

Abstract
In Leishmania major, the core of the abundant surface lipophosphoglycan (LPG) is structurally related to that of the smaller glycosylinositolphospholipids (GIPLs) in containing galactosylfuranose (Galf ) residues in a Galf (β1, 3)Man motif. However, deletion of the putative Galf-transferase (Galf T) LPG1 affected Galf incorporation in LPG but not GIPLs. We hypothesized that the presumptive GIPL Galf-transferases could be homologous to LPG1, and identified three related genes in the L. major genome. These were termed LPG1L, LPG1R, and LPG1G, the latter of which was found in three identical copies located at the telomeres of chromosomes 5, 19, and 32 based on Leishmania genome project data. Neither LPG1 nor its homologues LPG1L and LPG1R were involved in the biosynthesis of GIPLs, as an lpg1−/lpg1l−/lpg1r− triple knockout (the first such in Leishmania) grew normally and made wild-type levels of Galf-containing GIPLs. In contrast, overexpression of these three led to elevated galactose incorporation in glycoproteins. Galf-containing glycoproteins had not been described in Leishmania but occur at high levels in other closely related trypanosomatids including Trypanosoma cruzi, Crithidia, Leptomonas, and Endotrypanum, and LPG1L and LPG1R homologs were detected in these species. These data suggest that the glyco-synthetic capabilities of Leishmania and perhaps other trypanosomatids may be larger than previously thought, with some activities being ‘cryptic’ in different lineages and potentially serving as reservoirs for glycoconjugate variation during evolution. Future tests will address whether the LPG1G family encodes the hypothesized GIPL-specific Galf T.
Keywords: Glycosylinositolphospholipids, Lipophosphoglycan, Galactosylfuranose, Galactosylfuranose transferase
1. Introduction
Protozoan parasites of the genus Leishmania are the causative agent of the disease leishmaniasis, which infects more than 10 million people worldwide [1]. Leishmania parasites are transmitted through sand fly bites, where the flagellated promastigote cells are introduced into the mammalian host. Parasites are then taken up by macrophages and differentiate into the non-flagellated amastigotes. Depending on parasite species, Leishmania infections in humans cause manifestations from self-containing cutaneous lesions to lethal visceral infections.
The surface of the promastisgote stages of Leishmania is coated with a variety of interrelated glycoconjugates including lipophosphoglycan (LPG), glycosylinositolphospholipids (GIPLs), proteophosphoglycan (PPG), and GPI-anchored proteins [2–4]. In the sand fly, stage-specific modifications of LPG are responsible for the attachment and release of promastigotes from the midgut [5–7]. In the mammalian host, LPG confers resistance to complement-mediated lysis, oxidative stress, and inhibits phagolysosomal fusion [8,9]. However, LPG is down-regulated in amastigotes, whose surface is dominated instead by the abundant GIPLs, which suggested that these play critical roles in amastigote survival and virulence [10,11]. This view was recently called into question by studies of a Leishmania major mutant lacking the enzyme alkyldihydroxyacetone phosphate synthase (ADS1), which is required for the synthesis of ether phospholipids. The ads1− null mutant lacked LPG and GIPLs but maintain the presence of GPI-anchored proteins, probably bearing a modified GPI anchor [12]. Remarkably, ads1− mutants showed little phenotype beyond that attributable to loss of LPG (e.g. reduced ability to establish macrophage infections), and ads1− amastigotes can replicate in macrophages and cause disease in susceptible mice [12]. In L. mexicana, there are conflicting data about the role of GIPLs in parasite viability and/or virulence [13–15]. A potential drawback is that the various Leishmania mutants studied were pleiotropic and affected a number of glycoconjugates, making inferences about GIPL function indirect. Thus, the identification of genes that specifically affect GIPL synthesis would simplify and strengthen genetic studies of their function.
Leishmania GIPLs have different structures in different species, consisting of type I GIPLs whose structure resembles that of protein GPI anchors, type II GIPLs whose structure resembles that of the LPG core, and/or “hybrid” GIPLs [3,10,11]. L. major synthesize primarily type II GIPLs, which contain galactosylfuranose (Galf) in the Gal0–2Galf(β3)Man sequence, which is also found in the LPG core ([16,17], Fig. 1). We showed previously that LPG1, encoding a putative galactofuranosyl transferase (Galf T), was responsible for adding Galf to the LPG core structure, but that lpg1− mutants continued to synthesize the Galf-containing GIPLs [18]. Thus, there must be additional Galf Ts that are responsible for the synthesis of GIPLs. Given the similarity of the Galf T acceptors for LPG and GIPLs, it seemed reasonable to postulate that these GIPL-specific Galf Ts could be related to LPG1. In this report we describe the characterization of a family of LPG1 homologs in the L. major genome, and studied the effect of genetic inactivation of three of these singly or in combinations (LPG1, LPG1L, and LPG1R).
Fig. 1.
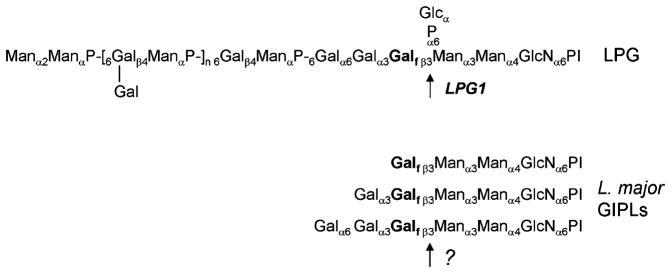
Structures of LPG and GIPLs in L. major. Abbreviations: Gal, galactose; Galf, galactosylfuranose; Man, mannose; Glc, glucose; GlcN, glucosamine; P, phosphate; PI, phosphatidylinositol anchor.
2. Materials and methods
2.1. Leishmania cultures
L. major LV39 clone 5 (Rho/SU/59/P) cells were grown in M199 medium supplemented with 10% heat inactivated fetal calf serum at 26 °C [19]. Selective drugs used in this study included G418 (10 μg/ml), hygromycin (50 μg/ml), puromycin (10 μg/ml), nourseothricin (80 μg/ml), blasticidin (10 μg/ml), and phleomycin (10 μg/ml).
2.2. Isolation of LPG1L and LPG1R open reading frames
At the time these studies were initiated the L. major genome was largely incomplete. Sequence data was obtained from the Sanger Institute website at http://www.sanger.ac.uk/Projects/Lmajor/. Searches with LPG1 first identified a 537-bp region from the Leishmania Genome Database; this region was PCR amplified and used to identify a cosmid (B4308) containing the full length gene from a cLHYG genomic cosmid library made from L. major Friedlin strain V1 (MHOM/IL/80/Friedlin). This gene was termed LPG1L (for LPG1-like) and its open reading frame and flanking regions were sequenced (GenBank accession number AY235572). A second gene termed LPG1R (LPG1-related) was similarly identified in the Leishmania Genome Database; this ORF and flanking sequences were recovered by PCR and their sequences were determined (AY235573). Preliminary sequence data from T. cruzi was obtained from The Institute of Genomic Research through their website at http://www.tigr.org.
2.3. Molecular constructs
To generate the C-terminal GFP tagged proteins, full length open reading frames lacking stop codons were PCR amplified from L. major FV1 genomic DNA using primers SMB1307 (5′-TTATCAggatccACCATGAAGGGCAGACTAC-3′, lower case letters indicate added restriction sites) and SMB1310 (5′-ATATAAgcggccgcCGGGCTGACAGCCTGCAG) for LPG1L and SMB1354 (5′-TATAGCggatccACCATGAAGCGCGGGCAGAGG) and SMB1405 (5′-ACAGCTgatatcCTTTCGCCAATCCGGCTCTG) for LPG1R. The resulting DNA fragments were digested with appropriate restriction enzymes (BamHI and NotI for LPG1L, BamHI and EcoRV for LPG1R) and cloned into the expression site of pXG−/GFP+ (a vector for making C-terminal GFP fusion proteins; strain B2863 [20]) to make pXG-LPG1L-GFP (B4400) and pXG-LPG1R-GFP (B4455).
