

Significance
Tumor-associated macrophages (TAMs) are cells of our innate immune system that have been associated with poor prognosis in many types of cancers. When polarized toward the anti-inflammatory state, TAMs promote immune evasion and angiogenesis, thereby driving tumor growth. Using a peptide library selection strategy, we identified a sequence, called M2pep, that preferentially binds to anti-inflammatory murine macrophages. We then used M2pep to carry a proapoptotic peptide to TAMs by i.v. delivery and demonstrated that selective reduction of TAMs resulted in improved survival in tumor-bearing mice. These results suggest that a molecular-targeting approach for TAM depletion is a promising adjunct strategy to add to the arsenal of anticancer therapies.
Keywords: peptide phage display, immunomodulation, tumor targeting, macrophage polarization, flow cytometry
Abstract
Most current cancer therapies focus on killing malignant cells, but these cells are often genetically unstable and can become resistant to chemotherapy. Tumor-associated macrophages (TAMs) facilitate disease progression by promoting angiogenesis and tumor cell growth, as well as by suppressing the adaptive immune response. TAMs are therefore potential targets for adjuvant anticancer therapies. However, resident macrophages are critical to host defense, and preferential ablation of TAMs remains challenging. Macrophage activation is broadly categorized as classically activated, or M1, and alternatively activated, or M2, and TAMs in the tumor microenvironment have been shown to adopt the anti-inflammatory, M2-like phenotype. To date, there are no methods for specific molecular targeting of TAMs. In this work, we report the discovery of a unique peptide sequence, M2pep, identified using a subtractive phage biopanning strategy against whole cells. The peptide preferentially binds to murine M2 cells, including TAMs, with low affinity for other leukocytes. Confocal imaging demonstrates the accumulation of M2pep in TAMs in vivo after tail vein injection. Finally, tail vein injection of an M2pep fusion peptide with a proapoptotic peptide delays mortality and selectively reduces the M2-like TAM population. This work therefore describes a molecularly targeted construct for murine TAMs and provides proof of concept of this approach as an anticancer treatment. In addition, M2pep is a useful tool for murine M2 macrophage identification and for modulating M2 macrophages in other murine models of disease involving M2 cells.
Macrophages are phagocytic cells of our immune system that are found in almost all tissues. Originating from progenitor cells in bone marrow, circulating blood monocytes extravasate into tissues, where they differentiate into macrophages and can subsequently be shifted to diverse functional phenotypes by local environmental cues (1). These polarization states are broadly categorized as classically activated M1 macrophages or alternatively activated M2 macrophages. The M1 macrophage phenotype is induced by mediators, such as IFN-γ or LPS, resulting in a proinflammatory and microbicidal functional phenotype (2). In contrast, M2 macrophages are activated by IL-4 and IL-13, and they possess functional roles in resolution of inflammation and tissue remodeling (3).
There are several chronic pathological findings associated with increased tissue levels of M2 macrophages, including cancer, late-stage atherosclerosis, fibrosis, and asthma (4). In cancer, populations of tumor-associated macrophages (TAMs) mediate immunosuppression and possess M2-like qualities, such as poor antigen presentation, promotion of angiogenesis, and tissue remodeling and repair, although there is heterogeneity in pathways and phenotypes in different tumors (5). TAMs also secrete the factors milk fat globule-EGF factor 8 and IL-6 that lead to tumorigenicity and drug resistance of cancer stem/initiating cells (6). The extent of TAM accumulation within tumors generally correlates with poor disease prognosis (7, 8). Indeed, the role of M2 macrophages in chronic inflammation and disease is increasingly appreciated, and the ability to modulate specific subsets of macrophage populations is a major focus of macrophage-targeted therapy (4). However, there is a dearth of available targeting entities that are specific for M2 macrophages.
Several ligands have been used to target macrophage populations. The small molecules mannose and folate, which are ligands for the mannose receptor and folate receptor β, respectively, have been conjugated to drugs or drug carriers for macrophage delivery (9–11). However, receptors for both molecules are highly expressed in other cell types. Mannose receptor is a pathogen recognition receptor that is also used by dendritic cells (12). In addition to activated macrophages, folate binds to receptors on normal epithelial cells and tumor cells (13). Segers et al. (14) reported a peptide that binds the scavenger receptor-A on macrophages found within atherosclerotic plaques, but scavenger receptor-A is also expressed on all dendritic cells.
Our goal is to develop an M2-targeted construct that can be used to ablate TAMs selectively but not other leukocytes. To achieve this goal, an M2-selective ligand was first identified by a library selection approach. Peptide phage library screening is a common method of identifying novel targeting ligands, and it has been used to select peptides for targeting tumor vasculature (15) and colon cancer (16) among several others (17). We therefore designed a subtractive phage panning strategy that uses bone marrow-derived, in vitro-activated macrophage subpopulations to increase binding specificity of a selected phage to the M2 macrophage subpopulation. We report here the discovery and evaluation of a peptide, called M2pep, which shows selective binding and internalization in M2 macrophages but with minimal M1 macrophage interaction. We demonstrate this peptide’s ability to identify murine M2-like TAMs within mixed cell populations isolated from mouse colon carcinoma tumors and to accumulate in TAMs after i.v. injection into tumor-bearing mice. We show that i.v. administration of M2pep fusion peptides with KLAKLAKKLAKLAK (KLA), a proapoptotic peptide, to tumor-bearing mice selectively reduces TAM populations and prolongs survival.
