initial template molecule. In LEA-Seq, the linear PCR primers are diluted to a concentration that generates ~150,000 amplicon reads at 20x coverage per amplicon on an Illumina HiSeq DNA sequencer (Fig. 1A, fig. S1).
Fig. 1. Multiplex bacterial 16S rRNA gene sequencing using LEA-Seq; comparison with previous methods using mock communities composed of sequenced gut bacterial species.

(A) Schematic of how the LEA-Seq method is used to redundantly sequence PCR amplicons from a set of linear PCR template extensions of bacterial 16S rDNA. This approach results in amplicon sequences with a higher precision than standard amplicon sequencing at lower abundance thresholds. (B) Performance of 16S rRNA amplicon sequencing methods assayed as the precision obtained for different sequence abundance thresholds. Standard methods for amplicon sequencing using the 454 pyrosequencer and the Illumina MiSeq instrument exhibit increased precision as less abundant reads are filtered out. By redundantly sequencing each amplicon with LEA-Seq, the precision of amplicon sequencing is increased at lower abundance thresholds for both the V1V2 region of the bacterial 16S rRNA gene (compare red and blue lines) and the V4 region (compare magenta and blue lines), thereby enabling detection of lower-abundance bacterial taxa at high precision.
To empirically test LEA-Seq against existing 16S rRNA amplicon sequencing methods, we first generated nine in vitro ‘mock’ communities composed of different proportions of strains from a 48-member collection of phylogenetically diverse, cultured human gut bacteria whose genomes had been characterized (see Supplemental Methods, Supplemental Results and table S1). To calculate precision, we compared amplicons generated using two sequencing platforms (Illumina MiSeq and 454 FLX instruments), targeting different variable (V) regions of the 16S rRNA gene with different PCR primers. We defined a TruePositive sequence as 100% identical across 100% of its length to the 16S rRNA gene sequence(s) in the reference genome. We calculated precision at different abundance thresholds by including only those sequences representing at least a minimal portion of the total sequencing reads (0.5%, 0.1%, 0.05%, 0.01%, or 0.005%). LEA-Seq produced amplicon sequences with higher precision from taxa present at lower abundance thresholds in the mock communities than existing standard approaches (Fig. 1B). For 16S rRNA sequences representing ≥ 0.01% of the reads, LEA-Seq enabled a precision of 0.83±0.02 (V4) and 0.63±0.03 (V1V2) versus 0.08±0.064 and 0.09±0.005 for the same regions with standard amplicon sequencing (table S2). These performance improvements are dependent on generating the consensus sequence from the redundant amplicon reads (table S2; Method = “LEA-Seq without consensus”). LEA-Seq also produced slower saturation in performance (precision of >0.7 for reads representing 0.001% of the total; fig. S2; table S2). Similar results were obtained using the several different mock communities (for additional details of the analysis, including V1V2 versus V4 comparisons, see ‘Optimization of bacterial 16S rRNA amplicon sequencing’ in Supplemental Results). Based on this assessment of its attributes, we used LEA-Seq to quantify the stability of the gut microbiota within individuals as a function of time and change in body mass index while consuming controlled monotonous and free diets.
Applying LEA-Seq to Define the Stability of the Fecal Microbiota of 37 Healthy Adults
Stability of a microbiota best fits a power law function
We used LEA-Seq to characterize the microbiota in 167 fecal samples obtained from 37 healthy adults residing throughout the USA; 33 of these donors were sampled 2–13 times up to 296 weeks apart (1, 10) (table S3). The remaining four individuals were sampled on average every 16 days for up to 32 weeks while consuming a monotonous liquid diet as part of a controlled in-patient weight-loss study (see Methods) (11–13). None of the individuals took antibiotics for at least two months prior to sampling. All fecal samples were frozen at −20°C immediately after they were produced and then at −80°C within 24h. DNA was isolated from all samples by bead beating in phenol/chloroform.
Employing an Illumina HiSeq2000 instrument to sequence amplicons from the V1V2 region of bacterial 16S rRNA genes, we generated 108,677±60,212 (mean ± SD) LEA-Seq reads per fecal DNA sample. Reads were then filtered using a minimum sequence abundance threshold cutoff of eight reads (i.e., to detect strains present in the fecal microbiota at an average relative abundance of 0.007%). Based on our mock community data, the precision at this threshold for the V1V2 region is 0.63. We defined the number of strains in a sample as the number of unique amplicon sequences and the number of species-level OTUs in the sample as the number of clusters with 97% shared sequence identity. To correct for false-positives, the number of strains was multiplied by the precision (i.e., if we detect 100 unique sequences, we expect 63 of them to be true). For individuals sampled over multiple time points, we calculated the number of species and strains for each sample individually and averaged them. The results indicated that individuals in this cohort harbored 195±48 bacterial strains in their fecal gut microbiota, representing 101±27 species.
