

Abstract
Alport syndrome, hereditary glomerulonephritis with hearing loss, results from mutations in type IV collagen COL4A3, COL4A4, or COL4A5 genes. The mechanism for delayed glomerular disease onset is unknown. Comparative analysis of Alport mice and CD151 knockout mice revealed progressive accumulation of laminin 211 in the glomerular basement membrane. We show mesangial processes invading the capillary loops of both models as well as in human Alport glomeruli, as the likely source of this laminin. l-NAME salt–induced hypertension accelerated mesangial cell process invasion. Cultured mesangial cells showed reduced migratory potential when treated with either integrin-linked kinase inhibitor or Rac1 inhibitor, or by deletion of integrin α1. Treatment of Alport mice with Rac1 inhibitor or deletion of integrin α1 reduced mesangial cell process invasion of the glomerular capillary tuft. Laminin α2–deficient Alport mice show reduced mesangial process invasion, and cultured laminin α2–null cells showed reduced migratory potential, indicating a functional role for mesangial laminins in progression of Alport glomerular pathogenesis. Collectively, these findings predict a role for biomechanical insult in the induction of integrin α1β1–dependent Rac1-mediated mesangial cell process invasion of the glomerular capillary tuft as an initiation mechanism of Alport glomerular pathology.
Alport syndrome is characterized by delayed-onset progressive glomerulonephritis associated with sensorineural hearing loss and retinal flecks.1 The most common form (80%) is X-linked and caused by mutations in the type IV collagen COL4A5 gene.2 The two autosomal forms of the disease account for the remaining 20% of Alport patients, and result from mutations in the COL4A3 and COL4A4 genes.3 The α3(IV), α4(IV), and α5(IV) proteins form a heterotrimer that is assembled into a subepithelial network in the glomerular basement membrane (GBM) that is physically and biochemically distinct from a subendothelial type IV collagen network comprising α1(IV) and α2(IV) heterotrimers.4 Mutations in any one of the three type IV collagen genes that cause Alport syndrome result in the absence of all three proteins in the GBM due to an obligatory association to form functional heterotrimers.5 Thus, the net result for all genetic forms of Alport syndrome is the absence of the α3(IV) α4(IV) α5(IV) subepithelial collagen network, resulting in a GBM type IV collagen network comprising only α1(IV) and α2(IV) heterotrimers.
This change in basement membrane composition does not result in immediate pathology. The GBM appears to function adequately for the first few years of life and sometimes past the first decade.6 This delayed onset predicts a triggering mechanism for glomerular disease initiation and a theoretical window for therapeutic intervention that may arrest or significantly ameliorate Alport renal disease in its earliest stages. The activation of genes encoding GBM matrix molecules, matrix metalloproteinases (MMPs), and proinflammatory cytokines have all been linked to the progression of Alport glomerular disease. These, however, are events that occur after the onset of proteinuria, and therefore, downstream of disease initiation events.7–11 Consistent with this notion, experiments aimed at blocking these pathways have offered only limited therapeutic benefit in mouse models for Alport syndrome.8–10,12 One of the earliest events we have documented is the appearance of an irregular deposition of laminin 211 in the GBM of Alport mice,8 an observation confirmed in both Alport dogs and human patients with the disease.13 This laminin is normally found only in the mesangium of the glomerulus, and is not expressed in the GBM at any stage of embryonic development.14 Indeed, several other mesangial matrix proteins appear in the GBM of Alport mice, including laminin 111 and fibronectin.15,16
In the Alport glomerulus, the podocytes are exposed to GBM that has an embryonic type IV collagen composition.17,18 This could result in altered cell signaling that may trigger the onset of the disease. It has been proposed that this type of mechanism may account for the reactivation of laminin 111 expression in podocytes,19 because laminin 111 is found in the GBM during development.14 Because the α1(IV)/α2(IV) collagen network contains significantly fewer interchain disulfide crosslinks,20 and the Alport GBM is thinner than normal,21 the Alport GBM is likely to be more elastic, resulting in elevated biomechanical strain on the glomerular cells at their points of contact with the GBM. Consistently, glomeruli from Alport mice have been shown to have elevated deformability relative to wild-type glomeruli,22 and salt-induced hypertension has been shown to accelerate glomerular disease progression in Alport mice.23
In this work, we show that the cellular origin of GBM laminin 211 in Alport glomeruli is mesangial cell process invasion, and that deletion of laminin 211 in Alport mice ameliorates the mesangial process invasion of the glomerular capillary loops in Alport mice. Salt-mediated hypertension exacerbates this mesangial process invasion. A knockout mouse for the integrin α3β1 coreceptor CD151 also develops mesangial process invasion of the capillary loops with GBM deposition of laminin 211, demonstrating the same phenotype for a completely unrelated component of the capillary structural barrier. The CD151 knockout mouse model also shows accelerated glomerular disease progression in response to hypertension.24 We show that biomechanical stretching of cultured mesangial cells induces promigratory cytokines transforming growth factor-β1 (TGF-β1) and connective tissue growth factor (CTGF), both known to be induced in Alport glomeruli.7,12 Inhibitor studies indicate that mesangial cell migration is mediated by integrin α1β1 signaling through the Rho GTPases RAC1 and CDC42. Consistently, integrin α1 deletion in Alport mice was previously shown to ameliorate glomerular disease progression and slow the accumulation of laminin 211 in Alport GBM.8 Here, we show that mesangial process invasion of the capillary loops is ameliorated in integrin α1–null Alport mice. These data define a role for biomechanical strain-mediated induction of mesangial cell process invasion as a key aspect of Alport glomerular disease initiation, and set the stage for defining novel therapeutic targets aimed at blocking this process.
Materials and Methods
Mice
All mice used in these studies were on pure genetic backgrounds. Autosomal recessive Alport mice were on 129/Sv background and developed in our laboratory.15 X-linked Alport mice were on C57Bl/6 background (acquired from Jackson Laboratories, Bar Harbor, ME), laminin α2–deficient mice were on 129/Sv background (acquired from Jackson Laboratories), integrin α1–null mice were on 129/Sv background,25 and CD151 knockout mice were on FVB background.26 All experiments were performed using strain/age-matched control mice. All animal studies were conducted in accordance with standards approved by the US Department of Agriculture and under the approval of the institutional animal care and use committee. Every effort was made to minimize pain and discomfort.
l-NAME Hypertension
Nω-nitro-l-arginine methyl ester (l-NAME; Sigma-Aldrich, St. Louis, MO) salt–induced hypertension was performed as previously described.23 Wild-type and X-linked Alport mice on C57Bl/6 background were given l-NAME salts at a concentration of 36 mg/100 mL of water (50 mg/kg/day) from 5 to 10 weeks of age. Blood pressure was monitored on a weekly basis using a CODA2 (Kent Scientific, Torrington, CT) noninvasive tail cuff monitoring system.
Treatment of Mice with Rac1 Inhibitor
129/Sv autosomal Alport mice and age/strain-matched wild-type mice were injected i.p. once daily with 2 mg/kg of the Rac1 inhibitor NSC 23766 (Tocris Bioscience, Ellisville, MO). Animals were treated from 2 weeks of age until 6 weeks of age.