Results
Identification of a Peptide Selective for M2 Macrophages by Subtractive Phage Biopanning.
First, we used a peptide phage screening strategy to identify motifs for targeting M2 macrophages. Although M1 and M2 macrophage phenotypes are observed in diseased tissues, cultured macrophages polarized toward the M1 or M2 phenotype in vitro show a more homogeneous response. Therefore, to generate M1 and M2 cells for biopanning, we polarized murine bone marrow-derived macrophages with IFN-γ and LPS and with IL-4, respectively. Immunophenotyping confirms that two distinct macrophage populations are generated: F4/80+CCR2+CCR9+ M1 cells and F4/80+CD301+CD11c+ M2 cells (Fig. S1). These markers have been previously reported for their respective subsets (18).
M2 macrophage-binding peptides were identified using three successive rounds of subtractive phage panning to select for phage that bind preferentially to M2 cells over M1 cells. The titer of phage eluted from M2 cells increased in each successive selection round (Fig. S2A). Sequencing of 10 random clones from the third round reveals two unique sequences: YEQDPWGVKWWY (M2pep), appearing in 8 of 10 clones, and HLSWLPDVVYAW, appearing in 2 of 10 clones. Selectivity of the phage clone displaying M2pep, hereafter called “M2pep Phage,” for M2 macrophages was confirmed by titering (Fig. 1A) and whole-cell ELISA (Fig. S2B). By both assays, M2pep Phage demonstrates higher affinity for M2 compared with M1 macrophages and dendritic cells (P < 0.05).
Fig. 1.
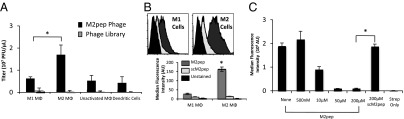
M2pep phage-displayed peptide and biotinylated peptide demonstrate specificity for M2 macrophages. (A) Cells were incubated with M2pep Phage, and bound phage was eluted and titered. (B) M2pep or scM2pep (200 μM) was incubated with M1 and M2 macrophages and analyzed by flow cytometry. AU, arbitrary units. (C) Bound M2pep Phage was competed with M2pep or scM2pep peptide. The remaining bound phage was quantified by flow cytometry. Binding is expressed as median fluorescence intensity (n = 3 for all experiments). *P < 0.05.
M2pep and a control peptide with a scrambled M2pep sequence (scM2pep) were synthesized with a Ser-Gly3-Lys3-Biotin C terminal tag and tested for selectivity of M2 macrophage binding by flow cytometry (Fig. 1B). M2pep exhibits significantly higher binding to M2 macrophages over M1 macrophages (P = 0.001). Furthermore, scM2pep exhibits negligible binding to both cell types, demonstrating the sequence selectivity of M2pep for M2 macrophages. M2pep exhibits 10.8-fold higher binding to M2 macrophages over scM2pep, as well as 5.7-fold higher binding to M2 over M1 macrophages. The equilibrium dissociation constant (Kd) of the M2pep with an oligoethyleneglycol-biotin tag for M2 macrophage was determined from a binding saturation curve to be ∼90 μM (Fig. S3). We were not able determine the Kd of the untagged M2pep by competitive binding experiments due to the solubility limits of this peptide.
To demonstrate sequence-specific binding of M2pep further, we performed competitive binding experiments for M2pep Phage with M2pep and scM2pep. Competition with M2pep results in concentration-dependent inhibition of M2pep phage binding with ∼50% reduction in binding obtained with 10 μM peptide (∼105 M excess over phage-displayed peptide) (Fig. 1C). Furthermore, competition with 200 μM scM2pep exhibits no knockdown of M2pep Phage binding (P = 0.887). Together, the direct and competitive binding studies demonstrate that M2pep exhibits both selective and specific binding to M2 macrophages.
M2pep Binds M2 Macrophages Within Mixed Populations of Cells.
To test binding of M2pep to mixed populations of primary cells, mice were administered the sterile irritant thioglycollate by i.p. injection and peritoneal cells were harvested after 4 d for flow cytometric analysis. CD45+ leukocytes (Fig. 2 A, i) were analyzed for expression of F4/80, CD11c (Fig. 2 A, ii) and CD301 (Fig. 2 A, iii), as well as binding to M2pep. M2pep exhibits higher binding to CD45+F4/80+ cells that were CD11c+ (gate 1, P = 0.00001) or CD301+ (gate 3, P = 0.002). M2pep exhibits 3.5-fold higher binding to CD45+F4/80+CD11c+ M2 cells over CD45+F4/80+CD11c− cells, as well as 9.1-fold higher binding to CD45+F4/80+CD301+ M2 cells over CD45+F4/80+CD301− cells (Fig. 2B).
