

Abstract
The pentraxins are an ancient family of proteins with a unique architecture found as far back in evolution as the Horseshoe crab. In humans the two members of this family are C-reactive protein and serum amyloid P. Pentraxins are defined by their sequence homology, their pentameric structure and their calcium-dependent binding to their ligands. Pentraxins function as soluble pattern recognition molecules and one of the earliest and most important roles for these proteins is host defense primarily against pathogenic bacteria. They function as opsonins for pathogens through activation of the complement pathway and through binding to Fc gamma receptors. Pentraxins also recognize membrane phospholipids and nuclear components exposed on or released by damaged cells. CRP has a specific interaction with small nuclear ribonucleoproteins whereas SAP is a major recognition molecule for DNA, two nuclear autoantigens. Studies in autoimmune and inflammatory disease models suggest that pentraxins interact with macrophage Fc receptors to regulate the inflammatory response. Because CRP is a strong acute phase reactant it is widely used as a marker of inflammation and infection.
1. Introduction
In this review, I focus on the two major, classical pentraxins: C-reactive protein (CRP) and serum amyloid P component (SAP). The pentraxins are serum proteins with a relatively uncommon pentameric structure. They function as pattern recognition molecules recognizing foreign antigens and altered self-antigens and tag these molecules for activation of the innate immune system. This property is characteristic of innate recognition molecules that preceded the development of the immunoglobulins. Pentraxins also interact with the complement system and Fc receptors to activate immune responses. It is likely that the interaction of pentraxins with the receptors for the Fc region of immunoglobulins preceded the development of immunoglobulins.
2. History
The pentraxins appeared very early in evolution with several different forms present in the horseshoe crab, which has been referred to as a living fossil having persisted 250–300 million years [1]. Despite this long lineage, our understanding of the function of these proteins remained obscure until very recently. The discovery of CRP in man was achieved serendipitously in the blood of a patient with severe Streptococcus pneumoniae pneumonia. The protein appeared in the blood when the patient was systemically ill and was not detectable before the infection or after the infection had been eradicated [2]. These clinical investigators at the Rockefeller University went on to characterize this protein biochemically. The protein was present in very high concentration in acute phase sera and it would induce precipitation of pneumococcal cell wall extracts but only in the presence of calcium.
3. Pentraxin Structure
The molecular mass of CRP and SAP is 115,135 daltons and 127,310 daltons, respectively. Both proteins are composed of five tightly arranged subunits (protomers) in planar symmetry. Using electron microscopy, it was determined that the molecule appeared as a doughnut-shaped ring [3]. Although it was a long held belief that CRP was composed of a single pentamer whereas SAP existed as a decamer, it was later determined that SAP, like CRP, circulates in blood as a single pentamer [4]. The pentameric structure of CRP imparts a high degree of stability to the molecule and resistance to enzymatic attack [5]. SAP shares many structural and biological characteristics with CRP. They are both cyclic pentamers that react with ligands in a calcium-dependent fashion. They share 51% amino acid identity and very similar structures [6]. Unlike CRP, SAP is glycosylated and the carbohydrate moiety has been defined [7]. See Table 1 for a comparison of CRP and SAP.
Table 1.
Comparison of the properties of the pentraxins: C-reactive protein (CRP) and serum amyloid P (SAP).
| C-reactive protein (CRP) | Serum amyloid P (SAP) | |
|---|---|---|
| Fc receptor binding | Yes | Yes |
| Calcium-dependent ligand binding | Yes | Yes |
| Complement activation through C1q | Yes | Yes |
| Ligands | Phosphocholine snRNP (Sm, RNP) Histones Apoptotic cells Oxidized LDL |
Phosphoethanolamine DNA, chromatin Heparin Apoptotic cells Amyloid fibrils |
| Major synthetic site | Liver | Liver |
| Inducers | IL-6 (acute phase reactant) | Constitutive |
| Structure | Cyclic pentamer 115,135 Da Each subunit 23,027 Da 206 amino acids |
Cyclic pentamer 127,310 Da Each subunit 25,462 Da 204 amino acids |
| Glycosylation | No | Yes |
| Chromosomal location | 1q23.2 | 1q23.2 |
The first crystallographic structure of the pentraxin family was solved for SAP, which revealed the five-fold symmetry of the molecule and the calcium-dependent binding site for the 4,6-cyclic pyruvate acetal of β-D galactose and phosphoethanolamine [8]. The structure of CRP was definitively determined when the first crystallographic model was reported [9]. Each pentraxin protomer consists of a conserved β-sandwich fold with two opposing β-sheets. The binding site for phosphocholine (PC) was proposed to be a hydrophobic pocket on one face of the protomer, and similarities and differences between the CRP and SAP binding sites were observed. Shortly thereafter a crystallographic solution of the interaction between CRP and the PC ligand was produced [10]. Figure 1 shows the ligand binding sites on CRP with two calcium ions forming part of the site. On the other face of the protomer was found a three-turn alpha helix, termed the ridge helix, and a deep groove of uncertain function. This face of the CRP pentamer was shown by mutational analysis to contain a single C1q binding site [11, 12] and by mutational analysis and cocrystallization to contain a single Fc receptor binding site [13, 14] (Figure 2).