To make deletion constructs for LPG1L, a 700-bp region immediately upstream of the start codon and a 900-bp region immediately downstream of the stop codon of LPG1L were PCR amplified from L. major genomic DNA and cloned into vector pX63PAC (B1129) [21] so that the end of the upstream region is linked to the beginning of the downstream region. Next a 909-bp NEO marker gene (confers resistance to G418) was excised from vector pXG (B4090) [20] and inserted in between the upstream and downstream regions of LPG1L to make pX63PAC-KO-LPG1L:NEO (B4369). Similarly, a 1052-bp HYG marker gene (conferring resistance to hygromycin) was excised from vector pX63HYG (B617) [22] using SpeI and BamHI and inserted in between the upstream and downstream regions of LPG1L to make pX63PAC-KO-LPG1L:HYG (B4370). Linear deletion cassettes containing NEO and HYG markers flanked by the upstream and downstream regions of LPG1L were generated by SmaI digestion.
To make deletion constructs for LPG1R, first a 2.4-kb DNA fragment containing the LPG1R ORF (plus sequences 550 bp from upstream and 440 bp from downstream) was PCR amplified from L. major genomic DNA and cloned into vector pUC18 to give pUC-LPG1R (B4413). Then a SacII/SphI fragment containing the first 500 bp of LPG1R ORF was replaced by drug markers (PAC or SAT, conferring resistance to puromycin or nourseothricin, respectively) amplified by PCR. The resulting constructs, pUC-KO-LPG1R:PAC (B4451) and pUC-KO-LPG1R:SAT (B4513) were digested with EcoRI and HindIII to generate linear deletion cassettes for LPG1R.
Previously, a 4.2-kb fragment containing the LPG1 ORF and flanking sequences was cloned into the BamHI site of vector pUC18 (pUC-LPG1-Bam, B2880). To make the deletion construct for LPG1, the first 700 bp from LPG1 ORF was replaced by the BSD (confers resistance to blasticidin) marker gene. The resulting construct, pUC-KO-LPG1:BSD (B4573), was digested with BamHI to generate a linear knockout cassette for LPG1.
An overexpression construct for LPG1, LPG1L, and LPG1R was made in three steps. First, the full-length LPG1L gene was cloned in the BglII site of vector pIR1PHLEO (B4054) derived from pIR1SAT (B3541; [23]) to make pIR1PHLEO-LPG1L (B4587). Second, the full-length LPG1R gene was cloned in the BamHI site of pIR1PHLEO-LPG1L (B4589). The resulting construct, pIR1PHLEO-LPG1L-LPG1R, was linearized with NsiI and blunt ended, followed by ligation with the 4.2-kb LPG1 fragment (BamHI digestion of pUC-LPG1-Bam then blunt ended) to generate pIR1PHLEO-LPG1-LPG1L-LPG1R (B4789).
2.4. Genetic manipulations
Leishmania were transfected by electroporation as described [24]. Typically, 5–10 μg of DNA was used for each transfection. Transfected cells were plated on medium containing 1% noble agar and appropriate concentrations of selective drugs. To study the localization of LPG1L and LPG1R, L. major cells containing pX63HYG-LPG2-HA (a marker of the parasite Golgi apparatus; strain B1878 [22]) were transfected with pXG-LPG1L-GFP (B4400) or pXG-LPG1R-GFP (B4455) and selected with hygromycin and G418. The lpg1−lpg1l−/lpg1r− triple knockout parasites were created using a loss-of-heterozygosity (LOH) approach [25]. Briefly, the first allele of LPG1 in the lpg1l−/lpg1r− double knockout was replaced by the BSD marker. After two passages in the presence of 50 μg/ml of blasticidin, cells were incubated in media containing 10 μg/ml of galactose-binding lectin ricin RCA120 (Sigma) at 107 cells/ml. Cells that failed to agglutinate were plated, and ~80% of the resulting colonies had lost the second allele of LPG1. All knockout strains were confirmed by Southern-blot analysis.
2.5. Molecular biology techniques
Southern-blot and Northern-blot analyses were performed as previously described [26]. The Southern blot shown in Fig. 9 was subsequently washed first in 2× SSPE/0.5% SDS for 15 min at room temperature, and then three times in 0.2× SSPE/0.5% SDS at 60 °C for 15 min each. RT-PCR analysis was used to map the mRNA splicing acceptor sites of LPG1L and LPG1R. Briefly, first strand synthesis was performed using gene-specific antisense primers SMB1228 (5′-GCGTGTCGCCATTTTGGGCGAG) for LPG1L and SMB1369 (5′-CCGCAGCGCGCAGCTTCGTC) for LPG1R. Reverse transcription was performed at 42 ′C with AMV reverse transcriptase (Sigma) and 2 μg of total RNA, followed by PCR amplification with the first strand primers plus the universal miniexon primer SMB936 (5′-AACGCTATATAAGTATCAGTTCTGTACTTTA) for L. major [27]. The resulting fragments were sequenced to map the mRNA splice acceptor sites.
Fig. 9.
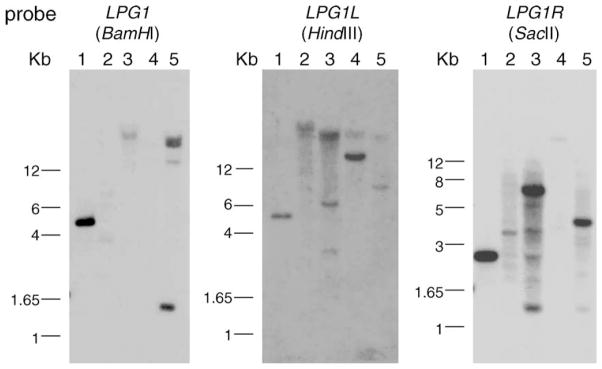
Homologues of LPG1L and LPG1R exist in other Kinetoplastida parasites. Genomic DNAs from L. major LV39 (lane 1), Trypanosoma cruzi (lane 2), Crithidia fasculata (lane 3), Leptomonas samuli (lane 4), and Endotypanum schaudinni (lane 5) were digested with restriction enzymes and hybridized with probes corresponding to the coding regions of LPG1, LPG1L, and LPG1R. The blots were washed under ‘moderately’ stringent conditions as described in the Section 2 (0.2× SSPE/0.5% SDS at 60 °C).
2.6. Western blots and fluorescence microscopy
Log phase (1–5 × 106 cells/ml) cell extracts and culture supernatants were resolved by SDS/PAGE and electroblotted onto Hybond ECL nitrocellulose membranes (Amersham Biosciences). Mouse monoclonal antibody WIC79.3 [28] was used to detect LPG and PPG (1:1000 dilution). Mouse anti-L. major GP63 monoclonal antibody [29] was used to detect GP63 (1:1000 dilution). Mouse monoclonal anti-α-tubulin antibody (T5168, Sigma) was used to detect α-tubulin (1:10000 dilution). An enhanced chemiluminescence (ECL) detection system (Amersham) was used to detect signals. For fluorescence microscopy, log phase cells were immobilized on polylysine coated cover slips, followed by fixation with 3.5% formaldehyde in PBS. After being permeated by 100% ethanol at 4 °C, cells were incubated with dilutions of primary and secondary antibodies as described [30].
2.7. Mouse footpad infections
Parasite virulence was evaluated by mouse footpad infections. Briefly, all parasite lines were first inoculated at high levels (2 × 107) into BALB/c mice in order to guard against effects arising from transfection/culture passage associated loss of virulence. They were recovered after 1 month prior to emergence of pathology, and converted into promastigotes in vitro. After three passages in culture, stationary phase parasites (day 3) were resuspended in DMEM media at 2 × 107 cells/ml and 106 were injected into the footpads of 8-week-old female BABL/c mice (5–6 mice per group). Lesion sizes were measured weekly with a Vernier caliper and parasite numbers in the infected foot pads were determined by limiting dilution assay [31].