Fig. 2.

M2pep (200 μM) specifically binds M2 macrophages in mixed populations of cells. (A) Among CD45+ cells isolated from the peritoneal cavity of thioglycollate-treated mice (i), M2pep binds preferentially to F4/80+CD11c+ cells (ii) or F4/80+CD301+ cells (iii). Max, maximum. Histograms are representative of three biological replicates. (B) Median fluorescence intensity of the peritoneal cell populations depicted in A. M2pep binding to CD11c+ and CD301+ cells relative to their negatively stained counterparts is shown (gate 1, P = 0.00001; gate 3, P = 0.002). (C) No specific binding of M2pep is observed to splenic B cells, T cells, or neutrophils (Neut), whereas bone marrow-derived M2 macrophages show selective binding (n = 3 for all experiments). *P < 0.01.
Binding of M2pep to mixed populations of cells isolated from the spleen and bone marrow was also tested. M2pep demonstrates higher binding to bone marrow-derived M2 macrophages relative to splenic B cells (P = 0.004) and T cells (P = 0.002), as well as bone marrow-derived neutrophils (P = 0.003) (Fig. 2C). B cells, T cells, and neutrophils also exhibit high nonspecific binding of scM2pep, whereas M2 macrophages do not.
M2pep Mediates Intracellular Delivery of Cargo.
Initial binding studies by flow cytometry indicate that M2pep binds selectively to M2 macrophages relative to both M1 macrophages and CT-26 cancer cells (P = 0.005) (Fig. S4A). To evaluate the ability of M2pep to mediate targeted intracellular delivery to M2 cells, Alexa Fluor 660-labeled M2pep and scM2pep were incubated at 37 °C with in vitro-derived M1 and M2 macrophages and CT-26 colon carcinoma cells (Fig. S4B). Confocal imaging demonstrates positive staining of M1 and M2 cells, but not CT-26 cells, for F4/80, a macrophage marker (red, Fig. S4B). M2pep (green, Fig. S4B) is efficiently internalized in M2 macrophages relative to M1 macrophages (P = 0.0002) or CT-26 cells (P = 0.000003). scM2pep is not internalized in any of the tested cell types. The mean fluorescence intensity of M2pep binding to M2 macrophages is 3.8-fold higher (P = 0.0002) and 5.3-fold higher (P = 0.000003) than that to M1 macrophages and CT-26 cells, respectively (Fig. S4C). We further demonstrate that M2pep facilitates the intracellular delivery of a model biological drug, streptavidin (Fig. S4D). These data thus illustrate the potential usefulness of M2pep as an M2-selective agent for intracellular delivery of cargo.
M2pep Binds CD11b+Ly6G-F4/80high TAMs Extracted from CT-26 Tumor-Bearing Mice.
To test whether M2pep binds TAMs, biotinylated M2pep was incubated with dissociated cells harvested from CT-26 syngeneic tumors in BALB/c mice (Fig. 3A). M2pep exhibits high binding to CD11b+Ly6G−F4/80high M2-like TAMs compared with CD11b+Ly6G−F4/80int/lo M1-like macrophage (P = 0.01), per markers defined previously by Spence et al. (19). Binding of M2pep to CD11b+Ly6G−F4/80high M2-like TAMs is also significant relative to CD11b+Ly6G+ neutrophils (P = 0.003) and CD11b−FSClow cells (P = 0.007) (Fig. 3 B and D). However, M2pep also exhibits binding to CD11b−FSChigh cells, which are likely the CT-26 tumor cells due to their high frequency in the overall population (∼70%).
Fig. 3.

M2pep binds TAMs harvested from tumor-bearing mice. (A) CT-26 tumors from BALB/c mice were dissociated and stained for flow cytometry analysis using 200 μM M2pep. Dot plots and histograms are representative of three biological replicates. (B) M2pep exhibits higher binding ex vivo to CD11b+Ly6G−F4/80high TAMs compared with other CD11b+ cells, but it also binds CD11b−FSChigh cells. (C) Gated cell populations displayed on a CD11b vs. F4/80 dot plot. (D) Median fluorescence intensity of populations depicted in B (n = 3). ns, not significant. *P < 0.05.
M2pep Binds F4/80+ Cells, but Not Tumor Cells, in CT-26 Tumors in Vivo.