Figure 1.
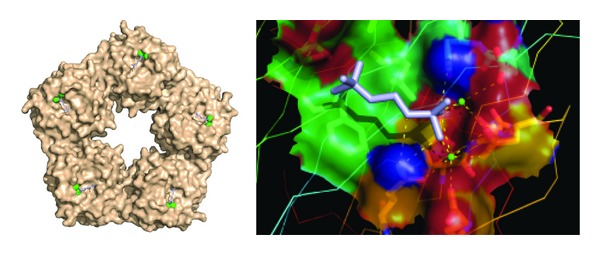
Surface view of the ligand binding (B) face of C-reactive protein. Each protomer contains a binding site, which is shown occupied by 2 calcium (green) and 1 PC molecule (blue). On the magnified view on the right the major interactions with bound calcium ions and specific amino acids can be seen more clearly. The structure is taken from structure file PDB ID: 1B09 from the NCBI.
Figure 2.
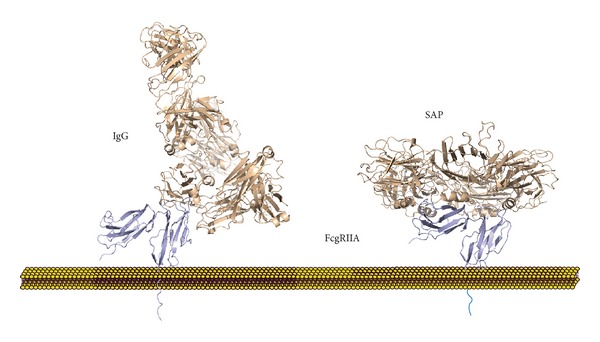
Comparison of the binding of SAP and IgG to the FcγRIIA molecule. The structure of the IgG-FcγRIIA is shown on the left and is based on the NCBI entry 3RY6. The structure of the SAP-FcγRIIA complex taken from the NCBI entry 3D5O is shown on the right [14]. The FcR interaction with SAP engages the ridge helices of two nonadjacent protomers, resulting in a one-to-one stoichiometry.
Another group of related proteins was described more recently and is known as the long pentraxins. The long pentraxins share a strong homology with the pentraxins in the C-terminal region but have a long N-terminal domain that is unrelated to the so-called short pentraxins or other known proteins. These “long pentraxins” are not as well structurally characterized yet and their functions less defined. Unlike the classical pentraxins, the long pentraxins are produced locally in response to inflammatory stimuli like TNF-α. The long pentraxins include guinea pig apexin, neural pentraxin I (NPTXI) and II (NPTXII), and long pentraxin 3 (PTX3). PTX3, the best studied of these, activates complement, binds to FcγRIII [14], protects from some fungal infections [15, 16], and may play a role in wound healing. The long pentraxins have been reviewed recently [17, 18].
4. Ligands Recognized
Much of the early work on CRP biology focused on its interaction with ligands expressed on bacteria and damaged tissue. CRP was initially identified and named for its interaction with the C-polysaccharide, a major component of the cell wall of S. pneumoniae [19]. CRP binding to the C-polysaccharide was shown very early on to occur through PC moieties, which are found on the cell wall teichoic acid and lipoteichoic acid [20]. Also see Figure 1. The binding to PC was calcium dependent. PC is expressed on a variety of pathogenic organisms to which CRP has been shown to bind. PC is also the polar head group of phosphatidylcholine, a component of the mammalian cell membrane. This PC head group of phosphatidylcholine is not exposed on normal healthy cells. However, damage to cell membranes by enzymatic action or complement attack leads to extensive binding of CRP to the damaged membrane [21, 22]. This was first demonstrated in vivo by injecting typhoid vaccine into rabbit muscle and examining CRP deposition [23]. Subsequently, similar results were obtained when coronary artery ligation was used to induce myocardial infarction [24]. Thus CRP can target dead and damaged cells for processing by the innate immune system. CRP also binds to PC exposed on oxidized LDL, which may account for its presence in atherosclerotic lesions [25, 26].