Immunofluorescence Microscopy
Fresh frozen kidneys were sectioned at 8 μm and acetone fixed. Sections were incubated overnight at 4°C with 0.3% PBST (Triton X-100), 5% fetal bovine serum, and with two of the following antibodies: rat anti-mouse laminin α2 antibody (Sigma-Aldrich) at 1:200, goat anti-mouse integrin α8 antibody (R & D Systems, Minneapolis, MN) at 1:100, rabbit anti-mouse laminin α5 antibody (a generous gift from Dr. Jeff Miner, Washington University) at 1:200, rabbit anti-human laminin α5 antibody (GeneTex, Irvine, CA) at 1:500. Slides were rinsed with 1× PBS and incubated with the appropriate Alexa Fluor donkey secondary antibodies at 1:300 for 1 hour at room temperature. They were then rinsed again with 1× PBS and mounted with Vectashield Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA). De-identified human Alport renal necropsy tissue was a gift from Dr. Raghu Kalluri and described previously.9
Transmission Electron Microscopy
Transmission electron microscopy was performed as described previously.8
Mesangial Cell Migration
Primary mesangial cell cultures were prepared from isolated glomeruli. Cells were isolated and propagated as described previously.11 Eight-micron, 24-well plate control inserts (BD Bioscience, Bedford, MA) were coated overnight at 4°C with 100 μL of 0.1% gelatin/PBS then washed 1× with PBS. Mesangial cell cultures were incubated in 1% fetal calf serum (FCS) overnight, then 0.05% bovine serum albumin–containing medium for at least 8 hours, washed 1× with PBS, and carefully trypsinized to ensure a single-cell suspension and limited clumping of cells. After serum neutralization and subsequent centrifugation, approximately 100,000 cells were resuspended in 1.5 mL of 0.05% bovine serum albumin medium containing inhibitors. The wells of a 24-well plate were filled with 0.75 mL of 10% FCS–containing medium with inhibitors (excluding the 0.05% bovine serum albumin control well). The cell suspension (0.05 mL) was loaded into a gelatin-coated insert and the insert placed in a well. Wells were visually inspected for bubbles beneath the insert and for equal distribution of cell suspension. Cells were allowed to migrate overnight (approximately 18 hours). Using a moistened cotton swab, nonmigrated cells were liberated from the apical side of the insert by gentle, but firm, rubbing. A second swab repeated the removal and was followed by a single wash with PBS. Inserts were fixed, stained, and then washed (2×) in companion 24-well plate(s) containing 0.5 mL of methanol, 0.5 mL of 1% toluidine blue in 1% borax, and 0.5 mL of distilled water, respectively. Inserts were air dried and counted at ×100 magnification. Five fields were counted on each insert, including one center and four periphery areas. Data were expressed as relative to the 10% FCS control well (set equal to one). Inhibitors included 10 μmol/L ILK inhibitor QLT-0267 (Valocor Therapeutics, Vancouver, BC, Canada), 100 μmol/L Rac1 inhibitor NSC 23766 (Tocris Bioscience), or 1 μmol/L pan-AKT inhibitor, GSK 690693 (Tocris Bioscience).
Scratch Wound Migration Assay
For basal lamina studies, Superfrost Plus (VWR International, Radnor, PA) microscope slides were coated with the following: 100 ng/mL Merosin (Millipore, Billerica, MA), 100 ng/mL human placental laminin (Sigma-Aldrich), 20 ng/mL human rlaminin-211 (BioLamina, Stockholm, Sweden), or 20 ng/mL human rlaminin-521 (BioLamina) per the manufacturer’s suggestion. Slide(s) were placed in a tissue culture dish and an 8 × 8-mm cloning ring (Bellco Glass, Vineland, NJ) placed on the coated area. A 100-μL cell suspension (approximately 30,000 cells) in 1% fetal bovine serum–containing medium was added to the cloning ring, and the cells were allowed to attach for approximately 8 hours; PBS was placed in the dish, and the ring was then removed. An approximately 0.3- to 0.5-mm swath of cells was removed by running a serological pipette at an approximately 45° angle through the monolayer. After capturing images of removed cells, slides were incubated for 24 hours in 1% fetal bovine serum–containing medium, washed with PBS, fixed in methanol for 5 minutes, air dried, and then stained for 30 minutes with modified Giemsa stain (Sigma-Aldrich). Images of previously photographed fields were captured using a Leica MZ10F microscope fitted with a Leica DFC310FX camera (Leica, Wetzlar, Germany).
Biomechanical Stretching of Cultured Mesangial Cells
Low-passage, subconfluent, primary mesangial cells were trypsinized and seeded onto Bioflex 6-well plates (Flexcell International Corp., Hillsborough, NC) coated with rat tail type I collagen (BD Biosciences). Cells were plated in 5% FCS–containing medium at densities that resulted in 20% to 40% confluence. The 0.5% FCS medium was placed on the cells the next day. Forty-eight hours later, the medium was changed and the cultures exposed to a regimen of 60 cycles of stretch and relaxation per minute with amplitude of 10% radial surface elongation. The Flexercell Strain Unit FX4000 (Flexcell International Corp.) was used to induce stretch/relaxation for 18 hours according to the manufacturer’s directions. Cells grown identically, but not exposed to stretch, served as controls.
Real-Time Quantitative RT-PCR
Total RNA was reverse transcribed using SuperScript III (Invitrogen; Life Technologies, Grand Island, NY) with Oligo(dT)20 Primer (Invitrogen). Real-time PCR was performed using TaqMan Gene Expression Master Mix (Applied Biosystems; Life Technologies), and quantified using ABI Prism 7000 sequence detection system (Applied Biosystems). Samples were normalized to Mouse GAPDH Endogenous Control VIC Probe (Applied Biosystems catalog #4352339E), which was run alongside the CTGF (Catalog #4331182, ID# Mm01192933_g1) and TGF-β1 (Catalog #4331182, ID# Mm01178820_m1) TaqMan Gene Expression Assay Probes (Applied Biosystems). Each of the samples was run in triplicate with a final reaction volume of 50 μL and with the following cycling parameters: 50°C for 2 minutes, 95°C for 10 minutes, followed by 40 cycles of a two-step PCR consisting of 95°C for 15s and 60°C for 1 minute. Relative changes in gene expression were determined by calculating the fold change using the comparative CT method of 2-ΔΔCT following manufacturer’s instructions.
Activation of Mesangial Cell Cultures by Treatment with Lipopolysaccharide
Subconfluent mesangial cells were trypsinized; plated at low density on rat tail type 1 collagen (BD Biosciences) coated cytology slides (VWR International) and incubated overnight in 1% FCS–containing medium. One hour after the addition of serum-free medium, 50 μmol/L CDC42 inhibitor (ML141) or 10 μmol/L Rac-1 inhibitor NSC 23766 (Tocris Bioscience) were added to individual slides and allowed to incubate for an additional hour. Lipopolysaccharide (LPS) (10 ng/mL; Sigma-Aldrich) was added to cells, incubated 1 hour, fixed in ice-cold acetone for 5 minutes, and then allowed to air dry approximately 2 hours. Cells were stained with a 1:100 dilution of antibodies specific for CDC42 (10155-1-AP; Proteintech, Chicago, IL), and/or phalloidin (Molecular Probes; Life Sciences) washed, and then imaged. Untreated, LPS alone and LPS plus inhibitor treatments were repeated on two different derivations of primary mesangial cells with qualitatively consistent results.
Pull-Down Assay
Pull-down experiments for Rac1 in mesangial cells were done using the Rac1 Activation Assay Bicochem Kit (BK035; Cytoskeleton, Denver, CO) and according to the manufacturer’s instructions with minor modifications. Briefly, 500 to 800 μg of protein lysates were incubated with 20 μL of PAK-PBD beads for 1 hour at 4°C. Pull-down samples and total protein lysates (30 to 50 μg of protein) were run in a 12% SDS-PAGE gel, transferred to polyvinylidene difluoride membranes, and then blocked in 5% milk for 30 minutes at room temperature. Rac-1 antibody incubation was done overnight at 4°C with rocking. After secondary antibody incubation and several washes, membranes were developed using the ECL Plus kit (32134; Pierce Biotechnology, Rockford, IL) for pull-down experiments or the SuperSignal West Femto kit (#34094; Pierce Biotechnology) for total lysates. Films were exposed for 40 minutes and 5 minutes, respectively, and developed using a film processor.
Confocal Microscopy
Slides were coverslipped using Vectashield mounting medium containing DAPI to counterstain the nuclei (Vector Laboratories) and confocal images captured using a Zeiss AxioPlan 2IF MOT microscope interfaced with a LSM510 META confocal imaging system, using a 63× NA:1.4 oil objective (Carl Zeiss, Oberkochen, Germany). Final figures were assembled using Adobe Photoshop and Illustrator CS6 software (Adobe Systems, San Jose, CA).
Statistical Analysis
Data were analyzed using the Student’s t-test with Bonferroni correction.