2.8. GIPL analysis
Exponentially growing cells were metabolically labeled with [3H]galactose (50 μCi for 8 × 108 cells) for 16 h, and [3H]GIPLs were purified as described [32]. The [3H]GIPLs were treated with 0.04 M trifluoroacetic acid for 1 h at 100 °C to cleave the labile galactofuranosidic bonds, and the [3H-Gal]fragments were chromatographed on paper as described [33]. Other aliquots of unlabeled GIPLs were subjected to nitrous acid deamination [33] to remove the lipid anchors, tagged at the reducing end with the fluorophore 9-aminonaphthalene 1,3,6-trisulfate, and analyzed by GLYKO-FACE electrophoresis according to the manufacturer’s specifications (GLYKO, Novato, CA).
2.9. LPG and glycoprotein analysis for the LPG1-LPG1L-LPG1R overexpressors
Parasites (5 × 106 cells/ml) were metabolically labeled for 16 h with [6-3H]galactose (50 μCi) in 5 ml of M199 culture medium supplemented with 10% fetal calf serum and 1 μg/ml of biopterin. [3H]LPG was solubilized by differential organic solvent extraction and removed by chromatography on phenyl-Sepharose as described [38]. The insoluble residue containing [3H]glycoproteins was converted to [3H]glycopeptides by digestion with Pronase as described [34] and desalted by chromatography on Sephadex G25. Maltohexose (1 μmole) was added to the protein residue prior to Pronase digestion as an internal standard and was quantitated by capillary electrophoresis using conditions: 20 °C, 20 psi, and 20 min.
3. Results
3.1. Identification of a family of six LPG1-related genes in L. major
Search for sequences with similarity to L. major LPG1 revealed the presence of fragments from related sequences in the L. major (Friedlin V1 strain) genome project data, which was then incomplete. The sequence for two of these genes, designated LPG1L (LPG1-like) and LPG1R (LPG1-related), was determined. Southern-blot analysis and gene deletion studies showed that these genes occurred in a single copy (data not shown or below). As these studies were being finished, the emerging L. major genome sequence revealed three additional related homologous genes that were named LPG1G, whose predicted amino acid sequences were identical and located on chromosomes 5, 19, and 32 (LmjF05.1230, LmjF19.650, and LmjF32.3900; sequence information is available at http://www.genedb.org/genedb/leish/index.jsp). Southern-blot analysis confirmed the presence of multiple LPG1G genes in L. major strain LV39 clone 5 as well (data not shown). Interestingly, all three of the L. major Friedlin strain LPG1G genes appear to be located adjacent to telomeres. LPG1 has been described in several Leishmania species previously [35,36]; preliminary analysis of data arising from shotgun sequence of L. infantum suggest that this species also has homologs of LPG1L, LPG1R, and at least one copy of LPG1G, all of which show >90% identity to the homologous gene of L. major (data not shown).
3.2. Properties of predicted LPG1-family proteins
The predicted open reading frames for LPG1, LPG1L, LPG1R, and LPG1G contained 434, 592, 460, and 599 amino acids, respectively. All four genes (LPG1, LPG1L, LPG1R, and LPG1G) were predicted to encode type-II transmembrane proteins with short, basic cytoplasmic tails (~20 amino acids) at the N-termini, followed by single transmembrane domains (Fig. 2A; predictions were made using the TMHMM Server at http://www.cbs.dtu.dk/services/TMHMM/). Relative to LPG1, there were insertions in LPG1L (encoding amino acid 43–161) and LPG1G (encoding amino acid 43–149) immediately after the transmembrane domain, that were not seen in LPG1 or LPG1R (Fig. 2A and B). The overall similarity among these four genes was modest (~20% amino acid identity; Fig. 2B). Nonetheless, the predicted lumenal domains of all four genes contained several highly conserved regions as shown in the alignment in Fig. 2B. One of these regions (G-F/Y-F/L-D-E-N-F/Y-Y/F-P-A/I-Y/L-G/Y/F/M-E/D-D-H/T/Y/I-D-Y/W/L) is likely to represent the catalytic site, since it contained the metal-binding DXD motif conserved amongst glycosyltransferases (Fig. 2A and B).
Fig. 2.

LPG1 gene family. (A) Schematic representations of LPG1, LPG1L, LPG1R, and LPG1G. Numbers indicate amino acids. Positions of the conserved DXD catalytic motifs are indicated. CT, cytoplasmic tail; TM, transmembrane domain. Compared to LPG1 and LPG1R, LPG1L and LPG1G contain additional “inserts” (from amino acid 43–161 for LPG1L and 43–149 for LPG1G) after the transmembrane domain, as indicated. Our analysis indicates that the three copies of LPG1G on chromosomes 5, 19, and 32 are identical. (B) Alignment of LPG1, LPG1L, LPG1R, and LPG1G from L. major (~20% identity among these genes at amino acid level) and a preliminary partial open reading frame (contig) from T. cruzi (T. cruzi ctg). Alignments were performed using the ClustaIW algorithm as implemented in the program Megalign (DNA Star). Transmembrane domains were highlighted. The underlined region contains the DXD catalytic motif (marked by asterisks).
In all following studies we focused exclusively on the characterization of LPG1L and LPG1R.
3.3. Detection of LPG1L and LPG1R mRNA in L. major
To test whether LPG1L and LPG1R were expressed in L. major, total RNA was prepared from early log phase, late log phase, stationary phase, metacyclic phase, and amastigote stage cells and subjected to Northern-blot analysis using the coding regions of LPG1L and LPG1R as probes. The 2.8-kb LPG1L and 2.0-kb LPG1R mRNAs were expressed at similar levels throughout parasite life cycle (Fig. 3). RT-PCR analysis mapped the site of trans-splicing sites for LPG1L and LPG1R to positions 129 and 160 nt upstream of the ATG start codon for LPG1L and LPG1R, respectively (data not shown).
Fig. 3.
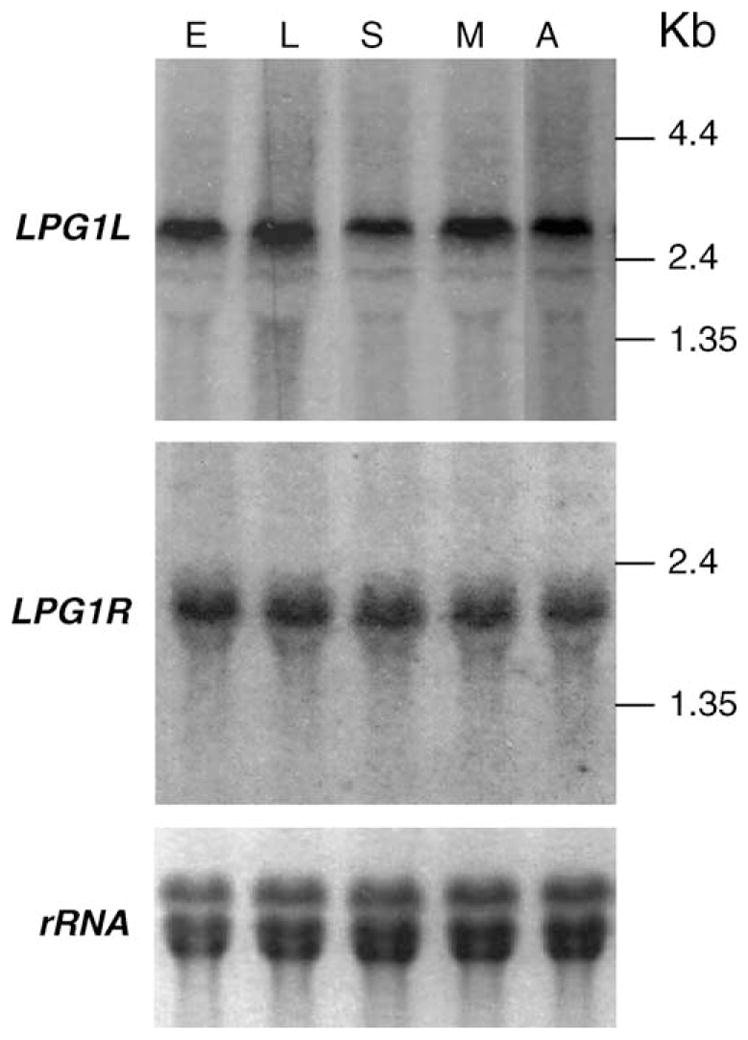
LPG1L and LPG1R are expressed throughout L. major life cycle. Northern-blot analysis was performed as described in Section 2. Probes were made from coding regions of LPG1L and LPG1R. E, early log phase; L, late log phase; S, stationary phase; M, metacyclic phase (prepared by negative selection for binding with peanut agglutinin; [53]); A, amastigotes (from infected mouse foot pads). Expression of rRNA was used as controls for loading.