The damaged cell can present and/or release various nuclear antigens that can stimulate the immune system and some of these are the targets of autoantibodies in connective tissue diseases. The most notable of these is systemic lupus erythematosus (SLE) in which patients develop high-titered antibodies to native DNA and ribonucleoprotein complexes [27]. CRP and SAP bind to these nuclear antigens and affect their clearance and antigenic processing. In cells CRP binds primarily to the small nuclear ribonucleoproteins (snRNPs) [28] and SAP binds to chromatin and native DNA [29, 30].
CRP binding to polycations has been reported and characterized [31–34]. CRP binding to polycations differs from binding to prototypic ligands such as PC in that it is inhibited by calcium and not by PC. No physiological role for CRP binding to polycations has been described. However, polyvalent binding to either type of ligand leads to complement activation through C1q [35, 36].
SAP has similar ligand binding sites on the B face of the molecule, but whereas it binds well to PE, it fails to bind to PC due to differences in the hydrophobic pocket [6, 8, 37]. SAP binds to DNA as well although the affinity is much stronger for human SAP than for mouse SAP [38]. This difference may complicate the study of the role of pentraxins in mouse models of SLE. SAP binds to other polyanions, including heparin, to carbohydrates on bacteria including Streptococcus pyogenes and Neisseria meningitidis, and to lipopolysaccharide (LPS) [39, 40]. The SAP ligand in agarose was identified as the 4,6-cyclic pyruvate acetal of β-D-galactose [41]. SAP binds to and is a constituent of all types of amyloid fibrils [42]. This ability to bind to amyloid is the basis of an assay to localize amyloid deposits in patients with amyloidosis [43].
5. Pentraxins and Complement
One of the first breakthroughs in pentraxin biology was the finding that CRP could activate the classical cascade of complement [36, 44]. This finding suggested an important biological function for CRP as the complement system has a broad range of activities in biological defense and regulation of inflammation [45]. CRP activates the classical cascade of complement through direct binding of C1q, the first component of the classical pathway. Each CRP pentamer has a single binding site for C1q and a minimum of two CRP molecules are required for C1 activation, similar to IgG [46]. No crystallographic solution of the CRP-C1q interaction has been produced to date. It was originally reported that CRP interacted with the collagen-like stalk of the A chain of C1q [47, 48]. However, more recently CRP interactions with C1q globular head groups were reported [49]. A molecular model has been presented in which one globular head group of C1q interacts through the central pore of CRP on the A face of the pentamer [50]. This model is based on site-directed mutagenesis studies of the CRP binding site for C1q [11, 12]. SAP either chemically cross-linked or bound to polyvalent ligands also binds C1q and activates the classical complement pathway [51, 52].
Complement activation by CRP is, at first glance, very similar to complement activation by IgM or IgG immune complexes. However, a more detailed comparison reveals that CRP activation does not efficiently proceed to generation of the membrane attack complex, whereas antibody activation does [53]. See Figure 3. CRP activates early steps in the classical pathway, with nearly complete consumption of C1, C4, and C2 and partial consumption of C3, but produces only minimal activation of C5–C9 and no cell lysis. Since C5a and C5b-9 are the strongest inflammatory mediators produced during complement activation, this restricted complement activation is likely to favor opsonization without a strong inflammatory response. Consistent with this hypothesis, CRP was shown to prevent lysis of apoptotic cells by complement, promoting opsonization and increasing anti-inflammatory cytokines [54].
Figure 3.
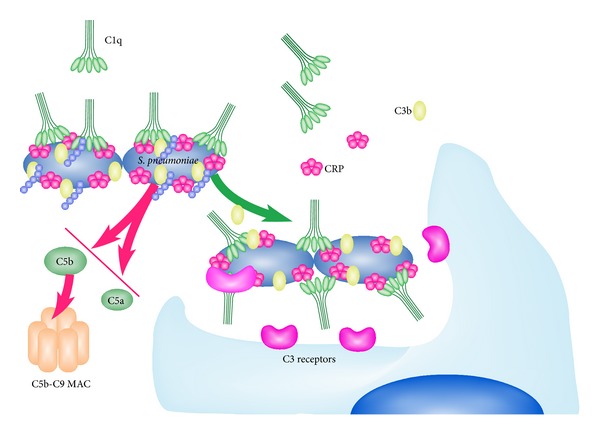
Activation of the complement cascade by CRP complexes. CRP interacts with bacteria that express repeating ligands like PC on the Gram-positive pathogen S. pneumoniae. A single CRP pentamer interacts with one globular head group of a C1q molecule. Interaction of C1q with multiple CRP molecules leads to C1 activation, C4 and C2 cleavage, and the formation of a C3 convertase. The cleavage of C3 in turn forms a C5 convertase. This step is limited by CRP recruitment of the inhibitory protein fH. Thus the cleavage of C5 resulting in C5a generation and formation of the MAC is blocked.
