

Abstract
Galectin-3 is a β-galactoside–binding animal lectin with diverse functions, including regulation of T helper (Th) 1 and Th2 responses. Current data indicate that galectin-3 expressed in dendritic cells (DCs) may be contributory. Th17 cells have emerged as critical inducers of tissue inflammation in autoimmune disease and important mediators of host defense against fungal pathogens, although little is known about galectin-3 involvement in Th17 development. We investigated the role of galectin-3 in the induction of Th17 immunity in galectin-3–deficient (gal3−/−) and gal3+/+ mouse bone marrow–derived DCs. We demonstrate that intracellular galectin-3 negatively regulates Th17 polarization in response to the dectin-1 agonist curdlan (a β-glucan present on the cell wall of fungal species) and lipopolysaccharide, agents that prime DCs for Th17 differentiation. On activation of dectin-1, gal3−/− DCs secreted higher levels of the Th17-axis cytokine IL-23 compared with gal3+/+ DCs and contained higher levels of activated c-Rel, an NF-κB subunit that promotes IL-23 expression. Levels of active Raf-1, a kinase that participates in downstream inhibition of c-Rel binding to the IL23A promoter, were impaired in gal3−/− DCs. Modulation of Th17 by galectin-3 in DCs also occurred in vivo because adoptive transfer of gal3−/− DCs exposed to Candida albicans conferred higher Th17 responses and protection against fungal infection. We conclude that galectin-3 suppresses Th17 responses by regulating DC cytokine production.
IL-17–producing CD4+ T helper 17 (Th17) cells play an essential role in the clearance of extracellular bacterial and fungal pathogens1,2 and promote inflammatory responses involved in autoimmune disease.3,4 The factors associated with Th17 cell development have been well characterized and include cytokines such as transforming growth factor-β (TGF-β) and IL-6, which promote Th17 differentiation in mice,5,6 and IL-23, which is required for Th17 cell expansion and effector functions.4 Dendritic cells (DCs) express germline-encoded pattern recognition receptors (PRRs) that recognize conserved molecular components expressed on microbial pathogens7 and are capable of generating the appropriate co-stimulatory molecules and cytokines that support Th17 development. Among the receptors known to induce Th17-promoting factors is dectin-1, a C-type lectin receptor that recognizes β-glucans expressed on cell walls of fungi.8,9 Engagement of dectin-1 with the β-glucan curdlan or Candida albicans has been shown to stimulate IL-23 production in DCs and promote Th17 cell responses in vitro and in vivo.10,11 Members of the Toll-like receptor (TLR) family, specifically TLR2, TLR3, TLR4, and TLR9, have also been reported to elicit Th17 responses through the induction of Th17-promoting cytokines.6,12
Galectin-3 is a β-galactoside–binding animal lectin, a pleiotropic protein capable of participating in a variety of cellular processes through intracellular and extracellular mechanisms.13,14 Extracellular galectin-3 has been shown to modulate cell adhesion, cell activation, and cell migration,15 whereas intracellular galectin-3 has been implicated in the regulation of cell survival,16 pre-mRNA splicing,17 and phagocytosis.18 We previously demonstrated that intracellular galectin-3 translocates to lipid raft microdomains in mouse bone marrow–derived DCs (BMDCs) on chemokine receptor activation and positively regulates DC migration.19 Increasing evidence suggests that galectin-3 may also play a role in the regulation of Th1/Th2 differentiation by affecting IL-12 production in DCs.20–22 However, the role of galectin-3 in Th17 development remains largely undefined. Regarding innate immunity, galectin-3 has been shown to be involved in the recognition of several microbial species and innate defense.23 Its expression is essential for the recognition of C. albicans by macrophages,24,25 and it has fungicidal activity against Candida species,26 indicating a direct role for the lectin in antifungal immunity. Whether galectin-3 plays a role in antifungal defense through the Th17 response has not been investigated.
Using an ovalbumin (OVA)-specific T-cell activation model, we compared antigen-specific Th17 polarization induced by gal3+/+ and gal3−/− BMDCs. Gal3−/− DCs primed with curdlan or high-dose lipopolysaccharide (LPS) produced higher levels of IL-17–axis cytokines and induced higher Th17 responses compared with gal3+/+ DCs. These findings were also observed in the presence of lactose in culture media, suggesting that galectin-3 affects DC functions through an intracellular mechanism. In addition to differences in cytokine secretion, gal3−/− DCs treated with either curdlan or high-dose LPS exhibited impaired mitogen-activated protein kinase extracellular signal-regulated kinase (ERK) 1/2 phosphorylation, suggesting that galectin-3 may regulate DC cytokine expression in response to dectin-1 and TLR4 stimulation through a common mechanism. Furthermore, curdlan-stimulated gal3−/− DCs expressed higher levels of c-Rel, a critical transcription factor involved in IL-23 production. On closer examination of dectin-1 signaling pathways, we found that curdlan-stimulated gal3−/− DCs expressed lower levels of phosphorylated Raf-1, a serine-threonine kinase that negatively regulates IL-23. In agreement with our in vitro studies, the adoptive transfer of C. albicans–exposed gal3−/− DCs into mice induced higher Th17 responses and promoted greater fungal clearance than gal3+/+ DCs, indicating that galectin-3 in DCs plays a pivotal role in the induction of antifungal immunity in vivo. Taken together, these findings reveal a novel role for endogenous galectin-3 in the regulation of DC cytokine expression involved in Th17 induction. Thus, targeting galectin-3 in DCs may be a means to modulate adaptive immune responses to fungal infections and in autoimmune disease.
Materials and Methods
Mice
Gal3−/− mice were developed as previously described27 and were backcrossed to C57BL/6 mice for nine generations. Gal3+/+ and gal3−/− littermates obtained from heterozygous breeders were used to prepare BMDCs. OVA-TCR transgenic mice (OT-II) on the C57BL/6 background were obtained from The Jackson Laboratory (Bar Harbor, ME). C57BL/6 mice used in adoptive transfer experiments were bred in-house or were purchased from The Jackson Laboratory. All the experiments were approved by the Institutional Animal Care and Use Committee of the University of California, Davis (Sacramento, CA) or the National Taiwan University College of Medicine (Taipei, Taiwan).
Reagents
LPS from Escherichia coli O111:B4 was from List Biological Laboratories (Campbell, CA). Heat-killed C. albicans (HKCA) was from InvivoGen (San Diego, CA). β-Lactose, sucrose, and curdlan were from Sigma-Aldrich (St. Louis, MO). Recombinant granulocyte-macrophage colony-stimulating factor, IL-1β, IL-6, IL-10, IL-12, IL-17A, and interferon γ (IFN-γ) were from PeproTech (Rocky Hill, NJ) and recombinant IL-23 was from eBioscience Inc. (San Diego, CA). All capture and detection antibodies for enzyme-linked immunosorbent assays (ELISAs) and neutralizing antibodies against IL-6 (MP5-20F3), IL-23 p19 (G23-8), and IL-12/23 p40 (C17.8) were from eBioscience Inc. Anti–phospho-p44/p42 (ERK1/2) mitogen-activated protein kinase, total p44/p42, and anti–phospho–Raf-1 (Ser 338) (56A6) were from Cell Signaling Technology Inc. (Danvers, MA), and anti–phospho–Raf-1 (Tyr340/341) (44-506G) was from Invitrogen (Carlsbad, CA). Total Raf-1 antibody was from BD Transduction Laboratories (San Jose, CA).
