

Abstract
The overexpression of the serine/threonine specific polo-like kinase 1 (Plk1) has been detected in various types of cancer, and thus has fast become an attractive therapeutic target for cancer therapy. BI 2536 is the first selective inhibitor of Plk1 that inhibits cancer cell proliferation by promoting G2/M cell cycle arrest at nanomolar concentrations. Unfortunately, alike most chemotherapeutic agents, the development of acquired resistance to BI 2536 is prone to present a significant therapeutic challenge. One of the most common mechanisms for acquired resistance in cancer chemotherapy is associated with the overexpression of ATP-binding cassette (ABC) transporters ABCB1, ABCC1 and ABCG2. Here, we discovered that overexpressing of either ABCB1 or ABCG2 is a novel mechanism of acquired resistance to BI 2536 in human cancer cells. Moreover, BI 2536 stimulates the ATPase activity of both ABCB1 and ABCG2 in a concentration-dependent manner, and inhibits the drug substrate transport mediated by these transporters. More significantly, the reduced chemosensitivity and BI 2536-mediated G2/M cell cycle arrest in cancer cells overexpressing either ABCB1 or ABCG2 can be significantly restored in the presence of selective inhibitor or other chemotherapeutic agents that also interact with ABCB1 and ABCG2, such as tyrosine kinase inhibitors nilotinib and lapatinib. Taken together, our findings indicate that in order to circumvent ABCB1 or ABCG2-mediated acquired resistance to BI 2536, a combined regimen of BI 2536 and inhibitors or clinically active drugs that potently inhibit the function of ABC drug transporters, should be considered as a potential treatment strategy in the clinic.
Keywords: ABC transporter, multidrug resistance, Polo-like kinase 1, BI 2536
1. Introduction
Polo-like kinase 1 (Plk1) is a serine/threonine kinase that is known to regulate multiple key steps of mitosis [1–3], including mitotic entry [4], centrosome maturation [5], formation of the bipolar spindle [6], mitotic exit [7], and a target of the DNA damage checkpoint [8]. Overexpression of Plk1 has been detected in multiple human cancers and was found associated with poor prognosis in various cancers [3], thus providing a rationale for Plk1 as a valid target for anti-cancer treatment [3, 9, 10]. Small molecule BI 2536 has been shown to be highly effective against the activity of Plk1, causing significant cell cycle arrest and apoptosis in many cancers, including Plk1-overexpressing acute myeloid leukemia (AML) [11], chronic myeloid leukemia (CML) [12], triple-negative breast cancer (TNBC) [13], cervical carcinoma [14], multiple myeloma [15] and inhibits the proliferation of osteosarcoma cells [16], neuroblastoma cells [17], melanoma cells [18] and medulloblastoma cells [19]. Therefore, several inhibitors targeting the Plk1 pathway, including BI 2536, have now entered clinical development [20–22].
In contrast to an overwhelming number of encouraging studies and reports on the anticancer activity of BI 2536, very little is known about the mechanisms of acquired resistance to BI 2536 [23–25]. The development of multidrug resistance (MDR) remains as a major obstacle in modern cancer chemotherapy. While multiple mechanisms are known to contribute, the most common mechanism for acquired resistance in cancer chemotherapy is associated with the overexpression of three major ATP-binding cassette (ABC) drug transporters ABCB1, ABCC1 and ABCG2 [26, 27]. These transporters are capable of effluxing a wide range of functionally and structurally unrelated anticancer agents directly out of cancer cells, causing MDR, relapse and death [27]. In recent years, the efficacy of many therapeutic protein kinase inhibitors, including nilotinib and vemurafenib, has been questioned due to the emergence of MDR that is associated with the overexpression of ABC drug transporters in cancers [28–30]. Therefore, it is crucial to study the pharmacological and biochemical interactions between BI 2536 and these three major ABC drug transporters, as well as exploring the potential beneficial use of combined therapy of BI 2536 and modulators of ABC drug transporters.
In the present study, we discovered that the overexpression of human ABCB1 and ABCG2 proteins, but not ABCC1, can lead to substantial acquired drug resistance to BI 2536 in cancer cells. We demonstrated that BI 2536 potently inhibits efflux mediated by either ABCB1 or ABCG2, and that BI 2536 has higher affinity towards ABCB1 than ABCG2. Moreover, the stimulation of ABCB1 and ABCG2 ATPase hydrolysis by BI 2536 is consistent with BI 2536 being a transport substrate of both ABCB1 and ABCG2. In addition, we showed that the chemosensitivity and BI 2536-mediated G2/M cell cycle arrest in ABCB1 and ABCG2-overexpressing MDR cancer cells can be restored significantly by inhibiting the function of ABCB1 and ABCG2. Together, these results reveal a novel resistance mechanism for Plk1 inhibitor BI 2536 in cancer cells, as well as provides a rationale for investigating combination co-administration of BI 2536 and modulators of ABC drug transporters in cancer patients to increase the efficiency of chemotherapy.
2. Materials and methods
2.1. Cell cultures and compounds
KB-3-1, KB-8-5-11, KB-C-1, KB-V-1, OVCAR-8, NCI-ADR-RES, NIH3T3, NIH3T3-G185, pcDNA3.1-HEK293, R482-HEK293, MDR19-HEK293, MRP1-HEK293, MCF7 and MCF7-FLV1000 cells were cultured in DMEM (Gibco, Invitrogen, Carlsbad, CA, USA), supplemented with 10 % fetal calf serum (FCS), 2 mM L-glutamine and 100 units of penicillin/streptomycin/mL at 37 °C in 5 % CO2 humidified air. All HEK293 and HEK293 transfected lines were maintained in 2 mg/mL G418 [29]; MCF7-FLV1000 cells were cultured in the presence of 1 μg/mL flavopiridol [31, 32], whereas 1 μg/μL vinblastine was added to KB-V-1 cells [33]. CORL-23/P, CORL-23/R, S1 and S1-M1-80 cells were cultured in RPMI-1640 (Gibco, Invitrogen, CA, USA), supplemented with 10 % FCS, 2 mM L-glutamine and 100 units of penicillin/streptomycin/mL at 37 °C in 5 % CO2 humidified air. The CORL-23/R cells were maintained in the presence of 0.2 μg/mL of doxorubicin, whereas S1-M1-80 cells were cultured in 80 μM of mitoxantrone as described previously [29]. NIH3T3-G185 cells were maintained in the presence of 60 ng/mL colchicine [34]. Mitoxantrone, MTT dye, Cell Counting Kit-8 (CCK-8), doxorubicin and all other chemicals were purchased from Sigma (St. Louis, MO, USA), unless stated otehrwise. Fumitremorgin C (FTC) and tariquidar were generous gifts from Dr Susan Bates (National Cancer Institute, NIH, Bethesda, MD). BI 2536 (99% purity by HPLC, Chiral HPLC) was purchased from ChemieTek (Indianapolis, IN, USA).
2.2. Cytotoxicity assay
CCK-8 and MTT assays were used to determine the general sensitivities of cells to the tested chemicals as described previously [29].
