

Abstract
The Hedgehog (Hh) family of secreted signaling proteins is a master regulator of cell fate determination in metazoans, contributing to both pattern formation during embryonic development and postembryonic tissue homeostasis. In a universally used mode of action, graded distribution of Hh protein induces differential cell fate in a dose-dependent manner in cells that receive Hh. Though much of this pathway has been elucidated from genetically based studies in model organisms, such as Drosophila and mice, the importance of Hh-mediated signaling in humans is clearly evident from malformations and a broad range of cancers that arise when the pathway is corrupted.
The goal of this Perspective and the two Connections Maps (1, 2) is to highlight recent insights into the unconventional methods by which Hh proteins normally function and how this pathway is implicated in pathological contexts such as cancer. Production of active Hh protein begins with autocatalytic cleavage of a precursor molecule to yield a cholesterol-modified amino-terminal signaling domain (HhN). Subsequent palmitoylation of HhN results in a dually lipidated molecule that is restricted to the cell membrane. Release of HhN from the cell membrane is mediated by Dispatched (Disp/Disp1, hereafter Disp), a 12-transmembrane protein that is structurally similar to the Hh receptor, Patched (Ptc/Ptch1, hereafter Ptc) (Fig. 1A). Both proteins belong to the Resistance Nodulation Division (RND) superfamily of proteins that in prokaryotes function to transport small molecules across membranes. Disp and Ptc likely act as small-molecule transporters, as their activity in the Hh pathway is dependent on residues important to the function of RND protein family members. In Drosophila, HhN released by Disp appears to be incorporated into particles scaffolded by the lipid-transporting lipophorin proteins (3). This previously unknown role for lipoprotein complexes in Hh signaling may represent a universal mechanism for distributing other lipophilic signaling molecules in animals, such as the Wnt proteins (3).
Fig. 1.
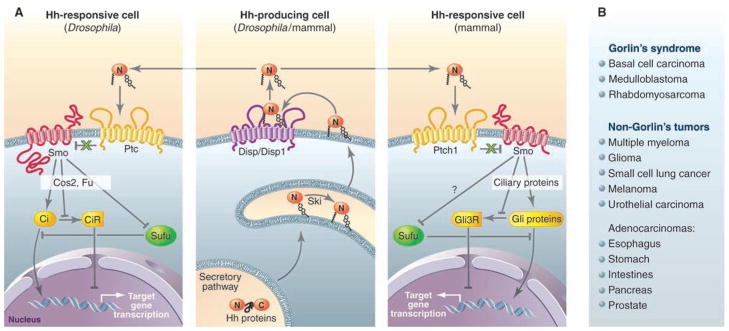
Hh signaling in animals. (A) Hh production is highly conserved between Drosophila and mammals (middle). Autocatalytic cleavage of the Hh precursor protein yields a cholesterol-modified signaling peptide (HhN), which is further palmitoylated by Skinny Hedgehog (Ski), then released from the cell by Disp (Drosophila) or Disp1 (mammals). In Drosophila (left), the released lipophilic HhN is incorporated into lipophorin complexes (not shown) and distributed to other cells with the help of heparan sulfate proteoglycans (Dally and Dlp; also not shown). The suppressive action of Ptc or Ptch1 on Smo is conserved in Hh-responsive cells from Drosophila (left) and mammals (right). Members of the CDO receptor family (not shown), including Ihog, facilitate Hh binding and inhibition of Ptc or Ptch1, allowing activation of Smo. In Drosophila, Dlp also appears to facilitate Hh response. Smo-mediated regulation of Ci (Drosophila) or Gli (mammals) nuclear localization and proteolytic processing into a repressor (CiR or Gli3R) depends on Cos2, Fu, and Su(fu) in Drosophila, and proteins that function in the primary cilium in mammals. The mechanism by which Smo inhibits the pathway suppressor Su(fu) in mammals is unknown. (B) Tumors with aberrant Hh pathway activity in Gorlin’s syndrome, as well as their sporadic counterparts, frequently harbor mutations in Ptch1. Other tumors for which molecular lesions have not been defined also exhibit aberrant Hh pathway response. References for most of the tumors described here can be found in (16).