Preparation of Endotoxin-Free OVA
Egg whites from organic eggs were extracted under sterile conditions and were lyophilized using a SpeedVac concentrator (Thermo Fisher Scientific Inc., Rockford, IL). Lyophilized egg whites were reconstituted in sterile PBS, and the protein concentrations were determined by bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL). Endotoxin levels were below the level of detection, as determined by Pyrogent gel clot limulus amebocyte lysate assay (Lonza Inc., Walkersville, MD).
In Vitro Generation and Activation of BMDCs
BMDCs were generated from gal3+/+ and gal3−/− mice by culturing bone marrow cells for 8 to 10 days as described previously28 in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (HyClone, Thermo Fisher Scientific Inc.), penicillin/streptomycin (Invitrogen), 2ME (Gibco, Carlsbad, CA), HEPES (Mediatech Inc., a Corning subsidiary, Manassas, VA), minimal essential medium nonessential amino acids (Invitrogen), and 20 ng/mL of granulocyte-macrophage colony-stimulating factor (PeproTech). For measuring cytokine production, 2 × 105 DCs per well were stimulated with 0.1 or 100 ng/mL of LPS; 1, 10, or 100 μg/mL of curdlan; or 105, 106, 107 cells/mL of HKCA in 96-well plates at 200 μL per well for 24 hours.
In Vitro Activation of CD4+ T Cells by DCs
CD4+ cells were purified from OT-II splenocytes using biotinylated anti-mouse CD4+ and a biotin selection kit (STEMCELL Technologies Inc., Vancouver, BC, Canada) according to the manufacturer's instructions. For OT-II activation, DCs were pulsed overnight (approximately 16 hours) with 100 μg/mL of endotoxin-free OVA alone or in the presence of 0.1 ng/mL of LPS, 100 ng/mL of LPS, or 10 μg/mL of curdlan. DCs were washed and co-cultured with CD4+ OT-II cells (100,000 CD4+ cells per well and 20,000 DCs per well) in 96-well round-bottomed plates. In some conditions, neutralizing antibodies were added at 10 μg/mL on day 0. Cell culture supernatants were harvested after 3 days, and cytokines were measured by ELISA.
Cell Signaling
DCs were plated in media without fetal bovine serum at 2 × 106 cells per well in 12-well plates for 2 hours and then were left untreated or stimulated with curdlan or LPS for 5, 10, 30, or 60 minutes. Cell lysates were prepared by resuspending cell pellets in lysis buffer [20 mmol/L Tris (pH 7.4), 137 mmol/L NaCl, 2 mmol/L EDTA (pH 7.4), 1% Triton X-100 (Roche Diagnostics GmbH, Mannheim, Germany), 25 mmol/L β-glycerophosphate, 2 mmol/L sodium pyrophosphate, 10% glycerol, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L Na3VO4, 5 mmol/L NaF, and 1× protease inhibitor cocktail] before centrifugation at 12,000 rpm, 4°C for 20 minutes. Twenty micrograms of protein were loaded per lane onto a 10% SDS-PAGE gel, and the separated proteins were transferred onto polyvinylidene difluoride membranes. Membranes were incubated with the indicated antibodies, and bands were detected by using SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific Inc.).
Flow Cytometry
For cell surface staining of co-stimulatory molecules, DCs were incubated with anti–CD80-PE, anti–CD86-PE, and anti–major histocompatibility complex II–fluorescein isothiocyanate (eBiosciences Inc.) in PBS containing 1% fetal bovine serum and 0.09% sodium azide for 30 minutes and were analyzed by flow cytometry (BD FACScan; BD Biosciences, San Jose, CA). To compare cell viability in DCs after activation, cells were first stained with anti–CD11c–fluorescein isothiocyanate (eBiosciences Inc.) and were subsequently stained with 7-aminoactinomycin D (Sigma-Aldrich) in PBS on ice for 30 minutes. After washing, cells were fixed in 1% paraformaldehyde supplemented with 50 μg/mL of actinomycin D before flow cytometry analysis. To compare regulatory T cell (Treg) induction by curdlan-OVA–stimulated DCs, T-cell–DC co-cultures were harvested after 3 days and were stained using anti–CD4-PE-Cy5 (eBioscience Inc.) and anti–CD25-PE (BD Pharmingen, San Diego, CA). Cells were fixed and permeabilized using a BD Cytofix/Cytoperm kit before intracellular staining with anti–Foxp3–fluorescein isothiocyanate (BD Pharmingen).
Detection of Raf-1 Phosphorylation
DCs were stimulated with curdlan for 15 minutes and then were lysed with buffer containing 50 mmol/L Tris-HCl (pH 8.0), 150 mmol/L NaCl, 1% Nonidet P-40 (Caledon Laboratories Ltd., Georgetown, ON, Canada), 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L Na3VO4, 5 mmol/L NaF, and protease inhibitor cocktail (Sigma-Aldrich). Cell lysates were immunoprecipitated with mouse anti–Raf-1 and recombinant Protein G-Sepharose 4B (Zymed-Invitrogen, Carlsbad, CA). Phosphorylated Raf-1 was detected by immunoblotting with rabbit anti–phospho–Raf-1 (Ser338) and rabbit anti–phospho–Raf-1 (Tyr340/341).
Transcription Factor Activation Assays
1 × 106 DCs per milliliter were stimulated with curdlan for 2 hours. Nuclear extracts from DCs were prepared using an NE-PER nuclear and cytoplasmic extraction reagents kit (Thermo Fisher Scientific Inc.). NF-κB DNA binding was performed by using a TransAM NF-κB family kit (Active Motif, Carlsbad, CA).
Immunization with Antigen-Pulsed DCs
The protocol for immunization with antigen-pulsed DCs was adapted from that of d’Ostiani et al29 with slight modifications. DCs were stimulated with 5 × 105 HKCA cells/mL overnight. Cells were washed in sterile PBS and were s.c. injected twice, a week apart, into C57BL/6 mice at a concentration of 5 × 105 cells per mouse in 50 μL of PBS. Seven days after the last injection of DCs, splenocytes were harvested and restimulated with HKCA at 1 × 105 cells/mL. CD4+ cells were also purified from splenocytes and were restimulated with HKCA-treated antigen-presenting cells. After 72 hours, supernatants were harvested for cytokines.
Adoptive Transfer of DCs and Fungal Infection
The following protocol for adoptive transfer of DCs was followed: 1 × 106 DCs/mL were co-cultured with 1 × 106 viable C. albicans cells/mL in RPMI 1640 medium at room temperature for 1 hour. The mixture was subsequently i.v. injected into wild-type (gal3+/+) mice (500 μL per mouse). Brain and kidney were harvested 3 and 6 days after infection. To determine the cytokine levels, brain and kidney tissues were homogenized in lysis buffer (RayBiotech Inc., Norcross, GA). Homogenate supernatants were collected after centrifugation, and cytokine concentrations were measured by ELISA. For fungal burden determination, tissues were homogenized in RPMI 1640 medium using a tissue grinder. Homogenates were serially diluted and plated on YPD agar. Yeast colonies were counted 3 days after incubation at 30°C. CD4+ T cells from the spleen were purified using an EasySep mouse CD4+ selection kit (STEMCELL Technologies Inc.).
Real-Time Quantitative PCR
Total RNA was extracted from purified CD4+ T cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was synthesized with Moloney Murine Leukemia Virus High-Performance Reverse Transcriptase (Epicentre Biotechnologies, Madison, WI) according to the manufacturer's instructions. Real-time quantitative PCR (qPCR) was performed by using the iCycler real-time PCR detection system (Bio-Rad Laboratories, Hercules, CA). The primers (forward and reverse) used for qPCR are as follows: mIl17a: 5′-AGGCAGCAGCGATCATCC-3′ and 5′-TGGAACGGTTGAGGTAGTCTG-3′; mIl-17f: 5′-CTGGAGGATAACACTGTGAGAGT-3′ and 5′-TGCTGAATGGCGACGGAGTTC-3′; RORγt: 5′-CGCCTCACCTGACCTACC-3′ and 5′-TTGCCTCGTTCTGGACTATAC-3′; RORα: 5′-TCTCCCTGCGCTCTCCGCAC-3′ and 5′-TCCACAGATCTTGCATGGA-3′; and mβ-actin: 5′-TGTATGAAGGCTTTGGTCTCCCT-3′ and 5′-AGGTGTGCACTTTTATTGGTCTCAA-3′. Each sample was analyzed in triplicate. The relative cytokine mRNA expression level of each sample was normalized against β-actin expression.