Results
GBM Laminin 211 in Alport Mice Is of Mesangial Origin
In the glomerulus, laminin 211 is normally found only in the mesangial matrix. Figure 1, A–C, demonstrates mesangial distribution of laminin 211 in 7-week-old 129/Sv wild-type mice, which is distinct from the glomerular basement membrane (laminin α5). In Alport glomeruli (Figure 1, D–F), irregular distribution of laminin 211 in the GBM, which appears to accumulate preferentially in irregularly thickened regions of the GBM (here, the GBM is marked by immunostaining with antibodies specific for laminin α5), is observed. The cellular source of the GBM laminin 211 has never been determined. Dual immunofluorescence labeling with antibodies against laminin α2 and integrin α8 shows mesangial specific immunostaining in wild-type glomeruli (Figure 2, A–C), as reported previously.27 In Alport glomeruli (at 7 weeks of age), immunostaining for both laminin α2 and integrin α8 appears to have spread into the capillary loops, consistent with mesangial cell process invasion of the capillary loops (Figure 2, D–F). Dual immunofluorescence immunostaining using the basement membrane marker laminin α5 with the mesangial marker integrin α8 confirms that integrin α8 immunostaining, although absent from the GBM in wild-type mice (Figure 2, G–I), is clearly present in most of the GBM of Alport mice (Figure 2, J–L). Transmission electron microscopy occasionally showed the presence of mesangial processes in the glomerular capillary loops of Alport mice (Figure 2N), which are not observed in the capillary loops from wild-type mice (Figure 2M). Collectively, these data support that GBM laminin 211 arises from a mesangial cell process invasion of the capillary loops, and thus is of mesangial cell origin.
Figure 1.
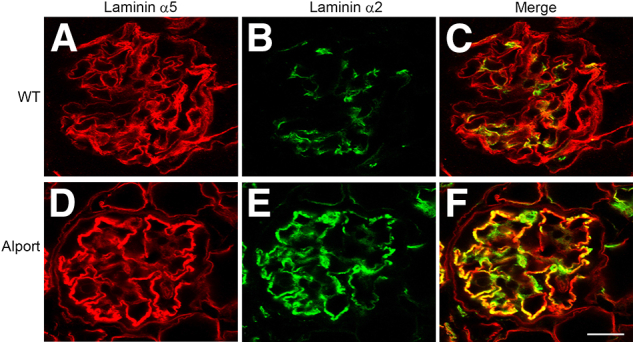
Laminin 211 localizes to the GBM in Alport glomeruli. Dual immunofluorescence immunostaining was performed on wild-type (WT) (A–C) and Alport (D–F) glomeruli from 7-week-old 129/Sv mice. GBM was labeled with anti–laminin α5 antibodies (red) and anti–laminin α2 immunostaining (green). Note the irregular deposits of laminin 211 in the Alport GBM, especially in the thickened regions of the GBM. Scale bar = 15 μm.
Figure 2.
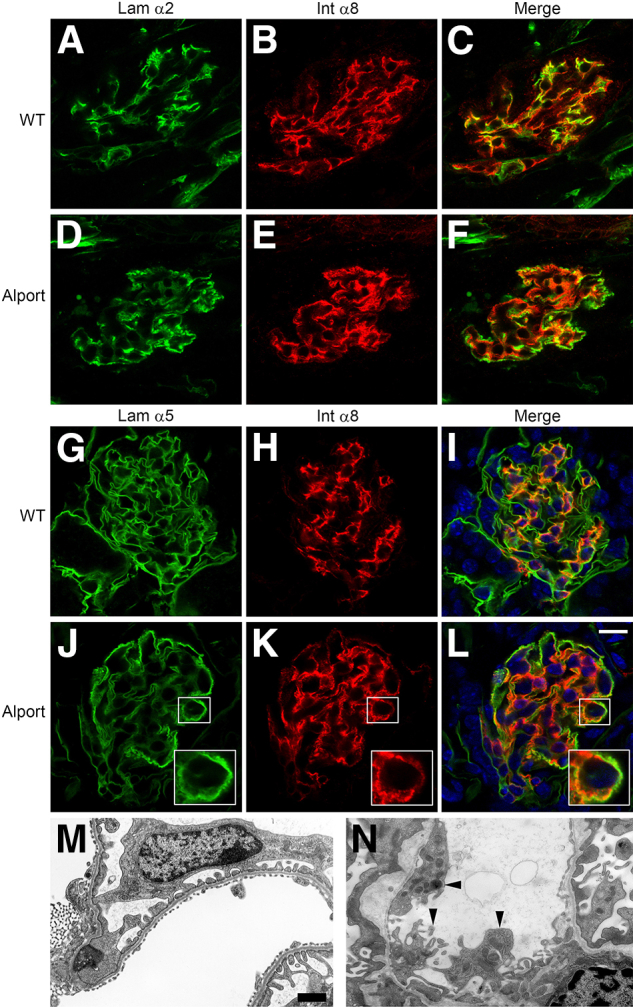
Mesangial processes invade the capillary loops of Alport glomeruli where they colocalize with laminin 211 deposits. Dual immunofluorescence immunostaining was performed on wild-type (A–C and G–I) or Alport (D–F and J–L) kidney sections from 7-week-old 129/Sv mice. Panels show localization of laminin α2 (Lam α2) and integrin α8 (Int α8) (a mesangial cell marker), and laminin α5 (a GBM marker) and integrin α8. Note circumferential colocalization of laminin α2 and integrin α8 in the Alport glomerulus in D–F, and the colocalization of integrin α8 and laminin α5 in J–L, indicating invasion of the glomerular capillary tufts with mesangial processes. Scale bar = 10 μm (J–L); 5 μm (insets). Transmission electron micrograph of a capillary loop from a 7-week-old wild-type (WT) mouse (M) and an age-matched Alport mouse (N). Arrowheads denote extensions coming from the interface of the mesangial cell with the glomerular capillary loop, consistent with mesangial process invasion. Scale bar = 500 nm.
To determine the relevance of this observation to human Alport syndrome, we stained cryosections from human Alport necropsy kidney sections with antibodies specific for integrin α8 and laminin α5. The results in Figure 3 show that mesangial processes are clearly present adjacent to the laminin α5-immunopositive GBM in the human specimen.
Figure 3.

Mesangial processes invade the capillary loops of human Alport glomeruli where they localize adjacent to laminin 521. Cryosections from normal (A–C) and Alport (D–F) human kidneys were stained with antibodies specific for laminin α5 (Lam α5) (green) and integrin α8 (Int α8) (red). The integrin α8–labeled mesangial processes localize adjacent to the laminin α5–positive GBM in human Alport, but not in normal human glomeruli, consistent with mesangial process invasion. Scale bar = 15 μm.
Mesangial Process Invasion of the Capillary Loops Is Exacerbated by Elevated Biomechanical Strain
In an earlier report, we demonstrated that hypertension exacerbated the progression of Alport glomerular disease.23 Hypertension accelerated several aspects of glomerular disease progression, including proteinuria and induction of matrix metalloproteinases. The accumulation of GBM laminin 211 was also accelerated. In Figure 4, we show that salt-induced hypertension clearly accelerates the inundation of the glomerular capillary loops by mesangial processes, as evidenced by the presence of integrin α8 immunopositivity in the GBM (Figure 4, D–F).
Figure 4.

Hypertension exacerbates mesangial invasion of the glomerular capillary tufts in Alport mice. The X-linked Alport mouse model (on C57Bl/6 background) was made hypertensive by providing l-NAME salts in the drinking water from 5 weeks to 10 weeks of age. Control Alport mice were given normal drinking water. Glomeruli were analyzed by dual immunofluorescence immunostaining using antibodies against laminin α2 (Lam α2) and integrin α8 (Int α8). Extensive mesangial process invasion of the capillary tuft was observed in the glomeruli from the salt-treated mice (D–F) relative to the mice given normal drinking water (A–C). Scale bar = 10 μm.
We presume the increased biomechanical stress on the glomerular capillary tuft in Alport glomeruli is due to the change in GBM type IV composition from dual networks of α1(IV)/α2(IV) and α3(IV)/α4(IV)/α5(IV) collagen to one comprising only α1(IV)/α2(IV) collagen. The latter is thinner and known to contain fewer interchain disulfide crosslinks,20 which would intuitively be expected to result in increasing the elasticity of the glomerular filtration barrier. To provide independent validation of this assumption, we examined a completely different model that would also be expected to affect the elastic integrity of the glomerular filtration barrier, the CD151 knockout mouse. CD151 is a tetraspanin co-receptor for integrin α3β1 that functions to increase the affinity of integrin α3β1 for its GBM ligand, laminin α5.28 Deletion of CD151 results in glomerular disease with morphological changes in the GBM, strikingly similar to Alport syndrome.27 Therefore, we examined glomeruli from the CD151 knockout mouse for mesangial process invasion and laminin 211 deposition in the GBM. The results in Figure 5 show a near complete inundation of the glomerular capillary tufts with integrin α8 and laminin α2 immunopositivity in the CD151 knockout mouse, demonstrating mesangial process invasion and deposition of mesangial laminins in the GBM in this genetically unrelated model.