2.3. Fluorescent drug accumulation assay
ABCB1 and ABCG2-mediated efflux assays were carried out using a FACSort flow cytometer equipped with Cell Quest software (Becton-Dickinson) as described previously [35]. The effect of BI 2536, tariquidar, MK-571 and FTC on ABCB1-mediated calcein-AM efflux or ABCC1-mediated calcein efflux or ABCG2-mediated efflux of pheophorbide A (PhA), was measured and analyzed as described previously [35].
2.4. ATPase assay of ABCB1 and ABCG2
ATPase activity of ABCB1 and ABCG2 in High-Five cell crude membranes was measured by endpoint Pi assay as described previously [36]. ABCB1- and ABCG2-specific ATPase activities were recorded as vanadate (Vi)-sensitive ATPase activity as described previously [36].
2.5. Immunoblotting
Antibodies C219 (1:1000), BXP-21 (1:500) and anti-α-tubulin (1:2000) were used to detect ABCB1, ABCG2 and tubulin as positive control for Western blotting, respectively. The secondary antibody used was the Horseradish peroxidase-conjugated goat anti-mouse IgG (1:10,000). Signals were detected as described previously [37].
2.6. Cell cycle analysis
Cell cycle experiments were carried out using standard propidium iodide (PI) staining method and analyzed using a FACSort flow cytometer equipped with Cell Quest software (Becton-Dickinson). Briefly, cells were treated with indicated regimens for 24 hrs before harvested in PBS and fixed in ethanol overnight. Cells were washed once with PBS, then treated with 0.5 % TritonX-100 and 0.05 % RNase in PBS at 37 °C for 1 hr. Cells were washed, propidium iodide (50 μg/mL) added, then incubated the cells at 4 °C for at least 20 min before analysis.
2.7. Statistical analysis
Data are presented as mean ± S.E.M, whereas IC50 values were calculated as mean ± SD from at least three independent experiments. Differences between any mean values were analyzed by two-sided Student’s t-test and results were considered statistically significant at P < 0.05.
3. Results
3.1. The chemosensitivity to BI 2536 is significantly reduced in cells overexpressing ABCB1or ABCG2
Based on many reports of BI 2536 inhibits the proliferation of human cancer cells, we first examined the BI 2536-induced toxicity in multiple drug sensitive and resistant human cancer cell lines. The resistance factor (RF) is used here to compare the relative toxicity of BI 2536 to cells overexpressing a given ABC drug transporter. RF represents the degree of resistance caused by the presence of a particular ABC transporter, which is calculated by dividing the IC50 value of ABC transporter overexpressing subline by the IC50 value of the respective parental line (Table 1). We observed straight away that KB-8-5-11, KB-C-1 and KB-V-1, three ABCB1-positive sublines of human epidermal KB-3-1 cell line, were highly resistant to BI 2536 when compared to the drug-sensitive parental cell lines (Table 1). Moreover, the levels of resistance correlated with the amount of ABCB1 protein expression amongst the KB sublines (Fig. 1A). Subsequently, we found that ABCB1-overexpressing human ovary NCI-ADR-RES cells were also highly resistant to BI 2536 (Fig. 1B). In order to investigate whether the resistance of BI 2536 is organ or transporter-specific, we continued to examine several other cell lines with origins from the breast, the colon and lung, which overexpress other ABC transporters such as ABCG2 and ABCC1 (summarized in Table 1). We found that sublines with ABCG2 overexpression were also substantially more resistant to BI 2536, with the calculated RF values of approximately 77 and 11 for MCF7-FLV1000 and S1-M1-80 cells, respectively compared to the parental cell lines. In contrast, drug sensitive CORL-23/P and ABCC1-overexpressing CORL-23/R cells were equally sensitive to BI 2536. It is worth noting that human colon S1 cancer cells appeared to be intrinsically less sensitive to BI 2536 than the other drug sensitive human cancer cells used in the study. In order to affirm the cytotoxicity of BI 2536 is mediated by the overexpression of ABC drug transporters, we examined and compared the relative toxicity of BI 2536 in cells transfected with either ABCB1, ABCG2 or ABCC1 to their respective parental cells. Similar to the results observed in human cancer cell lines, the ABCB1-transfected mouse fibroblast NIH3T3-G185 and human MDR19-HEK293 cells (Fig. 1C) were significantly more resistant to BI 2536 than the parental cells, with calculated RF values of 59 and 31, respectively. The ABCG2-transfected R482-HEK293 cells were almost 10-fold more resistant to BI 2536 than the parental cells (Fig. 1D), whereas the ABCC1-transfected MRP1-HEK293 cells were equally sensitive to BI 2536 as the parental cells (Fig. 1E).
Table 1.
Sensitivity of various cell lines to polo-like kinase 1 inhibitor BI 2536
| Cell line | Cancer origin | Transporter expressed | IC50 (nM)a | R.Fb |
|---|---|---|---|---|
| KB-3-1 | epidermal | - | 1.48 ± 0.53 | - |
| KB-8-5-11 | epidermal | ABCB1 | 5.01 ± 2.01* | 3.39 |
| KB-C-1 | epidermal | ABCB1 | 16.76 ± 4.51** | 11.32 |
| KB-V-1 | epidermal | ABCB1 | 281.24 ± 31.57*** | 190.03 |
| OVCAR-8 | ovary | - | 7.19 ± 3.60 | - |
| NCI-ADR-RES | ovary | ABCB1 | 1273.30 ± 191.20*** | 177.09 |
| MCF7 | breast | - | 5.11 ± 1.47 | - |
| MCF7-FLV1000 | breast | ABCG2 | 393.18 ± 74.83*** | 76.94 |
| S1 | colon | - | 24.30 ± 4.75 | - |
| S1-M1-80 | colon | ABCG2 | 261.06 ± 48.25** | 10.74 |
| CORL-23/P | lung | - | 4.38 ± 1.25 | - |
| CORL-23/R | lung | ABCC1 | 5.16 ± 1.83 | 1.18 |
| NIH3T3 | - | - | 40.60 ± 4.97 | - |
| NIH3T3-G185 | - | ABCB1 | 2413.40 ± 423.66*** | 59.44 |
| pcDNA-HEK293 | - | - | 2.75 ± 0.67 | - |
| MDR19-HEK293 | - | ABCB1 | 85.50 ± 13.67*** | 31.09 |
| R482-HEK293 | - | ABCG2 | 25.97 ± 4.95** | 9.44 |
| MRP1-HEK293 | - | ABCC1 | 2.99 ± 1.49 | 1.09 |
Abbreviation: R.F, resistance factor.
IC50 values are mean ± SD calculated from dose-response curves obtained from three independent experiments using cytotoxicity assay as described in Materials and Methods.
RF were calculated by dividing IC50 values of ABC transporter overexpressing cells by IC50 values of respective parental
cells.
P < 0.05;
P < 0.01;
P < 0.001
Fig. 1.