Histologic Staining
H&E and PAS staining of tissue sections was performed in the Pathology Core Laboratory at the Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan. Enumeration of infiltrated neutrophils in H&E slides was performed by a certified veterinary pathologist at the Institute of Biomedical Sciences Pathology Core Laboratory based on the presence of polymorphic nuclei and appropriate cell size.
Statistical Analysis
Statistical analysis of experimental groups was performed by unpaired Student’s t-tests using GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA). P < 0.05 was considered significant.
Results
Galectin-3 in DCs Negatively Regulates Th17 Polarization in Response to Dectin-1 Agonists
β-Glucans present on the cell wall of fungal species activate dectin-1 and induce the production of Th1- and Th17-polarizing cytokines in DCs.10 To study the effect of galectin-3 on DC cytokine production in response to dectin-1 activation, we compared cytokine profiles in gal3−/− and gal3+/+ DCs after stimulation with the β-glucan curdlan. Curdlan-stimulated gal3−/− DCs secreted higher levels of IL-23 compared with gal3+/+ DCs. In contrast, levels of IL-1β, IL-6, IL-10, and IL-12 were comparable (Figure 1), and levels of TGF-β were undetectable (data not shown). Differences in cytokine levels were not due to differences in cell death after stimulation (Supplemental Figure S1A). Furthermore, curdlan induced surface expression of co-stimulatory molecules on gal3−/− and gal3+/+ DCs to a similar extent (Supplemental Figure S1, B and C).
Figure 1.
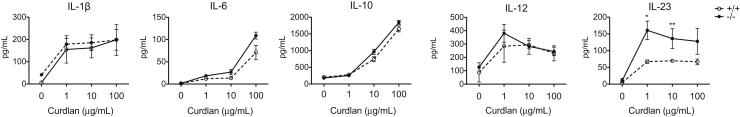
Curdlan-stimulated gal3−/− DCs produced higher levels of IL-23 compared with gal3+/+ DCs. ELISA of cytokines produced by gal3+/+ and gal3−/− DCs after stimulation with various amounts of curdlan overnight. Data are given as means ± SD and are representative of at least four independent experiments. ∗P < 0.05, ∗∗P < 0.01.
To investigate the ability of galectin-3 to influence DC-mediated Th17 polarization in response to β-glucan, we pulsed gal3−/− or gal3+/+ DCs with endotoxin-free OVA in the presence of curdlan, co-cultured the cells with OT-II CD4+ cells, and measured T-cell cytokines in co-culture supernatants. Gal3−/− DCs induced more antigen-specific IL-17 production relative to gal3+/+ DCs after curdlan treatment (Figure 2A). Apart from Th17 cells, curdlan-stimulated DCs also promote the induction of Th1 cells.10 Curdlan-primed gal3−/− DCs induced higher IFN-γ secretion in T cells relative to gal3+/+ DCs; however, the differences were not statistically significant. As expected, DCs pulsed with endotoxin-free OVA alone failed to activate OT-II cells, presumably owing to relatively low levels of co-stimulatory molecule expression.30 Using a different approach, we cultured gal3−/− and gal3+/+ DCs with curdlan for 16 hours and tested Th17-polarizing activity in the presence of the superantigen Staphylococcus Enterotoxin B. In agreement with the findings from the OVA-specific T-cell activation model described in Figure 2A, curdlan-primed gal3−/− DCs induced higher IL-17 production in CD4+ cells compared with gal3+/+ DCs (Figure 2B).
Figure 2.
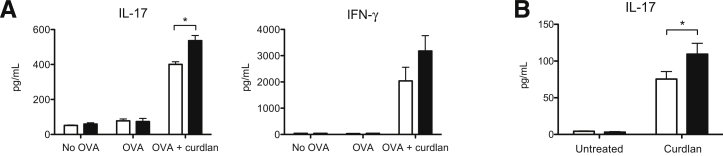
Galectin-3 in DCs negatively regulated Th17 responses. A: ELISA of cytokines in supernatants of CD4+ OT-II T cells that had been cultured with OVA-pulsed gal3+/+ (white bars) or gal3−/− (black bars) DCs in the presence or absence of curdlan for 3 days. B: ELISA of IL-17 in supernatants of CD4+ cells cultured with curdlan-primed gal3+/+ (white bars) or gal3−/− (black bars) DCs for 3 days in the presence of the superantigen Staphylococcus enterotoxin B. Data are given as means ± SD. Each experiment was performed at least two times. ∗P < 0.05.
Tregs have been reported to act as a source of TGF-β to drive Th17 differentiation.6 To determine whether galectin-3 in DCs indirectly affected Th17 responses by altering Treg development, we compared the number of CD4+CD25+Foxp3+ cells induced by curdlan-primed gal3−/− and gal3+/+ DCs. The frequencies of Foxp3+ Treg cells induced by both DC genotypes were comparable (Supplemental Figure S2), suggesting that the higher Th17-polarized responses by gal3−/− DCs were independent of Treg induction.
Differential IL-23 Production Accounts for Enhanced Th17 Induction by gal3−/− DCs
Curdlan-induced Th17 cell differentiation was shown to depend on IL-23,10 suggesting that the enhanced Th17 induction by gal3−/− DCs in this model may be due to differential IL-23 secretion by gal3−/− and gal3+/+ DCs (Figure 1). To confirm the importance of IL-23 in Th17 induction by curdlan-primed DCs, we performed the OVA-specific T-cell activation experiment in the presence of IL-23–neutralizing antibodies. Neutralization of IL-23 p19 decreased Th17 differentiation induced by gal3−/− and gal3+/+ DCs. Unexpectedly, neutralization of IL-23 p40 did not have a significant effect (Figure 3). On the other hand, it reduced Th1 development induced by gal3−/− and gal3+/+ DCs, likely due to the targeting of IL-12 by antibody, which shares the p40 chain with IL-23. As expected, blocking p19 had no effect on Th1-polarized responses.
Figure 3.

Differential IL-23 production by gal3−/− (black bars) and gal3+/+ (white bars) DCs contributed to differences in Th17 induction. ELISA of cytokines in OVA-curdlan DC–T-cell (OT-II) co-cultures after the addition of neutralizing anti–IL-23p19, anti–IL-12/23p40, or isotype control antibodies on day 0. The co-cultures were maintained for 3 days. Data are given as means ± SD and are representative of at least four independent experiments. ∗P < 0.05, ∗∗P < 0.01.
Galectin-3 in DCs Negatively Regulates Th17 Polarization in Response to High-Dose LPS
To determine whether galectin-3 in DCs also influences Th17 responses to other stimuli, we tested other Th17-priming agents on DC-mediated Th17 induction. LPS derived from Gram-negative bacteria activates TLR4 and is commonly associated with Th1 cell differentiation; however, recent studies indicate that high concentrations of LPS can also induce Th17 responses.30 After stimulation with LPS at high concentrations, gal3−/− DCs produced higher levels of the Th17-associated cytokines IL-6 and IL-23 compared with gal3+/+ DCs (Figure 4). In contrast, gal3−/− and gal3+/+ DCs secreted similar cytokine levels after low-dose LPS treatment (Figure 4).