3.4. Cellular localization of LPG1L and LPG1R
Previous data showed that an LPG1::GFP fusion protein was localized in the Golgi apparatus in Leishmania [20]. To examine the cellular localization of LPG1L and LPG1R gene products, we created similar fusion constructs joining the green fluorescent protein (GFP) to the C-termini of LPG1L and LPG1R. When expressed in Leishmania from episomal pXG vectors, both LPG1L::GFP and LPG1R::GFP fusion proteins showed strong green fluorescence in a region close to the kinetoplast, the location of the Golgi apparatus. To confirm Golgi localization, we introduced a construct expressing a hemagglutinin (HA)-tagged LPG2 gene, which encodes a Golgi GDP-mannose transporter [22,37]. Immunofluorescence analysis of the LPG2::HA showed co-localization with the LPG1L::GFP and LPG1R::GFP signals (Fig. 4). This result was further confirmed by immuno-EM study using anti-GFP antibody (data not shown). Therefore, similar to LPG1, the gene products of LPG1L and LPG1R were also localized at the Golgi apparatus, consistent with the cellular location of most glycosyltransferases.
Fig. 4.
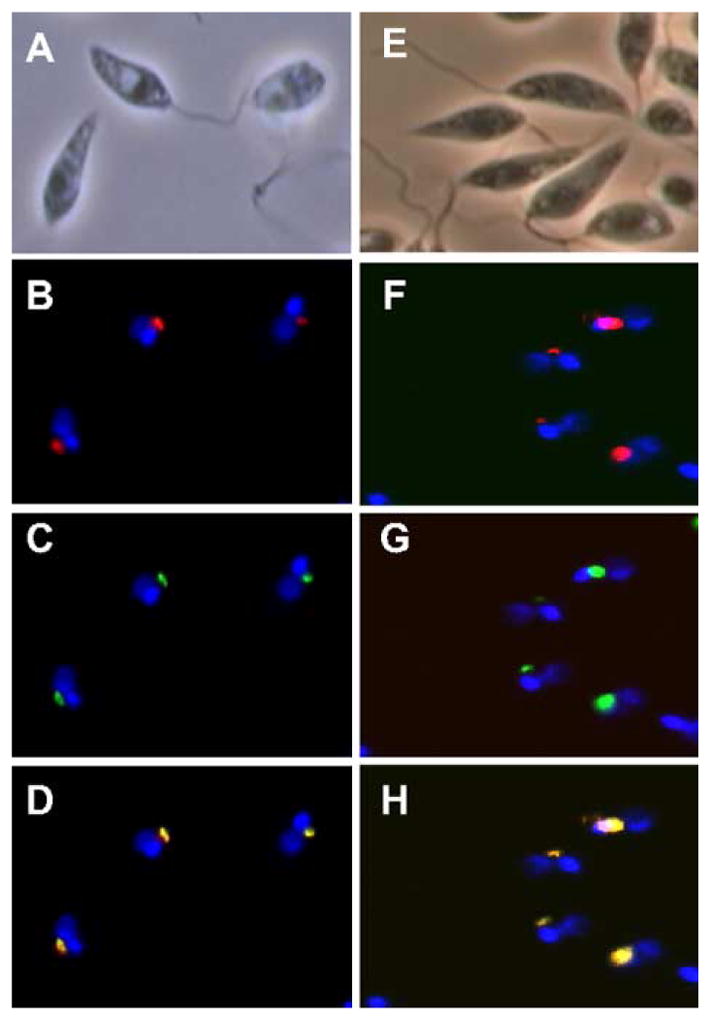
LPG1L and LPG1R are localized at the Golgi apparatus in L. major. LV39 WT cells were cotransfected with pX63HYG-LPG2-HA (Golgi marker) and pXG-LPG1L-GFP (A–D) or pXG-LPG1R-GFP (E–H). Localizations of LPG1L and LPG1R were determined by GFP fluorescence (C and G, respectively). The golgi apparatus was revealed by staining cells with rabbit anti-HA polyclonal antibody, followed by Texas Red-labeled mouse anti-rabbit monoclonal antibody (B and F). Hoechst 33258 dye was used to stain DNA (both nuclear and kinetoplast). D: merge of B and C; H: merge of F and G.
3.5. Generation of double and triple LPG1, LPG1L and LPG1R knockouts
Because Leishmania has a diploid genome, two rounds of replacement are required to inactivate any given gene. This presents an obstacle in genetically assessing the function of all six members of the LPG1 gene family, as currently just six suitable markers exist. Here we studied the effects of inactivation of LPG1L and LPG1R, singly, together, or in combination with LPG1. An outline of the gene inactivation protocol is shown in Fig. 5A. Briefly, LPG1L alleles were replaced by HYG and NEO markers (as lpg1l−), while LPG1R alleles were replaced by PAC and SAT markers (as lpg1r−), and all four alleles were replaced in the double lpg1r−/lpg1l− mutant. The triple knockout parasites (lpg1−/lpg1l−/lpg1r−) were generated through a LOH approach [25], by first replacing one LPG1 allele of the lpg1l−/lpg1r− mutant with the BSD marker, then selecting for the loss of LPG. In all lines, Southern-blot analysis confirmed that the planned replacements had occurred; data for the lpg1−/lpg1l−/lpg1r− triple knockout parasite is shown in Fig. 5B.
Fig. 5.

Deletions of LPG1L, LPG1R, and LPG1 in L. major. (A) Procedure to generate the lpg1−/lpg1l−/lpg1r− triple knockout parasites. (B) Southern-blot analysis to confirm the lpg1−/lpg1l−/lpg1r− triple knockout parasites (nos. 4-1 and 5-1 are two independent clones). Genomic DNA was digested with HindIII and hybridized with probes made from the coding regions of LPG1, LPG1L, and LPG1R.
The single, double and triple knockout parasites grew at similar rates as L. major wild-type cells in culture (data not shown). Western-blot analysis showed that all lines tested synthesized wild-type levels of PPG and GP63, and that the lpg1l−, lpg1r−, and lpg1l−/lpg1r− mutants synthesized WT levels of LPG (Fig. 6 and data not shown). These data indicated that LPG1L and LPG1R were not involved in the synthesis of large surface glycoconjugates such as LPG, PPG, or the GPI-anchored protein GP63. Additionally, expression of LPG1L and LPG1R from episomal vectors did not restore the synthesis of LPG in the lpg1− mutant, arguing that LPG1L and LPG1R could not substitute for the function of LPG1 even when overexpressed (data not shown).
Fig. 6.
