

Significance
Biological nitrogen reduction is a fascinating transformation whose mechanism remains uncertain. Recently, an interstitial carbon has been identified within the FeMo-cofactor (FeMoco) of nitrogenase whose role is unknown and warrants model studies. In this report we disclose a series of five-coordinate Fe complexes bound to an ancillary ligand featuring a central C atom. This model system coordinates N2 trans to the C atom, and displays unusual Fe–C bonding motifs that may shed light on a possible role of the interstitial carbon in FeMoco.
Keywords: nitrogenase, nitrogen fixation, Fe–N2 complexes, small molecule activation, ammonia production
Abstract
We report here a series of four- and five-coordinate Fe model complexes that feature an axial tri(silyl)methyl ligand positioned trans to a substrate-binding site. This arrangement is used to crudely model a single-belt Fe site of the FeMo-cofactor that might bind N2 at a position trans to the interstitial C atom. Reduction of a trigonal pyramidal Fe(I) complex leads to uptake of N2 and subsequent functionalization furnishes an open-shell Fe–diazenido complex. A related series of five-coordinate Fe–CO complexes stable across three redox states is also described. Spectroscopic, crystallographic, and Density Functional Theory (DFT) studies of these complexes suggest that a decrease in the covalency of the Fe–Calkyl interaction occurs upon reduction and substrate binding. This leads to unusually long Fe–Calkyl bond distances that reflect an ionic Fe–C bond. The data presented are contextualized in support of a hypothesis wherein modulation of a belt Fe–C interaction in the FeMo-cofactor facilitates substrate binding and reduction.
MoFe-nitrogenase catalyzes the fascinating but poorly understood conversion of nitrogen to ammonia at its iron-molybdenum cofactor (FeMoco) (1, 2). The core of the FeMoco was originally thought to be vacant (3). Later work on Azotobacter vinelandii indicated the presence of a light interstitial atom coordinated to six central, so-called “belt Fe atoms” (4). Crystallographic and spectroscopic studies (5, 6), in addition to studies mapping the biosynthetic pathway of C-atom incorporation (7, 8), establish that carbon is the interstitial atom, as shown in Fig. 1.
Fig. 1.

A hypothetical N2-binding event at a belt iron in FeMoco illustrating a proposed Fe–C elongation. The degree and positions of protonation are unknown under electron loading, but the inorganic sulfides are plausible candidate positions.
Although the site(s) of N2 reduction remain(s) uncertain, a body of evidence that includes biochemical, spectroscopic, and computational studies on FeMoco point to a belt Fe center as a plausible candidate (2, 9–13). In a scenario in which N2 binds terminally to one of the belt Fe centers, the N2 ligand would initially be coordinated trans to the interstitial C atom (Fig. 1) (10). This hypothesis calls for model complexes that depict such an arrangement to explore factors that might govern substrate coordination and subsequent reduction. Structurally faithful models of the FeMoco that include an Fe6C unit stabilized by sulfide or other sulfur-based ligands present a formidable synthetic challenge (14). Moreover, N2 coordination to synthetic iron–sulfur clusters has yet to be established (15). Model complexes featuring a single Fe site with a C-atom anchor positioned trans to an N2 binding site are unknown, but would provide a useful tool to evaluate how an Fe–C interaction might respond to N2 binding and the Fe redox state. Model compounds of this type may facilitate the evaluation of theoretical (10) and spectroscopic studies (16) on FeMoco that suggest a single, flexible Fe–C interaction is observed under turnover conditions.
It is in this context that we have pursued mononuclear Fe complexes supported by tripodal, tetradentate ligands featuring three phosphine donor arms tethered to a tertiary alkyl anchor. Although a number of such ligands featuring a central C atom has been described (17–19) we reasoned that an alkyl ligand featuring only electropositive substituents adjacent to the C-atom anchor would provide a crude model of the interstitial carbide of the FeMoco and permit a high degree of ionic bonding to a single Fe–N2 binding site. To achieve this goal, the C-atom anchor of the auxiliary ligand described is surrounded by three electropositive Si centers, in addition to the Fe site. This model system successfully coordinates N2 trans to the C-atom anchor and shows how the local Fe geometry and the Fe–C interaction respond as a function of such binding. Using CO instead of N2, this Fe system also presents an opportunity to study the Fe–C interaction as a function of the Fe redox state. As described below, unusually long Fe–C distances can be accessed that are consistent with a much higher degree of ionic character at the Fe–C interaction than would be anticipated for a prototypical Fe alkyl.
Results and Discussion
A suitable ligand framework for the present study, whose lithium salt was previously reported by Avent et al. (20), is (Ph2PCH2SiMe2)3CH. This ligand, denoted herein as  (1) and depicted in Fig. 2, features a tri(silyl)methyl core tethered to three soft phosphines. Given that the interstitial carbide in FeMoco is surrounded by six electropositive Fe centers, we speculated that an Fe5C subunit would stabilize partial negative charge on the C atom as the sixth Fe–C interaction elongates upon reduction/N2 binding. Relative to methane, the three electropositive silyl substituents of the tri(silyl)methane subunit reduce the basicity of the central carbon (pKa[(Me3Si)3CH]: 36.8 in THF) by ∼20 orders of magnitude (21, 22). We anticipated that the tempered basicity of the tri(silyl)methyl subunit would translate to a flexible Fe–C interaction.
(1) and depicted in Fig. 2, features a tri(silyl)methyl core tethered to three soft phosphines. Given that the interstitial carbide in FeMoco is surrounded by six electropositive Fe centers, we speculated that an Fe5C subunit would stabilize partial negative charge on the C atom as the sixth Fe–C interaction elongates upon reduction/N2 binding. Relative to methane, the three electropositive silyl substituents of the tri(silyl)methane subunit reduce the basicity of the central carbon (pKa[(Me3Si)3CH]: 36.8 in THF) by ∼20 orders of magnitude (21, 22). We anticipated that the tempered basicity of the tri(silyl)methyl subunit would translate to a flexible Fe–C interaction.
Fig. 2.

Qualitative d-orbital splitting diagrams reflective of experimentally measured spin states for Fe–Cl complexes of  and
and 
Benzyl potassium was found to effect rapid and quantitative deprotonation of 1 as evidenced by 31P NMR spectroscopy. Subsequent salt metathesis with FeCl2 (Fig. 3) afforded the Fe halide complex  (2), as a pale yellow powder. Complex 2 exhibits broad 1H NMR signatures from δ = −1 to 30 ppm and a solution magnetic moment of 4.9 μB (CD2Cl2, 21 °C), consistent with an S = 2 species. This high spin state is noteworthy given the strong ligand field strength that might be expected from a tris(phosphine)alkyl donor set and attests to the comparatively weak ligand field arising from the
(2), as a pale yellow powder. Complex 2 exhibits broad 1H NMR signatures from δ = −1 to 30 ppm and a solution magnetic moment of 4.9 μB (CD2Cl2, 21 °C), consistent with an S = 2 species. This high spin state is noteworthy given the strong ligand field strength that might be expected from a tris(phosphine)alkyl donor set and attests to the comparatively weak ligand field arising from the  ligand. The structurally similar and isoelectronic five-coordinate Fe(II) chloride complexes supported by the tris(phosphine)silyl ligand
ligand. The structurally similar and isoelectronic five-coordinate Fe(II) chloride complexes supported by the tris(phosphine)silyl ligand 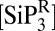 (23, 24) display an intermediate S = 1 spin state as a consequence of the strong and highly covalent Fe–Si bond. Zero-field Mössbauer data (SI Appendix, Fig. S14) collected on 2 is consistent with its S = 2 assignment (25).