Figure 5.

Extensive mesangial process invasion of the glomerular capillary tufts is observed in CD151 knockout (KO) mice. Kidney cryosections from 8-week-old wild-type (WT) (A–C) and CDC151 KO mice (D–F) (on the FVB background) were analyzed by dual immunofluorescence immunostaining using antibodies against laminin α2 and integrin α8. Extensive mesangial process invasion of the capillary tuft was observed in the glomeruli from CD151 knockout mice relative to wild-type mice. Note that the extent of mesangial process invasion in CD151 knockout mice is much greater than that observed in Alport mice (Figure 2F). Scale bar = 15 μm.
If biomechanical strain can induce the activation of mesangial process invasion of the capillary tuft, we should be able to activate promigratory responses in vitro by mechanically stretching cultured primary mesangial cells. We subjected primary cultured mesangial cells, derived from 129/Sv mice, to cyclic cell stretching using the Flexcell system for 18 hours. Expression of several promigratory cytokines was quantified by real-time RT-PCR. The results in Figure 6 demonstrate that expression of both TGF-β1 and CTGF are significantly elevated in cells subjected to biomechanical stretching relative to cells cultured under identical conditions, but not subjected to stretch. The data represent two independent derivations of mesangial cells with three independent stretch experiments where individual treatment groups were run in triplicate.
Figure 6.
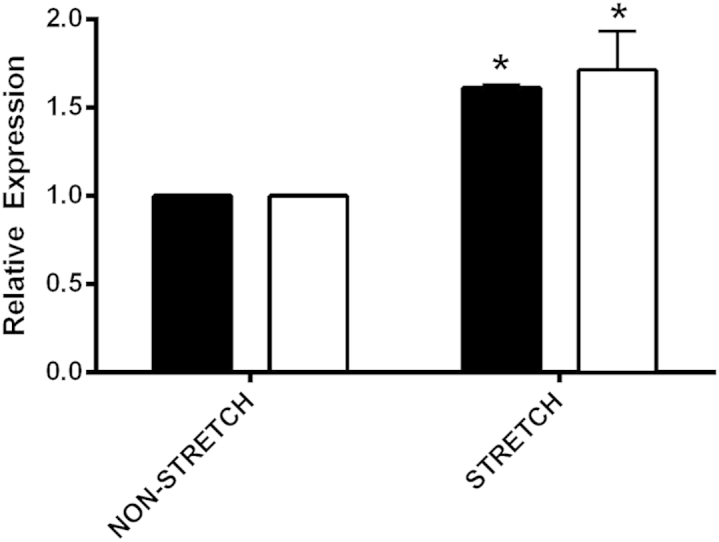
Biomechanical stretching of cultured primary mesangial cells induces expression of promigratory cytokines, CTGF (white bars) and TGF-β1 (black bars) mRNA. Two independent derivations of primary mesangial cell cultures from wild-type mice were subjected to cyclic biomechanical stretching for 18 hours. RNA from multiple stretch experiments, which were run in triplicate, were analyzed by quantitative real-time RT-PCR for CTGF and TGF-β1 mRNA. Statistically significant increases in expression for both cytokines were observed. ∗P < 0.05.
In Vitro Mesangial Cell Migration and in Vivo Mesangial Process Invasion of the Glomerular Capillary Loops Are Regulated by Integrin α1β1–Mediated Rac1/CDC42 Activation
In earlier work, we demonstrated that deletion of integrin α1 markedly attenuated the progression of glomerular disease in Alport mice.8 Although it is highly likely that disease attenuation in integrin α1–null Alport mice emanates from the mesangial compartment where integrin α1β1 is highly expressed, the molecular mechanism underlying this effect has remained unclear. In Figure 7, we show that deletion of integrin α1 reduces the dynamics of mesangial process invasion of the capillary tufts in Alport mice, consistent with the reduction in GBM laminin 211 deposition shown here and previously.8
Figure 7.

Integrin α1 deletion in Alport mice results in markedly reduced mesangial process invasion of the glomerular capillary tufts. Glomeruli from 7-week-old integrin α1–null mice (A–C), Alport mice (D–F), and integrin α1–null Alport mice (DKO) on 129/Sv background mice (G–I) were analyzed by dual immunofluorescence immunostaining using antibodies against laminin α2 (Lam α2) and integrin α8 (Int α8). The degree of mesangial process invasion of the glomerular capillary tufts is greatly reduced in the integrin α1–null Alport mice relative to age/strain-matched Alport mice. Scale bars: 10 μm (A–I); 5 μm (insets, D–F).
Because it is well established that the formation of filopodia and lamellipodia requires the concerted action of the small GTPases Rac1 and CDC42,29 we performed cell migration assays using the Boyden chamber approach to determine whether such a functional connection was evident in cultured wild-type and integrin α1–null mesangial cells. The results in Figure 8A show that integrin α1–null mesangial cells show a significant reduction in migratory potential relative to wild-type mesangial cells. Migration of wild-type cells was significantly reduced when cells were treated with either the integrin-linked kinase inhibitor QLT-0267, or the Rac1 inhibitor NSC 23766. Cell migration of wild-type cells was not affected by treatment with the pan AKT inhibitor GSK 690693. Integrin α1–null mesangial cell migration was not affected by treatment of cells with Rac1 inhibitors (data not shown), suggesting that deletion of integrin α1 abrogates Rac1-dependent cell migration.
Figure 8.

Integrin α1β1–dependent Rac1/CDC42 activation mediates dynamic remodeling of the actin cytoskeleton in cultured primary mesangial cells (MES). A: Migration of primary cultured mesangial cells is significantly reduced under conditions of integrin α1 deletion, integrin-linked kinase inhibition, Rac1 inhibition, but not AKT inhibition. Migration was measured by Boyden chamber assay in the presence or absence of the ILK inhibitor QLT-0267 (QLT), the Rac1 inhibitor NSC 23766, or the pan-AKT inhibitor GSK 690693, and for integrin α1–null mesangial cells (Iα1 Null MES). Multiple replicate experiments were performed on multiple independent derivations of mesangial cells and the data analyzed by Student’s t-test. ∗P < 0.05 versus 10% FCS alone. BSA, bovine serum albumin. B–E: Treatment of cultured mesangial cells with LPS induced cytoskeletal rearrangement with numerous actin spikes (asterisks in C), as determined by phalloidin staining (untreated cells, B; LPS-treated cells, C), and these morphological changes were blocked by treatment of cells with either Rac1 inhibitors (D) or CDC42 inhibitors (E). Untreated integrin α1–null cells did not respond to LPS treatment (data not shown). F–I: Treatment of cultured mesangial cells with LPS results in polarized localization of CDC42 (red) and is associated with filopodia (phalloidin in green) (G, inset), compared to Golgi and cytosolic localization of CDC42 in wild-type cells (F). Pretreatment of cells with the Rac1 inhibitor NSC 23766 abolished LPS-activated polarized localization of CDC42 (H), indicating crosstalk between Rac1 and CDC42. I: GTP-Rac1 pull-down assay confirms LPS-mediated activation of Rac1 in cultured mesangial cells is blocked by pretreatment with Rac1 inhibitors, but not by CDC42 inhibitor. Scale bars: 12 μm (B–H); 6 μm (inset, G). Cont, control; inh, inhibitor; lys, lysates.