To test M2pep homing to TAMs in vivo, Alexa Fluor 660-labeled M2pep and scM2pep were injected into the tail vein of syngeneic tumor-bearing BALB/c mice. Tissues harvested 30 min postinjection were imaged using a Xenogen imager. Tumors were cryosectioned, stained with an anti-F4/80 antibody and DAPI, and imaged. Confocal images from mice treated with M2pep show high fluorescence within the TAMs, but low signal in other cells, and tumors from mice treated with scM2pep and PBS controls have low fluorescence in all cells (Fig. 4A). The mean fluorescence intensity per F4/80+ cell is 4.1-fold (P = 0.002) and 3.4-fold (P = 0.003) higher, respectively, in tumors from M2pep-injected mice compared with scM2pep- or PBS-injected mice (Fig. S5A). Additional images from M2pep-injected mice confirm the uptake into F4/80+ cells (Fig. S5B). These results demonstrate that M2pep does not bind tumor cells in vivo.
Fig. 4.

M2pep binds F4/80+ cells in vivo. BALB/c mice bearing 2-wk CT-26 tumors were injected via the tail vein with 50 μg of Alexa Fluor 660-labeled M2pep, scM2pep, or PBS injection buffer. At 30 min, live anesthetized mice were perfused with PBS, and tumors and organs were harvested. (A) Tumors were cryosectioned, stained, and imaged on a confocal microscope using the same settings (blue, DAPI; red, F4/80; green, peptide). Images are representative of three biological replicates. (B) Xenogen images of organs excised from three mice injected with Alexa Fluor 660-labeled M2pep demonstrate uptake in tumors. sr, steradian. (C) Trend toward greater uptake of M2pep by tumors is observed compared with the heart, lung, and spleen, but the trend does not reach statistical significance.
Xenogen images of tissues demonstrate greater accumulation of M2pep in tumor tissue compared with heart, lung, and spleen tissue in three mice injected with peptide (Fig. 4 B and C). The liver and kidneys show high fluorescence, likely due to clearance of peptide and fluorophore from these organs. Sulfonated cyanine dyes like the Alexa Fluors have been shown to accumulate in and be cleared from the liver (20). We therefore imaged liver sections of treated mice. Diffuse fluorescence was seen throughout the liver in M2pep and scM2pep-injected mice, with less fluorescence in PBS-injected mice, suggesting that liver fluorescence is due to dye clearance and not to Kupffer cell accumulation of peptide (Fig. S5C). Spleen confocal images show no peptide accumulation in any cell type, including F4/80+ cells (Fig. S5D). Thus, M2pep accumulates in TAM populations but not in resting tissue macrophages.
M2pepKLA Mediates Delayed Mortality in Tumor-Bearing Mice.
The KLA peptide D(KLAKLAK)2 is a proapoptotic motif first used by Pasqualini and coworkers (21) to initiate apoptosis in mammalian cells via disruption of the mitochondrial membrane. KLA is not plasma membrane-permeable and requires facilitated intracellular delivery for optimal activity. KLA fusion peptides with a targeting sequence have been used previously to kill tumor cells selectively (21, 22). Because TAMs facilitate immune evasion of cancer, many have hypothesized that their selective ablation can assist anticancer therapy (23–25). We synthesized M2pepKLA and scM2pepKLA fusion peptides to test if these constructs can mediate selective ablation of TAMs and delay tumor growth in mice.
Tumor-bearing mice received four injections of peptides (2.5 nmol of peptide per gram of body weight per dose of M2pepKLA, scM2pepKLA, or M2pep) every other day. Multiple doses of peptide were administered due to the rapid elimination of peptides with a molecular mass <5 kDa (26). Mouse tumor size was measured every 2–3 d by caliper measurements, and mouse weight was recorded. Mice were killed when ulceration occurred or tumor volume reached 10% of an animal’s body weight. The untreated mouse groups tended to have the greatest number of tumors that ulcered: injection buffer (6 of 8 mice tumors ulcered), M2pep (3 of 10 mice tumors ulcered), scM2pepKLA (5 of 8 mice tumors ulcered), and M2pepKLA (1 of 10 mice tumors ulcered). The fast-growing nature of the untreated groups in this immunocompetent animal model may have contributed to the increased ulceration rate. Due to the high rates of ulceration in the injection buffer and scM2pepKLA groups, we compared tumor growth rates in the M2pep and M2pepKLA groups and found slower tumor growth in the M2pepKLA groups (P < 0.04 on days 9 and 10; Fig. S6A). Furthermore, survival of mice receiving M2pepKLA is significantly longer than that of those receiving other treatments (P = 0.0032) (Fig. 5A). M2pepKLA is well tolerated, with little weight change in the treated mice over the course of the study. The trends in reduced tumor growth rate and prolonged survival from M2pepKLA treatment were reproducible in a second independent study that used three doses spread over 7 d (Fig. S6 B and C). The improved outcome in tumor-bearing mice receiving M2pepKLA strongly supports the potential use of M2pep-targeted agents as an adjuvant to anticancer therapies.
Fig. 5.