To study each individual’s microbiota over time, we took all possible pairs of samples from the time series of each individual (table S3) and calculated the time in weeks between the sample dates as well as the fraction of shared strains between them, as measured by the binary Jaccard Index (an unweighted metric of community overlap).
Control experiments using mock communities (table S1), established that LEA-Seq of V1V2 16S rRNA amplicons produced highly accurate estimates of the Jaccard Index (correlation between known and measured Jaccard Index = 0.996; see Supplemental Results). To characterize the stability of an individual’s microbiota, fecal samples were binned into intervals (<3 weeks, 3–6 weeks, 6–9 weeks, 9–12 weeks, 12–32 weeks, 32–52 weeks, 52–104 weeks, 104–156 weeks, 156–208 weeks, 208–260 weeks, and >260 weeks) and Jaccard Index values were averaged for each bin. The results disclosed that the bacterial composition of each individual’s fecal microbiota changed over time, with more strains shared between closer time intervals compared with long intervals (Fig. 2A). Nonetheless, overall the set of microbial strains was remarkably stable, with over 70% of the same strains remaining after one year and few additional changes occurring over the following four years. The stability of a microbiota best fits a power law function (R2 = 0.96; Fig. 2A blue line; table S4) where large differences in community composition occur on shorter time scales, while a stable core set of strains persists at longer time scales.
Fig. 2. Measuring the stability of an individual’s fecal microbiota over time with LEA-Seq.
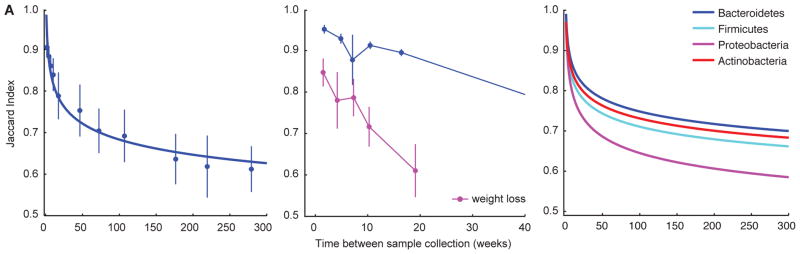
(A) The Jaccard Index (fraction of shared strains) was calculated between all possible pairwise combinations of fecal samples collected from each individual, where bacterial strains were considered shared if the nucleotide sequence was 100% identical across 100% of the length of the V1V2 region of their 16S rRNA genes. Jaccard Indexes were binned into intervals of <3 weeks, 3–6 weeks, 6–9 weeks, 9–12 weeks, 12–32 weeks, 32–52 weeks, 52–104 weeks, 104–156 weeks, 156–208 weeks, 208–260 weeks, and >260 weeks apart (mean±SE for each bin is shown). The decay in the Jaccard Index as a function of time between two samples best fits a power law (blue line). (B) Four individuals losing 10% of their body weight in the study involving consumption of a monotonous low calorie liquid diet (magenta) had significantly less stable microbiota than the mean of the 33 remaining individuals (blue). Mean±SE for the Jaccard Index are plotted. (C) At the phylum level, Bacteroidetes (blue) and Actinobacteria (red) were more stable components of the microbiota than the Proteobacteria and Firmicutes (hypergeometric distribution).
To define the stability of a given strain as a function of its relative abundance in the microbiota, we used all pairwise combinations of fecal samples obtained from each individual to calculate (i) the mean abundance of the strains shared by two or more samples, and (ii) the mean abundance of strains that were not shared between any two samples. Strains that were shared across two time points were roughly three-fold more abundant than those that were not shared [0.030±0.013 fraction of the community versus 0.011±0.011 (mean ± SD); p-val = 2.2×10−9 (t-test) fig. S3A]. We also binned the strain abundances for each donor using five fractional abundance thresholds of 0.1, 0.01, 0.001, 0.0001, and <0.0001 (e.g., bin 0.01 contains all strains ≤0.1 and >0.01) and calculated the probability that strains in a given bin were shared between samples. We found the higher the fractional abundance of a strain, the more likely the strain was shared between samples (r = 0.96, p<0.0087; fig. S3B). Together, these results suggest that the more stable components of the microbiota are also the most abundant members.
Effects of a monotonous low calorie diet and associated weight loss on diversity
To explore the role of weight loss on the microbiota, we applied LEA-Seq to the fecal microbiota of four individuals sampled over the course of a 8- to 32-week period in a three phase study that used different caloric intakes of a defined monotonous liquid diet to first stabilize initial weight, then to decrease weight by 10%, and finally maintain weight at the 10% reduced level (Fig. 2B; table S3). Daily caloric intake was 2988±290, 800, and 2313±333 kcal for the three phases of the study, respectively (13,14). While on this diet, these four individuals experienced significantly reduced stability of their microbiota, as measured by the Jaccard Index (Fig. 2B). For each individual, we found no significant correlation between time and diversity/richness (i.e., number of strains in a sample; minimum p-value = 0.17). Additionally, we found no significant correlation between the change in composition of the microbiota (Jaccard Index between two samples) and the change in diversity/richness (absolute difference in the number of species/strains between two samples) (p-values = 0.09 and 0.44 for strains and species, respectively). Considering family-level taxonomic bins, there were several groups whose abundance was strongly correlated with time during the weight loss period including Clostridiaceae [average correlation (r) across donors during weight loss = 0.60], Coriobacteriaceae (r=0.53), Bifidobacteriaeceae (r=0.55), and Enterobacteriaceae (r=0.58), Lachnospiraceae (r=−0.65), Oscillospiraceae (r=−0.53), and Oxalobateraceae (r=−0.74).