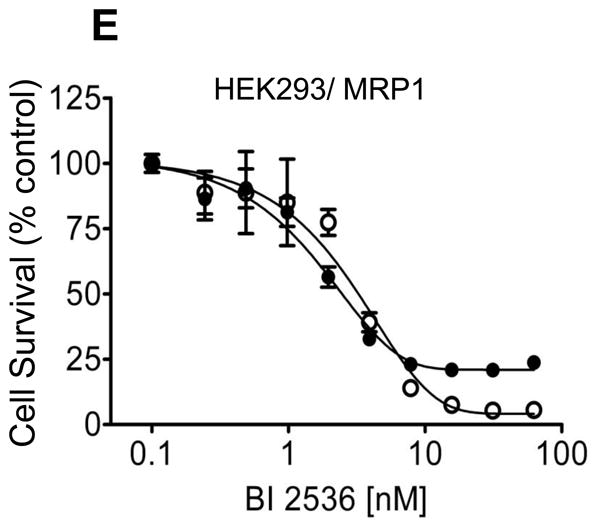
The impact of human ABCB1, ABCC1 and ABCG2 overexpression on cellular sensitivity to BI 2536. (A) The respective cytotoxicity of BI 2536 in KB-3-1 (○) and ABCB1-overexpressing sublines KB-8-5-11 (△), KB-C-1 (□) and KBV-1 (●) cells; (B) OVCAR8 (○) and NCI-ADR-RES (●); (C) parental HEK293 (○) and ABCB1-transfected MDR19-HEK293 cells (●); (D) parental HEK293 (○) and ABCG2-transfected R482-HEK293 cells (●); (E) parental HEK293 (○) and ABCC1-transfected MRP1-HEK293 cells (●), was determined as described previously [29]. Points, mean from at least three independent experiments; bars, SEM.
3.2. BI 2536 inhibits substrate transport mediated by ABCB1 and ABCG2
In order to examine the interactions between BI 2536 and the three major ABC drug transporters, we first studied how BI 2536 directly affects the functions of ABCB1, ABCC1 and ABCG2. We evaluated the inhibitory effect of BI 2536 on ABCB1, ABCC1 and ABCG2-mediated drug efflux in short-term drug accumulation assays. Calcein-AM is a known substrate of ABCB1 and ABCC1, whereas PhA is a known fluorescent substrate of ABCG2, thus were used to track ABCB1, ABCC1 or ABCG2-mediated efflux, respectively [29]. The efflux assays were carried out in the absence (solid lines) or presence of BI 2536 (shaded, solid lines) or the benchmark inhibitor (dotted lines) of ABCB1 (tariquidar, 3 μM) or ABCC1 (MK-571, 25 μM) or ABCG2 (FTC, 5 μM) as described in Materials and Methods. As shown in Fig. 2A and 2B, the ABCB1-mediated calcein-AM efflux was fully inhibited by BI 2536 at 10 μM in KB-V-1 and MDR19-HEK293 cells. In contrast, at the same concentration, BI 2536 had no significant effect on ABCC1-mediated calcein efflux in MRP1-HEK293 cells (Fig. 2C) or ABCG2-mediated PhA efflux in MCF7-FLV1000 (Fig. 2D), S1-M1-80 (Fig. 2E) or R482-HEK293 (Fig. 2F) cells. However, we discovered that BI 2536 does in fact inhibit ABCG2-mediated efflux of PhA, but only at higher concentrations. BI 2536 inhibits ABCB1-mediated calcein-AM efflux from KB-V-1 cells (Fig. 2G, left panel) and ABCG2-mediated PhA efflux from MCF7-FLV1000 cells (Fig. 2G, right panel) in a concentration dependent manner, with calculated IC50 values of 5.81 ± 3.76 and 44.92 ± 12.62 μM, respectively. On the other hand, ABCC1-mediated calcein efflux was not inhibited by BI 2536 even at higher concentrations (data not shown).
Fig. 2.

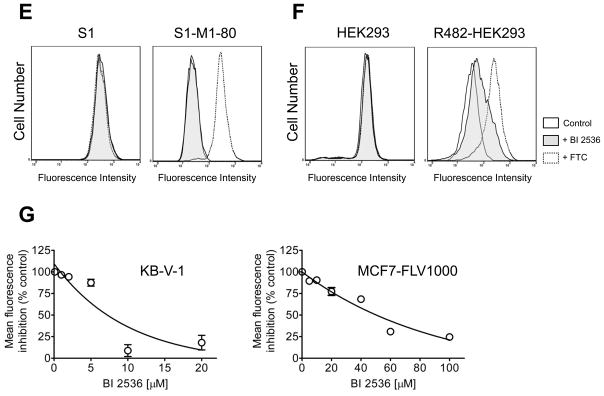
Effect of BI 2536 on the fluorescent substrate transport mediated by ABCB1, ABCG2 or ABCC1. The accumulation of fluorescent calcein in KB-3-1 (A, left panel) and KB-V-1 (A, right panel); HEK293 (B, left panel) and MDR19-HEK293 (B, right panel); HEK293 (C, left panel) and MRP1-HEK293 (C, right panel) cells, as well as the accumulation of fluorescent substrate Pheophorbide A (PhA) in MCF7 (D, left panel) and MCF7-FLV1000 (D, right panel); S1 (E, left panel) and S1-M1-80 (E, right panel); HEK293 (F, left panel) and R482-HEK293 (F, right panel) cells was measured in the presence or absence of BI 2536 or known inhibitors and analyzed immediately by flow cytometry as described in Materials and Methods. Representative histograms of three independent experiments are shown; cells in the absence of 10 μM BI 2536 or inhibitors (solid lines), shaded solid lines represent cells in the presence of 10 μM BI 2536 (shaded solid lines), whereas dotted lines represent cells in the presence of known inhibitors (3 μM tariquidar for ABCB1, 25 μM MK-571 for ABCC1 and 5 μM FTC for ABCG2). (G) Concentration-dependent inhibition of ABCB1-mediated calcein-AM efflux (left panel) or ABCG2-mediated PhA efflux (right panel) by BI 2536 in KB-V-1 cells or MCF7-FLV1000 cells. Data points represent the mean ± SEM from three independent experiments. The IC50 values were calculated as the concentration that inhibited the efflux to 50 % of the control values.
3.3. BI 2536 stimulates ATPase activity of ABCB1 and ABCG2
To obtain biochemical information on the interactions between BI 2536 and ABCB1 or ABCG2, we studied the effect of BI 2536 on the Vi-sensitive ATPase activity in crude membranes isolated from insect cells expressing either ABCB1 or ABCG2, as described in Materials and Methods. BI 2536 stimulates Vi-sensitive ATPase activity of both ABCB1 and ABCG2 in a concentration dependent manner, with maximum stimulation of approximately 250 % (Fig. 3A) and 200 % (Fig. 3B) higher than the respective basal levels of ABCB1 and ABCG2, respectively. The concentration for BI 2536 to obtain 50 % stimulation of ATPase activity of ABCB1 and ABCG2 was 0.7 μM and 0.9 μM, respectively (Fig. 3). The data here showed that BI 2536 stimulates ABCB1 and ABCG2 ATP hydrolysis in the same manner as other well-established substrates.