Additional studies established that the characteristic early classical pathway activation by CRP is due to inhibition of the alternative pathway convertase, which provides an essential amplification loop for both the classical and lectin pathways [55, 56]. This feedback loop is especially important for forming the C5 convertase, generating inflammatory mediators, C5a and C5b-9, and contributing to complement-mediated pathology [57]. The inhibitory effect of CRP on alternative pathway activation required the complement regulatory protein, factor H (fH), and CRP was shown to recruit fH to the activating surface [55]. CRP binding to the related regulatory protein, C4b binding protein has also been reported [58].
More recent investigations have identified at least two binding sites on fH for CRP [59–61]. One of these has received particular attention because it includes the polymorphic residue (Y402H) in short consensus repeat 7 of fH that is genetically linked to the risk of developing age-related macular degeneration [62–65]. Several groups have reported that CRP binds with lower affinity to the fH variant (H402) that is associated with the disease [66, 67]. These findings suggest that although elevated serum CRP levels are associated with the chronic inflammatory process in age-related macular degeneration [68], the ability of CRP to bind fH may have a protective role in this disease.
An important role for CRP activation of complement was shown in S. pneumoniae infection models where CRP activation of complement contributed substantially to protection from lethal infection and clearance of bacteria [69–72]. There is also some evidence for complement activation by CRP in acute injury, such as myocardial infarction [73]. Complement activation by CRP was reported to contribute to ischemia-reperfusion injury in a rat model of myocardial infarction although the findings are difficult to interpret because in this model endogenous rat CRP played no role and infusion of human CRP was required to activate rat complement [74].
6. Receptor Binding
Another mechanism by which CRP interacts with the innate immune system is through its interaction with Fcγ receptors (FcγRs). FcγRs are a family of membrane receptors found on myeloid cells, B lymphocytes, NK cells, and platelets. In man, they exist in three major classes with multiple subtypes [75] (Table 2). For many years it was thought that CRP could interact with cells of the immune system by binding to a specific “CRP receptor” [76]. A great deal of effort was spent searching for the specific CRP receptor. It was first proposed that CRP might interact with an Fc receptor or a receptor that was somehow associated with the Fc receptors. It was later concluded by these investigators that CRP did not bind to Fc receptors but to its own specific receptor [76]. However, numerous attempts to clone this receptor by others and by us failed to produce meaningful results. Reexamination of the cells to which CRP bound and inhibition studies suggested to us that FcγR could indeed be the direct receptors for CRP. Final identification of the receptors for CRP on leukocytes was made possible through the cloning and expression of the Fc receptors on transfected cell membranes. Using transfected cell lines we first determined that CRP was capable of binding to cells through the high affinity receptor for IgG, FcγRI [77]. Other laboratories confirmed the interaction of CRP with FcγRI [78, 79]. It was further shown that the interaction of CRP with FcγRI on transfected cells was markedly increased by the cotransfection of the cells with γ-chain [80]. Thus surface plasmon resonance (SPR) studies of CRP interaction with FcγRI in the absence of the γ-chain may underestimate the true affinity for FcγRI.
Table 2.
Overview of pentraxin receptors.