(23, 24) display an intermediate S = 1 spin state as a consequence of the strong and highly covalent Fe–Si bond. Zero-field Mössbauer data (SI Appendix, Fig. S14) collected on 2 is consistent with its S = 2 assignment (25).
Fig. 3.

Synthesis of iron complexes supported by a tris(phosphino)alkyl  tripod.
tripod.  .
.
The X-ray crystal structure of 2 exhibits a rigorously trigonal bipyramidal geometry about the Fe center as exemplified by a τ5 value of 1.00 (SI Appendix), and confirms ligation of the tri(silyl)methyl carbon to Fe, albeit at an unusually long distance [2.263(2) Å]. Reported high-spin Fe(II)–Calkyl distances range from 2.00 to 2.21 Å (26). The Fe–P distances are also quite long [2.5696(3) Å] as a consequence of the high-spin state. The Fe atom is displaced just 0.006 Å from the plane defined by the three crystallographically equivalent phosphine ligands. This structure contrasts the geometric parameters imposed by phenylene- and ethylene-linked tetradentate tripodal phosphine ligands that force the metal center to protrude out of the equatorial phosphine plane away from the apical ligand (27, 28). The incorporation of Si and P atoms in each five-membered chelate ring of  likely provides the flexibility required for the metal complex to adopt such a symmetric geometry.
likely provides the flexibility required for the metal complex to adopt such a symmetric geometry.
Na/Hg amalgam reduction of 2 under an N2 atmosphere affords the Fe(I) complex, (3) (Fig. 3), as a brick-red powder. The absence of IR absorptions in the range of 2,100 to 1,700 cm−1 (KBr pellet) rules out terminal N2 coordination to a mononuclear Fe center in the solid state. A solution magnetic moment of 3.8 μB (C6D6) is observed at room temperature, and EPR measurements of 3 recorded at 10 K display g values of 4.66, 3.77 and 2.02 arising from Ms = 1/2 transitions, in addition to a feature at g = 6.22 stemming from transitions within the Ms = 3/2 manifold, consistent with a quartet ground state (29). The solid-state structure of 3 (Fig. 4) reveals a four-coordinate Fe complex. The Fe center is positioned below the P3 plane defined by the phosphine donors [d(Fe-Pplane): 0.322 Å], and displays a shortened Fe–C distance [2.153(2) Å; Fig. 3]. The local Fe geometry is intermediate between tetrahedral and trigonal monopyramidal (τ4 = 0.68) (30). Its geometry is topologically similar, although more pyramidalized, to that of the belt Fe centers in the structurally characterized resting state of FeMoco (τ4 = 0.46), where the Fe atoms are directed toward the interstitial C atom and resting below the S3 plane of the surrounding sulfides (5).
Fig. 4.

X-ray crystal structures of 3, 4, 5, and 6 with thermal ellipsoids set at 50% probability. Hydrogen atoms and solvent molecules have been removed. The phenyl rings have been rendered transparent to aid in visualization of the inner coordination sphere of iron. The {K(benzo-15-crown- 5)2} countercation of 4 has been removed for clarity. Only one independent molecule of 6 is shown.
In hopes of preparing an Fe–N2 complex supported by this ligand scaffold, we investigated the chemical reduction of 3. Compound 3 cleanly reacts with KC8, and encapsulation of the potassium countercation of the resulting species with two equivalents of benzo-15-crown-5 affords a dark purple powder displaying an intense IR ν(NN) absorption at 1,927 cm−1 for the terminal Fe–N2 complex {K(benzo-15-crown-5)2} (4). A Toepler pump analysis (SI Appendix) of 4 analyzed for 0.9 molar equivalents of N2 upon oxidation with ferrocenium trifluoromethanesulfonate in THF. Complex 4 is diamagnetic in THF solution (31P NMR, d8-THF, δ = 43.6 ppm). Its solid-state crystal structure (Fig. 4) confirms axial coordination of dinitrogen trans to the Fe-Calkyl donor and shows that, upon coordination, the Fe center slides into the P3 plane of the phosphines [d(Fe-Pplane): 0.012 Å] and away from the apical carbon [d(Fe-C) = 2.254(5) Å].
(4). A Toepler pump analysis (SI Appendix) of 4 analyzed for 0.9 molar equivalents of N2 upon oxidation with ferrocenium trifluoromethanesulfonate in THF. Complex 4 is diamagnetic in THF solution (31P NMR, d8-THF, δ = 43.6 ppm). Its solid-state crystal structure (Fig. 4) confirms axial coordination of dinitrogen trans to the Fe-Calkyl donor and shows that, upon coordination, the Fe center slides into the P3 plane of the phosphines [d(Fe-Pplane): 0.012 Å] and away from the apical carbon [d(Fe-C) = 2.254(5) Å].