Modeling the relationship between time, body composition, and microbiota stability
Given the correlation between weight loss and changes in the microbiota of individuals consuming a monotonous 800 kcal/day diet, we took a broader view across all 37 individuals in our study to determine if this correlation was due to the monotonous diet that the four individuals had consumed, or if there is a generalizable and quantifiable relationship between weight stability and microbiota stability. To explore this question, we not only calculated the time (Δtime) and Jaccard Index between all pairs of fecal samples collected from an individual (Fig. 2), but also the absolute value of the change in log(BMI) (abbreviated ΔlnBMI) between all pairs. We found a significant negative correlation between ΔlnBMI and Jaccard Index (Fig. 3A; r = −0.68; p-val = 2.98×10−73) that was even greater than Δtime and Jaccard Index (Fig. 3B; r = −0.42; p-val = 1.45×10−43). These relationships held when we removed the data generated from the four individuals on the monotonous diet (ΔlnBMI: r = −0.69; p-val = 3.27×10−54; Δtime: r = −0.65; p-val = 9.05×10−46).
Fig. 3. Relationship between weight stability, time, and fecal microbiota stability.

(A) The microbiota sampled from a given individual during periods of weight loss or gain has decreased stability (lower Jaccard Index). (B) The Jaccard Index decreased as the time between samples increased (also see Fig. 2). (C) Across samples from 37 individuals, a linear model of microbiota stability as a function of changes in lnBMI and changes in time explained 46% of the variation in the stability of the microbiota (Jaccard Index). Note that changes in lnBMI explained more of the variation in microbiota stability than did the passage of time. Color changes correspond to the Jaccard Index values in the color bar on the right. Blue dots show the change in Jaccard Index, time, and lnBMI between two samples from a given individual.
To quantify the relationship between Δtime, ΔlnBMI, and the Jaccard Index between samples (Fig. 3C), we fit the following model:
where microbiota_stability is the Jaccard Index between samples, XlnBMI is the change in lnBMI between any two samples collected from the individual (ΔlnBMI), Xtime is the time between the two samples being compared (Δtime), β0 is the estimated parameter for the intercept; and βlnBMI and βtime are the linear regression estimated parameters for ΔlnBMI and Δtime, respectively. Remarkably, this model explained 46% of the variance in the stability of the microbiota (Jaccard Index) within the individuals over time (R2 = 0.46; p-val = 1.94×10−72 and R2 = 0.51; p-val = 1.40×10−58 when the monotonous dieters were excluded). Once again the weight stability of an individual (ΔlnBMI; ANOVA p-val = 1.18×10−51) was a better predictor of fecal microbiota stability than the time between samples (Δtime; ANOVA p-val = 0.09), with Δtime only being a significant predictor of stability when the monotonous dieters were excluded (ANOVA p-val = 2.82×10−7). Together, these relationships between time, BMI, and the stability of an individual’s microbiota highlight the role that longitudinal surveys of a microbiota could play in health diagnostics.
Sequenced Collections of Fecal Bacteria Obtained from Individuals over Time
As in previous studies (1,15–18), we found that each individual’s microbiota at a given time point was most similar to their own at other time points (Jaccard Index 0.82±0.022), followed by their family members (Jaccard Index 0.38±0.020), and then unrelated individuals (Jaccard Index 0.30±0.005). The accuracy of the Jaccard Index estimates with LEA-Seq suggests that on average any two unrelated individuals share ~30% of strains in their microbiota. However, it is possible that unrelated individuals on average share no strains in their microbiota and this 30% represents the lower resolving limit of 16S rRNA amplicon sequencing of the targeted variable region (V1V2) and currently available maximum read lengths on the Illumina HiSeq 2000 instrument (paired-end 101bp).
Whole genome alignments between bacteria isolated and sequenced from different samples provide many orders of magnitude of additional resolving power to determine which strains (now defined at the level of whole genome sequence identity rather 16S rRNA identity) remain in an individual’s microbiota over time, or reside in two unrelated individuals. Isolation and sequencing of extensive collections of organisms from the human gut microbiota (19) provides a practical method to look at the plasticity and evolution of the gene content of microbial strains harbored in individuals’ intestines over time. Therefore, adapting a high-throughput method we had developed for generating clonally arrayed collections of anaerobic bacteria in multi-well format from frozen fecal samples (19), we produced draft genome sequences for 444 bacterial isolates recovered from the frozen fecal microbiota of five donors who had been sampled across periods from 7–69 weeks apart (n=1–4 time points/donor; 11 total samples; mean coverage/microbial genome = 118x; see tables S5, S6 and Supplemental Methods). These genomes span a broad phylogenetic range within the four dominant bacterial phyla that comprise the human gut microbiota (Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria; table S6).