Treatment of cells with the bacterial endotoxin LPS activates both Rac1 and CDC42 GTPases,30,31 and is known to induce the formation of both lamellipodia and filopodia in cultured mesangial cells.32 We treated cultured wild-type mesangial cells with LPS, stained the actin filaments with phalloidin, and examined the cultures for morphological changes. As shown in Figure 8, B–E, after 30 minutes, treated cells underwent a stark morphological change: about half of the cells sprouted numerous filopodia (Figure 8C) that were easily identified by a blinded observer (D.C.) in at least five replicate experiments. Cells treated with LPS in combination with either the Rac1 inhibitor NSC 23766 or the CDC42 inhibitor ML 141 could not be distinguished in blinded experiments from untreated wild-type mesangial cells (Figure 8, D and E, respectively). Interestingly, treatment of integrin α1–null mesangial cells with LPS had no discernible effect on cell morphology (data not shown). To further validate these findings, we stimulated either wild-type or α1-null mesangial cell cultures with LPS in the presence or absence of either Rac1 or CDC42 inhibitors and performed immunofluorescence analysis for CDC42 localization and pull-down assays for activated Rac1. As shown in Figure 8, F–I, treatment of cells with LPS resulted in polarized localization of CDC42 associated with staining in adjacent filopodia (Figure 8G), an established characteristic of CDC42 activation.33,34 Treatment of these cells with Rac1 inhibitor abolished this polarized activation, indicating crosstalk between Rac1 and CDC42 (Figure 8H). Integrin α1–null mesangial cells did not respond to LPS activation with polarized CDC42 localization (data not shown). Pull down assays demonstrate that LPS treatment does indeed activate Rac1, and that pretreatment of cells with the Rac1 inhibitor abolishes its activation (Figure 8I). Interestingly, pretreatment of cells with CDC42 inhibitors did not block LPS-mediated Rac1 activation, suggesting that, whereas Rac1 inhibitors block LPS-mediated CDC42 activation (Figure 8H), CDC42 inhibitors do not block Rac1 activation (Figure 8I).
To examine the effect of Rac1 inhibitors on Alport glomerular disease progression, we treated Alport mice with inhibitors by i.p. injection from 2 weeks to 6 weeks of age. Glomeruli were examined for mesangial process invasion of the capillary tufts by dual immunofluorescence microscopy using antibodies specific for either integrin α8 or the GBM marker laminin α5. The results in Figure 9 demonstrate that saline-injected mice show significant colocalization of integrin α8 and laminin α5 throughout many of the glomerular capillary tufts, whereas mice injected with the Rac1 inhibitor showed very little mesangial process invasion. Combined, the data in Figures 7, 8, and 9 confirm that mesangial process invasion of the glomerular capillaries is a Rac1-dependent process, and is attenuated by integrin α1 deletion in vivo. Furthermore, LPS activation of filopodia in wild-type mesangial cells (but not in α1-null mesangial cells) involves both Rac1 and CDC42 activation, suggesting integrin α1β1–dependent crosstalk between the two small GTPases in the signaling complex.
Figure 9.

Treatment of Alport mice with Rac1 inhibitors partially ameliorates mesangial cell process invasion of the glomerular capillary tufts. Alport mice on 129/Sv background were injected once daily with either saline (A–C) or the Rac1 inhibitor NSC 23766 (D–F) from 2 weeks to 6 weeks of age. Kidney cryosections were analyzed by dual immunofluorescence immunostaining using antibodies against either laminin α2 (Lam α2) or integrin α8 (Int α8). The degree of mesangial process invasion of the glomerular capillary tufts is ameliorated in the Rac1 inhibitor–treated mice relative to mice injected with saline. Scale bar = 15 μm. Original magnification, 7.5 μm (insets).
Laminin 211 Enhances Mesangial Cell Migration and Mesangial Process Invasion of the Capillary Loops
We crossed laminin α2–deficient mouse with the Alport mouse to produce a double knockout. One effect of laminin α2 deficiency was a marked reduction of mesangial process invasion of the capillary loops (Figure 10, A–C). We interpreted this to mean that laminin 211 might facilitate mesangial process invasion of the capillary loops. To test this theory, we performed cell migration assays on either laminin 211 or laminin 521 (GBM laminin). Two different laminin preparations were used. One was extracted laminin from either placenta (primarily laminin 511) or muscle (primarily laminin 211); the other preparation, commercially available purified recombinant laminin heterotrimers. A scratch wound assay was used as opposed to the Boyden chamber because here, we are looking at the role of specific extracellular matrix in potentiating mesangial cell migration. As shown in Figure 10, D–G, wild-type mesangial cells migrate much more efficiently on laminin 211 compared to laminin 521. The effect was more pronounced on the muscle laminin preparation relative to the placental laminin preparation; it was also clear on the pure recombinant laminin substrates. To more directly confirm the role of laminin α2 in migratory potential, we measured the relative migration of wild-type mesangial cells to mesangial cells derived from laminin α2–deficient mice, using the Boyden chamber approach. The results in Figure 10H demonstrate a statistically significant reduction in the migratory potential of laminin α2–deficient mesangial cells relative to wild-type mesangial cells, based on analysis of three independent derivations run in triplicate (P < 0.001). Collectively, the data in Figure 10 suggest that laminin 211 deposition by the mesangial processes functionally contributes to the process invasion of the capillary tuft in Alport (and CD151-knockout) glomeruli.
Figure 10.

Laminin 211 potentiates mesangial process invasion of the glomerular capillary loops in Alport mice and promotes mesangial cell migration in vitro. A–C: Laminin α2–deficient Alport mice on 129/Sv background show reduced mesangial process invasion of the glomerular capillary tufts. Cryosections of kidney tissue from 8-week-old laminin α2–deficient Alport mice were analyzed by dual immunofluorescence immunostaining using antibodies against laminin α5 and integrin α8. The degree of mesangial process invasion of the glomerular capillary tufts is greatly reduced in the laminin α2–null Alport mice relative to Alport mice (compare with Figure 2, J–L). D–G: Wild-type mesangial cells migrate more robustly on laminin 211 compared to laminin 521 (GBM laminin). Wound scratch assays were performed using wild-type mesangial cells cultured on either recombinant purified laminins or commercially available laminins extracted from either placenta (primarily laminin 511) or muscle (primarily laminin 211). Images shown are representative of multiple replicates. H: Primary mesangial cells from laminin α2–deficient mice (1° dY MES) show impaired migratory potential relative to wild-type mesangial cells (1° WT MES). Boyden chamber assays were performed. Blinded cell counts from multiple replicates were analyzed. ∗∗∗P < 0.001. Scale bar = 10 μm.
Discussion
Earlier studies of Alport mouse, dog, and humans reported the presence of abnormal laminins in the GBM, including laminin 211 and laminin 111.8,13,19 These laminins tend to accumulate in areas of irregular thickening of the GBM, and these thickened areas have been shown to be more permeable to ferritin, suggesting that they comprise loosely assembled or partially degraded extracellular matrix.35 In addition to the abnormal laminins, fibronectin has also been reported to accumulate in the GBM of Alport mice.15 These glomerular ECM components are normally found in the mesangial matrix,36 suggesting that the abnormal GBM matrix molecules that progressively accumulate in the Alport GBM may be of mesangial cell origin. Here, we used integrin α8 as a specific mesangial cell surface marker to demonstrate that mesangial processes invade the capillary tufts and colocalize with laminin 211, a mesangial laminin. Integrin α8 is expressed in mesangial cells, but not in other glomerular cell types,27 and its expression is generally restricted to smooth muscle cells and neuronal cell types.37,38 Mesangial process invasion of the glomerular capillary tufts was exacerbated by hypertension, suggesting that the mechanism triggering this event was mediated by biomechanical stress, likely at the interface between the mesangial processes and the subendothelial interface with the glomerular capillaries, an area known to provide important structural support for the capillary loops.36 The Alport mutations result in the absence of the collagen α3(IV)/α4(IV)/α5(IV) network from the GBM. The consequence is a thinner GBM comprising only α1(IV) and α2(IV) collagens, which have been shown to contain fewer interchain disulfide crosslinks.20 This structural change would be predicted to alter the biomechanical properties of the capillary tuft, resulting in stresses on the cells comprising the tuft even under normal glomerular blood pressures. We examined a second model, the CD151 knockout mouse, which would also be expected to show enhanced strain on the capillary tufts. In this model, enhanced strain arises as a result of reduced adhesion of the podocyte pedicles to the GBM due to reduced affinity for the podocyte integrin α3β1 for its GBM ligand laminin 521.28 Mesangial process invasion of the glomerular capillary tufts in the CD151 mouse was even more robust than that for the Alport model. Like the Alport model,23 glomerular pathology in the CD151 mouse model, which shows ultrastructural lesions in the GBM strikingly similar to Alport syndrome,39,40 is significantly exacerbated under hypertensive conditions.24 Collectively, this evidence supports the notion that mutations affecting structural integrity of the glomerular capillary tuft result in unnatural stresses on the cells in contact with the tuft. In the mesangial cell compartment, this results in mesangial process invasion into the tuft and deposition of matrix proteins in the GBM that are of mesangial cell origin.