Figure 4.

Gal3−/− (filled circles) DCs produced higher levels of the Th17-axis cytokines IL-6 and IL-23 compared with gal3+/+ (open circles) DCs after high-dose LPS stimulation. ELISA of cytokines produced by gal3+/+ and gal3−/− DCs after stimulation with low- or high-dose LPS overnight. Data are given as means ± SD and are representative of at least three independent experiments. ∗P < 0.05.
To determine the effect of galectin-3 on LPS-mediated Th17 priming, we studied Th17 induction by gal3−/− and gal3+/+ DCs after treatment with LPS at different concentrations. Compared with gal3+/+ DCs, gal3−/− DCs primed with high-dose LPS induced significantly more IL-17 production in OT-II CD4+ cells (Figure 5A). In accordance with previous reports that galectin-3 deficiency promotes Th1 responses,21,22 we also observed higher IFN-γ production in T cells cultured with low-dose LPS-primed gal3−/− DCs.
Figure 5.
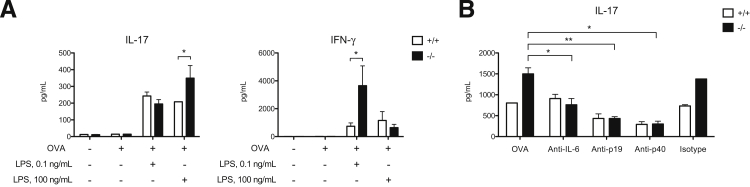
Gal3−/− DCs primed with high-dose LPS induced higher Th17 responses compared with gal3+/+ DCs. A: ELISA of cytokines in supernatants of CD4+ OT-II T cells that had been cultured with OVA-pulsed gal3+/+ or gal3−/− DCs for 3 days in the presence of low-dose (0.1 ng/mL) or high-dose (100 ng/mL) LPS. B: ELISA of IL-17 in OVA-LPS (100 ng/mL) DC–T-cell (OT-II) co-cultures after the addition of anti–IL-6, anti–IL-23p19, anti–IL-12/23p40, or isotype control antibodies on day 0. The co-cultures were maintained for 3 days. Data are given as means ± SD and are representative of at least three independent experiments. ∗∗P < 0.01, ∗∗∗P < 0.001.
Neutralization of either IL-6 or IL-23 subunits significantly reduced Th17-polarized responses induced by gal3−/− DCs, supporting the notion that higher production of these cytokines contributed to differences in Th17 induction (Figure 5B).
Galectin-3 Negatively Regulates Activation of c-Rel in DCs after Curdlan Stimulation
These data suggest that galectin-3 negatively regulates IL-23 production in DCs after dectin-1 or TLR4 stimulation. Dectin-1 signals through a spleen tyrosine-kinase and caspase recruitment domain protein 9 (Syk-CARD9)–dependent pathway and leads to the activation of NF-κB subunits involved in cytokine production.11 Regarding IL-23 expression, several groups have shown that c-Rel is a critical NF-κB subunit that promotes IL-23 p19 transcription.11,31 To investigate whether galectin-3 influenced IL-23 by affecting c-Rel activation, we compared nuclear translocation and DNA binding of c-Rel in curdlan-stimulated gal3−/− and gal3+/+ DCs. Consistent with higher IL-23 production, gal3−/− DCs also expressed higher levels of active c-Rel relative to gal3+/+ DCs after curdlan treatment (Figure 6A). Significantly higher levels of active p50 and p52 were also observed in gal3−/− DCs.
Figure 6.

Galectin-3 negatively regulates c-Rel transcription factor activation through the Raf-1 signaling pathway. A: Nuclear extracts from unstimulated or curdlan-stimulated gal3+/+ (white bars) and gal3−/− (black bars) DCs were added to plates coated with oligonucleotide containing an NF-κB consensus-binding site. Binding of activated NF-κB subunits was detected using antibodies against c-Rel, RelB, p50, p52, and p65. B: Raf-1 phosphorylation at Ser338 and Tyr340/341 as determined by immunoblot (IB) analysis in unstimulated and curdlan-stimulated gal3+/+ and gal3−/− DCs. Data are representative of at least two independent experiments. IP, immunoprecipitate. C: Densitometry analysis of phosphorylated Raf-1. Percentage of activation was calculated as the ratio of phosphorylated Raf-1 to total Raf-1. Data are given as means ± SD and are representative of two independent experiments. ∗P < 0.05, ∗∗P < 0.01.
Galectin-3 Promotes Raf-1 Activation in Dectin-1–Activated DCs
Galectin-3 had no influence on cell surface expression of dectin-1, as we observed no differences in receptor levels between gal3−/− and gal3+/+ DCs (data not shown). In addition to the Syk-CARD9 pathway, dectin-1 also activates a second signaling cascade mediated by the serine-threonine kinase Raf-1. Raf-1 modulates Syk-induced signaling at the point of NF-κB activation by inducing the phosphorylation and acetylation of p65, the latter of which increases its DNA binding affinity.11 The enhanced DNA-binding affinity of acetylated p65 enables it to compete with c-Rel for binding to the IL-23A promoter, thereby decreasing IL-23 expression.
We compared Syk and Raf-1 activation in curdlan-stimulated gal3−/− or gal3+/+ DCs. Syk activation was comparable in curdlan-treated gal3−/− and gal3+/+ DCs (data not shown), indicating that galectin-3 does not affect c-Rel activation through Syk-induced signaling. We then compared Raf-1 activation in gal3−/− and gal3+/+ DCs after curdlan treatment. A requirement for Raf-1 activation is phosphorylation at Ser338 by p21-activated kinases and at Tyr340/341 by Src kinases.32,33 Cell lysates from curdlan-stimulated gal3−/− or gal3+/+ DCs were immunoprecipitated with anti–Raf-1, and Raf-1 phosphorylation was determined by immunoblotting with anti–phospho–Raf-1 Ser338 and anti–phospho–Raf-1 Tyr340/341. Phosphorylation of Raf-1 at Ser338 in gal3−/− DCs after curdlan stimulation was twofold to threefold less compared with gal3+/+ DCs, suggesting that galectin-3 promotes Raf-1 activation (Figure 6, B and C).
In agreement with lower Raf-1 activation, gal3−/− DCs stimulated with curdlan had impaired activation of ERK, a downstream signaling molecule of the Raf-1 signaling pathway, 5 and 10 minutes after stimulation but recovered to normal levels at later time points (Figure 7, A and B). gal3−/− DCs stimulated with LPS also exhibited impaired ERK activation as early as 10 minutes after stimulation and failed to sustain activation after 1 hour (Figure 7, C and D). The trend in defective ERK signaling by gal3−/− DCs after dectin-1 and TLR4 stimulation suggests that galectin-3 may modulate a common factor shared by the signaling pathways initiated by these two distinct receptors.
Figure 7.

Defective ERK signaling in gal3−/− DCs stimulated with curdlan or LPS. A: Immunoblot analysis of ERK activation in gal3−/− (black bars) and gal3+/+ (white bars) DCs stimulated with 10 μg/mL of curdlan. B: Densitometric analysis of ERK activation in curdlan-stimulated DCs. Data are representative of three experiments performed. C: Immunoblot analysis of ERK activation in gal3−/− and gal3+/+ DCs stimulated with 100 ng/mL of LPS. D: Densitometric analysis of ERK activation in LPS-stimulated DCs. Data from one of two experiments are represented. Densitometric analyses were performed using ImageJ software version 1.45s (NIH, Bethesda, MD). Values shown are levels of phospho-ERK divided by total ERK.