Fig. 3.

Effect of BI 2536 on Vi-sensitive ABCB1 and ABCG2 ATPase activity. Effect of 0 to 10 μM BI 2536 (A) and 0 to 1 μM (A, insert) on Vi-sensitive ABCB1 ATPase activity, as well as the effect of 0 to 30 μM BI 2536 (B) and 0 to 1 μM (B, insert) on Vi-sensitive ABCG2 ATPase activity was measured as described previously [37]. Points, mean from three independent experiments; bars, SD.
3.4. BI 2536 has no effect on ABCB1 or ABCG2 protein expression level
Several studies have shown that some compounds which interact directly with ABC drug transporters can potentially cause up-regulation or down-regulation of ABC proteins [38–40]. Therefore, we examined the effect of BI 2536 on the protein expression level of ABCB1 in KB-3-1 and ABCB1-overexpressing KB-V-1 cells (Fig. 4A), as well as ABCG2 in MCF7 and ABCG2-overexpressing MCF7-FLV1000 cells (Fig. 4B). Cells were treated with increasing concentrations of BI 2536 for 72 hrs before processed for immunodetection of ABCB1 or ABCG2 as described in Materials and Methods. We found that BI 2536 has no significant effect on the protein expression level of either ABCB1 or ABCG2 in these cancer cell lines. Our data indicated that even though BI 2536 directly inhibits the function of ABCB1 and ABCG2, it does not induce up-regulation or down-regulation of these transporters.
Fig. 4.

Effect of BI 2536 on expression level of ABCB1 and ABCG2 proteins in human MDR cancer cells. The averaged relative protein expression levels obtained from immunoblot detection of (A) ABCB1 in total cell lysate protein (10 μg) in parental KB-3-1 and ABCB1-overexpressing subline KB-V-1 cells, treated with increasing concentrations (0 – 0.2 nM) or (0 – 100 nM) of BI 2536, and (B) ABCG2 in parental MCF7 and ABCG2-overexpressing subline MCF7-FLV1000 cells, treated with increasing concentrations (0 – 5 nM) or (0 – 200 nM) of BI 2536, respectively for 72 h as described previously [37]. α-Tubulin was used as an internal control for equal loading. Values are presented as mean ± SD calculated from three independent experiments.
3.5. The chemosensitivity of ABCB1 and ABCG2-overexpressing cancer cells to BI 2536 can be restored by using a substrate or modulator of ABCB1 and ABCG2
Knowing that ABCB1 and ABCG2 can mediate resistance to BI 2536, we next evaluated the effect of a substrate or modulator of ABC drug transporters on BI 2536-induced cell cycle arrest and toxicity in cells overexpressing either ABCB1 or ABCG2. We first examined the effect of ABCB1 substrate nilotinib (5 μM) and inhibitors tariquidar (1 μM) and lapatinib (5 μM) on BI 2536-induced G2/M cell cycle arrest in human epidermoid carcinoma KB-3-1 (Fig. 5A) and KB-V-1 cells (Fig. 5B). Substrate or inhibitor alone at the tested concentrations had no effect on G2/M arrest in all of the cell lines used in this study (Fig. 5A–D, left panels). At 10 nM, BI 2536-mediated significant G2/M arrest in KB-3-1 cells (from ≈ 10 % to 86 %) in the absence or presence of above compounds (Fig. 5A, right panels). In contrast, in the absence of nilotinib, tariquidar or lapatinib, BI 2536 at 10 nM had no effect on G2/M arrest in ABCB1-overexpressing KB-V-1 cells, while in the presence of inhibitors, the level of BI 2536-induced G2/M arrest was restored to the same extent as in ABCB1-negative KB-3-1 cells (Fig. 5B, right panels). Similarly, we examined the effect of ABCG2 substrate nilotinib and inhibitors FTC (3 μM) and lapatinib (5 μM) on BI 2536-induced G2/M arrest in human colon carcinoma S1 (Fig. 5C) and S1-M1-80 cells (Fig. 5D). Since S1 cells are less sensitive to BI 2536 than KB cells (Table 1), a higher concentration of BI 2536 was used to treat S1 cells in this experiment. At 100 nM, BI 2536 induced significant G2/M arrest (from ≈ 8 % to 90 %) in S1 cells in the absence or presence of inhibitors (Fig. 5C, left panels), but had no significant effect on G2/M arrest in ABCG2-overexpressing S1-M1-80 cells. However, in the presence of FTC, nilotinib or lapatinib, the level of BI 2536-mediated G2/M arrest was restored in S1-M1-80 cells (Fig. 5D, right panels). The overall effect of BI 2536 and inhibitors on the phases of cell cycle in these cancer cells is summarized in Table 2. Next, we examined the reversal effect of inhibitors on the sensitivity of cells overexpressing either ABCB1 or ABCG2 to BI 2536. As shown in Table 3, in the presence of ABCB1 benchmark inhibitor tariquidar (1 μM), the sensitivity of ABCB1-overexpressing KB-V-1 cells to BI 2536 was restored completely. Moreover, the sensitivity of these cells was also significantly restored by nilotinib (0.5 μM) and lapatinib (0.3 μM), two tyrosine kinase inhibitors that have been shown to inhibit the function of both ABCB1 and ABCG2 [41, 42]. Similarly, the sensitivity of ABCG2-overexpressing S1-M1-80 and MCF7-FLV1000 cells to BI 2536 was significantly restored by ABCG2 benchmark inhibitor FTC (3 μM), as well as by nilotinib and lapatinib. Moreover, we observed the same reversal effect by these drugs in cell transfected with either ABCB1 (MDR19-HEK293) or ABCG2 (R482-HEK293), suggests that inhibition of ABCB1 or ABCG2 function is sufficient enough to reverse ABCB1- or ABCG2-mediated drug resistance to BI 2536 (Table 3).
Fig. 5.




Effect of ABCB1 and ABCG2 inhibitors on BI 2536-mediated G2/M cell cycle arrest in sensitive KB-3-1 (A), resistant ABCB1-overexpressing KB-V-1(B), sensitive S1 (C) and resistant ABCG2-overexpressing S1-M1-80 (D) cells. Cells were plated and maintained in the absence (left panels) or presence of BI 2536 (right panels, 10 nM for KB cells and 100 nM for S1 cells) in combination with tariquidar (1 μM), FTC (3 μM), nilotinib (5 μM) or lapatinib (5 μM) alone (left panels) for 24 h before harvest for cell cycle analysis. Representative histograms of three independent experiments are shown.
Table 2.