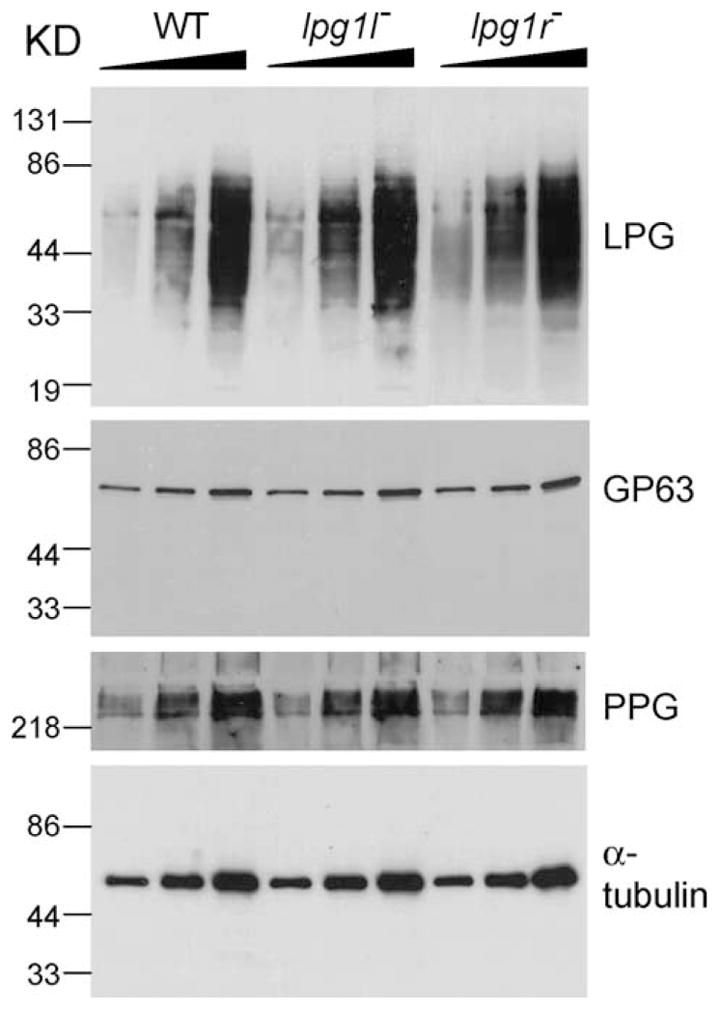
LPG1L and LPG1R are not required for the synthesis of LPG, PPG, or GPI-anchored proteins. Western blots of cell extracts (for LPG, GP63, and α-tubulin) and culture supernatants (for PPG) were performed as described in Section 2. For each cell type (WT, lpg1l−, and lpg1r−), materials from 2.5 × 105, 5 × 105, and 106 cells (from left to right) were loaded.
3.6. Deletion of LPG1L and/or LPG1R did not affect the biosynthesis of GIPLs
The effect of LPG1-family mutants on GIPL synthesis was evaluated in two ways. First, total GIPL fractions from wild type and mutant cells were extracted, dilipidated, fluorophore-labeled, and analyzed by FACE (fluorophore-assisted carbohydrate electrophoresis). GIPLs from single (lpg1l−, lpg1r−) or double (lpg1l−/lpg1r−) knockouts showed a pattern similar to the GIPL pattern seen for wild type (Fig. 7A and data not shown). In contrast, the GIPL pattern for the triple knockout (lpg1−/lpg1l−/lpg1r−) containing the deletion of LPG1 showed a pattern similar to the lpg1− knockout alone (Fig. 7A). Accumulation of the expected LPG biosynthetic intermediate (Glc-P-Man2-GlcN) was observed in all lines bearing the homozygous lpg1− deletions. Second, cells were metabolically labeled with [3H]galactose and total GIPLs were extracted and treated with trifluoroacetic acid to cleave the Galf-Man linkage in GIPLs with the structure Gal0–2-Galf-Man-Man-GlcN. After separation by paper chromatography, similar profiles were seen in WT and all mutant lines (Fig. 7B and data not shown). Therefore, it is unlikely that any of genes (LPG1, LPG1L, and LPG1R) were involved in the biosynthesis of the Galf-containing type-II GIPLs in L. major.
Fig. 7.
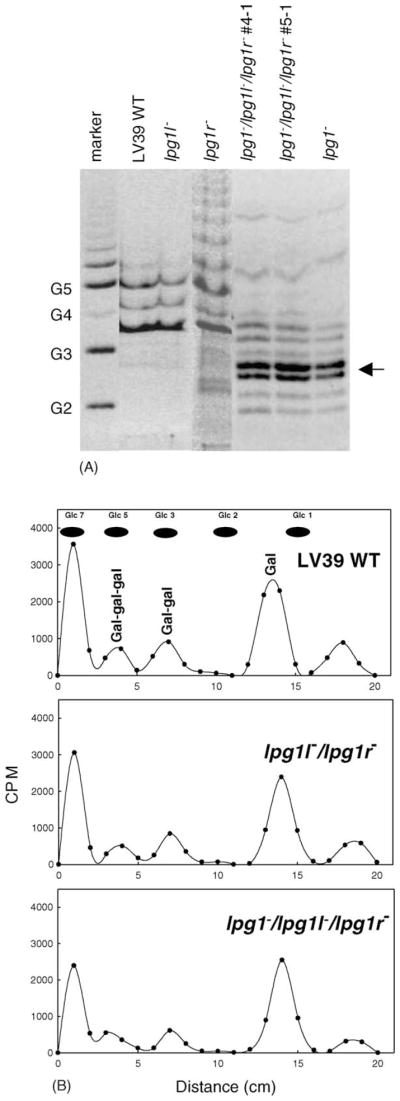
Triple knockout cells (lpg1−/lpg1l−/lpg1r−) still synthesize Galf-containing GIPLs. (A). Total GIPLs from LV39 WT, lpg1−, lpg1l−, lpg1r−, and lpg1−/lpg1l−/lpg1r− cells were extracted and analyzed by a GLYCO-FACE gel electrophoresis as described in Section 2. The arrow indicates material derived from truncated form of LPG (GlcP-Man2-GlcN-PI), which is minimal in LV39 WT cells. The marker (G2–G5) contains oligomeric derivatives of glucose. (B) WT (top panel), lpg1l−/lpg1r− (middle panel), and lpg1−/lpg1l−/lpg1r− (lower panel) cells were metabolically labeled with [3H]galactose and [3H]GIPLs were purified. The [3H]GIPLs were treated with TFA to cleave galactofuranosidic bonds and the fragment were separated by paper chromatography. Migration of unlabeled glucose standards is indicated.
3.7. LPG1L and LPG1R were not required for metacyclogenesis nor mouse infectivity
The percent metacyclic promastigotes (infectious form) present in stationary phase cultures showed very little difference amongst the wild type and various knockout cells (data not shown). To test if LPG1L and/or LPG1R were implicated in virulence, stationary phase parasites were inoculated in susceptible BABL/c mice. There was no significant difference in lesion formation between wild type and various knockout cells (lpg1l−, lpg1r−, and lpg1l−/lpg1r−), arguing that LPG1L and LPG1R were not required for virulence in the mammalian host (Fig. 8).
Fig. 8.

LPG1L and LPG1R are not required for L. major virulence. Mouse footpad infections were performed as described in Section 2. Stationary (WT, lpg1−, lpg1r−, and lpg1l−/lpg1r−) cells (106) were injected into each mouse. Five to six mice were used in each experiment. Error bars represent standard deviations.
3.8. Potential role of LPG1L and LPG1R in adding Galf to glycoproteins
Since neither LPG1L nor LPG1R appeared to function in GIPL biosynthesis, we asked whether these genes might participate in other glycoconjugate synthetic pathways. Galf residues are not synthesized by the mammalian hosts, but are found in glycoconjugates synthesized by a number of pathogens including Mycobacterium, T. cruzi, and Aspergillus [38–40]. In Leishmania, Galf residues have only been found in glycolipids [41]. In contrast, other Kinetoplastid species such as T. cruzi, Crithidia fasculata, Leptomonas samueli, and Endotrypanum schaudinni contain abundant amounts of Galf residues in their glycoproteins, often β1, 3-linked to mannose as in the LPG and GIPLs in L. major [38,42–44]. Southern-blot analysis was performed on the genomic DNA from the species mentioned above using the coding regions of LPG1, LPG1L, and LPG1R from L. major as probes. After several washes at moderate stringy (Section 2), strong signals were seen for several of these LPG1 family members in all species (Fig. 9). Additionally, LPG1-related genes have been identified in the emerging genome sequence of T. cruzi (Fig. 2).