Silylation of M–N2 complexes has been used to gauge the reactivity of the bound N2 ligand toward electrophiles (31–33) and we canvassed related reactions with the present system. Whereas trimethylsilyl trifluoromethanesulfonate reacts productively with 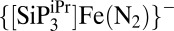 to afford
to afford  (24), Fe-N2−
4 required the use of a highly encumbered silylating agent, triisopropylsilyl trifluoromethanesulfonate, at −78 °C to furnish a tractable diazenido product, (5). Characterization data for 5 include a ν(NN) vibration at 1,719 cm−1, a solution magnetic moment of 2.75 μB (C6D6, 23 °C), and a broad room temperature 1H NMR spectrum. Upon cooling to −80 °C in d8-toluene, the 1H NMR signals of 5 resolve into 14 distinguishable resonances between δ = 32 and −10 ppm, suggestive of an unsymmetrical paramagnetic species. An X-ray Diffraction (XRD) study of 5 (Fig. 4) accounts for this asymmetry at low temperature by verifying silylation of the beta N-atom of the N2 ligand and also establishing a dechelated phosphine donor arm. The Fe geometry of 5 in the solid state is thus four-coordinate with a short Fe–Calkyl distance of 2.116(1) Å, an Fe–N(1) distance of 1.713(1) Å, and two Fe–P bonds with an Fe–Pavg of 2.332 Å. The diazenido ligand features an elongated N(1)–N(2) bond length of 1.203(2) Å, consistent with substantial activation of the N≡N triple bond in free N2 (1.098 Å). The diazenido ligand is severely bent at N(2) with a contracted N(1)-N(2)–Si (4) angle of 135.11°. Similar bond angles found in (TPB)Fe(N2SiMe3) (33) [TPB = tris(phosphine)borane] and
(24), Fe-N2−
4 required the use of a highly encumbered silylating agent, triisopropylsilyl trifluoromethanesulfonate, at −78 °C to furnish a tractable diazenido product, (5). Characterization data for 5 include a ν(NN) vibration at 1,719 cm−1, a solution magnetic moment of 2.75 μB (C6D6, 23 °C), and a broad room temperature 1H NMR spectrum. Upon cooling to −80 °C in d8-toluene, the 1H NMR signals of 5 resolve into 14 distinguishable resonances between δ = 32 and −10 ppm, suggestive of an unsymmetrical paramagnetic species. An X-ray Diffraction (XRD) study of 5 (Fig. 4) accounts for this asymmetry at low temperature by verifying silylation of the beta N-atom of the N2 ligand and also establishing a dechelated phosphine donor arm. The Fe geometry of 5 in the solid state is thus four-coordinate with a short Fe–Calkyl distance of 2.116(1) Å, an Fe–N(1) distance of 1.713(1) Å, and two Fe–P bonds with an Fe–Pavg of 2.332 Å. The diazenido ligand features an elongated N(1)–N(2) bond length of 1.203(2) Å, consistent with substantial activation of the N≡N triple bond in free N2 (1.098 Å). The diazenido ligand is severely bent at N(2) with a contracted N(1)-N(2)–Si (4) angle of 135.11°. Similar bond angles found in (TPB)Fe(N2SiMe3) (33) [TPB = tris(phosphine)borane] and  (24) of 166.64° and 165.55°, respectively, are far more obtuse. Dechelation of the phosphine donor arm in 5 can be attributed, at least in part, to the necessity of installing such a large silyl substituent.
(24) of 166.64° and 165.55°, respectively, are far more obtuse. Dechelation of the phosphine donor arm in 5 can be attributed, at least in part, to the necessity of installing such a large silyl substituent.
To discern how the Fe–Calkyl interaction changes as a function of Fe redox state, we pursued structurally related carbonyl complexes of the  scaffold. The advantage of the
scaffold. The advantage of the  (n = +1, 0, −1) series of complexes is that a five-coordinate, approximately trigonal bipyramidal geometry is conserved across the three redox states with the strong-field CO ligand remaining bound at the axial position. This contrasts the situation when N2 is the terminally bonded ligand, where probing the Fe–Calkyl interaction across redox states is not possible due to a variety of other geometric and electronic changes, most notably the loss of the N2 ligand that occurs upon oxidation of 4 to 3.
(n = +1, 0, −1) series of complexes is that a five-coordinate, approximately trigonal bipyramidal geometry is conserved across the three redox states with the strong-field CO ligand remaining bound at the axial position. This contrasts the situation when N2 is the terminally bonded ligand, where probing the Fe–Calkyl interaction across redox states is not possible due to a variety of other geometric and electronic changes, most notably the loss of the N2 ligand that occurs upon oxidation of 4 to 3.
Preparation of this series (Fig. 3) begins with exposure of 3 to 1 atm of CO to afford the neutral and vacuum-stable carbonyl adduct  (6). Carbonyl 6 features a solid-state IR absorption at 1,865 cm−1 and is obtained in near-quantitative yield as a red-orange powder. Its room-temperature solution magnetic moment is 1.5 μB (C6D6) and it features a nearly axial signal in the frozen glass EPR spectrum (10 K, 2-MeTHF). An XRD study showed the presence of three independent molecules of 6 in the unit cell and confirms coordination of CO to a distorted (τ5,
avg = 0.68) trigonal bipyramidal Fe center (Fig. 4). The presence of slightly nonlinear Calkyl–Fe–Ccarbonyl angles (174.38°–178.13°) and marked asymmetry of the three P–Fe–P angles in the pseudoequatorial plane likely results from the orbitally-degenerate, Jahn–Teller active doublet ground state (SI Appendix, Table S2).
(6). Carbonyl 6 features a solid-state IR absorption at 1,865 cm−1 and is obtained in near-quantitative yield as a red-orange powder. Its room-temperature solution magnetic moment is 1.5 μB (C6D6) and it features a nearly axial signal in the frozen glass EPR spectrum (10 K, 2-MeTHF). An XRD study showed the presence of three independent molecules of 6 in the unit cell and confirms coordination of CO to a distorted (τ5,
avg = 0.68) trigonal bipyramidal Fe center (Fig. 4). The presence of slightly nonlinear Calkyl–Fe–Ccarbonyl angles (174.38°–178.13°) and marked asymmetry of the three P–Fe–P angles in the pseudoequatorial plane likely results from the orbitally-degenerate, Jahn–Teller active doublet ground state (SI Appendix, Table S2).
Reversible oxidation and reduction events at −0.62 and −1.85 V, respectively, are observed for 6 (SI Appendix, Fig. S12). Reduction of 6 with KC8 in THF and subsequent encapsulation of the potassium ion with two equivalents of benzo-15-crown-5 furnishes, (7), as a dark purple powder. An IR absorption at 1,782 cm−1 (KBr pellet) is indicative of considerable π-backbonding from the formally Fe(0) center into the C-O π* manifold, but to a lesser extent than in the isoelectronic complex {Na(12-crown-4)2} in which isopropyl instead of phenyl substituents decorate the phosphines (34). Compound 7 is diamagnetic in solution and gives rise to a single resonance in the 31P NMR spectrum. Inspection of the XRD structure of 7 (SI Appendix, Fig. S15) indicates a substantial lengthening of the Fe–Calkyl distance upon reduction (Table 1, Fig. 4) to greater than 2.30 Å. This fact highlights the effect of placing a flexible Fe-C subunit trans to a substrate-binding site.
in which isopropyl instead of phenyl substituents decorate the phosphines (34). Compound 7 is diamagnetic in solution and gives rise to a single resonance in the 31P NMR spectrum. Inspection of the XRD structure of 7 (SI Appendix, Fig. S15) indicates a substantial lengthening of the Fe–Calkyl distance upon reduction (Table 1, Fig. 4) to greater than 2.30 Å. This fact highlights the effect of placing a flexible Fe-C subunit trans to a substrate-binding site.
Table 1.