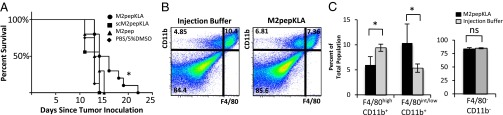
M2pepKLA fusion peptide mediates selective reduction of TAMs and delays mortality. (A) Seven days after tumor inoculation, mice were administered 2.5 nmol/g of M2pepKLA (n = 10), scM2pepKLA (n = 8), M2pep (n = 10), or PBS/5% DMSO (n = 8) four times every other day. Mice receiving M2pepKLA had significantly longer survival compared with other groups (P = 0.0032). (B) Following three injections of M2pepKLA or injection buffer, tumors were harvested and dissociated, and cells were stained for flow cytometry analysis. M2pepKLA administration results in a reduction of CD11b+F4/80high TAMs from 64% to 38% of all CD11b+ cells (P = 0.018). Dot plots are representative of mice (n = 5) from each group. (C) M2pepKLA administration results in a decrease in CD11b+F4/80high cells (P = 0.007), an increase in CD11b+F4/80int/low cells (P = 0.042), and no change in CD11b−F4/80− cells (P = 0.308). *P < 0.05.
M2pepKLA Mediates Selective Elimination of TAMs.
To confirm our hypothesized mechanism of M2pepKLA efficacy, we analyzed tumors for selective elimination of TAMs after M2pepKLA treatment. Tumor-bearing mice received three total injections of 2.5 nmol of peptide per gram or injection buffer every other day. The day following the last injection, mice were killed, their tumors were excised, and cell composition was analyzed by flow cytometry. M2-like, CD11b+F4/80hi TAMs decreased from 64% in the injection buffer mice to 38% in the M2pepKLA mice (P = 0.018) (Fig. 5B). CD11b+F4/80hi cells decreased from 9.40% to 5.97% of the total population (P = 0.007), whereas CD11b+F4/80int/lo cells increased from 5.33% to 10.33% (P = 0.042) and CD11b− cells remained unchanged at ∼84% (P = 0.308) when treated with M2pepKLA (Fig. 5C). The CD11b+F4/80lo population consists primarily of CD11b+Ly6G+ neutrophils. Therefore, the ratio of M2- to M1-like cells was determined as the ratio of CD11b+F4/80hiLy6G− to CD11b+F4/80int/loLy6G−. Compared with injection buffer-treated mice, the M2/M1 ratio is significantly reduced by M2pepKLA treatment (M2/M1 = 1.6 for buffer vs. M2/M1 = 0.6 for M2pepKLA; P = 0.0017; n = 5). This demonstrates the ability of the M2pepKLA fusion peptide to reduce TAMs selectively in vivo.
Discussion
In this study, we develop a unique construct for molecular targeting of murine TAMs and demonstrate the potential of this approach for anticancer therapy. We used a peptide library selection approach because peptide ligands generally offer higher binding affinity and specificity for their receptors compared with small molecules and are typically less immunogenic and cheaper to scale and manufacture compared with antibodies.
The lower binding affinity and faster blood clearance of most peptides compared with antibodies can be improved by peptide engineering approaches, such as cyclization, artificial amino acids, multivalency, and polymer conjugation (26). Although the binding affinity of M2pep is ∼90 μM, similar to other peptides identified by library selection, in vivo targeting and activity of this peptide were observed. We hypothesize that the rapid internalization of M2pep by TAMs (Fig. 4A) enhances accumulation in these target cells. Two examples of successful in vivo cell targeting for peptides identified by phage panning with similar target-binding affinities are DUP-1, which binds to PC-3 prostate cancer cells with micromolar affinity and LyP-1, which binds to MDA-MB-435 cells with an apparent Kd in the hundreds of micromolar range (27, 28). Both of these peptides are also rapidly internalized by their target cell.
Importantly, we provide in vivo evidence that M2pep preferentially binds to and accumulates in murine TAMs compared with other cells (e.g., resting macrophages, tumor epithelial cells, neutrophils, stromal cells) and can be used to deliver a therapeutic payload. Although binding to tumor isolates unexpectedly shows M2pep binding to a CD11b− population that is likely CT-26 tumor cells (Fig. 3D), in vitro binding studies show no binding to or accumulation in CT-26 cells (Fig. S4 A–C). Furthermore, we observe no peptide accumulation in F4/80− tumor cells after i.v. injection (Fig. 4A and Fig. S5B) and no change in the percentage of CD11b− cells after M2pepKLA administration in vivo (Fig. 5C). Taken together, these results suggest that M2pep-targeted constructs do not accumulate in CD11b− cells in vivo.
Associated with a shift to the M2 phenotype, TAMs have been shown to promote tumor cell growth, angiogenesis, metastasis, and immune evasion through the recruitment of regulatory T cells (29, 30). Furthermore, TAMs can contribute to chemotherapy resistance in tumors (31, 32). Work by Zeisberger et al. (25) showed that the combination of anti-VEGF therapy and clodronate-encapsulated liposomes for selective knockout of macrophages resulted in delayed tumor progression in mice. Although the work demonstrated the utility of macrophage depletion in cancer therapy, the researchers noted that the ability to target liposomes to TAMs better could decrease systemic toxic side effects.