Since Galf had never been observed previously in Leishmania proteins, we undertook an overexpression approach to determine whether LPG1 and its homologues (LPG1L and LPG1R) had any effect on protein galactosylation. In these studies, we generated a construct which simultaneously expressed LPG1, LPG1L, and LPG1R and integrated it into the ribosomal RNA locus of L. major, a position which gives high levels of expression [23]. WT and transfectants were metabolically labeled with [3H]galactose, and incorporation of the radiolabel into glycoproteins was measured. The results showed a small but reproducible increase in protein galactosylation (Fig. 10A). Efforts to assign this activity to either LPG1, LPG1R, or LPG1L in single gene overexpressors were inconclusive, possibly due to the low level of incorporation.
Fig. 10.

(A) Overexpression of LPG1, LPG1L, and LPG1R caused increased incorporation of [3H]galactose in glycoproteins. LV39 WT cells and cells overexpressing LPG1, LPG1L, and LPG1R (triple OE: triple overexpressor) were metabolically labeled with [3H]galactose and glycopeptides were extracted as described in Section 2. Radioactivity in these glycoproteins was determined using a scintillation counter; several experiments were done and the results of one with three replicas is shown; error bars represent standard deviation. (B) Demonstration of terminal Galf residues in L. major glycoproteins. [3H-Gal]Glycoproteins from LV39 WT cells were digested with pronase and desalted [25], and 10,000 cpm were incubated in the presence or absence of exo-β-galactofuranosidase [47]. After 24 h of incubation at 37 °C, the samples were applied to a column (2 ml) of Dowex AG1-X8 (acetate) over Dowex AG50-X8 (hydrogen) and the eluent was subjected to descending chromatography on paper using the solvent n-butanol:pyridine:water (6:4:3). Radioactivity was measured and plotted for samples without (upper panel) or with galactofuroanosidase (lower panel) treatment, along with migration of glucose oligomeric standards.
Unlike several other protozoans, the presence of Galf in N-linked carbohydrate chains in L. mexicana has been investigated and was not detected [45,46], possibly due to the low abundance of the monosaccharide. To examine whether L. major might possess Galf-containing proteins, [3H-Gal]glycopeptides were prepared by Pronase digestion of glycoproteins from wild-type cells and aliquots were incubated in the presence or absence of exo-β-galactofuranosidase [47], desalted by anion exchange that removes most of the glycopeptides, and analyzed by paper chromatography. No radioactive material that co-migrated with standard galactose was present in the chromatogram from the untreated [3H]glycopeptides (Fig. 10B, upper panel) whereas galactofuranosidase treatment to the release of low but measurable levels of [3H]galactose (Fig. 10B, lower panel), indicating the presence of terminal Galf in Leishmania glycopeptides. The large majority of radiolabeled glycopeptides was not susceptible to exo-β-galactofuranosidase digestion, which could be due to carbohydrate chains with internal Galf residues and/or could be glycopeptides containing galactopyranose. Efforts to further characterize [3H]glycopeptides using treatment with dilute trifluoracetic acid to release Galf-containing fragments were inconclusive, possibly because of the low level of incorporation.
4. Discussion
In this study we described the LPG1 gene family of L. major, and reported detailed characterization of two of the genes, LPG1L and LPG1R. The five new genes predict proteins showing strong similarity to the active LPG-specific Galf T encoded by LPG1, in being type II transmembrane glycoproteins with a short N-terminal cytoplasmic domain and a lumenal C-terminal domain bearing a canonical glycosyltransferases DXD motif (Fig. 2A). Both LPG1R and LPG1L are constitutively expressed in stable mRNA throughout the parasite life cycle (Fig. 3). Furthermore, episomally expressed LPG1L and LPG1R-GFP fusion proteins localized to the parasite Golgi apparatus (Fig. 4). Thus, these genes were reasonable candidates to be the GIPL-specific Galf transferase postulated previously [9], and their modest degree of overall sequence conservation (~20% identity) provides the requisite opportunity for divergence in Galf T acceptor specificity.
Due to the late emergence of the three LPG1G gene sequences and the availability of only six selectable genetic markers, we focused functional studies on the LPG1, LPG1L, and LPG1R genes. Single and double mutants of LPG1L and LPG1R were made, and a mutant lacking all three was also constructed, the first such triple knockout mutant reported in Leishmania (Fig. 5). These studies showed collectively that LPG1L and LPG1R had no effect on parasite growth, differentiation or the ability to induce disease in animals (Fig. 8). Analysis of glycoconjugate synthesis showed similarly that loss of LPG1L and LPG1R had no effect on the synthesis of the abundant glycoconjugates GP63, PPG, or LPG (Fig. 6). Lastly, while the triple mutant showed the expected adverse effect on LPG synthesis due to deletion of the LPG1 gene, there was no effect on GIPL biosynthesis (Fig. 7). These data suggest that none of these three genes are essential for the GIPL-specific Galf T. Obviously, the proposal that the LPG1G genes encode this activity is attractive, but the possibility that a completely unrelated gene encodes this activity cannot be excluded. It is interesting that L. infantum appears to contain an LPG1G homolog; while the GIPL composition of this species isolate has not been investigated, its close relative L. donovani lacks Galf-containing GIPLs [11]. While we were unable to test the effect of LPG1G knockouts in these studies, our repertoire of six selectable markers will be sufficient to permit the eventual inactivation of all three LPG1G genes simultaneously in the future.
In L. major Galf residues are present almost exclusively in glycolipids such as LPG and GIPLs, whereas in other trypanosomatid species other glycoconjugates contain Galf similarly linked to Man [38,42–44]. This suggested that perhaps one or more of the LPG1 family members participate in Galf T reactions involving other acceptors including the carbohydrate chains of proteins. Although Parodi and coworkers reported that L. mexicana lacks Galf-containing glycoproteins [45,46], a low abundance of this substituent was not excluded. Consistent with the possibility of Galf in Leishmania glycoproteins, several species known to express Galf-containing proteins possess genes homologous to LPG1, LPG1L, and LPG1R (Fig. 9), and the T. cruzi genome project also has revealed a family of LPG1-homologs (http://www.tigr.org/tdb/e2k1/tca1/). Significantly, simultaneous overexpression of LPG1, LPG1R, and LPG1L resulted in increased incorporation of galactose into L. major proteins (Fig. 10), at least some of which was in the form of terminal Galf. Although we were unable to assign this function to a single gene, it is likely that one or more of the LPG1-family genes encode proteins with protein-glycoconjugate Galf T activity. This would be especially relevant to studies of T. cruzi, where the abundant mucins are highly modified by Galf and are thought to contribute to parasite virulence [48,49].
One scenario consistent with our data is that Leishmania LPG1L and LPG1R may be cryptic, relatively “silent counterparts” of genes highly active in other trypanosomatids. This further suggests that the potential genomic glyco-synthetic repertoire of these parasites may be larger than revealed by biochemical assays, which typically focus on the most abundant molecules or subsets thereof. A similar finding was reached in studies of the SCG family of LPG side chain galactosyltransferases, as only a subset of the seven genes of this family have shown significant GalT activity [6]. The presence of a number of silent but potentially active glycosyltransferases genes is intriguing and raises a number of speculative possibilities. Potentially, these genes represent a source for the emergence of novel or modified glycoconjugate synthesis during evolution, which may contribute in any number of ways to parasite virulence and host range. In protozoa such as Plasmodium or Trypanosoma variant surface proteins represent an interesting analogy, as gene families in these organisms comprise a group of cryptic genes that are expressed in various ways amongst strains and species and contribute to pathology [50–52]. In these species many such genes are located at telomeres and it is interesting to note that the three LPG1G genes appear to be located at telomeres in L. major and L. infantum (unpublished data; Leishmania Genome Project). Presumably during the evolutionary divergence of trypanosomatids, different lineages have chosen to emphasize the synthesis of particular glycoconjugates relevant to their biological niche within the mammalian and insect hosts. This raises the possibility that this cryptic glyco-synthetic capability may be reactivated and expressed at higher levels in the future, with consequences best understood at present by the parasite.