Selected bond distances (in Å) for  and
and  compounds
compounds
| Compound | d(Fe-Calkyl/Si) | d(Fe-CCO) | d(Fe-Pavg) |
| 8 | 2.138(2) | 1.786(2) | 2.387 |
| 6 | 2.236† | 1.734† | 2.301† |
| 7 | 2.303‡ | 1.738‡ | 2.177‡ |
| 11¶ | 2.3245(7) | 1.842(3) | 2.390 |
| 9¶ | 2.2942(4) | 1.769(2) | 2.276 |
| 10¶ | 2.2586(8) | 1.732(3) | 2.186 |
Average from three molecules in the unit cell.
Average from two molecules in the unit cell.
Ref. 34.
Oxidation of 6 with [Cp2Fe][B(3,5-(CF3)2-C6H3)4] leads to a lightening of the solution and growth of an intense ν(CO) stretch at 1,937 cm−1 arising from the cationic carbonyl complex  {B(3,5-(CF3)2-C6H3)4}, (8). The solution magnetic moment for 8 (2.79 μB, CD2Cl2, 20 °C) is consistent with the expected S = 1 spin state. Attempts to obtain combustion analysis data on 8 were frustrated by its instability to vacuum. An XRD study confirms its structure (SI Appendix, Fig. S15) and reveals a mildly distorted (τ5 = 0.85) trigonal bipyramidal Fe center with an Fe–Calkyl bond distance of 2.138(2) Å that is appreciably shortened by comparison with that in 6 and 7. Cationic 8 displays the shortest Fe–Calkyl bond of all of the five-coordinate compounds detailed herein.
{B(3,5-(CF3)2-C6H3)4}, (8). The solution magnetic moment for 8 (2.79 μB, CD2Cl2, 20 °C) is consistent with the expected S = 1 spin state. Attempts to obtain combustion analysis data on 8 were frustrated by its instability to vacuum. An XRD study confirms its structure (SI Appendix, Fig. S15) and reveals a mildly distorted (τ5 = 0.85) trigonal bipyramidal Fe center with an Fe–Calkyl bond distance of 2.138(2) Å that is appreciably shortened by comparison with that in 6 and 7. Cationic 8 displays the shortest Fe–Calkyl bond of all of the five-coordinate compounds detailed herein.
Mössbauer measurements on compounds 6–8 were undertaken and indicate a decrease in the isomer shift (δ) upon reduction from 8 to 6 to 7 (SI Appendix, Fig. S15). This behavior is consistent with increasing backdonation into unfilled ligand orbitals upon reduction, and thus a higher degree of overall metal-ligand covalency and an increase in the s electron density at Fe (35). The δ range observed for compounds 6–8 (0.13 mm/s) is small and suggests that reduction occurs in diffuse orbitals, minimizing the electronic impact at the Fe nucleus. The trend (or lack thereof) in quadrupole splittings (ΔEQ) is not readily explained.
The substantial reductive elongation of the Fe–Calkyl bond that is observed across the  (n = +1, 0, −1) series from 8 to 6 to 7 differs markedly from the corresponding and isoelectronic series
(n = +1, 0, −1) series from 8 to 6 to 7 differs markedly from the corresponding and isoelectronic series  (n = +1, 0, −1), for which modest shortening of the Fe–Si distances is instead observed upon reduction (34). On reduction, the Fe–Si bond in
(n = +1, 0, −1), for which modest shortening of the Fe–Si distances is instead observed upon reduction (34). On reduction, the Fe–Si bond in  {B(3,5-(CF3)2-C6H3)4} (11) contracts from 2.3245(7) to 2.2942(4) Å in
{B(3,5-(CF3)2-C6H3)4} (11) contracts from 2.3245(7) to 2.2942(4) Å in  (9), to 2.2586(8) Å in
(9), to 2.2586(8) Å in  —Na(THF)3 (10) (Table 1).
—Na(THF)3 (10) (Table 1).
In compounds 6–8, the Fe–Ccarbonyl and Fe–P bonds generally shorten on reduction just as they do in 9–11. In contrast, the Fe–Calkyl bond lengthens (Table 1), and in 7 the central C atom forms three short Si–Calkyl bonds (Table 2). The stability of tri(silyl)methyl carbanions has been attributed to the electropositivity of Si and negative hyperconjugation into adjacent Si-C σ* orbitals (36, 37), resulting in shorter Si–Calkyl bonds. Thus, the structural data suggests the formation of a partially dissociated tri(silyl)methyl carbanion subunit upon reduction.
Table 2.
Selected results from NBO analysis
| Compound | d(Fe-Calkyl), Å | d(Si-Calkyl),† Å | Fe-Calkyl σ-bond occupancy | q(Calkyl)‡ | q(Fe)‡ | Mean q(Si)‡ |
 ¶ ¶
|
| 4 | 2.254(5)§ | 1.845(5)§ | 1.76 (22.7% Fe, 77.3% C) | −1.76 | −1.39 | 1.79 | 102 |
| 7 | 2.305(2)|| | 1.840(3)|| | 1.75 (22.7% Fe, 77.3% C) | −1.76 | −1.78 | 1.78 | 101 |
| 12 | 2.144** | Not relevant | 1.78 (39.7% Fe, 60.3% C) | −0.74 | −1.96 | Not relevant | 191 |
| 13 | 2.251** | 1.866** | 1.76 (31.6% Fe, 68.4% C) | −1.51 | −1.87 | 0.96 | 114 |
Average, central C–Si bonds.
Computed natural charge.
Donor–acceptor interaction energies (in kcal/mol) from the second-order perturbation analysis, nC denotes a lone pair centered on Calkyl,  denotes an unoccupied orbital centered on Fe.
denotes an unoccupied orbital centered on Fe.
Distance obtained from XRD structure.
Distance obtained from one molecule in the asymmetric unit of the XRD structure.
Distance obtained from geometry optimization.
The Fe–Calkyl distances in anions 4 and 7 are remarkably long, especially given that they are diamagnetic species. To aid in explaining this observation, we undertook natural bond orbital (NBO) analyses of these species as a means to compare the localized Fe–C bonding orbitals. NBO has been used previously to assess the differences in M–CH3 and M–CF3 bonding (38). Single-point calculations were performed at the B3LYP/6–31++G** level of theory using the crystallographically determined coordinates of the heavy atoms in 4 and 7. The calculations locate a highly polarized σ-interaction between the Calkyl anchor of the  ligand and the coordinated Fe atom (Fig. 5). The disparate contributions from Fe (22.7% 4, 22.7% 7) and C (77.3% 4, 77.3% 7) suggest a comparatively ionic σ bond (Fig. 5, Table 2) that is atypical of a mid-to-late transition metal alkyl (39). The dative Fe–P σ-bonds display an average of 22.9 and 25.7% Fe character in 4 and 7, respectively. The degree of ionicity in the Fe–Calkyl interaction contrasts that calculated for the Fe–Si bond in the isoelectronic
ligand and the coordinated Fe atom (Fig. 5). The disparate contributions from Fe (22.7% 4, 22.7% 7) and C (77.3% 4, 77.3% 7) suggest a comparatively ionic σ bond (Fig. 5, Table 2) that is atypical of a mid-to-late transition metal alkyl (39). The dative Fe–P σ-bonds display an average of 22.9 and 25.7% Fe character in 4 and 7, respectively. The degree of ionicity in the Fe–Calkyl interaction contrasts that calculated for the Fe–Si bond in the isoelectronic  complex, which displays a far more covalent σ-bond between the Ar3Si subunit and the Fe center (53.3% Fe and 46.7% Si character).
complex, which displays a far more covalent σ-bond between the Ar3Si subunit and the Fe center (53.3% Fe and 46.7% Si character).