The percentage distribution of cells in cycle phases
| Cell cycle phase | |||
|---|---|---|---|
|
| |||
| KB-3-1 | G1 (%) | S (%) | G2/M (%) |
| Control | 64.06 ± 4.34 | 25.46 ± 0.64 | 10.49 ± 0.74 |
| + Tariquidar | 65.08 ± 3.66 | 23.64 ± 0.26 | 11.28 ± 0.95 |
| + Nilotinib | 70.63 ± 1.80 | 19.13 ± 0.49 | 10.24 ± 0.82 |
| + Lapatinib | 68.57 ± 5.94 | 20.71 ± 0.72 | 10.72 ± 2.26 |
| + BI 2536 | 2.94 ± 2.54 | 10.70 ± 2.34 | 86.36 ± 4.45 |
| + BI 2536 + tariquidar | 2.91 ± 2.06 | 10.16 ± 0.33 | 86.93 ± 3.05 |
| + BI 2536 + nilotinib | 7.22 ± 3.43 | 17.88 ± 0.62 | 74.9 ± 2.26 |
| + BI 2536 + lapatinib | 6.56 ± 6.08 | 15.62 ± 0.42 | 77.82 ± 4.23 |
|
| |||
|
KB-V-1
|
|||
| Control | 59.01 ± 1.62 | 29.70 ± 1.28 | 11.29 ± 1.32 |
| + Tariquidar | 63.24 ± 2.18 | 26.22 ± 0.98 | 10.54 ± 0.61 |
| + Nilotinib | 63.81 ± 1.19 | 24.98 ± 1.40 | 11.21 ± 0.65 |
| + Lapatinib | 58.89 ± 2.42 | 28.63 ± 0.72 | 12.48 ± 1.80 |
| + BI 2536 | 59.61 ± 6.13 | 27.63 ± 5.08 | 12.76 ± 1.39 |
| + BI 2536 + tariquidar | 4.74 ± 2.44 | 21.88 ± 6.34 | 73.38 ± 8.26 |
| + BI 2536 + nilotinib | 10.45 ± 1.84 | 31.24 ± 7.09 | 58.31 ± 7.91 |
| + BI 2536 + lapatinib | 6.42 ± 4.71 | 17.51 ± 1.18 | 76.07 ± 5.84 |
|
| |||
|
S1
|
|||
| Control | 48.32 ± 6.10 | 43.34 ± 5.76 | 8.34 ± 3.01 |
| + FTC | 47.64 ± 4.27 | 44.98 ± 5.48 | 7.38 ± 1.50 |
| + Nilotinib | 52.31 ± 7.11 | 36.17 ± 3.87 | 11.52 ± 4.88 |
| + Lapatinib | 52.11 ± 5.49 | 39.05 ± 5.61 | 8.84 ± 3.96 |
| + BI 2536 | 3.57 ± 0.47 | 5.69 ± 0.71 | 90.74 ± 0.42 |
| + BI 2536 + FTC | 3.13 ± 0.27 | 5.32 ± 0.82 | 91.56 ± 0.68 |
| + BI 2536 + nilotinib | 2.33 ± 0.44 | 5.67 ± 0.40 | 92.00 ± 0.04 |
| + BI 2536 + lapatinib | 2.20 ± 0.67 | 5.20 ± 0.48 | 92.60 ± 0.71 |
|
| |||
|
S1-M1-80
|
|||
| Control | 48.06 ± 9.67 | 31.94 ± 4.55 | 20.01 ± 6.14 |
| + FTC | 48.05 ± 8.45 | 32.06 ± 1.59 | 19.89 ± 8.40 |
| + Nilotinib | 49.31 ± 4.52 | 31.33 ± 3.91 | 19.37 ± 0.98 |
| + Lapatinib | 47.28 ± 5.31 | 30.28 ± 3.29 | 22.44 ± 2.26 |
| + BI 2536 | 43.06 ± 6.75 | 32.78 ± 2.14 | 24.16 ± 6.42 |
| + BI 2536 + FTC | 1.39 ± 0.16 | 4.78 ± 0.42 | 93.84 ± 0.58 |
| + BI 2536 + nilotinib | 5.15 ± 1.61 | 17.07 ± 3.26 | 77.78 ± 4.79 |
| + BI 2536 + lapatinib | 1.64 ± 0.59 | 5.81 ± 1.36 | 92.56 ± 1.89 |
The percentage values were calculated from three independent experiments.
Table 3.
Effect of ABC transporter modulators on the chemosensitivity of BI 2536 in ABCB1- or ABCG2-overexpressing cell lines
| Cell lines | IC50 (nM)a | ||||
|---|---|---|---|---|---|
|
| |||||
| BI 2536 | BI 2536 + tariquidar (1 μM) | BI 2536 + FTC (3 μM) | BI 2536 + nilotinib (0.5 μM) | BI 2536 + lapatinib (0.3 μM) | |
| KB-3-1 | 1.48 ± 0.53 | 1.35 ± 0.49 | N.D | 0.83 ± 0.40 | 0.89 ± 0.41 |
| KB-V-1 | 281.24 ± 31.57 | 2.09 ± 0.77*** | N.D | 11.44 ± 1.87*** | 11.83 ± 2.41*** |
| S1 | 24.30 ± 4.75 | N.D | 27.18 ± 2.54 | 13.94 ± 2.69* | 25.14 ± 8.11 |
| S1-M1-80 | 261.06 ± 48.25 | N.D | 32.34 ± 12.73** | 18.53 ± 3.03*** | 13.21 ±4.20*** |
| MCF7 | 5.11 ± 1.47 | N.D | 8.37 ± 2.59 | 4.24 ± 1.36 | 9.68 ± 3.19 |
| MCF7-FLV1000 | 393.18 ± 74.83 | N.D | 41.99 ± 28.36** | 124.21 ± 9.57** | 128.31 ± 8.49** |
| pcDNA-HEK293 | 2.75 ± 0.67 | 1.47 ± 0.50 | 2.62 ± 0.72 | 2.13 ± 0.81 | 1.95 ± 0.59 |
| MDR19-HEK293 | 85.50 ± 13.67 | 1.30 ± 0.46*** | N.D | 7.78 ± 1.77*** | 29.61 ± 8.25** |
| R482-HEK293 | 25.97 ± 4.95 | N.D | 3.95 ± 1.43** | 6.26 ± 1.83** | 7.02 ± 2.31** |
Abbreviation: N.D, not determined.
IC50 values are mean ± SD in the presence and absence of BI 2536 or other tested compounds. The IC50 values were calculated from dose-response curves obtained from three independent experiments.
P < 0.05;
P < 0.01;
P < 0.001
4. Discussion
The serine/threonine-specific Plk1 is a key regulator of mitosis [2]. The fact that Plk1 is overexpressed in multiple human cancer types, and it is not known to regulate any major cellular functions other than mitosis, makes Plk1 a good therapeutic target for cancer treatment [9, 11, 12, 17, 43]. BI 2536 is a dihydropteridinone compound discovered by Steegmaier et al. in a drug screening study for compounds that selectively inhibited the catalytic activity of Plk1. It is the first potent and selective Plk1 inhibitor that induces all hallmarks of Plk1 inhibition and inhibits the proliferation of multiple human cancer cell lines in nanomolar concentrations [24]. Recently, many promising results have been reported by independent groups showing BI 2536 being highly effective against various type of cancers, including AML, CML, TNBC and cervical carcinoma [11–13], as well as medulloblastoma and melanoma [18, 19]. However, very little is known about the potential mechanism of acquired resistance to BI 2536, which could present a significant therapeutic challenge to cancer patients receiving BI 2536 treatment. Given that the overexpression of ABC drug transporters is one of the most common mechanisms of acquired resistance to anticancer agents, we examined whether ABCB1, ABCC1 or ABCG2 overexpression could lead to acquired BI 2536 resistance in human cancer cells.