In earlier work, we showed that deletion of the mesangial integrin α1β1 in Alport mice resulted in a marked attenuation in the progression of the glomerular pathology, with reduced proteinuria and a near doubling of lifespan.8 The mechanism underlying the influence of mesangial integrin α1β1 on Alport renal disease progression has, until now, remained unclear. In this study, we show that mesangial process invasion is markedly attenuated in integrin α1–null Alport mice relative to strain/age-matched Alport mice. This observation suggested that the signaling pathway that activates actin cytoskeletal rearrangements is perturbed in the absence of integrin α1β1. Consistent with this notion, we observed decreased migratory potential for primary cultures of α1-null mesangial cells relative to wild-type mesangial cells from strain/age-matched mice (Figure 8A). LPS, which activates both Rac1 and CDC42 in wild-type mesangial cells (Figure 8, B–E), failed to activate Rac1 or CDC42, and failed to activate actin cytoskeletal rearrangements in cultured α1-null mesangial cells (data not shown). Collectively, these data support a mechanism that might explain why deletion of integrin α1 results in attenuation of Alport glomerular pathogenesis, where integrin α1β1 is a key sensor of biomechanical strain at the glomerular capillary tuft, and participates in the adhesive signaling mechanism that links to the Rho GTPases Rac1 and CDC42, which activate actin polymerization dynamics required to process invasion of the glomerular capillary tufts. In addition to integrin α1β1, the collagen receptor discoidin domain receptor tyrosine kinase 1 (DDR1) has been functionally linked to mesangial cell migration and adhesion of mesangial cells,41 and deletion of DDR1 slows the progression of renal disease in Alport mice.42 These observations suggest that DDR1 may have functions in the mesangial cell compartment that are similar to that of integrin α1β1. The link between integrin adhesion, ILK signaling, and activation of Rac1-mediated cytoskeletal rearrangements has been described previously.43 In an earlier report, Gross et al44 demonstrated a strong nephroprotective effect for the angiotensin-converting enzyme inhibitor ramipril in the 129/Sv autosomal Alport mouse model, an observation that has recently been extended to humans with Alport syndrome.45 These investigators were not able to fully explain the nephroprotective effect of angiotensin-converting enzyme inhibition by its antihypertensive and antiproteinuric influences. It is possible that the effect of angiotensin-converting enzyme inhibition on the mesangial compartment might confer this additional therapeutic benefit.
Classically, Rac1 activation is associated with lamellipodia formation, and CDC42 activation is associated with filopodia formation.46 Recently, evidence for crosstalk between the two Rho GTPases has emerged.47 This phenomenon is likely regulated through the guanine nucleotide exchange factor β1pix, which contains binding sites for both CDC42 and Rac1.48,49 Here, we provide evidence for crosstalk between Rac1 and CDC42 in cultured mesangial cells regulating actin cytoskeletal rearrangement including: i) treatment of mesangial cells with LPS, known to activate rapid actin cytoskeletal rearrangement,32 activates Rac1 in wild-type mesangial cells (Figure 8I); ii) membrane localization of CDC42, a known prerequisite for its activation,50,51 is blocked by addition of RAC1 inhibitor coincident with LPS stimulation (Figure 8, F–I); and iii) inclusion of either Rac1 inhibitor or CDC42 inhibitor on stimulation of mesangial cell cultures with LPS blocks actin cytoskeletal rearrangements (Figure 8, B–E).
Mesangial cell cultures subjected to cyclic biomechanical strain expressed elevated levels of the promigratory cytokines CTGF and TGF-β1, providing further evidence that biomechanical strain could activate actin cytoskeletal dynamics required for mesangial process invasion. Both CTGF and TGF-β1 signaling have been shown to activate CDC42,52,53 and both cytokines have been shown to be induced in Alport glomeruli,7,13 suggesting that activation of these signaling pathways might be an important underlying mechanism for the activation of mesangial process invasion of glomerular capillary tufts in Alport syndrome. Indeed, earlier work from our laboratory showed that inhibition of TGF-β1 in the Alport mouse resulted in abrogation of GBM thickening, in support of this notion.8 We also showed that when TGF-β1 was inhibited in integrin α1–null Alport mice, a synergistic improvement in glomerular disease was observed, suggesting that TGF-β1 and integrin α1 are working through distinct pathways. On the basis of the current study, these pathways may converge on strain-mediated activation of Rac1/CDC42 in the mesangial cell compartment. Although the deposition of laminin 211 in the GBM of Alport mice was described more than 10 years ago,8,13 a functional role for laminin 211 in Alport glomerular pathology has not been described. We show reduced mesangial process invasion of the glomerular capillary loops in Alport mice that are also lacking laminin α2, suggesting that laminin 211 itself promotes the migration of processes into the glomerular capillary loops (Figure 10, A–C). Consistently, we show that wild-type mesangial cells migrate more robustly when cultured on laminin 211 compared to laminin 521, and that primary mesangial cells from laminin α2–deficient mice show impaired migration relative to primary wild-type mesangial cells from age/strain-matched mice (Figure 10, D–H). Although modulation of mesangial cell migration by ECM has been described previously,54 these data suggest that the strain-mediated mesangial process invasion of the capillary loops is enhanced by mesangial cell–secreted laminin 211, which may explain why laminin 211 accumulates in the patchy, irregularly thickened regions of the Alport GBM (see Figure 1).
Our data suggest that the changes in the biophysical properties of the Alport glomerular capillary tuft result in biomechanical stresses that result in the induction of pathological processes. Parallel observations in Alport and CD151 mouse models, including mesangial process invasion of the glomerular capillary tufts and deposition of laminin 211, provide strong support for this notion, because the two mouse models arise from mutations that would be expected to relax the structural integrity of the glomerular capillary tufts, but are otherwise mechanistically unrelated to each other. Recent studies of the biophysical properties of Alport glomeruli from pre-proteinuric mice reported increased deformability.22 Collectively, our current work suggests a model where biomechanical stresses on the glomerular capillary tufts activate a promigratory signaling cascade in mesangial cells involving integrin α1β1–mediated activation of Rac1/CDC42. This activation culminates in the invasion of the capillary loops by mesangial processes. These processes clearly deposit laminin 211, which further exacerbates the mesangial process invasion. In addition to laminin 211, other mesangial matrix molecules are likely deposited in the GBM, and local action of mesangial cytokines (TGF-β1 and CTGF, for example) and MMPs might also contribute to the structural and functional properties of the Alport GBM (irregular thickening, splitting, permeability, and so on). In addition, all of these events are very likely to influence podocyte cell health. Therefore, we conclude that mesangial process invasion of the GBM is an important early event that precipitates glomerulosclerosis in Alport syndrome. The fact that we observe mesangial process invasion of glomerular capillary loops in human Alport glomeruli provides relevance for these observations to the human disease. A better understanding of the activation process might reveal novel targets capable of preventing this event and arresting the Alport glomerular pathogenesis in its pre-initiated state.
Acknowledgments
We thank Skip Kennedy for help in figure preparation, Marrtin Hemmler (Harvard University) for the gift of CD151 knockout mice, and the Tissue Science Facility Core (University of Nebraska Medical Center) for the normal human tissue.
Footnotes
Supported by NIH grant R01 DK055000. Confocal microscopy was conducted at the Integrative Biological Imaging Facility, Creighton University, Omaha, NE (supported by 5P20RR016469, National Center for Research Resources, NIH, and 8P20GM103427, National Institute of General Medical Sciences, NIH).