Fig. 5.

(A) Isocontour plot of the Fe–Calkyl sigma bond of 7, and (B) the Fe–Si sigma bond of  located from NBO analyses. Arrows indicate the location of highest electron density. Refer to methods for complete calculation details.
located from NBO analyses. Arrows indicate the location of highest electron density. Refer to methods for complete calculation details.
To better understand the unusually long bond lengths found in 4 and 7, we turned to computations on simple alkyl- and tri(silyl)alkyl-ligated five-coordinate Fe complexes. The structure of the S = 0, trigonal bipyramidal Fe alkyl complex [(Ph3P)2N][(CO)4Fe-(n-propyl)] has been previously determined, and it features an n-propyl ligand in an axial site with a long Fe–Calkyl distance of 2.20(2) Å. As such, it was of interest to us to compare the Fe–Calkyl bonding of complexes of this framework to that in 4 and 7. To this end, NBO analyses were performed on the hypothetical model complexes 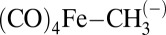 , (12), and
, (12), and  , (13). The optimized geometries of 12 and 13 also display axial coordination of the alkyl substituent to Fe. Notable structural parameters and NBO-derived data for 12 and 13 are tabulated in Table 2. Collectively, the data suggest that the introduction of electropositive Si substituents on carbon in 13 markedly increases the Fe–Calkyl bond length relative to 12. The uncorrected energy of the Fe–Calkyl interaction in 13 is substantially less than that of 12 (38). In addition, the Fe–Calkyl σ-bond displays a decrease in covalency upon introduction of the electropositive Si substituents. As reflected in the natural charges (Table 2) and seen in the electrostatic potential maps of 12 and 13 (Fig. 6), the polarizable Si substituents in 13 display a comparatively positive charge, leading to a compensatory increase in the natural negative charge on the Calkyl carbon relative to 12. As the Fe sites also display negative natural charges, an enhanced electrostatic repulsion between Fe and Calkyl may explain the long Fe–Calkyl distances in 13, as well as 4 and 7, relative to 12. Finally, an augmented electrostatic attraction between the electropositive Si atoms and the highly charged central carbon explains the short Si–Calkyl bonds in 13, 4, and 7 [the Si–C bond length in Me4Si is 1.875(2) Å] (40, 41).
, (13). The optimized geometries of 12 and 13 also display axial coordination of the alkyl substituent to Fe. Notable structural parameters and NBO-derived data for 12 and 13 are tabulated in Table 2. Collectively, the data suggest that the introduction of electropositive Si substituents on carbon in 13 markedly increases the Fe–Calkyl bond length relative to 12. The uncorrected energy of the Fe–Calkyl interaction in 13 is substantially less than that of 12 (38). In addition, the Fe–Calkyl σ-bond displays a decrease in covalency upon introduction of the electropositive Si substituents. As reflected in the natural charges (Table 2) and seen in the electrostatic potential maps of 12 and 13 (Fig. 6), the polarizable Si substituents in 13 display a comparatively positive charge, leading to a compensatory increase in the natural negative charge on the Calkyl carbon relative to 12. As the Fe sites also display negative natural charges, an enhanced electrostatic repulsion between Fe and Calkyl may explain the long Fe–Calkyl distances in 13, as well as 4 and 7, relative to 12. Finally, an augmented electrostatic attraction between the electropositive Si atoms and the highly charged central carbon explains the short Si–Calkyl bonds in 13, 4, and 7 [the Si–C bond length in Me4Si is 1.875(2) Å] (40, 41).
Fig. 6.

(A) Model compounds 12 and 13 used as in silico models of an Fe(0) site interacting with an alkyl ligand. Partial charges on the C(SiH3)3 ligand of 13 are illustrated to emphasize the enhanced negative charge found on C as a result of three electropositive Si substituents. (B) Electrostatic potential maps of 12 (Left) and 13 (Right). Blue and red colors represent areas of negative and more positive potentials, respectively.
With the above structural, spectroscopic, and computational data in hand, it is of interest to return to the issue of the interstitial C atom of the cofactor and to consider whether the data presented herein can be of use as a model. In the resting state of FeMoco, the six belt Fe–C bonds are all relatively short [d(Fe-C) ∼2.0 Å] (5), imparting a pseudotetrahedral geometry to each Fe center. Such a geometry is observed for complex 3, albeit with an appreciably longer Fe–C distance [2.153(2) Å]. Our hypothesis, as suggested previously and depicted in Fig. 1 (11, 42) is that under electron-loading conditions the interaction between a belt Fe center and the interstitial carbon atom is weakened, allowing the Fe center to slide into a position closer to, or within, the plane of the three adjacent sulfide ligands (which may be protonated) concomitant with or before substrate coordination. This geometric change would serve to favor a terminal Fe–N2 π-backbonding interaction, as observed for the transformation of 3 to 4.
Further reduction of the substrate-bound cluster (and possibly substrate protonation) could additionally weaken and thereby elongate the Fe–Cinterstitial interaction. Such elongation is modeled by the carbonyl complexes 6 and 7, where additional Fe–Calkyl lengthening occurs on reduction. Upon reduction, the buildup of negative charge on Calkyl is compensated by shorter Si–Calkyl bonds observed in 4, 7, and hypothetical 13 (Table 2). As the Pauling electronegativities of Fe (χFe = 1.83) and Si (χSi =1.90) are quite similar (43), the behavior of the Si atoms in  may crudely model that proposed for the remaining five Fe atoms within FeMoco. These electropositive Fe atoms may stabilize an interstitial carbide bearing an increasing degree of negative charge as it moves away from the substrate-bound Fe site, while maintaining overall structural integrity of the cofactor. This hypothesis finds some theoretical support in computational studies performed by Dance (44), Nørskov and coworker (10), and Noodleman and coworkers (45) on the FeMoco. Their studies, performed before (10, 45) and after (44) the assignment of the light interstitial atom (X) as C, suggest that substrate coordination to Fe at a site trans to X is favorable, and induces an elongation of the Fe–X interaction. Likewise, these studies suggest that an increase in charge density at X, caused by the singular elongated Fe–X interaction, is compensated by shorter Fe–X bonds to the five additional belt Fe atoms in what has been described as coordinative allosterism (44). It is interesting to note that George et al. (16) have recently communicated Nuclear Resonance Vibrational Spectroscopy (NRVS) and Extended X-ray Absorption Fine Structure (EXAFS) results that suggest elongation of a single Fe–C bond in the FeMoco under catalytic conditions in the presence of propargyl alcohol.