In this report, M2pep was used to target and deliver a proapoptotic peptide to murine TAMs in vivo. TAM-targeted delivery of the proapoptotic peptide alone, without an anticancer agent, was sufficient to delay mortality. Although selective killing of TAMs may aid in cancer regression, it is unclear whether macrophage-targeted therapy alone will limit cancer growth. In fact, other work has demonstrated that macrophage depletion alone may not effectively eliminate cancer (25). However, our studies demonstrating a trend toward slower tumor growth in the M2pepKLA treatment group are promising, indicating that targeted TAM depletion in conjunction with other anticancer agents may improve cancer therapy.
Increased macrophage polarization to the M2 phenotype is associated with the progression of a number of other pathological findings, including asthma, allergic inflammation, atherosclerosis, and fibrosis (4, 33). For example, in a mouse model of chronic inflammatory lung disease, Kim et al. (34) demonstrated the role of M2 macrophages in the progression of mucous cell metaplasia. The researchers also reported increased numbers of IL-13–producing, alternatively activated macrophages in human subjects with severe asthma compared with healthy patients. The M2pep sequence identified in this work may be valuable for studying macrophage-targeted treatment strategies for other conditions in which the balance between M1 and M2 macrophages is disturbed. In fact, we identify M2 macrophages in cells directly isolated from mouse atherosclerotic lesions using M2pep and verify the identity of these cells using another M2 marker (Fig. S7). Thus, M2pep-based diagnostics may be developed to identify M2 macrophages in other disease states and offer insight into disease progression. Additionally, M2pep may be conjugated to alternative drugs for macrophage-targeted therapies. Together, these observations suggest a broad spectrum of applications for M2pep.
The biopanning performed in this work identified a unique targeting ligand for murine M2 macrophages, and this strategy can easily be extended to identify similar ligands for alternatively activated human macrophages. Although we fail to detect M2pep binding to human M1 and M2 cells generated by differentiation and activation of human peripheral mononuclear cells, this is not unexpected. Previous work has demonstrated differences between human and mouse macrophage gene expression even though murine and human TAMs share many functional characteristics. For example, markers commonly used to identify murine-activated macrophages, such as arginase I and inducible nitric oxide synthase, do not exhibit similar up-regulation of homologs in humans (33). The data presented here demonstrate a method to identify novel targeting ligands for alternatively activated M2 macrophage phenotypes. In addition, we confirm that a macrophage-binding motif identified using an in vitro selection strategy is relevant for in vivo applications despite the heterogeneous and plastic nature of these cells. Therefore, application of a similar selection strategy to human macrophages has the potential to identify targeting ligands for human TAMs, and thus to fulfill the need for a well-tolerated TAM-targeting approach for anticancer treatment.
Materials and Methods
Generation of Macrophage Cell Types.
All protocols for animal handling were approved by the University of Washington Institutional Animal Care and Use Committee. Bone marrow-derived macrophages were generated as previously described using mouse macrophage colony-stimulating factor (Shenandoah Biotech) (35). Seven-day differentiated macrophages were activated for 48 h in media containing cytokines as follows: M1 cells use 25 ng/mL IFN-γ (R&D Systems) and 100 ng/mL LPS (InvivoGen), and M2 cells use 25 ng/mL IL-4 (R&D Systems).
Subtractive Phage Panning.
The first round of panning was conducted only on M2 macrophages to select for all M2 binders. All phage incubations were done for 1 h at 4 °C, rotating. First, a 2-h receptor-clearing period was performed by removing media from M2 cells and replacing the media with warmed RPMI. Next, cells were lifted and incubated with PhD C7 and PhD12 libraries (2 × 1011 pfu; New England Biolabs) in PBS/1% (wt/vol) BSA. Cells were washed five times using PBS/1% (wt/vol) BSA and then resuspended in glycine⋅HCl on ice for 15 min. The cell solution was neutralized with 20 mM Tris (pH 9.1) and then underwent three freeze–thaw cycles. The cell solution was then titered and amplified according to a New England Biolabs protocol. For subsequent subtractive pans, 2 × 1011 pfu of the amplified eluate was first applied to receptor-cleared M1 cells. M1 cells were centrifuged, and the supernatant was applied to M2 cells. Phage was eluted from M2 cells and amplified. Subtractive panning was repeated three times, using the amplified eluate from the previous pan as the starting library for each round. Plaques from the third round of panning were selected for DNA sequencing.
Peptide Synthesis.
Peptides (M2pep with the sequence YEQDPWGVKWWY and scM2pep with the sequence WEDYQWPVYKGW) with a Lys3Gly3Ser linker and a C-terminal biotin tag were purchased from Elim Biopharmaceuticals at >95% purity. KLA materials were synthesized and purified at >95% purity as follows: M2pepKLA (YEQDPWGVKWWYGGGS-D[KLAKLAK]2), scM2pepKLA (WEDYQWPVYKGWSGGGS-D[KLAKLAK]2), and KLA (D[KLAKLAK]2).