To look for changes in bacterial genome content across time in each individual, we performed whole genome alignment with nucmer (20) and calculated the fraction of DNA sequence aligned between each pair of genomes ( ; where X and Y are the lengths of genome X and Y, respectively, and Xaln and Yaln are the number of aligned bases of genome X and Y respectively) (21) (see Supplemental Results). We found the shared genome content between isolates from unrelated individuals was broadly distributed for taxa from the same genus (coverage score = 0.30±0.20) or species (0.77±0.12), with a maximum of 0.956 (Fig. 4A, blue; fig. S4). We then compared the shared genome content between isolates within each fecal sample (i.e., self-versus-self at a single time point) and found isolates that shared a very high proportion of their content (0.965–0.999) (Fig. 4A, red). Remarkably, we found the same high proportion of shared genome content between isolates from a given donor between different time points (i.e., self-versus-self over time; Fig. 4A green), suggesting that the same strains of bacteria persisted in these individuals over the course of the sampling period.
Fig. 4. Comparison of genome stability in fecal bacterial isolates recovered from individuals over time.

The fraction of aligned nucleotides between any two microbial genomes was calculated using the coverage score (see text for definition). (A) Histogram of the fraction of aligned genome content between all sequenced bacterial isolates from unrelated individuals (blue; only coverage scores ≥ 0.01 are shown) shows that the alignable genome content never exceeded 96% (dotted line). However, highly conserved strains with coverage scores exceeding this threshold were readily detected in the microbiota of individuals at a single time point (red) or between samples from an individual taken up to 15 months apart (green). The y-axis “Counts” represent the number of times a sample fell into each coverage score bin. (B,C) Sequencing the genomes of M. smithii strains (panel B) and B. thetaiotaomicron strains (panel C) revealed that no two isolates from unrelated individuals had more than 96% shared (alignable) gene content (blue), while highly conserved strains above this threshold were found between isolates obtained from a single individual’s fecal microbiota at a single time point (red), as well as from isolates taken from different members of the same family (brown).
Defining replicate bacterial strains as those with a coverage score >0.96 and species as those with a coverage score >0.5 (fig. S4), we subsequently clustered the genome isolates by sample and by individual (table S5); this effort yielded a total of 165 strains and 69 species across the five donors (Table 1). Across the four donors with multiple time points, on average 36% of an individual’s bacterial strains were isolated from multiple time points. This fraction of shared bacterial strains across time at the level of the genome is lower than that measured by LEA-Seq; however, this likely reflects the increased sampling depth and culture independence of LEA-Seq [detecting isolates at depths of 1:10,000–1:100,000 (0.01-0.001%) compared with 0.14-0.06% for high-throughput culturing]. For the most deeply sampled individual (F3T1 in table S3), where isolates were sequenced from four samples taken over the course of ~16 months, over 60% of the strains were isolated from multiple samples.
Table 1.
Species composition of the sequenced arrayed culture collections from six donors.
| donor | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| species ID | species | alternative name | F3T1 | F58T1 | F60T1 | F60T2 | F61T1 | F61T2 |
| 1 | Alistipes indistinctus | Anaerococcus | + | + | ||||
| 2 | Anaerococcus vaginalis | + | + | + | + | |||
| 3 | Anaerofustis stercorihominis | + | + | |||||
| 4 | Anaerofustis stercorihominis | + | ||||||
| 5 | Bacteroides | + | ||||||
| 6 | Bacteroides caccae | + | + | + | ||||
| 7 | Bacteroides finegoldii | + | + | |||||
| 8 | Bacteroides fragilis | + | + | |||||
| 9 | Bacteroides intestinalis | Bacteroides cellulosilyticus | + | + | + | + | ||
| 10 | Bacteroides massiliensis | + | + | |||||
| 11 | Bacteroides ovatus | + | + | + | + | |||
| 12 | Bacteroides salyersiae | + | ||||||
| 13 | Bacteroides thetaiotaomicron | Bacteroides faecis | + | + | + | + | ||
| 14 | Bacteroides uniformis | Bacteroides acidifaciens | + | + | + | + | ||
| 15 | Bacteroides vulgatus | Bacteroides dorei | + | + | + | + | + | + |