In the present study, we discovered that cancer cells that overexpress either ABCB1 or ABCG2, are highly resistant to BI 2536. Moreover, ectopic expression of ABCB1 or ABCG2 also confers resistance to BI 2536 in HEK293 cells (Fig. 1, Table 1), implicating that ABCB1- or ABCG2-mediated transport alone can sufficiently reduce the intracellular concentration of BI 2536. To confirm our findings further, we carefully examined the interactions between BI 2536 and ABC drug transporters. First, we studied the effect of BI 2536 on the drug efflux function of ABCB1, ABCC1 or ABCG2. Our data showed that BI 2536 can inhibit ABCB1- and ABCG2-mediated transport of calcein-AM and PhA (Fig. 2), suggest that BI 2536 interacts directly with both ABCB1 and ABCG2. In contrast, BI 2536 appeared not to interact with ABCC1 at all. Interestingly, in a recent study aimed to develop a novel high-throughput assay identifying ABCB1 interacting compounds, BI 2536 was identified as a positive hit from a library 193 compounds [44]. Next, we measured the effect of BI 2536 on the vanadate-sensitive ATPase activity of ABCB1 and ABCG2 (Fig. 3). It has been shown that the stimulation of ATP hydrolysis is coupled with substrate transport by ABC transporters [45, 46]. Our data showed that BI 2536 elicited a 250 % stimulation on ABCB1-mediated ATP hydrolysis, and 200 % stimulation on ABCG2-mediated mediated ATP hydrolysis (Fig. 3). This result not only confirmed the interactions between BI 2536 and ABCB1 and ABCG2, it also showed that BI 2536 behaves as a transported substrate of both ABCB1 and ABCG2. This is further supported by the ability of ABCB1 and ABCG2 to confer resistance to this drug (Fig 1, Table 1). Given that several studies have reported that the protein expression of ABC drug transporters can be regulated by some ABC transporter-interacting drugs or compounds [38–40, 46], we examined the protein expression of ABCB1 and ABCG2 in human cancer cell lines after exposing these cells to BI 2536 for 72 hrs. In contrast to those studies, our data showed that BI 2536 has no significant effect on the protein expression of either ABCB1 or ABCG2 (Fig. 4).
In view of the fact that ABCB1- or ABCG2-mediated resistance to BI 2536 can potentially pose a significantly therapeutic problem in the future, and that functional inhibition of ABC drug transporters has been shown to reverse acquired drug resistance mediated by ABC transporters [37, 41, 47], we examined the effect of a combination therapy using BI 2536 and modulators of ABCB1 and ABCG2 in human cancer cell lines. First, we evaluated the effect of a known ABCB1 inhibitor tariquidar and a known ABCG2 inhibitor FTC, as well as nilotinib, a substrate for both ABCB1 and ABCG2 and lapatinib, a modulator that has been shown to restore drug sensitivity in ABCB1 and ABCG2-overexpressing cells [41], on the BI 2536-mediated G2/M cell cycle arrest and chemosensitivity to BI 2536 in resistant human cancer cell lines. A key characteristic of BI 2536 is the inhibition of human cancer cell growth through induction of G2/M cell cycle arrest with EC50 values in the range of 1 – 20 nM [24]. At 10 nM, BI 2536 was shown to cause cell cycle blockage at the G2 phase or mitosis stage in HeLa cells, thus, increasing G2/M arrest can be considered as a indication of the effectiveness of BI 2536 in various cell lines [24]. The reported EC50 values are in consistent with our findings. In our study, BI 2536 is highly effective against the proliferation of KB-3-1 human epidermoid carcinoma cells, OVCAR8 human ovarian cancer cells, MCF-7 human breast cancer cells, S1 human colorectal cancer cells and CORL-23/P human lung cancer cells, however, BI 2536 was significantly less effective against their ABCB1 and ABCG2-overexpressing sublines (Table 1). In order to examine the effect modulators and BI 2536 has on ABCB1- and ABCG2-overexpressing human cancer cells, we studied the BI 2536-mediated G2/M cell cycle arrest in drug sensitive KB-3-1 and S1 cancer cell lines, as well as in resistant KB-V-1 and S1-M1-80 sublines, in the absence or presence of modulators. At 10 nM and 100 nM, BI 2536 induced high levels of G2/M arrest in KB-3-1 and S1 cell lines, while had no significant effect in ABCB1-overexpressing KB-V-1 or ABCG2-overexpressing S1-M1-80 sublines (Fig. 5). However, in the presence of tariquidar, nilotinib or lapatinib, the BI 2536-mediated G2/M arrest in resistant KB-V-1 cells was restored to a comparable level as in KB-3-1 cells (Table 2), which is also reflected in the corresponding IC50 values listed in Table 3. Similarly, in ABCG2-overexpressing S1-M1-80 cells, the level of BI 2536-mediated G2/M arrest and IC50 value were significantly restored by FTC, nilotinib and lapatinib to a comparable levels as in drug sensitive S1 cells. These results are supported by the fact that the chemosensitivity of BI 2536 in cells with ectopic expression of either ABCB1 or ABCG2 can be significantly restored by inhibitors of ABCB1 and ABCG2 (Table 3). Overall, our data indicate that the overexpression of ABCB1 and ABCG2 will result in significant resistance to BI 2536, and by inhibiting the function of ABCB1 and ABCG2, the effectiveness of BI 2536 can be significantly restored.
In summary, our study indicates that BI 2536 interacts with and inhibits the function of ABCB1 and ABCG2, stimulates the ATPase activity of both ABCB1 and ABCG2, and more significantly, the overexpression of either ABCB1 or ABCG2 in human cancer cells can lead to substantial resistance to BI 2536. As a result, there is decreased effect on BI 2536-induced G2/M cell cycle arrest and thus, renders BI 2536 ineffective (Fig. 6). Taking into account the known effect of ABCB1 and ABCG2 on the distribution and oral bioavailability of many therapeutic drugs [47], it is likely that the overexpression of ABCB1 and ABCG2 at the intestinal walls and blood-brain barrier can reduce the overall absorption and distribution of BI 2536, which remains to be determined. In this study, although there is currently no inhibitor suitable to be used clinically, inhibitors or competitive substrates of ABCB1 and ABCG2 were used to demonstrate that both BI 2536 mediated G2/M cell cycle arrest and chemosensitivity of BI 2536 can be significantly restored by modulating the functions of ABCB1 and ABCG2 in cells overexpressing either ABCB1 or ABCG2. Therefore, further investigation is warranted for the use of combinations of BI 2536 and inhibitor of ABC drug transporters or clinically active drugs that can potentially increase the oral bioavailability and overall antitumor response in cancer patients treated with BI 2536.