Titering Phage-Binding Study.
A total of 2 × 1011 pfu of M2pep Phage was incubated with 106 cells of resting, M1, M2, and dendritic phenotype for 2 h at 4 °C. Cells were then washed and resuspended in Triton 1X, and then underwent one freeze–thaw cycle. Thawed cells were passed through a 25-gauge needle to facilitate cell lysis and were titered according to the New England Biolabs protocol.
Peptide Flow Cytometry Binding Studies.
Primary murine macrophage cell types were activated as described above, neutrophils were obtained from the bone marrow, and B and T cells were obtained from the spleen. Neutrophils, B cells, and T cells were identified by antibodies against Ly6G, B220, and CD3 antigens, respectively. Cells were also isolated from the peritoneal cavity of a thioglycollate-injected mouse and probed with antibodies against CD45, CD11c, CD301, and F4/80. A total of 105 cells were incubated with 200 μM of M2pep for 30 min at 4 °C, washed three times, and probed with either Streptavidin-FITC or Streptavidin-PEcy7 (eBioscience). Cells were analyzed on a FACSCanto (Becton Dickinson) or MACSQuant (Miltenyi) flow cytometer. Gating was selected based on the fluorescence of unstained cells, or gates were independently chosen by two scientists for differential F4/80 staining.
M2pep and scM2pep Competition Study with M2pep Phage.
M2 macrophages were incubated with 109 pfu of M2pep Phage. Unbound phage was then removed and replaced with M2pep or scM2pep at varying concentrations. The presence of phage was probed using rabbit anti-M13 Phage (Sigma) and goat anti-rabbit–FITC (Sigma) antibodies. Cells were analyzed using a MACSQuant flow cytometer.
Confocal Imaging of M2pep-Alexa Fluor 660-Labeled Cells.
Alexa Fluor 660-labeled M2pep and scM2pep (20 μM) were incubated with M1 and M2 cells as well as with CT-26 cells plated on glass coverslips. Cells were stained with DAPI and an anti-F4/80 antibody (clone BM8) with Alexa Fluor 555 secondary antibody. Coverslips were mounted onto glass slides and imaged using a Zeiss LSM 510 META confocal microscope.
CT-26 Flow Cytometry and Confocal Tumor Studies.
The s.c. tumors were formed by the injection of 106 CT-26 cells into the right flank of BALB/c mice. Tumor-bearing mice were used 2 wk postinoculation at a maximum tumor diameter of 1.5 cm. For flow cytometry analysis, tumors were harvested and single-cell suspensions were recovered using a Tumor Dissociation Kit (Miltenyi). A total of 105 cells of each type were incubated with antibodies (Table S1) and 200 μM of biotinylated peptide. Cells were analyzed with a MACSQuant Flow Cytometer and FlowJo Analysis software (Tree Star). For i.v. injections, 50 μg of Alexa Fluor 660-labeled M2pep, scM2pep, or injection medium (200 μL of saline) was injected in tumor-bearing mice. At 30 min, live anesthetized mice were perfused with PBS and tumors and organs were harvested. Tissues were imaged on a Caliper Xenogen Imager and fixed overnight using 4% (wt/vol) paraformaldehyde. Tissues were cryosectioned, stained using an anti-F4/80 antibody with Alexa Fluor 555 secondary antibody and DAPI, and imaged as described above.
Analysis of Tumor Growth and Survival.
The s.c. tumors were formed by injection of 106 CT-26 cells in RPMI and Matrigel into the right flank of BALB/c mice. After tumors reached ∼50 mm3 in volume, peptides (2.5 nmol/g) or injection buffer [PBS/5% (vol/vol) DMSO] was administered by tail vein injection four times over the course of 1 wk. Tumor volume was determined by caliper measurements [using volume = (a)(b2), where a is the longer of the two tumor dimensions] every 1–2 d, and mouse weight was recorded. Mice were killed when tumor volume exceeded 10% of body weight or ulceration occurred. For TAM elimination analysis by flow cytometry, mice were dosed with M2pepKLA or injection buffer three times every other day. Tumors were collected 1 d after the last injection for flow cytometry analysis using CyTRAK Orange as a nuclear dye to identify nucleated cells.
Statistics.
Differences between groups were examined using the Student’s paired t test, except for survival data, which was examined using the log-rank Mantel–Cox test. Error bars are reported as SDs except where noted.
Supplementary Material
Acknowledgments
We thank Leslie Chan for assistance in confocal imaging, Chayanon Ngambenjawong for assistance in binding studies, Cara Comfort for assistance in bone marrow harvesting, and Anne Manicone and Drew Sellers for helpful discussion. Flow cytometry was conducted, in part, at the University of Washington Cell Analysis Facility, and confocal microscopy was completed at the Keck Microscopy Facility. Tumor growth analysis was conducted with the help of In Vivo Services at the University of Washington. This work was supported by National Institutes of Health Grants R21 HL093385 (to S.H.P.) and P01 HL018645 and R01 HL067267 (to E.W.R.), as well as by a National Science Foundation Graduate Research Fellowship (to M.C.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312197110/-/DCSupplemental.