| 16 | Barnesiella intestinihominis | + | ||||||
| 17 | Bifidobacterium adolescentis | + | + | |||||
| 18 | Bifidobacterium bifidum | + | ||||||
| 19 | Bifidobacterium longum | + | + | + | ||||
| 20 | Bifidobacterium pseudocatenulatum | + | + | |||||
| 21 | Blautia | + | ||||||
| 22 | Blautia schinkii | + | + | |||||
| 23 | Butyricimonas virosa | + | + | + | + | |||
| 24 | Clostridiales | + | ||||||
| 25 | Clostridiales | + | ||||||
| 26 | Clostridiales | + | ||||||
| 27 | Clostridiales | + | ||||||
| 28 | Clostridiales | + | ||||||
| 29 | Clostridiales | + | ||||||
| 30 | Clostridiales | + | ||||||
| 31 | Clostridium | + | + | + | ||||
| 32 | Clostridium | + | ||||||
| 33 | Clostridium bolteae | + | ||||||
| 34 | Clostridium hylemonae | + | ||||||
| 35 | Clostridium leptum | + | + | |||||
| 36 | Clostridium scindens | + | + | |||||
| 37 | Clostridium scindens | + | ||||||
| 38 | Collinsella | + | ||||||
| 39 | Collinsella aerofaciens | + | + | + | ||||
| 40 | Coprococcus comes | + | + | + | ||||
| 41 | Dorea formicigenerans | + | + | + | + | |||
| 42 | Dorea longicatena | + | + | + | + | |||
| 43 | Dorea longicatena | + | ||||||
| 44 | Eggerthella lenta | Subdoligranulum variabile | + | |||||
| 45 | Escherichia coli | + | + | + | + | + | ||
| 46 | Eubacterium biforme | + | ||||||
| 47 | Eubacterium callanderi | + | ||||||
| 48 | Eubacterium contortum | + | ||||||
| 49 | Eubacterium eligens | + | ||||||
| 50 | Finegoldia magna | Dialister invisus | + | |||||
| 51 | Lactobacillus | + | ||||||
| 52 | Lactobacillus casei | + | ||||||
| 53 | Megasphaera elsdenii | + | ||||||
| 54 | Odoribacter splanchnicus | + | + | + | ||||
| 55 | Parabacteroides distasonis | + | + | + | + | + | ||
| 56 | Parabacteroides goldsteinii | + | + | |||||
| 57 | Parabacteroides merdae | + | + | |||||
| 58 | Peptoniphilus harei | + | + | |||||
| 59 | Roseburia intestinalis | + | ||||||
| 60 | Ruminococcaceae | + | ||||||
| 61 | Ruminococcus | Lachnospiraceae | + | + | ||||
| 62 | Ruminococcus albus | + | ||||||
| 63 | Ruminococcus bromii | + | + | |||||
| 64 | Ruminococcus gauvreauii | + | ||||||
| 65 | Ruminococcus gnavus | + | + | + | ||||
| 66 | Ruminococcus obeum | + | ||||||
| 67 | Ruminococcus sp CCUG 37327 A | + | + | |||||
| 68 | Ruminococcus sp DJF VR70k1 | + | ||||||
| 69 | Ruminococcus torques | + | + | |||||
| 70 | Streptococcus | + | ||||||
| 71 | Streptococcus gordonii | + | ||||||
| 72 | Streptococcus parasanguinis | + | ||||||
| 73 | Streptococcus thermophilus | + | ||||||
| 74 | Subdoligranulum variabile | + | + | + | + | + | ||
| 75 | Veillonella parvula | + | ||||||
Stability Viewed from the Perspective of Phylum-Level Membership
When we assigned phylum-level taxonomy to all LEA-Seq 16S rRNA amplicons from each of the 37 individuals in our study (22), we found that members of the Bacteroidetes and Actinobacteria were significantly more stable components of the microbiota than the population average (hypergeometric distribution comparing the total number of shared/not shared strains within a given phylum for all samples versus the total number of shared/not shared strains across all phyla, except the phylum of interest; p-value = 7.54 × 10−28 and 0.0068, respectively), while the Firmicutes and Proteobacteria were significantly less stable (Fig. 2C; p-values = 1.83 × 10−11 and 0.0015). The cultured bacterial strains manifested similar trends for the Bacteroidetes and Firmicutes, where 52% and 21%, respectively, of the strains were isolated and sequenced across multiple time points (table S7), thus demonstrating at a whole genome level the strain stability initially identified when just the 16S rRNA gene was targeted for analysis.
Strains Shared between Members of Human Families
The power law response of the Jaccard Index as a function of the time between sample collection makes it possible to extrapolate beyond the sampling time frame of the current study and suggests that the majority of strains in the microbiota represent a stable core that persists in an individual’s intestine for their entire adult life, and could represent strains acquired during childhood from parents or siblings (fig. S5). Therefore, we used LEA-Seq to measure the fraction of shared strains between family members (sister-sister or mother-daughter). As in previous studies (1), we found the microbiota of related individuals was more similar than unrelated ones with a significantly larger proportion of shared V1V2 16S rRNA sequences [Jaccard Index = 0.38±0.020 (related) and 0.30±0.005 (unrelated); p-val=0.00053].