C. albicans–Exposed gal3−/− DCs Adoptively Transferred into Mice Induce Higher Th17 Responses in Vivo
β-Glucans are expressed on the inner wall of fungal species. However, they can become exposed on the cell surface in budding or heat-killed yeasts and subsequently activate dectin-1.34,35 To study the effect of endogenous galectin-3 on DC cytokine expression in response to fungi, we stimulated DCs with HKCA and compared cytokine profiles in gal3−/− and gal3+/+ DCs. Gal3−/− DCs produced significantly higher levels of IL-23 but lower levels of IL-12 after stimulation with HKCA at 106 and 107 cells/mL compared with their wild-type counterparts (Figure 8A). Although there were no differences in IL-1β production, gal3−/− DCs produced higher levels of IL-6 in response to HKCA at 107 cells/mL. Thus, galectin-3 also affects DC cytokine production in response to C. albicans.
Figure 8.

Adoptive transfer of C. albicans–treated gal3−/− DCs conferred higher Th17 responses. A: ELISA of cytokines produced by gal3+/+ (open circles) and gal3−/− (closed circles) DCs after stimulation with HKCA overnight at various concentrations. Data are representative of at least three independent experiments. B: Untreated or HKCA-treated gal3−/− or gal3+/+ DCs were s.c. injected into mice twice, 1 week apart. A week after the last DC transfer, splenocytes were harvested and evaluated for cytokine expression. Cytokines in culture supernatants of splenic CD4+ cells restimulated ex vivo with HKCA-treated DCs. C: Cytokines in culture supernatants of unfractionated splenocytes restimulated ex vivo with HKCA. All culture supernatants were harvested after 3 days. Data are representative of at least two independent experiments. Data are given as means ± SD. ∗P < 0.05, ∗∗P < 0.01.
To assess the ability of galectin-3 in DCs to influence CD4+ priming in vivo, we immunized mice with HKCA-pulsed gal3−/− or gal3+/+ DCs by s.c. injection twice at 1-week intervals. A week after the last injection, we measured cytokine production in culture supernatants from purified splenic CD4+ cells, and splenocytes restimulated with HKCA. CD4+ cells and unfractionated splenocytes from mice that received C. albicans–pulsed gal3−/− DCs produced higher levels of IL-17 compared with those from mice that received C. albicans–pulsed gal3+/+ DCs or untreated DCs (Figure 8, B and C). Furthermore, we adoptively transferred gal3−/− or gal3+/+ DCs mixed with live C. albicans into wild-type mice by i.v. injection. Three and 6 days after DC transfer, we measured cytokine levels in the kidneys of infected mice and evaluated IL-17 expression in splenic CD4+ cells. Levels of IL-6, TGF-β, IL-23, and IL-17 in mice receiving gal3−/− DCs were significantly higher than in mice receiving gal3+/+ DCs on day 3 but not on day 6 after infection (Figure 9A). Purified CD4+ cells from C. albicans–infected mice receiving gal3−/− DCs expressed higher levels of IL-17A and the Th17-specific transcription factor RORγt compared with those from mice receiving gal3+/+ DCs on day 6 after infection (Figure 9B). Moreover, fungal burdens in the kidneys and brains of mice receiving gal3−/− DCs were lower compared with those of mice receiving gal3+/+ DCs on both days 3 and 6 after infection (Figure 9C).
Figure 9.

Galectin-3 in DCs regulated antifungal immunity in vivo. Gal3−/− (black bars) or gal3+/+ (white bars) DCs mixed with live C. albicans were adoptively transferred into wild-type mice by i.v. injection. A: Kidneys of C. albicans–infected mice were collected on days 3 and 6 after adoptive transfer of gal3+/+ and gal3−/− DCs and infection. Kidneys were homogenized, and cytokine levels in the supernatants of the homogenates were determined. The experiment was performed three times, with two to four mice included per group in each experiment. B: Real-time qPCR for indicated mRNAs in splenic CD4+ cells from mice that received C. albicans–exposed gal3−/− or gal3+/+ DCs. The relative mRNA fold induction was calculated based on the fold induction of nontreated samples. Three to four mice were included in each group. The experiment was repeated three times. C: Fungal burdens [colony-forming units (CFUs) per gram of tissue] in brains and kidneys of mice that received DCs were collected on days 3 and 6 after infection. Nine to 10 mice were included in each group. Data are given as means ± SD. ∗P < 0.05.
In agreement with higher IL-17A production, renal histologic analysis revealed higher cellular infiltrates in mice that received C. albicans–exposed gal3−/− DCs (Figure 10A). Consistent with the fact that cellular infiltration is essential for fungal clearance and survival,36 the number of pathogenic C. albicans hyphae was lower in kidneys of mice that received adoptively transferred gal3−/− DCs (Figure 10B). Furthermore, mice that received C. albicans–exposed gal3−/− DCs had significantly higher levels of infiltrating neutrophils, which may be explained by the higher levels of IL-17–axis cytokines that promote neutrophil recruitment (Figure 10C).37 Thus, galectin-3 expression in DCs is associated with suppression of cellular infiltration and ineffective fungal clearance after Candida infection. Because Th17 cells play a critical role in antifungal immunity, these results suggest that the ability of gal3−/− DCs to produce higher levels of IL-17–axis cytokines promotes differentiation of CD4+ T cells and maintenance of Th17 cells and protects mice against C. albicans dissemination to kidneys and brain. Collectively, these findings indicate that galectin-3 negatively regulates Th17 responses after C. albicans infection through a mechanism involving DC cytokine regulation.
Figure 10.

Renal histologic features in mice that received adoptively transferred gal3−/− or gal3+/+ DCs exposed to C. albicans. gal3−/− or gal3+/+ BMDCs were left untreated (naive) or were pulsed with C. albicans for 1 hour and then were adoptively transferred into gal3+/+ mice i.v. Six days after transfer (6 d.p.i.), kidneys were harvested and fixed in formalin. Tissue sections were stained with H&E (A) and PAS (B). A: Adoptive transfer of gal3−/− DCs promoted increased levels of cellular infiltration into kidneys compared with gal3+/+ DCs after infection. Sites of cellular infiltration are indicated by orange dashed lines. Objectives: 10× (top); 40× (bottom). Scale bars: 250 μm (top); 50 μm (bottom). B: Higher C. albicans burden in mice that received adoptively transferred gal3+/+ DCs. Fungi are indicated by orange dashed lines.Objective, 40×. Scale bar = 50 μm. Representative images are shown. C: Higher levels of neutrophil infiltration in kidneys of mice receiving adoptively transferred gal3−/− DCs. Neutrophils were identified by their polymorphic nuclei and cell size and were enumerated from five nonoverlapping high-power fields. Means are depicted by the horizontal lines.
Discussion
In the present study, we identified a novel role for endogenous galectin-3 as a regulator of cytokine expression in DCs. We demonstrate that endogenous galectin-3 negatively regulates the production of IL-17–axis cytokines in DCs stimulated with dectin-1 and TLR4 agonists. Gal3−/− DCs primed with either curdlan or LPS produced higher levels of IL-23 and promoted higher Th17 responses compared with gal3+/+ DCs. Neither the presence of lactose nor the addition of recombinant galectin-3 to DC cultures affected cytokine production (data not shown), supporting the notion that galectin-3 functions intracellularly to modulate DC cytokine expression.