may crudely model that proposed for the remaining five Fe atoms within FeMoco. These electropositive Fe atoms may stabilize an interstitial carbide bearing an increasing degree of negative charge as it moves away from the substrate-bound Fe site, while maintaining overall structural integrity of the cofactor. This hypothesis finds some theoretical support in computational studies performed by Dance (44), Nørskov and coworker (10), and Noodleman and coworkers (45) on the FeMoco. Their studies, performed before (10, 45) and after (44) the assignment of the light interstitial atom (X) as C, suggest that substrate coordination to Fe at a site trans to X is favorable, and induces an elongation of the Fe–X interaction. Likewise, these studies suggest that an increase in charge density at X, caused by the singular elongated Fe–X interaction, is compensated by shorter Fe–X bonds to the five additional belt Fe atoms in what has been described as coordinative allosterism (44). It is interesting to note that George et al. (16) have recently communicated Nuclear Resonance Vibrational Spectroscopy (NRVS) and Extended X-ray Absorption Fine Structure (EXAFS) results that suggest elongation of a single Fe–C bond in the FeMoco under catalytic conditions in the presence of propargyl alcohol.
Conclusions
In conclusion, we have prepared a series of Fe coordination complexes designed to explore a possible role of the interstitial C atom recently assigned for the FeMoco. We have shown that the  ligand can be installed on Fe to afford complexes featuring an unusually flexible Fe–Calkyl interaction owing to a high degree of ionicity in the Fe–Calkyl bond made possible by the three silyl substituents bonded to the C atom. A trigonal pyramidal
ligand can be installed on Fe to afford complexes featuring an unusually flexible Fe–Calkyl interaction owing to a high degree of ionicity in the Fe–Calkyl bond made possible by the three silyl substituents bonded to the C atom. A trigonal pyramidal  complex can be prepared that, upon reduction, exhibits Fe–Calkyl bond lengthening concomitant with N2 binding trans to the C atom. The resulting five-coordinate trigonal bipyramidal anion
complex can be prepared that, upon reduction, exhibits Fe–Calkyl bond lengthening concomitant with N2 binding trans to the C atom. The resulting five-coordinate trigonal bipyramidal anion  thus features an unusually long Fe–Calkyl bond distance and, according to an NBO analysis, an electron pair between Fe and Calkyl that is polarized toward the carbon atom (i.e., carbanion character). Silylation of the coordinated N2 ligand at the beta N-atom furnishes a silyldiazenido product
thus features an unusually long Fe–Calkyl bond distance and, according to an NBO analysis, an electron pair between Fe and Calkyl that is polarized toward the carbon atom (i.e., carbanion character). Silylation of the coordinated N2 ligand at the beta N-atom furnishes a silyldiazenido product  , wherein one phosphine arm has dissociated from Fe in the solid state. To further explore how the Fe–Calkyl interaction responds solely as a function of the formal Fe redox state, the series of geometrically similar trigonal bipyramidal carbonyl complexes
, wherein one phosphine arm has dissociated from Fe in the solid state. To further explore how the Fe–Calkyl interaction responds solely as a function of the formal Fe redox state, the series of geometrically similar trigonal bipyramidal carbonyl complexes  (n = +1, 0, −1) was characterized. Combined Mössbauer, structural, and DFT data collectively suggest a decreasing degree of covalency in the Fe–Calkyl bond and a corresponding increase in the covalency of the Fe–P and Fe–CCO bonds as the formal oxidation state at Fe is decreased from Fe(II) to Fe(I) to Fe(0). The presence of the three electropositive and polarizable silyl substituents allows the Calkyl carbanion to partially dissociate from the Fe center as electrons are added to the complex. The collection of data described has been considered as an inorganic model to explore the hypothesis that a belt Fe–Cinterstitial bond in FeMoco might be modulated as a means of facilitating N2 binding and reduction at a single Fe site.
(n = +1, 0, −1) was characterized. Combined Mössbauer, structural, and DFT data collectively suggest a decreasing degree of covalency in the Fe–Calkyl bond and a corresponding increase in the covalency of the Fe–P and Fe–CCO bonds as the formal oxidation state at Fe is decreased from Fe(II) to Fe(I) to Fe(0). The presence of the three electropositive and polarizable silyl substituents allows the Calkyl carbanion to partially dissociate from the Fe center as electrons are added to the complex. The collection of data described has been considered as an inorganic model to explore the hypothesis that a belt Fe–Cinterstitial bond in FeMoco might be modulated as a means of facilitating N2 binding and reduction at a single Fe site.
Materials and Methods
General Considerations.
All manipulations were carried out using standard Schlenk or glovebox techniques under an N2 atmosphere. Unless otherwise noted, solvents were deoxygenated and dried by thoroughly sparging with Ar gas followed by passage through an activated alumina column in the solvent purification system by SG Water. Nonhalogenated solvents were tested with a standard purple solution of sodium benzophenone ketyl in tetrahydrofuran to confirm effective oxygen and moisture removal. All reagents were purchased from commercial vendors and used without further purification unless otherwise stated.  (1), benzyl potassium, KC8, and [Cp2Fe][B(3,5-(CF3)2-C6H3)4] were synthesized following literature procedures. Elemental analyses were performed by Midwest Microlab. Deuterated solvents were purchased from Cambridge Isotope Laboratories, degassed, and dried over activated 3-Å molecular sieves before use. Deuterated THF was dried over NaK alloy before use. The 1H chemical shifts are reported in parts per million relative to tetramethylsilane, using residual solvent proton resonances as internal standards; 31P chemical shifts are reported in parts per million relative to 85% aqueous H3PO4. Solution phase magnetic measurements were performed by NMR line-shift analysis. IR measurements were obtained on samples prepared as KBr pellets on a Bio-Rad Excalibur FTS 3000 spectrometer. The X-band EPR spectra were obtained on a Bruker EMX spectrometer on 5-mM solutions prepared as frozen glasses in 2-MeTHF. Optical spectroscopy measurements were taken on a Cary 50 UV-Vis spectrophotometer using a 1-cm two-window quartz cell.
(1), benzyl potassium, KC8, and [Cp2Fe][B(3,5-(CF3)2-C6H3)4] were synthesized following literature procedures. Elemental analyses were performed by Midwest Microlab. Deuterated solvents were purchased from Cambridge Isotope Laboratories, degassed, and dried over activated 3-Å molecular sieves before use. Deuterated THF was dried over NaK alloy before use. The 1H chemical shifts are reported in parts per million relative to tetramethylsilane, using residual solvent proton resonances as internal standards; 31P chemical shifts are reported in parts per million relative to 85% aqueous H3PO4. Solution phase magnetic measurements were performed by NMR line-shift analysis. IR measurements were obtained on samples prepared as KBr pellets on a Bio-Rad Excalibur FTS 3000 spectrometer. The X-band EPR spectra were obtained on a Bruker EMX spectrometer on 5-mM solutions prepared as frozen glasses in 2-MeTHF. Optical spectroscopy measurements were taken on a Cary 50 UV-Vis spectrophotometer using a 1-cm two-window quartz cell.