Initiation of the pathway response entails Hh proteins binding to Ptc, which in turn derepresses the seven-transmembrane protein Smoothened (Smo). This process may be transduced through a small molecular intermediary because Ptc sub-stoichiometrically inhibits Smo (4). Several candidate small molecules have emerged, including cholesterol biosynthesis metabolites, such as oxysterols, that promote Smo function when exogenously added to cultured cells (5). Considering that the potent Smo antagonist cyclopamine, a naturally occurring teratogen, is structurally similar to sterols, there is growing evidence that Ptc gates interactions between Smo and specific sterols to regulate Smo function.
A number of receptors facilitate Hh binding to Ptc (1, 2), including members of the cell adhesion molecule–related/down-regulated by oncogenes (Cdo) family. Cdo and its Drosophila homolog Interference hog (Ihog) associate with Hh through a fibronectin type III (FnIII) repeat (6, 7), a motif with potential for binding sulfate ions (8). Indeed, dimerization of Ihog and its conversion from a weak to a high-affinity Hh-binding molecule can be induced by heparin, a protein with sulfated polysaccharide modifications. How Ihog and Cdo proteins promote Hh-mediated responses in coordination with Ptc and other Hh receptors, particularly the heparan sulfate–modified Dally-like protein (Dlp), which also contributes to the Hh response (1), remains to be addressed.
Ultimately, the concerted action of these receptors activates Smo by promoting Hh interaction with Ptc. Smo activation is best understood in Drosophila (9). Initial phosphorylation of cytosolic tail sequences in Smo corresponds with protein accumulation at the plasma membrane, thus favoring interactions with a cytoplasmic regulatory complex scaffolded by the kinesin-like molecule Costal-2 (Cos2). This complex also contains the serine/threonine kinase Fused (Fu) and the transciptional effector Cubitus interruptus (Ci) (Fig. 1A). The mechanism by which Smo stimulates the transcriptional activity of Ci and inhibits proteolytic processing of Ci to a repressor (CiR) through Cos2, Fu, and another cytoplasmic regulator, Suppressor of Fu [Su(fu)], was previously reviewed (9).
The ultimate target of Smo action in mammals is the Gli zinc finger family of proteins composed of three mammalian homologs of Ci (Gli1 to Gli3), with proteolytically processed Gli3 (Gli3R) predominantly functioning as a transcriptional repressor (Fig. 1A). Many studies support the hypothesis that Smo in Drosophila and mammals uses different mechanisms of action to activate Ci or Gli proteins, respectively (10). The inability to identify mammalian Cos2 and Fu homologs also likely exemplifies differences in Hh signaling between flies and mammals (2, 10).
Insight into the mammalian Hh pathway has come from forward genetic screens in mice with chemically induced mutations that have revealed genes essential to neural tube formation, a Hh-dependent process (11). Surprisingly, the majority of these genes are involved in the formation of the primary cilium, a microtubule-scaffolded organelle found in most cells (2). Subsequent studies revealed that components of the cilia, such as intraflagellar transport proteins, participate not only in the activation of Gli proteins but also in the processing of Gli3 (11). In this capacity, ciliary proteins appear to be the functional equivalent of Cos2 (Fig. 1A). Furthermore, almost all the known mammalian Hh components, including Ptch1, Smo, Su(fu), and Gli proteins, localize to primary cilia (11, 12). Ptch1 apparently inhibits the localization of Smo to cilia in a Hh-dependent manner, suggesting that this compartment is essential to Smo activation (12). Though it is conceivable that the cilium may simply represent an assembly point for pathway components, its requirement for both Hh pathway activation and suppression implicates a more direct role.