Fig. 6.
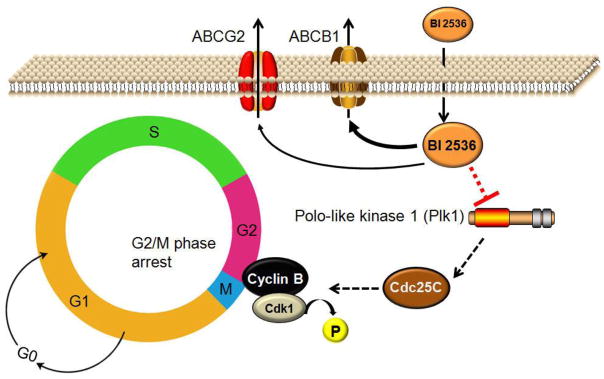
Schematic showing the role of ABCB1 or ABCG2 transporters in the decrease in effectiveness of BI 2536 in resistant cancer cells. Polo-like kinase 1 (Plk1) is known to play a central role in regulating cell division in human cells and the overexpression of Plk1in human cancer cells promotes cell growth [43]. BI 2536 is the first Plk1 inhibitor that selectively inhibits the catalytic activity of Plk1. Typically, BI 2536 blocks the multifunctional mitotic kinase activity of Plk1 with high efficiency, which leads to G2/M cell cycle arrest and apoptosis in cancer cells. However, the direct drug efflux function of ABCB1 and ABCG2 can potentially lead to reduced intracellular BI 2536 concentration in ABCB1- or ABCG2-expressing cancer cells, rendering BI 2536 ineffective.
Acknowledgments
The authors thank Dr Susan Bates (National Cancer Institute, NIH), for providing FTC and tariquidar. This work was supported by funds from the Chang Gung Medical Research Program (CMRPD190653) and grant to Chang Gung University from the Ministry of Education EMRPD1B0271. Drs H-M. Sim and S.V. Ambudkar were supported by the Intramural Research Program of the National Cancer Institute, NIH, Center for Cancer Research.
Abbreviations
- MDR
multidrug resistance
- ABC
ATP-binding cassette
- Plk-1
Polo-like kinase 1
- PhA
Pheophorbide A
- FTC
Fumitremorgin C
- MTT
3-(4,5-dimethylthiazol-yl)-2,5-diphenyllapatinibrazolium bromide
- Vi
sodium orthovanadate
Footnotes
Conflict of Interest
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Petronczki M, Lenart P, Peters JM. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev Cell. 2008;14:646–59. doi: 10.1016/j.devcel.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 2.van Vugt MA, Medema RH. Getting in and out of mitosis with Polo-like kinase-1. Oncogene. 2005;24:2844–59. doi: 10.1038/sj.onc.1208617. [DOI] [PubMed] [Google Scholar]
- 3.Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer. 2007;7:107–17. doi: 10.1038/nrc2049. [DOI] [PubMed] [Google Scholar]
- 4.Toyoshima-Morimoto F, Taniguchi E, Shinya N, Iwamatsu A, Nishida E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature. 2001;410:215–20. doi: 10.1038/35065617. [DOI] [PubMed] [Google Scholar]
- 5.Lane HA, Nigg EA. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J Cell Biol. 1996;135:1701–13. doi: 10.1083/jcb.135.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sumara I, Gimenez-Abian JF, Gerlich D, Hirota T, Kraft C, de la Torre C, et al. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol. 2004;14:1712–22. doi: 10.1016/j.cub.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 7.Eckerdt F, Strebhardt K. Polo-like kinase 1: target and regulator of anaphase-promoting complex/cyclosome-dependent proteolysis. Cancer Res. 2006;66:6895–8. doi: 10.1158/0008-5472.CAN-06-0358. [DOI] [PubMed] [Google Scholar]
- 8.Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol. 2000;2:672–6. doi: 10.1038/35023629. [DOI] [PubMed] [Google Scholar]
- 9.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–30. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 10.Nappi TC, Salerno P, Zitzelsberger H, Carlomagno F, Salvatore G, Santoro M. Identification of Polo-like kinase 1 as a potential therapeutic target in anaplastic thyroid carcinoma. Cancer Res. 2009;69:1916–23. doi: 10.1158/0008-5472.CAN-08-1693. [DOI] [PubMed] [Google Scholar]
- 11.Renner AG, Dos Santos C, Recher C, Bailly C, Creancier L, Kruczynski A, et al. Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood. 2009;114:659–62. doi: 10.1182/blood-2008-12-195867. [DOI] [PubMed] [Google Scholar]
- 12.Gleixner KV, Ferenc V, Peter B, Gruze A, Meyer RA, Hadzijusufovic E, et al. Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer Res. 2010;70:1513–23. doi: 10.1158/0008-5472.CAN-09-2181. [DOI] [PubMed] [Google Scholar]
- 13.Maire V, Nemati F, Richardson M, Vincent-Salomon A, Tesson B, Rigaill G, et al. Polo-like kinase 1: a potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res. 2013;73:813–23. doi: 10.1158/0008-5472.CAN-12-2633. [DOI] [PubMed] [Google Scholar]
- 14.Pezuk JA, Brassesco MS, Oliveira JC, Morales AG, Montaldi AP, Sakamoto-Hojo ET, et al. Antiproliferative in vitro effects of BI 2536-mediated PLK1 inhibition on cervical adenocarcinoma cells. Clin Exp Med. 2013;13:75–80. doi: 10.1007/s10238-011-0166-1. [DOI] [PubMed] [Google Scholar]
- 15.Stewart HJ, Kishikova L, Powell FL, Wheatley SP, Chevassut TJ. The polo-like kinase inhibitor BI 2536 exhibits potent activity against malignant plasma cells and represents a novel therapy in multiple myeloma. Exp Hematol. 2011;39:330–8. doi: 10.1016/j.exphem.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Choy E, Harmon D, Yang S, Yang C, Mankin H, et al. Inhibition of polo-like kinase 1 leads to the suppression of osteosarcoma cell growth in vitro and in vivo. Anticancer Drugs. 2011;22:444–53. doi: 10.1097/CAD.0b013e32834513f4. [DOI] [PubMed] [Google Scholar]
- 17.Ackermann S, Goeser F, Schulte JH, Schramm A, Ehemann V, Hero B, et al. Polo-like kinase 1 is a therapeutic target in high-risk neuroblastoma. Clin Cancer Res. 2011;17:731–41. doi: 10.1158/1078-0432.CCR-10-1129. [DOI] [PubMed] [Google Scholar]
- 18.de Oliveira JC, Brassesco MS, Pezuk JA, Morales AG, Valera ET, Montaldi AP, et al. In vitro PLK1 inhibition by BI 2536 decreases proliferation and induces cell-cycle arrest in melanoma cells. J Drugs Dermatol. 2012;11:587–92. [PubMed] [Google Scholar]
- 19.Harris PS, Venkataraman S, Alimova I, Birks DK, Donson AM, Knipstein J, et al. Polo-like kinase 1 (PLK1) inhibition suppresses cell growth and enhances radiation sensitivity in medulloblastoma cells. BMC Cancer. 2012;12:80. doi: 10.1186/1471-2407-12-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garland LL, Taylor C, Pilkington DL, Cohen JL, Von Hoff DD. A phase I pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase-1-interacting properties, in patients with advanced solid tumors. Clin Cancer Res. 2006;12:5182–9. doi: 10.1158/1078-0432.CCR-06-0214. [DOI] [PubMed] [Google Scholar]