X-Ray Crystallography.
XRD studies were carried out at the Beckman Institute Crystallography Facility on a Brüker Kappa Apex II diffractometer and Brüker Smart 1000 CCD diffractometer (Mo Kα radiation). Structures were solved using SHELXS and refined against F2 on all data by full-matrix least squares with SHELXL. The crystals were mounted on a glass fiber with Paratone N oil.
Electrochemistry.
Electrochemical measurements were carried out in a glovebox under an N2 atmosphere in a one-component cell using a CD instruments 600B electrochemical analyzer. A glassy carbon electrode was used as the working electrode and platinum wire was used as the auxiliary electrode. All reported potentials were referenced to the ferrocene couple Cp2Fe+/Cp2Fe. Solutions (THF) of electrolyte (0.3 M tetra-n-butylammonium hexafluorophosphate) and analyte were also prepared under an inert atmosphere.
Mössbauer Spectroscopy.
Spectra were recorded on a spectrometer from SEE Co. operating in the constant acceleration mode in a transmission geometry. Spectra were recorded with the temperature of the sample maintained at 80 K. The sample was kept in an SVT-400 Dewar from Janis, at zero field. Application of a magnetic field of 54 mT parallel to the γ-beam did not cause detectable changes in the spectra recorded at 80 K. The quoted isomer shifts are relative to the centroid of the spectrum of a metallic foil of α-Fe at room temperature. Samples were prepared by grinding polycrystalline material into a fine powder and then mounted in a cup fitted with a screw cap as a boron nitride pellet. Data analysis was performed using the program WMOSS (www.wmoss.org) and quadrupole doublets were fit to Lorentzian lineshapes.
DFT Calculations.
Single-point calculations and NBO analyses were performed using the Gaussian03 suite of programs with the RB3LYP level of theory and a 6–31++G** basis set for all atoms. The geometries of 12 and 13 were optimized with RB3LYP/6–31++G**. The geometries of 4 and 7 were obtained from the XRD coordinates. To obtain donor–acceptor interaction energies from the second-order perturbation analysis portion of NBO for the Fe–Calkyl sigma bonds, the occupancy threshold was manually adjusted. The total electron density described by these alternative Lewis structures is only slightly lower than that from the default options (percent electron densities for default/alternative Lewis structures: 4, 97.7/94.8; 7, 97.5/94.7; 12, 97.0 /95.9; 13, 97.7/97.1).
Supplementary Material
Acknowledgments
We thank Larry Henling and Charlene Tsay for crystallographic assistance as well as Dr. Angelo di Bilio for assistance with EPR measurements. This work was supported by the National Institutes of Health (GM 070757) and the Gordon and Betty Moore Foundation. J.R. was supported by a National Science Foundation graduate fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom (CSD reference nos. 909104–909108, 941163, and 941164).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1310153110/-/DCSupplemental.
References
- 1.Burgess BK, Lowe DJ. Mechanism of molybdenum nitrogenase. Chem Rev. 1996;96(7):2983–3012. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman BM, Dean DR, Seefeldt LC. Climbing nitrogenase: Toward a mechanism of enzymatic nitrogen fixation. Acc Chem Res. 2009;42(5):609–619. doi: 10.1021/ar8002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J, Rees DC. Structural models for the metal centers in the nitrogenase molybdenum-iron protein. Science. 1992;257(5077):1677–1682. doi: 10.1126/science.1529354. [DOI] [PubMed] [Google Scholar]
- 4.Einsle O, et al. Nitrogenase MoFe-protein at 1.16 A resolution: A central ligand in the FeMo-cofactor. Science. 2002;297(5587):1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 5.Spatzal T, et al. Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science. 2011;334(6058):940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lancaster KM, et al. X-ray emission spectroscopy evidences a central carbon in the nitrogenase iron-molybdenum cofactor. Science. 2011;334(6058):974–977. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lancaster KM, Hu Y, Bergmann U, Ribbe MW, DeBeer S. X-ray spectroscopic observation of an interstitial carbide in NifEN-bound FeMoco precursor. J Am Chem Soc. 2013;135(2):610–612. doi: 10.1021/ja309254g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiig JA, Hu Y, Lee CC, Ribbe MW. Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science. 2012;337(6102):1672–1675. doi: 10.1126/science.1224603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kästner J, Blöchl PE. Ammonia production at the FeMo cofactor of nitrogenase: Results from density functional theory. J Am Chem Soc. 2007;129(10):2998–3006. doi: 10.1021/ja068618h. [DOI] [PubMed] [Google Scholar]
- 10.Hinnemann B, Nørskov JK. Chemical activity of the nitrogenase FeMo cofactor with a central nitrogen ligand: density functional study. J Am Chem Soc. 2004;126(12):3920–3927. doi: 10.1021/ja037792s. [DOI] [PubMed] [Google Scholar]
- 11.Peters JC, Mehn MP. In: Activation of Small Molecules. Tolman WB, editor. Weinheim, Germany: Wiley-VCH; 2006. pp. 81–120. [Google Scholar]
- 12.Crossland JL, Tyler DR. Iron-dinitrogen coordination chemistry: Dinitrogen activation and reactivity. Coord Chem Rev. 2010;254(17-18):1883–1894. [Google Scholar]
- 13.Holland PL. Low-coordinate iron complexes as synthetic models of nitrogenase. Can J Chem. 2005;83(4):296–301. [Google Scholar]
- 14.Lee SC, Holm RH. The clusters of nitrogenase: Synthetic methodology in the construction of weak-field clusters. Chem Rev. 2004;104(2):1135–1158. doi: 10.1021/cr0206216. [DOI] [PubMed] [Google Scholar]
- 15.Takaoka A, Mankad NP, Peters JC. Dinitrogen complexes of sulfur-ligated iron. J Am Chem Soc. 2011;133(22):8440–8443. doi: 10.1021/ja2020907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.George SJ, et al. EXAFS and NRVS reveal a conformational distortion of the FeMo-cofactor in the MoFe nitrogenase propargyl alcohol complex. J Inorg Biochem. 2012;112:85–92. doi: 10.1016/j.jinorgbio.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halder P, Dey A, Paine TK. Molecular and electronic structure of a nonheme iron(II) model complex containing an iron-carbon bond. Inorg Chem. 2009;48(24):11501–11503. doi: 10.1021/ic901841w. [DOI] [PubMed] [Google Scholar]