| Receptor | Cells | Other ligands | Functions | |
|---|---|---|---|---|
| FcγRI (CD64) | Monocytes, macrophages, DC, inducible on PMN | Activating | High affinity for IgG | Antibody-dependent cell-mediated cytotoxicity Phagocytosis |
|
| ||||
| FcγRIIA/C (CD32A/C) | Monocytes, macrophages, DC neutrophils, platelets | Activating | IgG | Antibody-dependent cell-mediated cytotoxicity Phagocytosis Platelet activation |
|
| ||||
| FcγRIIB (CD32B) | B lymphocytes, macrophages, DC | Inhibitory | IgG | Regulation of responses through immunoreceptors |
|
| ||||
| FcγRIIIA (CD16A) | Macrophages, some monocytes, NK cells | Activating | IgG | Antibody-dependent cell-mediated cytotoxicity Phagocytosis |
|
| ||||
| FcγRIIIB (CD16B) | PMNs | GPI-linked | IgG | Immune complex binding Activation of FcγRIIA |
|
| ||||
| FcαRI (CD89) | Monocytes, macrophages, DC neutrophils, platelets | Activating/inhibitory | IgA | Phagocytosis Regulation of responses through other receptors |
Although it was shown that CRP did bind to FcγRI, it could not explain binding to cells in which FcγRI is not expressed, for example, K562 cells and platelets. Thus, a second receptor for CRP on leukocytes was sought. Ongoing studies of Fc receptor biology made it clear that the expression of Fc receptors varied among different leukocyte subsets and that different individuals could express different numbers of receptors with differing affinities. We were able to show that a large fraction of the remaining binding was due to CRP interaction with FcγRIIA, a receptor that is responsible for many of the functions induced by immune complexes [81]. The binding of CRP to FcγRIIA was also verified independently [82]. The affinity of this interaction could not be determined quantitative terms from the flow cytometry assays [81], but an equilibrium K D of 3.7 ± 1 μM was determined by confocal analysis of transfected cells [82]. This is in agreement with the K D of 1.9 ± 0.6 μM determined for CRP binding to FcγRIIA by SPR [14].
FcγRIIA is expressed in two forms in humans resulting from a single amino acid polymorphism at position 131, which may be either an arginine (R) or a histidine (H) [83]. This single amino acid difference results in a preferential binding of IgG2 to the H131 form of the receptor [84]. Using population studies it has been determined that this polymorphism is associated with an alteration of risk for a wide variety of human diseases including SLE, infection, myocardial infarction, and malaria [85–88]. Interestingly, CRP binds preferentially to the R131 form of FcγRIIA [14, 89, 90]. The differential binding of CRP to the R form of FcγRIIA results in much stronger responses in PMN and monocytes [89, 91]. Recently it was demonstrated that CRP stimulates neutrophil calcium signaling in an FcγRIIA allele-specific manner [92], which is consistent with previous findings [89].
One way in which investigators have sought to decipher the conflicting data concerning CRP's direct role at the cellular level is through the use of purified CRP, which may be purified from human fluids or recombinant protein production. In our experience and that of others [93] commercial recombinant CRP preparations are often contaminated with LPS and potentially other microbial products. This can lead to effects that are directly or indirectly related to toll-like receptor (TLR) activation. In addition, most of these studies are done with uncomplexed CRP. It is well known that FcγR aggregation by immune complexes is necessary for receptor activation [94]. If that were not the case, the levels of IgG in blood would constantly bind and activate cells. Thus, CRP, like IgG, is unlikely to activate receptors without crosslinking by ligands or aggregation due to storage conditions. Clearly, ligands that contain repeating determinants like PC on pathogenic bacteria would be an optimal platform for activation. Structural and isothermal titration calorimetry studies revealed a one-to-one stoichiometry between SAP or CRP and FcγR [14] (Figure 2). It has also been shown that the degree of receptor crosslinking affects the cytokine profile of the responding cells [95]. This very likely is the reason that some groups fail to demonstrate effects of CRP on cytokine or other responses using purified, uncomplexed CRP [93]. It is worth noting that CRP-mediated activation of complement also requires binding to multivalent ligands [46]. Like IgG in circulation even high concentrations of CRP do not activate cells or complement without a relevant ligand.
The interaction of CRP with cell surface receptors was expanded when SPR studies were performed with other related receptors. CRP did not react with FcRn, neonatal Fc receptor and no interaction was seen between CRP and the IgE receptor, FcεRI. However, it was determined that CRP bound to FcαRI (the IgA receptor also known as CD89) with an affinity that was comparable to its affinity for FcγR. The interaction with CD89 was functional as phagocytosis, and signaling and cytokine production was seen [96]. The in vivo functional consequences of this interaction await further studies.
After identification of FcγRIIA as the main receptor for CRP, signaling through this receptor was confirmed by Chi et al. [97] using HL-60 cells differentiated to a neutrophil type with DMSO. CRP induced tyrosine phosphorylation of human FcγRIIA and Syk, as well as inducing both phosphorylation and membrane localization of phospholipase Cγ2. This signaling pattern is what would be expected for IgG mediated signaling through ITAM bearing FcγR [75].