To determine if this increased similarity between family members manifested itself at the level of their gut microbial genome sequences, we used a targeted approach to look at genome content differences in (i) two families using previously sequenced Methanobrevibacter smithii isolates (23) from two sets of twin pairs and their mothers (six total donors; 19 genomes; table S3), and (ii) five families where 26 Bacteroides thetaiotaomicron strains were isolated with a species-specific monoclonal antibody (Supplemental Methods) (24) from nine donors including sister-sister and mother-daughter pairs (all isolates were from a single sample from each donor; table S3). M. smithii, a methanogen, is the dominant archaeon in the human gut microbiota and facilitates fermentation of polysaccharides by saccharolytic bacteria such as B. thetaiotaomicron by virtue of its ability to remove hydrogen (23). As with our untargeted large-scale genome sequencing of personal bacterial culture collections described above, we found that unrelated individuals had no pair of isolates of either species that shared >96% of their genome content. However, within an individual we once again found replicate isolates of the same strain (Fig. 4B,C; blue and red). Strikingly, we also found replicate strains of M. smithii or B. thetaiotaomicron shared across family members (Fig. 4B,C; brown and table S3).
In contrast with the results obtained using this taxon-targeted whole genome sequencing approach, our untargeted sequencing of the clonally arrayed personal bacterial culture collections had only involved two related individuals (female dizygotic co-twins 1 and 2 from family 60; F60T1 and F60T2; table S3) and had revealed no strains with >96% of their genomes aligned. Therefore, we isolated and sequenced an additional 89 genomes from two timepoints of the dizygotic twin sister (F61T2) of subject F61T1 (yielding a total of 188 strains and 75 species across the six donors). As with the previous donors, we were able to isolate numerous strains shared across the two time points (8 out of 25 = 32%). In addition, we were able to isolate two strains (B. thetaiotaomicron and Escherichia coli) in both of the sisters, showing that even non-targeted genome isolation and sequencing is capable of retrieving the same strain across family members. We did not explicitly sample members of our cohort of females during significant physiological transitions such as menarche and menopause. However, the presence of the same bacterial strain in mothers and their adult daughters who had progressed through one or both of these life cycle milestones suggests that components of the microbiota are retained during these events.
Prospectus
The objects we touch and consume during the course of our lives are covered with diverse microbial life. Despite this, we find with LEA-Seq that on average 60% of the approximately 200 microbial strains harbored in each adult’s intestine is retained in their host over the course of a five-year sampling period. Our results are supported by a microarray-based profiling of fecal microbiota collected from three males and two females over ~8 years (18), but differ from a similar analysis using standard 16S rRNA amplicon sequencing that found high variability in microbiota composition in two individuals sampled for up to 15 months (25). This difference likely reflects the fact that the sequencing depth and precision limitations of standard 16S rRNA amplicon sequencing are overcome to some extent with microarrays where amplicons are mapped/hybridized to a finite pool of target sequences (i.e., sacrificing resolution for precision). The differences could also be due to true differences in the stabilities in microbiota of the individuals, as both studies surveyed only a small number of individuals. Our findings are also supported by a recent report that mapped deep shotgun sequencing datasets of the fecal microbiome to a set of reference bacterial genomes (6) and found that the gut communities of these individuals were more similar to each other at the microbiome level than to unrelated individuals (average maximum time between samples = 32 weeks with two individuals sampled over a period >1 year). Applying LEA-Seq to longitudinal surveys of the fecal microbiota of 37 twins sampled for up to five years allowed us to identify that the stability of an individual’s microbiota follows a power-law function. Using this function, we could extrapolate the stability of the microbiota over decades. The resolution and accuracy of these predictions should improve as advances in sequencing chemistry enable longer regions of 16S rRNA genes to be characterized. LEA-Seq itself can be generalized to any application that requires deep amplicon sequencing with high precision (e.g., the VDJ regions of immunoglobulin and T-cell receptor genes, or targeted searches for variants in candidate or known disease-producing genes).
Our study also illustrates how a highly personalized analysis of the gut community, at strain-level microbial genome resolution can be conducted using collections of cultured bacteria (or archaea) generated from frozen fecal samples collected over time from a given subject. We demonstrate that this strain-level analysis can be part of a broad phylogenetic survey, or it can target a particular species.
The stability of the microbiota that we document in healthy individuals has important implications for future use of the microbiota (and microbiome) as a diagnostic tool as well as a therapeutic target for individuals of various ages. Our findings suggest that obtaining a routine fecal sample as part of a yearly physical examination designed to promote disease prevention would be sufficient to monitor changes in the composition and stability of an individual’s fecal microbiota. For example, in the case of inflammatory bowel diseases, the concordance for Crohn’s disease and ulcerative colitis among monozygotic twin pairs is only 38% and 15% respectively (26). Our results suggest that these twins likely share identifiable unique subsets of their microbiota that represent long term environmental exposures for their immune systems that should be considered when trying to predict disease risk, or infer which species/strains may have a causal role in disease initiation, progression, relapse and treatment responses. Moreover, the effects of travel, changes in diet, weight gain and loss, diarrheal disease, antibiotics, immunosuppressive therapy, or clinical trials designed to deliberately manipulate the microbiota (e.g., through administration of existing or new prebiotics, probiotics, synbiotics, antibiotics or transplantation of microbiota from healthy individuals to those with various diseases attributed to a dysfunctional microbiota) can be more accurately quantified by applying the methods we describe. Finally, the stability we document highlights the impact of early colonization events on our microbiota in later life; earlier colonizers, such as those acquired from our parents and siblings, have the potential to provide their metabolic products and exert their immunologic effects for our entire lives.