Neutralization of the IL-23 p19 subunit in DC–T-cell co-cultures abrogated differential cytokine responses between the two genotypes, thereby confirming the importance of IL-23. Neutralization of IL-23 p40 decreased Th17 induction by LPS-primed DCs but failed to suppress Th17 induction by curdlan-primed DCs. This discrepancy may be due to the neutralization of IL-12, which shares the p40 subunit in common with IL-23. Neutralization by anti-p40 antibodies and subsequent blockade of Th1 cells may also account for the retained production of IL-17, as Th1 cells have been reported to negatively regulate differentiation of Th17 cells.38 Because curdlan is a stronger inducer of IL-23 production than is LPS,10 it is also feasible that binding of p40 antibodies to IL-12 and IL-23 caused a depletion of antibody such that amounts remaining were insufficient to neutralize the persistent production of IL-23 by curdlan-stimulated DCs.
Although IL-23 is not required for Th17 cell differentiation from naive T cells, it induces Th17 cell expansion, is required for effector functions, and is also implicated in the terminal differentiation of Th17 cells in vivo.4,6,39 On stimulation by curdlan, gal3−/− and gal3+/+ DCs mainly differed in IL-23 production, suggesting that galectin-3 in DCs may influence Th17 cell expansion and/or effector functions after dectin-1 activation but have little effect on naive T-cell differentiation. In contrast to dectin-1 agonists, stimulation of DCs with high-dose LPS led to enhanced production of IL-6 and IL-23 in gal3−/− DCs, suggesting that galectin-3 in DCs negatively regulates Th17 cell differentiation and development in response to TLR4 agonists. It remains unclear why galectin-3 in DCs affects different stages of Th17 cell development in response to dectin-1 and TLR4 stimulation. A possible explanation may lie in the distinct signaling pathways mediated by dectin-1 and TLR4. As dectin-1 signals through Syk-dependent and Syk-independent (Raf-1) pathways, we show that galectin-3 promoted Raf-1 activation but seemed to have little effect on the Syk signaling pathway. Thus, galectin-3 seems to affect the Raf-1 arm of the dectin-1 signaling pathway in DCs.
The recognition of fungal pathogens by DCs is mediated by several PRRs. In addition to TLR4 and dectin-1, C. albicans activates TLR2, dectin-2, mannose receptor, and DC-SIGN.34 The adoptive transfer of gal3−/− DCs in systemic infection with C. albicans resulted in higher production of IL-6, IL-23, and TGF-β, suggesting that PRRs other than TLR4 and dectin-1 may also be involved in the negative regulation of IL-17–axis cytokines by galectin-3 in vivo. A potential PRR may be TLR2, as other studies have shown that galectin-3 in association with TLR2 or dectin-1 is required for the recognition of C. albicans in macrophages.24,25 Galectin-3 was not required for the endocytosis of yeasts but was found in macrophage phagocytic cups and phagosomes containing C. albicans.24 Thus, galectin-3 may not be necessary for the phagocytosis of yeasts but instead plays a role in modulating signaling pathways in response to fungal pathogens.
In contrast to the studies on galectin-3 in Th1/Th2 polarization, the literature on galectin-3 regarding Th17 regulation is scarce. Recently, Radosavljevic et al40 reported high serum levels of IFN-γ and IL-17 in gal3−/− mice inoculated with B16F1 melanoma cells. Although the role of IL-17 in protumor and antitumor responses remains controversial, their findings suggest that galectin-3 may negatively regulate Th1 and Th17 responses in tumor models. In animal models of autoimmune disease, where Th17 cells are known to contribute to inflammation, galectin-3 was shown to promote IL-17 production. Using an experimental autoimmune encephalomyelitis mouse model, Jiang and colleagues41 observed attenuated experimental autoimmune encephalomyelitis symptoms in gal3−/− mice accompanied by lower levels of IFN-γ and IL-17. They attributed Th1 and Th17 impairment in gal3−/− mice to an increased frequency of Foxp3+ Tregs present in the central nervous system and enhanced IL-10 production by gal3−/− DCs. We, however, observed higher Th1 and Th17 differentiation induced by gal3−/− DCs and no difference in Treg induction or IL-10 secretion. Such discrepancies may be due to differences in cell culture conditions or the experimental disease model.
We demonstrate that endogenous galectin-3 affects cytokine expression in DCs after stimulation of dectin-1 or TLR4, two different classes of PRRs. Although these receptors signal through different pathways, we observed impaired ERK mitogen-activated protein kinase signaling in gal3−/− DCs in response to both stimulants. Similar defects were reported in gal3−/− DCs after chemokine stimulation.19 Collectively, these studies indicate a common factor in receptor-mediated signal transduction that may be regulated by galectin-3. Dectin-1 and TLR4 have been reported to translocate to lipid raft microdomains on ligand stimulation,42,43 an event that initiates signal transduction. Disruption of lipid rafts using chemical agents resulted in the selective attenuation of ERK activation in LPS-stimulated macrophages44 and dectin-1–activated DCs,42 illustrating the importance of lipid raft integrity for ERK signaling. We previously showed that galectin-3 positively regulates DC migration by functioning in membrane structures. Compared with gal3+/+ DCs, gal3−/− DCs exhibited structural differences in membrane ruffles along with impaired chemotaxis and defective signal transduction in response to chemokine stimulation.19 Regarding the present study, impaired ERK activation in gal3−/− DCs suggests that galectin-3 may modulate the stability of lipid raft microdomains and contribute to efficient ERK signaling.
The association of galectin-3 with membrane domains may also be the basis for impaired Raf-1 activation in gal3−/− DCs after curdlan stimulation. Galectin-3 was previously identified as a binding partner of K-Ras,45 a member of the Ras family of guanine nucleotide-binding proteins present on the inner leaflet of the plasma membrane that also serves as an upstream activator for Raf-1.46–48 Interactions between galectin-3 and Ras promote Ras organization into nanoclusters. Although its role in DC signaling is yet to be fully delineated, galectin-3 may function as a scaffold for K-Ras–GTP nanocluster formation, providing a platform for Raf-1 recruitment to the plasma membrane and subsequent activation by phosphorylation.49 Because Raf-1 functions upstream of the mitogen-activated protein kinase/ERK signaling cascade,46 impaired Raf-1 activation in curdlan-stimulated gal3−/− DCs may correlate with our observations of defective ERK signaling.
A consequence of impaired Raf-1 activation in DCs is altered cytokine production. Recent studies have shown that Raf-1 inhibition increases IL-23 p19 mRNA expression in curdlan-stimulated DCs,11 indicating that Raf-1 negatively regulates production of this cytokine. The mechanism for Raf-1–mediated regulation of IL-23 is largely due to acetylated p65, which prevents c-Rel binding to the IL23A promoter. We show that galectin-3 promotes Raf-1 activation after dectin-1 stimulation, which may lead to increased levels of acetylated p65 and consequently less c-Rel activation. As c-Rel is a stronger activator of IL23A transcription than acetylated p65,11 enhanced Raf-1 activation induced by galectin-3 would, therefore, decrease IL-23 production. In support of this hypothesis, we show higher c-Rel nuclear translocation and DNA binding in curdlan-stimulated gal3−/− DCs compared with similarly treated gal3+/+ DCs. Significantly elevated levels of p50 and p52 were also observed in gal3−/− DCs, which may also contribute to IL23A promoter activation through heterodimer formation with NF-κB family members.50 In addition to IL-23, Raf-1–mediated acetylation of p65 regulates the expression of IL-6, IL-10, and IL-12.11 Although levels of p65 were slightly higher in curdlan-stimulated gal3−/− DCs, these levels did not reach statistical significance. In addition, the negative regulation of acetylated p65 on IL23A promoter activation may be offset by higher activation of c-Rel, p50, and p52 in curdlan-stimulated gal3−/− DCs.