References
- 1.Kashtan C.E., Michael A.F. Alport syndrome. Kidney Int. 1996;50:1445–1463. doi: 10.1038/ki.1996.459. [DOI] [PubMed] [Google Scholar]
- 2.Barker D.F., Hostikka S.L., Zhou J., Chow L.T., Oliphant A.R., Gerken S.C., Gregory M.C., Skolnick M.H., Atkin C.L., Tryggvason K. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. 1990;248:1224–1227. doi: 10.1126/science.2349482. [DOI] [PubMed] [Google Scholar]
- 3.Mochizuki T., Lemmink H.H., Mariyama M., Antignac C., Gubler M.C., Pirson Y., Verellen-Dumoulin C., Chan B., Schröder C.H., Smeets H.J. Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet. 1994;8:77–81. doi: 10.1038/ng0994-77. [DOI] [PubMed] [Google Scholar]
- 4.Kleppel M.M., Fan W.W., Cheong H.I., Michael A.F. Evidence for separate networks of classical and novel basement membrane collagen: characterization of alpha 3(IV)-Alport antigen heterodimer. J Biol Chem. 1992;267:4137–4142. [PubMed] [Google Scholar]
- 5.Kalluri R., Cosgrove D. Assembly of type IV collagen: insights from alpha3(IV) collagen-deficient mice. J Biol Chem. 2000;275:12719–12724. doi: 10.1074/jbc.275.17.12719. [DOI] [PubMed] [Google Scholar]
- 6.Kashtan C.E., Gubler M.C., Sisson-Ross S., Mauer M. Chronology of renal scarring in males with Alport syndrome. Pediatr Nephrol. 1998;12 doi: 10.1007/s004670050451. 269–227. [DOI] [PubMed] [Google Scholar]
- 7.Sayers R., Kalluri R., Rodgers K.D., Shield C.F., III, Meehan D.T., Cosgrove D.E. Role for transforming growth factor-β1 in Alport renal disease progression. Kidney Int. 1999;56:1662–1673. doi: 10.1046/j.1523-1755.1999.00744.x. [DOI] [PubMed] [Google Scholar]
- 8.Cosgrove D., Rodgers K., Meehan D., Miller C., Bovard K., Gilroy A., Gardner H., Kotelianski V., Gotwals P., Amatucci A., Kalluri R. Integrin α1β1 and transforming growth factor-β1 play distinct roles in Alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am J Pathol. 2000;157:1649–1659. doi: 10.1016/s0002-9440(10)64802-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao V.H., Meehan D., Delimont D., Nakajima M., Wada T., Gratton M.A., Cosgrove D. Role for MMP-12 in GBM damage associated with Alport syndrome. Am J Pathol. 2006;169:32–46. doi: 10.2353/ajpath.2006.050896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeisberg M., Khurana M., Sugimoto H., Cosgrove D., Rao V.H., Rougier J.-P., Werner M.C., Shield C.F., III, Werb Z., Kalluri R. Stage specific action of matrix metalloproteinases influence hereditary kidney disease. PLoS Med. 2006;3:e100. doi: 10.1371/journal.pmed.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cosgrove D., Meehan D.T., Pozzi A., Chen X., Rodgers K.D., Tempero R.M., Delimont D., Zallocchi M., Rao V.H. Integrin α1β1 regulates MMPs via p38 MAPkinase in mesangial cells: implications for Alport syndrome. Am J Pathol. 2008;172:761–773. doi: 10.2353/ajpath.2008.070473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koepke M.L., Weber M., Schulze-Lohoff E., Beirowski B., Segerer S., Gross O. Nephroprotective effect of the HMG-CoA-reductase inhibitor cerivastatin in a mouse model of progressive renal fibrosis in Alport syndrome. Nephrol Dial Transplant. 2007;22:1062–1069. doi: 10.1093/ndt/gfl810. [DOI] [PubMed] [Google Scholar]
- 13.Kashtan C.E., Kim Y., Lees G.E., Thorner P.S., Virtanen I., Miner J.H. Abnormal glomerular basement membrane laminins in murine, canine, and human Alport syndrome: aberrant laminin α2 deposition is species independent. J Am Soc Nephrol. 2001;12:252–260. doi: 10.1681/ASN.V122252. [DOI] [PubMed] [Google Scholar]
- 14.Miner J., Patton B.L., Lentz S.I., Gilbert D.J., Snider W.D., Jenkins N.A., Copeland N.G., Sanes J.R. The laminin alpha chains: expression, developmental transitions, and chromosomal locations of alpha 1-5, identification of heterotrimeric laminins 8-11, and cloning of a novel alpha3 isoform. J Cell Biol. 1997;137:685–701. doi: 10.1083/jcb.137.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cosgrove D., Meehan D.T., Grunkemeyer J.A., Kornak J.M., Sayers R., Hunter W.T., Samuelson G.C. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;10:2981–2992. doi: 10.1101/gad.10.23.2981. [DOI] [PubMed] [Google Scholar]
- 16.St John P.L., Abrahamson D.R. Glomerular endothelial cells and podocytes jointly synthesize laminin-1 and -11 chains. Kidney Int. 2001;60:1037–1046. doi: 10.1046/j.1523-1755.2001.0600031037.x. [DOI] [PubMed] [Google Scholar]
- 17.Kalluri R., Shield C.F., Todd P., Hudson B.G., Neilson E.G. Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest. 1997;99:2470–2478. doi: 10.1172/JCI119431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey S.J., Zheng K., Sado Y., Naito I., Ninomiya Y., Jacobs R.M., Hudson B.G., Thorner P.S. Role of distinct type IV collagen networks in glomerular development and function. Kidney Int. 1998;54:1857–1866. doi: 10.1046/j.1523-1755.1998.00188.x. [DOI] [PubMed] [Google Scholar]
- 19.Abrahamson D.R., Prettyman A.C., Robert B., St John P.L. Laminin-1 re-expression in Alport mouse glomerular basement membranes. Kidney Int. 2003;63:826–834. doi: 10.1046/j.1523-1755.2003.00800.x. [DOI] [PubMed] [Google Scholar]
- 20.Gunwar S., Ballester F., Noelken M.E., Sado Y., Ninomiya Y., Hudson B.G. Glomerular basement membrane. J Biol Chem. 1998;273:8767–8775. doi: 10.1074/jbc.273.15.8767. [DOI] [PubMed] [Google Scholar]
- 21.Kamenetsky I., Rangayan R.M., Benediktsson H. Analysis of the glomerular basement membrane in images of renal biopsies using the split-and-merge method: a pilot study. J Digital Imaging. 2010;23:463–474. doi: 10.1007/s10278-009-9233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wyss H.M., Henderson J.M., Byfield F.J., Bruggeman L.A., Ding Y., Huang C., Suh J.H., Franke T., Mele E., Pollak M.R., Miner J.H., Janmey P.A., Weitz D.A., Miller R.T. Biophysical properties of normal and diseased renal glomeruli. Am J Physiol Cell Physiol. 2011;300:C397–C405. doi: 10.1152/ajpcell.00438.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meehan D.T., Delimont D., Cheung L., Zallocchi M., Sansom S.C., Holzclaw J., Rao V., Cosgrove D. Biomechanical strain mediated maladaptive gene regulation as a contributing factor in Alport glomerular disease. Kidney Int. 2009;76:968–976. doi: 10.1038/ki.2009.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sachs N., Claessen N., Aten J., Kreft M., Teske G.J., Koeman A., Zuurbier C.J., Janssen H., Sonnenberg A. Blood pressure influences end-stage renal disease of Cd151 knockout mice. J Clin Invest. 2012;122:348–358. doi: 10.1172/JCI58878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gardner H., Kreidberg J., Kotelianski V., Jaenisch R. Deletion of integrin alpha 1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev Biol. 1996;175:301–313. doi: 10.1006/dbio.1996.0116. [DOI] [PubMed] [Google Scholar]
- 26.Takeda Y., Kazarov A.R., Butterfield C.E., Hopkins B.D., Benjamin L.E., Kaipainen A., Hemler M.E. Deletion of tetraspanin CD151 results in decreased pathologic angiogenesis in vivo and in vitro. Blood. 2007;109:1524–1532. doi: 10.1182/blood-2006-08-041970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartner A., Schöcklmann H., Pröls F., Müller U., Sterzel R.B. Alpha8 integrin in glomerular mesangial cells and in experimental glomerulonephritis. Kidney Int. 1999;56:1468–1480. doi: 10.1046/j.1523-1755.1999.00662.x. [DOI] [PubMed] [Google Scholar]