METHODS
Diet studies
Four obese (BMI > 30 kg/m2) female subjects with a mean (± SD) age of 26±3 years were admitted to the General Clinical Research Center at Columbia University Medical Center and remained as inpatients throughout the study. The protocol for recruitment, and the weight loss study was approved by the Institutional Review Board of the New York Presbyterian Medical Center and is consistent with guiding principles for research involving humans. Written informed consent was obtained from all subjects. The diet protocol has been described in detail previously (11, 12). Briefly, subjects were fed a liquid-formula diet with 40% of energy as fat (corn oil), 45% as carbohydrate (glucose polymer), and 15% as protein (casein hydrolysate). Diet composition but not quantity was constant throughout the study. The diet had a caloric density of 1.25 digestible kcal of energy/g and was supplemented with vitamins and minerals in quantities sufficient to maintain a stable weight, defined as an average daily weight variation of <10 g/d for ≥2 weeks. This weight plateau is designated as Wtinitial. The four individuals in this study consumed 2600–3300 Kcal/day of the diet to maintain Wtinitial. After a brief period at Wtinitial, subjects were provided 800 kcal energy/d of the same liquid-formula diet until they had lost ~10% of Wtinitial. The duration of the weight-loss phase ranged from 36 to 62 days (table S3). Once 10% weight loss had been achieved, intake was adjusted upward until subjects were again weight stable. Weight maintenance calories were disproportionately reduced (~22%) below those required to maintain initial weight and ranged from 2050–2800 Kcal/day for the four individuals. Subject F72 also received 25 μg/day triiodothyronine during this second weight stable period (table S3). Fecal samples were obtained throughout the study (table S3) and frozen at −80°C until processed for DNA extraction (1).
Twin Participants
Twins were selected from a general population cohort of female like-sex twin pairs, born in Missouri to Missouri-resident parents between July 1, 1975 and June 30, 1985, and first assessed at median age 15 with multiple waves of follow-up (27, 28). Selected twins were drawn from (i) a study, which included biological mothers where available, contrasting stably concordant lean twin pairs (both twins had BMIs in the range 18.5–24.9 by self-report at all completed assessments) and concordant obese twin pairs (both twins had BMI’s ≥ 30, but with pairs prioritized where at least one twin had BMI>35, to maximize separation from the concordant lean pairs) (1); (ii) a small-scale study of concordant lean MZ pairs contrasting free diet with free diet supplemented by twice daily consumption of a fermented milk product (10); and (iii) an ongoing study of twin pairs selected for BMI discordance (either discordant lean/obese, or quantitatively discordant).
Other Protocols
Procedures for (i) creating mock bacterial communities to benchmark standard methods for 16S rRNA sequencing and LEA-Seq, (ii) generating robotically arrayed personal bacterial culture collections from human fecal samples, (iii) isolating B. thetaiotaomicron strains from fecal samples collected over time from individuals and family members, and (iv) sequencing microbial genomes can be found in Supplemental Methods.
Supplementary Material
Acknowledgments
We thank Deborah Hopper and Stacey Marion for their contributions to the recruitment of twins from the MOAFTS cohort and for obtaining fecal samples for the present study, Jessica Hoisington-Lopez, Marty Meier, and Su Deng for technical assistance, plus members of the Gordon lab for their many helpful suggestions during the course of this study. This work was supported in part by grants from the NIH (DK30292, DK078669, DK70977, DK64774, and UL1TR000040), the Crohn’s and Colitis Foundation of America, by Danone (through partial support of the postdoctoral stipend of J.J.F), and by the Howard Hughes Medical Institute. LEA-Seq datasets have been deposited in the European Bioinformatics Institute (EBI) database under accession number ZZZ. Draft genome assemblies are available EBI under accession number PRJEB1925. Raw and processed LEA-Seq data as well as experimental protocols for LEA-Seq and phased 16S rRNA amplicon sequencing can be found at http://gordonlab.wustl.edu/microbiota_stability/.
Footnotes
The authors do not declare any conflicts of interest.
Author contributions: J.J.F. and J.I.G. designed the experiments; A.C.H, R.L.L and M.R. oversaw human studies; J.J.F. J.L.G., M.C., S.S. H.S., and A.L.G. generated the data; J.J.F., R.K., J.C.C. S.S., and J.I.G. analyzed data; J.J.F., and J.I.G. wrote the paper.