In summary, these data suggest that endogenous galectin-3 regulates DC cytokine expression and signal transduction in response to dectin-1 and TLR4 agonists. Based on published findings and the present observations, we propose that galectin-3 may be recruited from the cytosol to the plasma membrane in dectin-1– and TLR4-activated DCs and may participate in the organization of membrane components by acting as a protein scaffold or affecting lipid raft stability. Regarding dectin-1 signaling, galectin-3 may function as a scaffold for proteins such as Ras-GTP and subsequently promote efficient activation of Raf-1 (Figure 11). In turn, this may modulate downstream signaling events and, ultimately, influence cytokine expression.
Figure 11.

Proposed model for galectin-3 in the regulation of cytokine expression in DCs. On ligand-mediated DC activation, galectin-3 in the cell cytosol may be recruited to membrane components, as previously demonstrated with Ras-GTP.45,47 At the membrane, galectin-3 may participate in the reorganization of membrane proteins or may act as a protein scaffold and subsequently influence downstream signal transduction. In association with Ras-GTP, galectin-3 aids in the formation of Ras nanoclusters, which promotes the efficient activation of Raf-1. By means of p65 phosphorylation, Raf-1 negatively regulates IL-23 expression.
The finding that galectin-3 negatively regulates Th17-polarizing cytokines in DCs suggests that targeting this lectin in DCs may be useful in the development of new strategies to modulate Th17 responses. This could be applicable to antifungal immunity and autoimmune diseases.
Acknowledgments
We thank Yen-Hui Chen at the Pathology Core Laboratory (Institute of Biomedical Sciences, Academia Sinica) for expert technical assistance in the preparation and enumeration of inflammation in mouse tissues and Dr. Ching-Liang Chu for helpful discussion.
Footnotes
Supported by NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases grants AR056343 (F.-T.L.) and T32 HL07013 (A.F.L.).
Current address of A.F.L., John Wayne Cancer Institute, Santa Monica, CA.
Supplemental Data
Gal3−/− DCs stimulated with curdlan had no defects in cell activation. A: Cell viability in activated gal3−/− and gal3+/+ BMDCs was determined by trypan blue exclusion or 7-aminoactinomycin D (7-AAD) staining. B: Flow cytometry for surface expression of CD80, CD86, and major histocompatibility complex (MHC) II on gal3−/− and gal3+/+ BMDCs after overnight stimulation with curdlan. C: Percentages of cells expressing CD80, CD86, and MHC II as determined by flow cytometry.
Galectin-3 in DCs did not affect CD4+CD25+Foxp3+ Treg induction. Intracellular Foxp3 staining of CD4+CD25+ T cells co-cultured with OVA-curdlan–treated gal3−/− or gal3+/+ BMDCs for 3 days. FITC, fluorescein isothiocyanate; PE, phosphatidylethanolamine.
References
- 1.Huang W., Na L., Fidel P.L., Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 2.Conti H.R., Shen F., Nayyar N., Stocum E., Sun J.N., Lindemann M.J., Ho A.W., Hai J.H., Yu J.J., Jung J.W., Filler S.G., Masso-Welch P., Edgerton M., Gaffen S.L. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cua D.J., Sherlock J., Chen Y., Murphy C.A., Joyce B., Seymour B., Lucian L., To W., Kwan S., Churakova T., Zurawski S., Wiekowski M., Lira S.A., Gorman D., Kastelein R.A., Sedgwick J.D. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 4.Langrish C.L., Chen Y., Blumenschein W.M., Mattson J., Basham B., Sedgwick J.D., McClanahan T., Kastelein R.A., Cua D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettelli E., Carrier Y., Gao W., Korn T., Strom T.B., Oukka M., Weiner H.L., Kuchroo V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 6.Veldhoen M., Hocking R.J., Atkins C.J., Locksley R.M., Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Clark G.J., Angel N., Kato M., Lopez J.A., MacDonald K., Vuckovic S., Hart D.N.J. The role of dendritic cells in the innate immune system. Microbes Infect. 2000;2:257–272. doi: 10.1016/s1286-4579(00)00302-6. [DOI] [PubMed] [Google Scholar]
- 8.Ariizumi K., Shen G.-L., Shikano S., Xu S., Ritter R., Kumamoto T., Edelbaum D., Morita A., Bergstresser P.R., Takashima A. Identification of a novel dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J Biol Chem. 2000;275:20157–20167. doi: 10.1074/jbc.M909512199. [DOI] [PubMed] [Google Scholar]
- 9.Brown G.D., Gordon S. Immune recognition: a new receptor for β-glucans. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 10.LeibundGut-Landmann S., Grosz O., Robinson M.J., Osorio F., Slack E.C., Tsoni S.V., Schweighoffer E., Tybulewicz V., Brown G.D., Ruland J., Reis e Sousa C. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- 11.Gringhuis S.I., den Dunnen J., Litjens M., van der Vlist M., Wevers B., Bruijns S.C.M., Geijtenbeek T.B.H. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-κB activation through Raf-1 and Syk. Nat Immunol. 2009;10:203–213. doi: 10.1038/ni.1692. [DOI] [PubMed] [Google Scholar]
- 12.Acosta-Rodriguez E.V., Napolitani G., Lanzavecchia A., Sallusto F. Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 13.Liu F.-T., Patterson R.J., Wang J.L. Intracellular functions of galectins. Biochim Biophys Acta. 2002;1572:263–273. doi: 10.1016/s0304-4165(02)00313-6. [DOI] [PubMed] [Google Scholar]
- 14.Ochieng J., Furtak V., Lukyanov P. Extracellular functions of galectin-3. Glycoconj J. 2004;19:527–535. doi: 10.1023/B:GLYC.0000014082.99675.2f. [DOI] [PubMed] [Google Scholar]
- 15.Dumic J., Dabelic S., Flogel M. Galectin-3: an open-ended story. Biochim Biophys Acta. 2006;1760:616–635. doi: 10.1016/j.bbagen.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 16.Yang R.-Y., Hsu D.K., Liu F.-T. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc Natl Acad Sci U S A. 1996;93:6737–6742. doi: 10.1073/pnas.93.13.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dagher S.F., Wang J.L., Patterson R.J. Identification of galectin-3 as a factor in pre-mRNA splicing. Proc Natl Acad Sci U S A. 1995;92:1213–1217. doi: 10.1073/pnas.92.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sano H., Hsu D.K., Apgar J.R., Yu L., Sharma B.B., Kuwabara I., Izui S., Liu F.-T. Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest. 2003;112:389–397. doi: 10.1172/JCI17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu D.K., Chernyavsky A.I., Chen H.-Y., Yu L., Grando S.A., Liu F.-T. Endogenous galectin-3 is localized in membrane lipid rafts and regulates migration of dendritic cells. J Invest Dermatol. 2008;129:573–583. doi: 10.1038/jid.2008.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuberi R.I., Hsu D.K., Kalayci O., Chen H.-Y., Sheldon H.K., Yu L., Apgar J.R., Kawakami T., Lilly C.M., Liu F.-T. Critical role for galectin-3 in airway inflammation and bronchial hyperresponsiveness in a murine model of asthma. Am J Pathol. 2004;165:2045–2053. doi: 10.1016/S0002-9440(10)63255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernardes E.S., Silva N.M., Ruas L.P., Mineo J.R., Loyola A.M., Hsu D.K., Liu F.-T., Chammas R., Roque-Barreira M.C. Toxoplasma gondii infection reveals a novel regulatory role for galectin-3 in the interface of innate and adaptive immunity. Am J Pathol. 2006;168:1910–1920. doi: 10.2353/ajpath.2006.050636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saegusa J., Hsu D.K., Chen H.-Y., Yu L., Fermin A., Fung M.A., Liu F.-T. Galectin-3 is critical for the development of the allergic inflammatory response in a mouse model of atopic dermatitis. Am J Pathol. 2009;174:922–931. doi: 10.2353/ajpath.2009.080500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rabinovich G.A., Gruppi A. Galectins as immunoregulators during infectious processes: from microbial invasion to the resolution of the disease. Parasite Immunol. 2005;27:103–114. doi: 10.1111/j.1365-3024.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 24.Jouault T., El Abed-El Behi M., Martínez-Esparza M., Breuilh L., Trinel P.-A., Chamaillard M., Trottein F., Poulain D. Specific recognition of Candida albicans by macrophages requires galectin-3 to discriminate Saccharomyces cerevisiae and needs association with TLR2 for signaling. J Immunol. 2006;177:4679–4687. doi: 10.4049/jimmunol.177.7.4679. [DOI] [PubMed] [Google Scholar]