Acknowledgments
We thank A. Capul, D. Dobson, and K. Robinson for comments on the manuscript, M. Cunningham for pIR1PHLEO, W. Beatty for immuno-EM studies, everyone in our laboratories for helpful discussions, the Leishmania and T. cruzi Genome Projects for generously making available preliminary partial sequence of LPG1L, LPG1R, and the sequences LPG1G, and Maria Julia Manso Alves for the gift of exo-β-galactofuranosidase. This work was supported by NIH grant NIH AI 31078. Sequencing of the L. major genome was accomplished as part of the Leishmania Genome Network with support by The Wellcome Trust. Sequencing of T. cruzi was accomplished by TIGR with support from the NIAID.
Abbreviations
- LPG
lipophosphoglycan
- GIPL
glycosylinositolphospholipids
- PPG
proteophosphoglycan
- Galf
galactosylfuranose
- Galf T
galactofurranosyl transferase
- GFP
green fluorescent protein
- NEO
G418 resistance marker
- SAT
nourseothricin resistance marker
- PAC
puromycin resistance marker
- BSD
blasticidin resistance marker
- HYG
hygromycin resistance marker
- LOH
loss of heterozygosity
References
- 1.WHO Expert Committee. The Leishmaniasis. Geneva: WHO; 1984. [Google Scholar]
- 2.Turco SJ, Descoteaux A. The lipophosphoglycan of Leishmania parasites. Annu Rev Microbiol. 1992;46:65–94. doi: 10.1146/annurev.mi.46.100192.000433. [DOI] [PubMed] [Google Scholar]
- 3.McConville MJ, Ferguson MA. The structure, biosynthesis and function of glycosylated phosphatidylinositols in the parasitic protozoa and higher eukaryotes. Biochem J. 1993;294:5–24. doi: 10.1042/bj2940305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ilg T, Handman E, Stierhof YD. Proteophosphoglycans from Leishmania promastigotes and amastigotes. Biochem Soc Trans. 1999;27:518–25. doi: 10.1042/bst0270518. [DOI] [PubMed] [Google Scholar]
- 5.Dobson DE, Mengeling BJ, Cilmi S, Hickerson S, Turco SJ, Beverley SM. Identification of genes encoding arabinosyltransferases (SCA) mediating developmental modifications of lipophosphoglycan required for sand fly transmission of Leishmania major. J Biol Chem. 2003;278:28840–8. doi: 10.1074/jbc.M302728200. [DOI] [PubMed] [Google Scholar]
- 6.Dobson DE, Scholtes LD, Valdez KE, Sullivan DR, Mengeling BJ, Cilmi S, et al. Functional identification of galactosyltransferases (SCGs) required for species-specific modifications of the lipophosphoglycan adhesion controlling Leishmania major-sand fly interactions. J Biol Chem. 2003;278:15523–31. doi: 10.1074/jbc.M301568200. [DOI] [PubMed] [Google Scholar]
- 7.Sacks DL, Modi G, Rowton E, Spath G, Epstein L, Turco SJ, et al. The role of phosphoglycans in Leishmania-sand fly interactions. Proc Natl Acad Sci USA. 2000;97:406–11. doi: 10.1073/pnas.97.1.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Descoteaux A, Turco SJ. Functional aspects of the Leishmania donovani lipophosphoglycan during macrophage infection. Microbes Infect. 2002;4:975–81. doi: 10.1016/s1286-4579(02)01624-6. [DOI] [PubMed] [Google Scholar]
- 9.Spath GF, Garraway LA, Turco SJ, Beverley SM. The role(s) of lipophosphoglycan (LPG) in the establishment of Leishmania major infections in mammalian hosts. Proc Natl Acad Sci USA. 2003;100:9536–41. doi: 10.1073/pnas.1530604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McConville MJ, Collidge TA, Ferguson MA, Schneider P. The glycoinositol phospholipids of Leishmania mexicana promastigotes. Evidence for the presence of three distinct pathways of glycolipid biosynthesis. J Biol Chem. 1993;268:15595–604. [PubMed] [Google Scholar]
- 11.McConville MJ, Blackwell JM. Developmental changes in the glycosylated phosphatidylinositols of Leishmania donovani Characterization of the promastigote and amastigote glycolipids. J Biol Chem. 1991;266:15170–9. [PubMed] [Google Scholar]
- 12.Zufferey R, Allen S, Barron T, Sullivan DR, Denny PW, Almeida IC, et al. Ether phospholipids and glycosylinositolphospholipids (GIPLs) are not required for amastigote virulence nor for inhibition of macrophage activation by Leishmania major. J Biol Chem. 2003;278(45):44708–18. doi: 10.1074/jbc.M308063200. [DOI] [PubMed] [Google Scholar]
- 13.Garami A, Ilg T. Disruption of mannose activation in Leishmania mexicana: GDP-mannose pyrophosphorylase is required for virulence but not for viability. EMBO J. 2001;20:3657–66. doi: 10.1093/emboj/20.14.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garami A, Mehlert A, Ilg T. Glycosylation defects and virulence phenotypes of Leishmania mexicana phosphomannomutase and dolicholphosphate-mannose synthase gene deletion mutants. Mol Cell Biol. 2001;21:8168–83. doi: 10.1128/MCB.21.23.8168-8183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilgoutz SC, Zawadzki JL, Ralton JE, McConville MJ. Evidence that free GPI glycolipids are essential for growth of Leishmania mexicana. EMBO J. 1999;18:2746–55. doi: 10.1093/emboj/18.10.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McConville MJ, Homans SW, Thomas-Oates JE, Dell A, Bacic A. Structures of the glycoinositolphospholipids from Leishmania major A family of novel galactofuranose-containing glycolipids. J Biol Chem. 1990;265:7385–94. [PubMed] [Google Scholar]
- 17.McConville MJ, Thomas-Oates JE, Ferguson MA, Homans SW. Structure of the lipophosphoglycan from Leishmania major. J Biol Chem. 1990;265:19611–23. [PubMed] [Google Scholar]
- 18.Spath GF, Epstein L, Leader B, Singer SM, Avila HA, Turco SJ, et al. Lipophosphoglycan is a virulence factor distinct from related glycoconjugates in the protozoan parasite Leishmania major. Proc Natl Acad Sci USA. 2000;97:9258–63. doi: 10.1073/pnas.160257897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kapler GM, Coburn CM, Beverley SM. Stable transfection of the human parasite Leishmania major delineates a 30-kilobase region sufficient for extrachromosomal replication and expression. Mol Cell Biol. 1990;10:1084–94. doi: 10.1128/mcb.10.3.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ha DS, Schwarz JK, Turco SJ, Beverley SM. Use of the green fluorescent protein as a marker in transfected Leishmania. Mol Biochem Parasitol. 1996;77:57–64. doi: 10.1016/0166-6851(96)02580-7. [DOI] [PubMed] [Google Scholar]
- 21.Gueiros-Filho FJ, Beverley SM. Trans-kingdom transposition of the Drosophila element mariner within the protozoan Leishmania. Science. 1997;276:1716–9. doi: 10.1126/science.276.5319.1716. [DOI] [PubMed] [Google Scholar]
- 22.Descoteaux A, Luo Y, Turco SJ, Beverley SM. A specialized pathway affecting virulence glycoconjugates of Leishmania. Science. 1995;269:1869–72. doi: 10.1126/science.7569927. [DOI] [PubMed] [Google Scholar]