- 21.Jimeno A, Li J, Messersmith WA, Laheru D, Rudek MA, Maniar M, et al. Phase I study of ON 01910. Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors. J Clin Oncol. 2008;26:5504–10. doi: 10.1200/JCO.2008.17.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, et al. Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies. Clin Cancer Res. 2011;17:3420–30. doi: 10.1158/1078-0432.CCR-10-2946. [DOI] [PubMed] [Google Scholar]
- 23.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, et al. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–15. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 24.Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–22. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 25.Haupenthal J, Bihrer V, Korkusuz H, Kollmar O, Schmithals C, Kriener S, et al. Reduced efficacy of the Plk1 inhibitor BI 2536 on the progression of hepatocellular carcinoma due to low intratumoral drug levels. Neoplasia. 2012;14:410–9. doi: 10.1596/neo.111366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillet JP, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010;596:47–76. doi: 10.1007/978-1-60761-416-6_4. [DOI] [PubMed] [Google Scholar]
- 27.Wu CP, Hsieh CH, Wu YS. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol Pharm. 2011;8:1996–2011. doi: 10.1021/mp200261n. [DOI] [PubMed] [Google Scholar]
- 28.Shukla S, Skoumbourdis AP, Walsh MJ, Hartz AM, Fung KL, Wu CP, et al. Synthesis and characterization of a BODIPY conjugate of the BCR-ABL kinase inhibitor Tasigna (nilotinib): evidence for transport of Tasigna and its fluorescent derivative by ABC drug transporters. Mol Pharm. 2011;8:1292–302. doi: 10.1021/mp2001022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu CP, Sim HM, Huang YH, Liu YC, Hsiao SH, Cheng HW, et al. Overexpression of ATP-binding cassette transporter ABCG2 as a potential mechanism of acquired resistance to vemurafenib in BRAF(V600E) mutant cancer cells. Biochem Pharmacol. 2013;85:325–34. doi: 10.1016/j.bcp.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brozik A, Hegedus C, Erdei Z, Hegedus T, Ozvegy-Laczka C, Szakacs G, et al. Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin Drug Metab Toxicol. 2011;7:623–42. doi: 10.1517/17425255.2011.562892. [DOI] [PubMed] [Google Scholar]
- 31.Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001;61:6635–9. [PubMed] [Google Scholar]
- 32.Robey RW, Medina-Perez WY, Nishiyama K, Lahusen T, Miyake K, Litman T, et al. Overexpression of the ATP-binding Cassette Half-Transporter, ABCG2 (MXR/BCRP/ABCP1), in Flavopiridol-resistant Human Breast Cancer Cells. Clin Cancer Res. 2001;7:145–52. [PubMed] [Google Scholar]
- 33.Shen DW, Fojo A, Chin JE, Roninson IB, Richert N, Pastan I, et al. Human multidrug-resistant cell lines: increased mdr1 expression can precede gene amplification. Science. 1986;232:643–5. doi: 10.1126/science.3457471. [DOI] [PubMed] [Google Scholar]
- 34.Currier SJ, Kane SE, Willingham MC, Cardarelli CO, Pastan I, Gottesman MM. Identification of residues in the first cytoplasmic loop of P-glycoprotein involved in the function of chimeric human MDR1-MDR2 transporters. J Biol Chem. 1992;267:25153–9. [PubMed] [Google Scholar]
- 35.Gribar JJ, Ramachandra M, Hrycyna CA, Dey S, Ambudkar SV. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J Membr Biol. 2000;173:203–14. doi: 10.1007/s002320001020. [DOI] [PubMed] [Google Scholar]
- 36.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods in enzymology. 1998;292:504–14. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- 37.Wu CP, Shukla S, Calcagno AM, Hall MD, Gottesman MM, Ambudkar SV. Evidence for dual mode of action of a thiosemicarbazone, NSC73306: a potent substrate of the multidrug resistance linked ABCG2 transporter. Mol Cancer Ther. 2007;6:3287–96. doi: 10.1158/1535-7163.MCT-07-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuestas ML, Castillo AI, Sosnik A, Mathet VL. Downregulation of mdr1 and abcg2 genes is a mechanism of inhibition of efflux pumps mediated by polymeric amphiphiles. Bioorg Med Chem Lett. 2012;22:6577–9. doi: 10.1016/j.bmcl.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Lainey E, Sebert M, Thepot S, Scoazec M, Bouteloup C, Leroy C, et al. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle. 2012;11:4079–92. doi: 10.4161/cc.22382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Natarajan K, Bhullar J, Shukla S, Burcu M, Chen ZS, Ambudkar SV, et al. The Pim kinase inhibitor SGI-1776 decreases cell surface expression of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and drug transport by Pim-1-dependent and -independent mechanisms. Biochem Pharmacol. 2013;85:514–24. doi: 10.1016/j.bcp.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–14. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shukla S, Chen ZS, Ambudkar SV. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist Updat. 2012;15:70–80. doi: 10.1016/j.drup.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24:287–91. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- 44.Ansbro MR, Shukla S, Ambudkar SV, Yuspa SH, Li L. Screening Compounds with a Novel High-Throughput ABCB1-Mediated Efflux Assay Identifies Drugs with Known Therapeutic Targets at Risk for Multidrug Resistance Interference. PLoS One. 2013;8:e60334. doi: 10.1371/journal.pone.0060334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ambudkar SV, Cardarelli CO, Pashinsky I, Stein WD. Relation between the turnover number for vinblastine transport and for vinblastine-stimulated ATP hydrolysis by human P-glycoprotein. J Biol Chem. 1997;272:21160–6. doi: 10.1074/jbc.272.34.21160. [DOI] [PubMed] [Google Scholar]
- 46.Ludwig JA, Szakacs G, Martin SE, Chu BF, Cardarelli C, Sauna ZE, et al. Selective toxicity of NSC73306 in MDR1-positive cells as a new strategy to circumvent multidrug resistance in cancer. Cancer Res. 2006;66:4808–15. doi: 10.1158/0008-5472.CAN-05-3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shukla S, Ohnuma S, Ambudkar SV. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr Drug Targets. 2011;12:621–30. doi: 10.2174/138945011795378540. [DOI] [PMC free article] [PubMed] [Google Scholar]