REFERENCES AND NOTES
- 1.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ, et al. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci USA. 2010;107:7503–7508. doi: 10.1073/pnas.1002355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckburg PB, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitsuoka T. Intestinal flora and aging. Nutr Rev. 1992;50:438–446. doi: 10.1111/j.1753-4887.1992.tb02499.x. [DOI] [PubMed] [Google Scholar]
- 5.Qin J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schloissnig S, Arumugam M. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hiatt JB, Patwardhan RP, Turner EH, Lee C, Shendure J. Parallel, tag-directed assembly of locally derived short sequence reads. Nat Methods. 2010;7:119–122. doi: 10.1038/nmeth.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci USA. 2011;108:20166–20171. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kivioja T, et al. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2012;9:72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 10.McNulty NP, et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Science Translational Med. 2011;3:106ra106. doi: 10.1126/scitranslmed.3002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kissileff HR, et al. Leptin reverses declines in satiation in weight-reduced obese humans. Am J Clin Nutr. 2012;95:309–317. doi: 10.3945/ajcn.111.012385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenbaum M, et al. A comparative study of different means of assessing long-term energy expenditure in humans. Am J Physiol. 1996;270:R496–504. doi: 10.1152/ajpregu.1996.270.3.R496. [DOI] [PubMed] [Google Scholar]
- 13.Rosenbaum M, Nicolson M, Hirsch J, Murphy E, Chu F, Leibel RL. Effects of weight change on plasma leptin concentrations and energy expenditure. J Clin Endocrinol Metab. 1997;82:3647–3654. doi: 10.1210/jcem.82.11.4390. [DOI] [PubMed] [Google Scholar]
- 14.Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Eng J Med. 1995;332:621–628. doi: 10.1056/NEJM199503093321001. [DOI] [PubMed] [Google Scholar]
- 15.Zoetendal EG, Akkermans AD, De Vos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol. 1998;64:3854–3859. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costello EK, et al. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huttenhower C, et al. Human Microbiome Project Consortium, Stucture, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajilic-Stojanovic M, Heilig HGHJ, Tims T, Zoetendal EG, de Vos WM. Long-term monitoring of the human intestinal microbiota composition. Environ Microbiol. 2012 doi: 10.1111/1462-2920.12023. [DOI] [PubMed] [Google Scholar]
- 19.Goodman AL, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci USA. 2011;108:6252–6257. doi: 10.1073/pnas.1102938108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henz SR, Huson DH, Auch AF, Nieselt-Struwe K, Schuster SC. Whole-genome prokaryotic phylogeny. Bioinformatics. 2005;21:2329–2335. doi: 10.1093/bioinformatics/bth324. [DOI] [PubMed] [Google Scholar]
- 22.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansen EE, et al. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proc Natl Acad Sci USA. 2011;108:4599–4606. doi: 10.1073/pnas.1000071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peterson DA, McNulty NP, Guruge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host & Microbe. 2007;2:328–329. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 25.Caporaso JG, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halfvarson J. Genetics in twins with Crohn’s disease: less pronounced than previously believed? Inflammatory Bowel Dis. 2011;17:6–12. doi: 10.1002/ibd.21295. [DOI] [PubMed] [Google Scholar]
- 27.Slutske WS, Hunt-Carter EE, Nabors-Oberg RE, Sher KJ, Bucholz KK, Madden PA, Anokhin A, Heath AC. Do college students drink more than their non-college-attending peers? Evidence from a population-based longitudinal female twin study. J Abnorm Psychol. 2004;113:530–540. doi: 10.1037/0021-843X.113.4.530. [DOI] [PubMed] [Google Scholar]
- 28.Waldron M, Bucholz KK, Lynskey MT, Madden PA, Heath AC. Alcoholism and timing of separation in parents: findings in a midwestern birth cohort. J Stud Alcohol Drugs. 2013;74:337–348. doi: 10.15288/jsad.2013.74.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodman AL, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host & Microbe. 2009;6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zerbino DR, McEwen GK, Margulies EH, Birney E. Pebble and rock band: heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PloS One. 2009;4:e8407. doi: 10.1371/journal.pone.0008407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lozupone C, et al. Identifying genomic and metabolic features that can underlie early successional and opportunistic lifestyles of human gut symbionts. Genome Res. 2012;22:1974–1984. doi: 10.1101/gr.138198.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci USA. 1977;74:5088–5090. doi: 10.1073/pnas.74.11.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walters WA, et al. PrimerProspector: de novo design and taxonomic analysis of barcoded PCR primers. Bioinformatics. 2011;27:1159–1161. doi: 10.1093/bioinformatics/btr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muegge BD, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science. 2011;332:970–974. doi: 10.1126/science.1198719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007;35:e120. doi: 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caporaso JG, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choe SE, Boutros M, Michelson AM, Church GM, Halfon MS. Preferred analysis methods for Affymetrix GeneChips revealed by a wholly defined control dataset. Genome Biol. 2005;6:R16. doi: 10.1186/gb-2005-6-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.