- 28.Nishiuchi R., Sanzen N., Nada S., Sumida Y., Wada Y., Okada M., Takagi J., Hasegawa H., Sekiguchi K. Potentiation of the ligand-binding activity of integrin α3β1 via association with tetraspanin CD151. Proc Natl Acad Sci U S A. 2005;102:1939–1944. doi: 10.1073/pnas.0409493102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vicente-Manzanares M., Choi C.K., Horwitz A.R. Integrins in cell migration: the actin connection. J Cell Sci. 2009;122:199–206. doi: 10.1242/jcs.018564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanlioglu S., Williams C.M., Samavati L., Butler N.S., Wang G., McCray P.B., Jr., Ritchie T.C., Hunninghake G.W., Zandi E., Engelhardt J.F. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-α secretion through IKK regulation of NF-κB. J Biol Chem. 2001;276:30188–30198. doi: 10.1074/jbc.M102061200. [DOI] [PubMed] [Google Scholar]
- 31.Fessler M.B., Arndt P.G., Frasch S.C., Lieber J.G., Johnson C.A., Murphy R.C., Nick J.A., Bratton D.L., Malcolm K.C., Worthen G.S. Lipid rafts regulate lipopolysaccharide-induced activation of Cdc42 and inflammatory functions of the human neutrophil. J Biol Chem. 2004;279:39989–39998. doi: 10.1074/jbc.M401080200. [DOI] [PubMed] [Google Scholar]
- 32.Bursten S.L., Stevenson F., Torrano F., Lovett D.H. Mesangial cell activation by bacterial endotoxin. Am J Pathol. 1991;139:371–382. [PMC free article] [PubMed] [Google Scholar]
- 33.Etienne-Manneville S., Hall A. Integrin-mediated activation of CDC42 controls cell polarity in migrating astrocytes through PKCζ. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 34.Huang M., Satchell L., DuHadaway J.B., Prendergast G.C., Laury-Kleintop L.D. RhoB links PDGF signaling to cell migration by coordinating activation and localization of DCD42 and RAC. J Cell Biochem. 2011;112:1572–1584. doi: 10.1002/jcb.23069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abrahamson D.R., Isom K., Roach E., Stroganova L., Zelenchuk A., Miner J.H., St John P.L. Laminin compensation in collagen alpha3(IV) knockout (Alport) glomeruli contributes to permeability defects. J Am Soc Nephrol. 2007;18:2465–2472. doi: 10.1681/ASN.2007030328. [DOI] [PubMed] [Google Scholar]
- 36.Schlöndorff D., Banas B. The mesangial cell revisited: no cell is an island. J Am Soc Nephrol. 2009;20:1179–1187. doi: 10.1681/ASN.2008050549. [DOI] [PubMed] [Google Scholar]
- 37.Bossy B., Bossy-Wetzel E., Reichardt L.F. Characterization of the integrin alpha 8 subunit: a new beta 1-associated subunit which is predominantly expressed on axons and on cells in contact with basal lamina on chick embryos. EMBO J. 1991;10:2375–2385. doi: 10.1002/j.1460-2075.1991.tb07776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schnapp L.M., Breuss J.M., Ramos D.M., Sheppard D., Pytela R. Sequence and tissue distribution of the human integrin α8 subunit: a β1-associated α subunit expressed in smooth muscle cells. J Cell Sci. 1995;108:537–544. doi: 10.1242/jcs.108.2.537. [DOI] [PubMed] [Google Scholar]
- 39.Baleato R.M., Guthrie P.L., Gubler M.-C., Ashman L.K., Roselli S. Deletion of Cd151 results in a strain-dependent glomerular disease due to severe alterations of the glomerular basement membrane. Am J Pathol. 2008;173:927–937. doi: 10.2353/ajpath.2008.071149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sachs N., Kreft M., van den Bergh Weerman M.A., Beynon A.J., Peters T.A., Weening J.J., Sonnenberg A. Kidney failure in mice lacking the tetraspanin CD151. J Cell Biol. 2006;175:33–39. doi: 10.1083/jcb.200603073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curat C.A., Vogel W.F. Discoidin domain receptor 1 controls growth and adhesion of mesangial cells. J Am Soc Nephrol. 2002;13:2648–2656. doi: 10.1097/01.asn.0000032419.13208.0c. [DOI] [PubMed] [Google Scholar]
- 42.Gross O., Girgert R., Beirowski B., Kretzler M., Kang H.G., Kruegel J., Miosge N., Busse A.C., Segerer S., Vogel W.F., Muller G.A., Weber M. Loss of collagen receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010;29:346–356. doi: 10.1016/j.matbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Qian Y., Zhong X., Flynn D.C., Zheng J.Z., Qiao M., Wu C., Dedhar S., Shi X., Jiang B.H. ILK mediates actin filament rearrangements and cell migration and invasion through PI3K/Akt/Rac1 signaling. Oncogene. 2005;24:3154–3165. doi: 10.1038/sj.onc.1208525. [DOI] [PubMed] [Google Scholar]
- 44.Gross O., Beirowski B., Koepke M.L., Kuck J., Reiner M., Addicks K., Smyth N., Schulze-Lohoff E., Weber M. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63:438–446. doi: 10.1046/j.1523-1755.2003.00779.x. [DOI] [PubMed] [Google Scholar]
- 45.Gross O., Licht C., Anders H.J., Hoppe B., Beck B., Tonshoff B., Hocker B., Wygoda S., Ehrlich J.H., Pape L., Konrad M., Rascher W., Dotsch J., Muller-Wiefel D.E., Hoyer P., Study Group Members of the Gesellschaft fur Padriatrische Nephrologie, Knebelmann B., Pirson Y., Grunfeld J.P., Niaudet P., Cochat P., Heidet L., Lebbah S., Torra R., Friede T., Lange K., Muller G.A., Weber M. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012;81:494–501. doi: 10.1038/ki.2011.407. [DOI] [PubMed] [Google Scholar]
- 46.Nobes C.D., Hall A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 47.Zamudio-Meza H., Castillo-Alvarez A., González-Bonilla C., Meza I. Cross-talk between Rac1 and Cdc42 GTPases regulates formation of filopodia required for dengue virus type-2 entry into HMEC-1 cells. J Gen Bio. 2009;90:2902–2911. doi: 10.1099/vir.0.014159-0. [DOI] [PubMed] [Google Scholar]
- 48.Chahdi A., Sorokin A., Dunn M.J., Landry Y. The Rac/Cdc42 guanine nucleotide exchange factor beta1Pix enhances mastoparan-activated Gi-dependent pathway in mast cells. Biochem Biophys Res Commun. 2004;317:384–389. doi: 10.1016/j.bbrc.2004.03.062. [DOI] [PubMed] [Google Scholar]
- 49.Chahdi A., Miller B., Sorokin A. Endothelin 1 induces β(1)Pix translocation and Cdc42 activation via protein kinase A-dependent pathway. J Biol Chem. 2005;280:578–584. doi: 10.1074/jbc.M411130200. [DOI] [PubMed] [Google Scholar]
- 50.Gibson R.M., Gandi P.N., Tong X., Miyoshi J., Takai Y., Konieczkowsky M., Sedor J.R., Wilson-Delfosse A.L. An activating mutant of Cdc42 that fails to interact with Rho GDP-dissociation inhibitor localizes to the plasma membrane and mediates active reorganization. Exp Cell Res. 2004;301:211–222. doi: 10.1016/j.yexcr.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 51.Osamani N., Peglion F., Chavrier P., Etienne-Manneville S. Cdc42 localization and cell polarity depend on membrane traffic. J Cell Biol. 2010;191:1261–1269. doi: 10.1083/jcb.201003091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edlund S., Landström M., Heldin C.-H., Aspenström P. Transforming growth factor-β-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Bio Cell. 2002;13:902–914. doi: 10.1091/mbc.01-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crean J.K., Furlong F., Finlay D., Mitchell D., Murphy M., Conway B., Brady H.R., Godson C., Martin F. Connective tissue growth factor [CTGF]/CCN2 stimulates mesangial cell migration through integrated dissolution of focal adhesion complexes and activation of cell polarization. FASEB J. 2004;18:1541–1543. doi: 10.1096/fj.04-1546fje. [DOI] [PubMed] [Google Scholar]
- 54.Person J.M., Lovett D.H., Raugi G.J. Modulation of mesangial cell migration by extracellular matrix components. Am J Pathol. 1988;133:609–614. [PMC free article] [PubMed] [Google Scholar]