- 18.Allen OR, et al. Ruthenium complexes of CP3: A new carbon-centered polydentate podand ligand. Organometallics. 2011;30(23):6433–6440. [Google Scholar]
- 19.Ciclosi M, et al. A C3-symmetric palladium catalyst with a phosphorous-based tripodal ligand. Angew Chem Int Ed. 2006;45:6741–6744. doi: 10.1002/anie.200601084. [DOI] [PubMed] [Google Scholar]
- 20.Avent AG, et al. Crowded organometallic compounds of the alkali metals with diphenylphosphino substituents in the organic group. J Chem Soc, Dalton Trans. 2000:2183–2190. [Google Scholar]
- 21.Bordwell FG. Equilibrium acidities in dimethyl sulfoxide solution. Acc Chem Res. 1988;21(12):456–463. [Google Scholar]
- 22.Streitwieser A, Xie L, Wang P, Bachrach SM. Carbon acidity. 77. Ion pair carbon acidities of some silanes in tetrahydrofuran. J Org Chem. 1993;58(7):1778–1784. [Google Scholar]
- 23.Mankad NP, Whited MT, Peters JC. Terminal FeI-N2 and FeII...H-C interactions supported by tris(phosphino)silyl ligands. Angew Chem Int Ed Engl. 2007;46(30):5768–5771. doi: 10.1002/anie.200701188. [DOI] [PubMed] [Google Scholar]
- 24.Lee Y, Mankad NP, Peters JC. Triggering N2 uptake via redox-induced expulsion of coordinated NH3 and N2 silylation at trigonal bipyramidal iron. Nat Chem. 2010;2(7):558–565. doi: 10.1038/nchem.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakajima Y, et al. Electronic structure of four-coordinate iron(I) complex supported by a bis(phosphaethenyl)pyridine ligand. J Am Chem Soc. 2010;132(29):9934–9936. doi: 10.1021/ja102009n. [DOI] [PubMed] [Google Scholar]
- 26.Bart SC, Hawrelak EJ, Schmisseur AK, Lobkovsky E, Chirik PJ. Synthesis, reactivity, and solid state structures of four-coordinate iron(II) and manganese(II) alkyl complexes. Organometallics. 2004;23(2):237–246. [Google Scholar]
- 27.Mealli C, Ghilardi CA, Orlandini A. Structural flexibility and bonding capabilities of the ligand np3 toward the transition metals. Coord Chem Rev. 1992;120:361–387. [Google Scholar]
- 28.Dawsons JW, Lane BC, Mynott RJ, Venanzi LM. Some geometrical consideration on complexes of tripod-like ligands. Inorg Chim Acta. 1971;5:25–29. [Google Scholar]
- 29.Kostka KL, et al. High-valent transition metal chemistry: Mossbauer and EPR studies of high-spin (S = 2) iron(IV) and intermediate-spin (S = 3/2) iron(III) complexes with a macrocyclic tetraamido-N ligand. J Am Chem Soc. 1993;115(15):6746–6757. [Google Scholar]
- 30.Vela J, et al. Quantitative geometric descriptions of the belt iron atoms of the iron-molybdenum cofactor of nitrogenase and synthetic iron(II) model complexes. Inorg Chem. 2007;46(1):60–71. doi: 10.1021/ic0609148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hidai M, et al. Preparation and properties of molybdenum and tungsten dinitrogen complexes. XXI. Trimethylsilylation of coordinated dinitrogen. J Organomet Chem. 1984;272(2):155–167. [Google Scholar]
- 32.Kol M, Schrock RR, Kempe R, Davis WM. Synthesis of molybdenum and tungsten complexes that contain triamidoamine ligands of the type (C6F5NCH2CH2)3N and activation of dinitrogen by molybdenum. J Am Chem Soc. 1994;116(10):4382–4390. [Google Scholar]
- 33.Moret M-E, Peters JC. N2 functionalization at iron metallaboratranes. J Am Chem Soc. 2011;133(45):18118–18121. doi: 10.1021/ja208675p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee Y, Peters JC. Silylation of iron-bound carbon monoxide affords a terminal Fe carbyne. J Am Chem Soc. 2011;133(12):4438–4446. doi: 10.1021/ja109678y. [DOI] [PubMed] [Google Scholar]
- 35.Neese F. Prediction and interpretation of the 57Fe isomer shift in Mössbauer spectra by density functional theory. Inorg Chim Acta. 2002;337:181–192. [Google Scholar]
- 36.Mo Y, Zhang Y, Gao J. A simple electrostatic model for trisilylamine: Theoretical examinations of the n→σ* negative hyperconjugation, pπ→dπ bonding, and stereoelectronic interaction. J Am Chem Soc. 1999;121(24):5737–5742. [Google Scholar]
- 37.Brinkman EA, Berger S, Brauman JI. Alpha-silyl-substituent stabilization of carbanions and silyl anions. J Am Chem Soc. 1994;116(18):8304–8310. [Google Scholar]
- 38.Algarra AG, Grushin VV, Macgregor SA. Natural bond orbital analysis of the electronic structure of [LnM(CH3)] and [LnM(CF3)] complexes. Organometallics. 2012;31(4):1467–1476. [Google Scholar]
- 39.Weinhold F, Landis C. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective. Cambridge: Cambridge Univ Press; 2005. pp. 387–399. [Google Scholar]
- 40.Almansour AI, Eaborn C, Hawkes SA, Hitchcock PB, Smith JD. An alkyllithium compound with a free planar carbanion. The crystal structure of. Organometallics. 1997;16(26):6035–6037. [Google Scholar]
- 41.Beagley B, Monaghan JJ, Hewitt TG. Electron-diffraction studies of tetramethylsilane and hexamethyldisilane, and discussion of the lengths of the Si-C bonds. J Mol Struct. 1971;8(4):401–411. [Google Scholar]
-
42.MacBeth CE, Harkins SB, Peters JC. Synthesis and characterization of cationic iron complexes supported by the neutral ligands
 ,
,  , and
, and  . Can J Chem. 2005;83(4):332–340. [Google Scholar]
. Can J Chem. 2005;83(4):332–340. [Google Scholar] - 43.Allred AL. Electronegativity values from thermochemical data. J Inorg Nucl Chem. 1961;17(3-4):215–221. [Google Scholar]
- 44.Dance I. Ramifications of C-centering rather than N-centering of the active site FeMo-co of the enzyme nitrogenase. Dalton Trans. 2012;41(16):4859–4865. doi: 10.1039/c2dt00049k. [DOI] [PubMed] [Google Scholar]
- 45.Pelmenschikov V, Case DA, Noodleman L. Ligand-bound S = 1/2 FeMo-cofactor of nitrogenase: hyperfine interaction analysis and implication for the central ligand X identity. Inorg Chem. 2008;47(14):6162–6172. doi: 10.1021/ic7022743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.