References
- 1.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 2.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 4.Sica A, Mantovani A. Macrophage plasticity and polarization: In vivo veritas. J Clin Invest. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mantovani A, Sica A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 6.Jinushi M, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci USA. 2011;108(30):12425–12430. doi: 10.1073/pnas.1106645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steidl C, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362(10):875–885. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. 2011;19(4):541–555. doi: 10.1016/j.ccr.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Low PS, Henne WA, Doorneweerd DD. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc Chem Res. 2008;41(1):120–129. doi: 10.1021/ar7000815. [DOI] [PubMed] [Google Scholar]
- 10.Hashida M, Nishikawa M, Yamashita F, Takakura Y. Cell-specific delivery of genes with glycosylated carriers. Adv Drug Deliv Rev. 2001;52(3):187–196. doi: 10.1016/s0169-409x(01)00209-5. [DOI] [PubMed] [Google Scholar]
- 11.Yu SS, et al. Macrophage-specific RNA interference targeting via “click”, mannosylated polymeric micelles. Mol Pharm. 2013;10(3):975–987. doi: 10.1021/mp300434e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products. J Exp Med. 1995;182(2):389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross JF, Chaudhuri PK, Ratnam M. Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic and clinical implications. Cancer. 1994;73(9):2432–2443. doi: 10.1002/1097-0142(19940501)73:9<2432::aid-cncr2820730929>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 14.Segers FME, et al. Design and validation of a specific scavenger receptor class AI binding peptide for targeting the inflammatory atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2012;32(4):971–978. doi: 10.1161/ATVBAHA.111.235358. [DOI] [PubMed] [Google Scholar]
- 15.Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279(5349):377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 16. Hsiung P-L, et al. (2008) Detection of colonic dysplasia in vivo using a targeted heptapeptide and confocal microendoscopy. Nat Med 14(4):454–458, and erratum (2008) 14(5):585. [DOI] [PMC free article] [PubMed]
- 17.Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380(6572):364–366. doi: 10.1038/380364a0. [DOI] [PubMed] [Google Scholar]
- 18.Becker L, et al. Unique proteomic signatures distinguish macrophages and dendritic cells. PLoS ONE. 2012;7(3):e33297. doi: 10.1371/journal.pone.0033297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spence S, et al. Suppressors of cytokine signaling 2 and 3 diametrically control macrophage polarization. Immunity. 2013;38(1):66–78. doi: 10.1016/j.immuni.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 20.Hamann FM, Brehm R, Pauli J, Grabolle M. Controlled modulation of serum protein binding and biodistribution of asymmetric cyanine dyes by variation of the number of sulfonate groups. Mol Imaging. 2011;10(4):258–269. doi: 10.2310/7290.2011.00005. [DOI] [PubMed] [Google Scholar]
- 21.Ellerby HM, et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5(9):1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- 22.Mai JC, Mi Z, Kim SH, Ng B, Robbins PD. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001;61(21):7709–7712. [PubMed] [Google Scholar]
- 23.Luo Y, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116(8):2132–2141. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinha P, Clements VK, Ostrand-Rosenberg S. Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res. 2005;65(24):11743–11751. doi: 10.1158/0008-5472.CAN-05-0045. [DOI] [PubMed] [Google Scholar]
- 25.Zeisberger SM, et al. Clodronate-liposome-mediated depletion of tumour-associated macrophages: A new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95(3):272–281. doi: 10.1038/sj.bjc.6603240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollaro L, Heinis C. Strategies to prolong the plasma residence time of peptide drugs. MedChemCommun. 2010;1:319–324. [Google Scholar]
- 27.Mäkelä AR, Matilainen H, White DJ, Ruoslahti E, Oker-Blom C. Enhanced baculovirus-mediated transduction of human cancer cells by tumor-homing peptides. J Virol. 2006;80(13):6603–6611. doi: 10.1128/JVI.00528-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park J-H, et al. Cooperative nanomaterial system to sensitize, target, and treat tumors. Proc Natl Acad Sci USA. 2010;107(3):981–986. doi: 10.1073/pnas.0909565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 30.Lin EY, Pollard JW. Role of infiltrated leucocytes in tumour growth and spread. Br J Cancer. 2004;90(11):2053–2058. doi: 10.1038/sj.bjc.6601705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welford AF, et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest. 2011;121(5):1969–1973. doi: 10.1172/JCI44562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shree T, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25(23):2465–2479. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: An immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 34.Kim EY, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med. 2008;14(6):633–640. doi: 10.1038/nm1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): Isolation and applications. CSH Protoc. 2008 doi: 10.1101/pdb.prot5080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.