More complete understanding of cilia and their role in the Hh response awaits the identification of the immediate downstream effector of Smo and its subcellular localization. In Drosophila, the kinase Fu appears to contribute to most downstream events controlled by Smo, including suppression of Cos2 and Su(fu) (13, 14). The central role of Fu to Hh response in insects implies that a functional equivalent of Fu remains to be found in mammals. Whether or not a mammalian Fu exists, the mechanism of Smo action will likely have to account for Hh-dependent regulation of Su(fu), which appears to function as a major pathway suppressor in both insects and mammals (Fig. 1A) (15).
The role of the Hh pathway in tumorigenesis likely exemplifies its nearly universal participation in cell fate decision-making (16). Hh-related tumors can be broadly categorized based on whether or not they present as part of Gorlin’s (also called basal cell nevus) syndrome. Patients with this syndrome often harbor an inactivating mutation in Ptch1, which, independently of Hh ligand, promotes a broad range of tumors, most frequently basal cell carcinoma (Fig. 1B). Conversely, the oncogenic events that drive the Hh-dependent aberrant response often observed in non-Gorlin’s tumors have not been defined, despite the greater number of cancers that fall into this category (Fig. 1B). These tumors likely are sustained by Hh-dependent cell-autonomous signaling in populations of cancer stem cells (17), suggesting that therapeutics that attack the Hh pathway may offer specificity over strategies that generally block cell proliferation.
The wealth of mechanistic insight into how the Hh pathway functions has revealed both the sophistication of this signal transduction network and the challenges that remain in the treatment of Hh-related diseases. Of urgency is the development of rational therapeutic approaches using knowledge of the pathway to specifically target the events underlying aberrant pathway response. Success in this endeavor will require an understanding of how primary cilia, lipoproteins, and sterol biosynthesis contribute to Hh-related diseases.
Acknowledgments
Our research is supported by an endowment from Virginia Murchison Linthicum, the NIH, the American Cancer Society, and the Welch Foundation. We thank M. Dodge and other members of the Lum laboratory for useful discussions.
References and Notes
- 1.Jacob L, Lum L. Hedgehog signaling pathway. Sci STKE. (Connections Map, as seen October 2007) http://stke.sciencemag.org/cgi/cm/stkecm; CMP_19889.
- 2.Jacob L, Lum L. Hedgehog signaling pathway in Drosophila. Sci STKE. (Connections Map, as seen October 2007), http://stke.sciencemag.org/cgi/cm/stkecm; CMP_20386.
- 3.Panakova D, Sprong H, Marois E, Thiele C, Eaton S. Nature. 2005;435:58. doi: 10.1038/nature03504. [DOI] [PubMed] [Google Scholar]
- 4.Taipale J, Cooper MK, Maiti T, Beachy PA. Nature. 2002;418:892. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- 5.Corcoran RB, Scott MP. Proc Natl Acad Sci USA. 2006;103:8408. doi: 10.1073/pnas.0602852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tenzen T, et al. Dev Cell. 2006;10:647. doi: 10.1016/j.devcel.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Yao S, Lum L, Beachy P. Cell. 2006;125:343. doi: 10.1016/j.cell.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 8.McLellan JS, et al. Proc Natl Acad Sci USA. 2006;103:17208. doi: 10.1073/pnas.0606738103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lum L, Beachy PA. Science. 2004;304:1755. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- 10.Varjosalo M, Li SP, Taipale J. Dev Cell. 2006;10:177. doi: 10.1016/j.devcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Huangfu D, Anderson KV. Development. 2006;133:3. doi: 10.1242/dev.02169. [DOI] [PubMed] [Google Scholar]
- 12.Rohatgi R, Milenkovic L, Scott MP. Science. 2007;317:372. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 13.Claret S, Sanial M, Plessis A. Curr Biol. 2007;17:1326. doi: 10.1016/j.cub.2007.06.059. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Cao X, Jiang J, Jia J. Genes Dev. 2007;21:1949. doi: 10.1101/gad.1557407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Svard J, et al. Dev Cell. 2006;10:187. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 16.Beachy PA, Karhadkar SS, Berman DM. Nature. 2004;432:324. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 17.Peacock CD, et al. Proc Natl Acad Sci USA. 2007;104:4048. [Google Scholar]