- 23.Robinson KA, Beverley SM. Improvements in transfection efficiency and tests of RNA interference (RNAi) approaches in the protozoan parasite Leishmania. Mol Biochem Parasitol. 2003;128:217–28. doi: 10.1016/s0166-6851(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 24.Cruz A, Coburn CM, Beverley SM. Double targeted gene replacement for creating null mutants. Proc Natl Acad Sci USA. 1991;88:7170–4. doi: 10.1073/pnas.88.16.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gueiros-Filho FJ, Beverley SM. Selection against the dihydrofolate reductase-thymidylate synthase (DHFR-TS) locus as a probe of genetic alterations in Leishmania major. Mol Cell Biol. 1996;16:5655–63. doi: 10.1128/mcb.16.10.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual molecular cloning. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 27.Flinn HM, Smith DF. Genomic organisation and expression of a differentially-regulated gene family from Leishmania major. Nucleic Acids Res. 1992;20:755–62. doi: 10.1093/nar/20.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Ibarra AA, Howard JG, Snary D. Monoclonal antibodies to Leishmania tropica major: specificities and antigen location. Parasitology. 1982;85(Pt 3):523–31. doi: 10.1017/s0031182000056304. [DOI] [PubMed] [Google Scholar]
- 29.Connell ND, Medina-Acosta E, McMaster WR, Bloom BR, Russell DG. Effective immunization against cutaneous leishmaniasis with recombinant bacille Calmette-Guerin expressing the Leishmania surface proteinase gp63. Proc Natl Acad Sci USA. 1993;90:11473–7. doi: 10.1073/pnas.90.24.11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spath GF, Weiss MC. Hepatocyte nuclear factor 4 provokes expression of epithelial marker genes, acting as a morphogen in dedifferentiated hepatoma cells. J Cell Biol. 1998;140:935–46. doi: 10.1083/jcb.140.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Titus RG, Marchand M, Boon T, Louis JA. A limiting dilution assay for quantifying Leishmania major in tissues of infected mice. Parasite Immunol. 1985;7:545–55. doi: 10.1111/j.1365-3024.1985.tb00098.x. [DOI] [PubMed] [Google Scholar]
- 32.Orlandi PA, Jr, Turco SJ. Structure of the lipid moiety of the Leishmania donovani lipophosphoglycan. J Biol Chem. 1987;262:10384–91. [PubMed] [Google Scholar]
- 33.Huang C, Turco SJ. Defective galactofuranose addition in lipophosphoglycan biosynthesis in a mutant of Leishmania donovani. J Biol Chem. 1993;268:24060–6. [PubMed] [Google Scholar]
- 34.Turco SJ, Stetson B, Robbins PW. Comparative rates of transfer of lipid-linked oligosaccharides to endogenous glycoprotein acceptors in vitro. Proc Natl Acad Sci USA. 1977;74:4411–4. doi: 10.1073/pnas.74.10.4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilg T. Lipophosphoglycan is not required for infection of macrophages or mice by Leishmania mexicana. EMBO J. 2000;19:1953–62. doi: 10.1093/emboj/19.9.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan KA, Garraway LA, Descoteaux A, Turco SJ, Beverley SM. Isolation of virulence genes directing surface glycosylphosphatidylinositol synthesis by functional complementation of Leishmania. Proc Natl Acad Sci USA. 1993;90:8609–13. doi: 10.1073/pnas.90.18.8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma D, Russell DG, Beverley SM, Turco SJ. Golgi GDP-mannose uptake requires Leishmania LPG2. A member of a eukaryotic family of putative nucleotide-sugar transporters. J Biol Chem. 1997;272:3799–805. [PubMed] [Google Scholar]
- 38.Haynes PA, Ferguson MA, Cross GA. Structural characterization of novel oligosaccharides of cell-surface glycoproteins of Trypanosoma cruzi. Glycobiology. 1996;6:869–78. doi: 10.1093/glycob/6.8.869. [DOI] [PubMed] [Google Scholar]
- 39.Pan F, Jackson M, Ma Y, McNeil M. Cell wall core galactofuran synthesis is essential for growth of mycobacteria. J Bacteriol. 2001;183:3991–8. doi: 10.1128/JB.183.13.3991-3998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leal JA, Guerrero C, Gomez-Miranda B, Prieto A, Bernabe M. Chemical and structural similarities in wall polysaccharides of some Penicillium, Eupenicillium and Aspergillus species. FEMS Microbiol Lett. 1992;69:165–8. doi: 10.1016/0378-1097(92)90622-u. [DOI] [PubMed] [Google Scholar]
- 41.Pedersen LL, Turco SJ. Galactofuranose metabolism: a potential target for antimicrobial chemotherapy. Cell Mol Life Sci. 2003;60:259–66. doi: 10.1007/s000180300021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merello S, Xavier MT, Parodi AJ. The presence of galactofuranose and ribose units in asparagine-linked oligosaccharides of the digenetic trypanosomatid Endotrypanum schaudinni. Mol Biochem Parasitol. 1995;69:73–9. doi: 10.1016/0166-6851(94)00195-s. [DOI] [PubMed] [Google Scholar]
- 43.Moraes CT, Bosch M, Parodi AJ. Structural characterization of several galactofuranose-containing, high-mannose-type oligosaccharides present in glycoproteins of the trypanosomatid Leptomonas samueli. Biochemistry. 1988;27:1543–9. doi: 10.1021/bi00405a022. [DOI] [PubMed] [Google Scholar]
- 44.Mendelzon DH, Parodi AJ. N-linked high mannose-type oligosaccharides in the protozoa Crithidia fasciculata and Crithidia harmosa contain galactofuranose residues. J Biol Chem. 1986;261:2129–33. [PubMed] [Google Scholar]
- 45.Parodi AJ, Martin-Barrientos J, Engel JC. Glycoprotein assembly in Leishmania mexicana. Biochem Biophys Res Commun. 1984;118:1–7. doi: 10.1016/0006-291x(84)91058-1. [DOI] [PubMed] [Google Scholar]
- 46.Mendelzon DH, Previato JO, Parodi AJ. Characterization of protein-linked oligosaccharides in trypanosomatid flagellates. Mol Biochem Parasitol. 1986;18:355–67. doi: 10.1016/0166-6851(86)90092-7. [DOI] [PubMed] [Google Scholar]
- 47.Rietschel-Berst M, Jentoft NH, Rick PD, Pletcher C, Fang F, Gander JE. Extracellular exo-beta-galactofuranosidase from Penicillium charlesii: isolation purification and properties. J Biol Chem. 1977;252:3219–26. [PubMed] [Google Scholar]
- 48.Acosta-Serrano A, Almeida IC, Freitas-Junior LH, Yoshida N, Schenkman S. The mucin-like glycoprotein super-family of Trypanosoma cruzi: structure and biological roles. Mol Biochem Parasitol. 2001;114:143–50. doi: 10.1016/s0166-6851(01)00245-6. [DOI] [PubMed] [Google Scholar]
- 49.de Lederkremer RM, Colli W. Galactofuranose-containing glycoconjugates in trypanosomatids. Glycobiology. 1995;5:547–52. doi: 10.1093/glycob/5.6.547. [DOI] [PubMed] [Google Scholar]
- 50.Donelson JE. Antigenic variation and the African trypanosome genome. Acta Trop. 2003;85:391–404. doi: 10.1016/s0001-706x(02)00237-1. [DOI] [PubMed] [Google Scholar]
- 51.Barry JD, Ginger ML, Burton P, McCulloch R. Why are parasite contingency genes often associated with telomeres? Int J Parasitol. 2003;33:29–45. doi: 10.1016/s0020-7519(02)00247-3. [DOI] [PubMed] [Google Scholar]
- 52.Kyes S, Horrocks P, Newbold C. Antigenic variation at the infected red cell surface in malaria. Annu Rev Microbiol. 2001;55:673–707. doi: 10.1146/annurev.micro.55.1.673. [DOI] [PubMed] [Google Scholar]
- 53.Sacks DL, Hieny S, Sher A. Identification of cell surface carbohydrate and antigenic changes between noninfective and infective developmental stages of Leishmania major promastigotes. J Immunol. 1985;135:564–9. [PubMed] [Google Scholar]