- 25.Esteban A., Popp M.W., Vyas V.K., Strijbis K., Ploegh H.L., Fink G.R. Fungal recognition is mediated by the association of dectin-1 and galectin-3 in macrophages. Proc Natl Acad Sci U S A. 2011;108:14270–14275. doi: 10.1073/pnas.1111415108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohatsu L., Hsu D.K., Jegalian A.G., Liu F.-T., Baum L.G. Galectin-3 induces death of Candida species expressing specific beta-1,2-linked mannans. J Immunol. 2006;177:4718–4726. doi: 10.4049/jimmunol.177.7.4718. [DOI] [PubMed] [Google Scholar]
- 27.Hsu D.K., Yang R.-Y., Pan Z., Yu L., Salomon D.R., Fung-Leung W.-P., Liu F.-T. Targeted disruption of the galectin-3 gene results in attenuated peritoneal inflammatory responses. Am J Pathol. 2000;156:1073–1083. doi: 10.1016/S0002-9440(10)64975-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.d’Ostiani C.F., Del Sero G., Bacci A., Montagnoli C., Spreca A., Mencacci A., Ricciardi-Castagnoli P., Romani L. Dendritic cells discriminate between yeasts and hyphae of the fungus Candida albicans. J Exp Med. 2000;191:1661–1674. doi: 10.1084/jem.191.10.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters M., Dudziak K., Stiehm M., Bufe A. T-cell polarization depends on concentration of the danger signal used to activate dendritic cells. Immunol Cell Biol. 2010;88:537–544. doi: 10.1038/icb.2010.3. [DOI] [PubMed] [Google Scholar]
- 31.Carmody R.J., Ruan Q., Liou H.-C., Chen Y.H. Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J Immunol. 2007;178:186–191. doi: 10.4049/jimmunol.178.1.186. [DOI] [PubMed] [Google Scholar]
- 32.Gringhuis S.I., den Dunnen J., Litjens M., van het Hof B., van Kooyk Y., Geijtenbeek T.B.H. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kB. Immunity. 2007;26:605–616. doi: 10.1016/j.immuni.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 33.Wellbrock C., Karasarides M., Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 34.Netea M.G., Brown G.D., Kullberg B.J., Gow N.A.R. An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Micro. 2008;6:67–78. doi: 10.1038/nrmicro1815. [DOI] [PubMed] [Google Scholar]
- 35.Gow N.A.R., Netea M.G., Munro C.A., Ferwerda G., Bates S., Mora-Montes H.M., Walker L., Jansen T., Jacobs L., Tsoni V., Brown G.D., Odds F.C., Van der Meer J.W.M., Brown A.J.P., Kullberg B.J. Immune recognition of Candida albicans beta-glucan by dectin-1. J Infect Dis. 2007;196:1565–1571. doi: 10.1086/523110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romani L. Immunity to fungal infections. Nat Rev Immunol. 2004;4:11–24. doi: 10.1038/nri1255. [DOI] [PubMed] [Google Scholar]
- 37.Laan M., Cui Z.-H., Hoshino H., Lötvall J., Sjöstrand M., Gruenert D.C., Skoogh B.-E., Lindén A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- 38.Annunziato F., Cosmi L., Santarlasci V., Maggi L., Liotta F., Mazzinghi B., Parente E., Filì L., Ferri S., Frosali F., Giudici F., Romagnani P., Parronchi P., Tonelli F., Maggi E., Romagnani S. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGeachy M.J., Chen Y., Tato C.M., Laurence A., Joyce-Shaikh B., Blumenschein W.M., McClanahan T.K., O’Shea J.J., Cua D.J. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radosavljevic G., Jovanovic I., Majstorovic I., Mitrovic M., Lisnic V., Arsenijevic N., Jonjic S., Lukic M. Deletion of galectin-3 in the host attenuates metastasis of murine melanoma by modulating tumor adhesion and NK cell activity. Clin Exp Metastasis. 2011;28:451–462. doi: 10.1007/s10585-011-9383-y. [DOI] [PubMed] [Google Scholar]
- 41.Jiang H.-R., Al Rasebi Z., Mensah-Brown E., Shahin A., Xu D., Goodyear C.S., Fukada S.Y., Liu F.-T., Liew F.Y., Lukic M.L. Galectin-3 deficiency reduces the severity of experimental autoimmune encephalomyelitis. J Immunol. 2009;182:1167–1173. doi: 10.4049/jimmunol.182.2.1167. [DOI] [PubMed] [Google Scholar]
- 42.Xu S., Huo J., Gunawan M., Su I.H., Lam K.-P. Activated dectin-1 localizes to lipid raft microdomains for signaling and activation of phagocytosis and cytokine production in dendritic cells. J Biol Chem. 2009;284:22005–22011. doi: 10.1074/jbc.M109.009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakahira K., Kim H.P., Geng X.H., Nakao A., Wang X., Murase N., Drain P.F., Wang X., Sasidhar M., Nabel E.G., Takahashi T., Lukacs N.W., Ryter S.W., Morita K., Choi A.M.K. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med. 2006;203:2377–2389. doi: 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cuschieri J. Implications of lipid raft disintegration: enhanced anti-inflammatory macrophage phenotype. Surgery. 2004;136:169–175. doi: 10.1016/j.surg.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 45.Elad-Sfadia G., Haklai R., Balan E., Kloog Y. Galectin-3 Augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J Biol Chem. 2004;279:34922–34930. doi: 10.1074/jbc.M312697200. [DOI] [PubMed] [Google Scholar]
- 46.Moodie S.A., Willumsen B.M., Weber M.J., Wolfman A. Complexes of Ras. GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- 47.Shalom-Feuerstein R., Plowman S.J., Rotblat B., Ariotti N., Tian T., Hancock J.F., Kloog Y. K-Ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 2008;68:6608–6616. doi: 10.1158/0008-5472.CAN-08-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plowman S.J., Muncke C., Parton R.G., Hancock J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci U S A. 2005;102:15500–15505. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marais R., Light Y., Paterson H.F., Marshall C.J. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:3136–3145. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayden M.S., Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gal3−/− DCs stimulated with curdlan had no defects in cell activation. A: Cell viability in activated gal3−/− and gal3+/+ BMDCs was determined by trypan blue exclusion or 7-aminoactinomycin D (7-AAD) staining. B: Flow cytometry for surface expression of CD80, CD86, and major histocompatibility complex (MHC) II on gal3−/− and gal3+/+ BMDCs after overnight stimulation with curdlan. C: Percentages of cells expressing CD80, CD86, and MHC II as determined by flow cytometry.
Galectin-3 in DCs did not affect CD4+CD25+Foxp3+ Treg induction. Intracellular Foxp3 staining of CD4+CD25+ T cells co-cultured with OVA-curdlan–treated gal3−/− or gal3+/+ BMDCs for 3 days. FITC, fluorescein isothiocyanate; PE, phosphatidylethanolamine.