Most functional studies of CRP activation of FcγR have focused on innate immune cells, monocytes, macrophages, neutrophils, and dendritic cells. These are discussed in more detail below. An interesting example of CRP activation through FcγRIIA and FcγRIIC, which are activating forms of FcγRII, on myeloma cells was reported by Yang et al. [98]. These investigators found that primary myeloma cells and stressed myeloma cell lines bound CRP through FcγRIIA and FcγRIIC. CRP activated Akt, pERK, and NF-κB signaling pathways in these cells, led to increased IL-6 synthesis, and protected the myeloma cells from chemotherapy-induced apoptosis. The results were verified in vivo in myeloma SCID and SCID-human mouse models.
The structural basis of pentraxin-FcγR interaction was established when the crystal structure of SAP bound to FcγRIIA was solved [99] (Figure 2). This structure shows a single FcγR interacting with the ridge helices of two nonadjacent SAP protomers thus fixing the stoichiometry at one-to-one. Pentraxin and IgG binding sites for FcγR are partially overlapping and competitive binding is seen by SPR.
7. CRP and SAP Synthesis
Circulating CRP is synthesized primarily by the liver at very low levels constitutively [100]. The liver-specific transcription factor, hepatic NF-1 binding to its consensus sequence, regulates cytokine-independent CRP synthesis [101]. During the acute phase response, CRP transcription responds primarily to IL-6 and this response is enhanced by IL-1β. IL-6 induction is mediated by the main IL-6 activated transcription factors, STAT3 [102] and C/EBPβ [103], which bind to response elements in the CRP promoter. In clinical trials of a mAb directed against the IL6R there was a dramatic decrease in CRP levels, supporting the important role of IL-6 in acute phase CRP synthesis [104]. CRP and serum amyloid A (SAA) are the two human acute phase proteins that show the greatest dynamic range. CRP baseline serum concentrations average less than 1 μg/mL, but acute phase concentrations, are commonly in the range of 10–500 μg/mL. The increase in CRP levels following an acute phase stimulus is very rapid with blood levels peaking at 48 h [105]. More importantly, CRP levels also decrease rapidly after resolution of the inciting event has occurred. This makes it more useful than the widely measured erythrocyte sedimentation rate (ESR), which remains elevated long after the inflammatory state has resolved.
Human SAP is also produced in the liver at constitutive serum levels that average 33 μg/mL in women and 43 μg/mL in men [106]. Although SAP is not an acute phase protein in man, it is a very strong acute phase marker in the mouse where CRP is expressed at low levels (<5 μg/mL) constitutively [107]. Baseline levels of mouse SAP differ considerably among strains. Mouse SAP is induced by IL-6, similar to CRP in man [107]. Both CRP and SAP activate the complement cascade. Similarly SAP also binds to FcγR and this binding results in opsonization for phagocytosis by human or mouse phagocytes [14, 108].
8. CRP and SAP in Mouse Models
Several approaches have been used to investigate the function of CRP in vivo. Most studies have used mouse models of infection, inflammation, or autoimmune disease. Since mice express low levels of endogenous CRP, human or in some cases rabbit CRP has been used in these experiments. Fortunately both human and rabbit CRPs bind to mouse FcγR and activate mouse complement [108, 109]. In short-term disease models, injection of purified human CRP is effective. However, repeated injection is not possible because of the development of anti-CRP antibodies [110]. Another approach has been the development of transgenic mice expressing human CRP or rabbit CRP with expression controlled by either the human CRP promoter as an acute phase reactant or a diet-inducible promoter [109, 111]. Recently CRP-deficient mice were established in several laboratories although these have not yet been reported on extensively [112, 113]. For the most part studies with CRP transgenic (tg) mice and passively administered CRP have produced similar results. SAP is an acute phase protein in the mouse and SAP-deficient mice have also been studied in several disease models [40, 114–116]. These results in infection, inflammation, autoimmune, and cardiovascular disease models are summarized in the following sections.
9. Pentraxins and Protection from Infection
CRP binding to S. pneumoniae was the first indication that CRP might participate in protection from infection. Kindmark's group first showed the opsonic activity of CRP for S. pneumoniae and E. coli [117–119]. It was subsequently demonstrated that CRP could protect mice from experimental infection with S. pneumoniae [120] and that this effect was mediated in large part through activation of complement [121]. Protection from pneumococcal infection was also seen when human CRP was expressed from a transgene [71]. Similarly CRPtg mice were protected from infection by Salmonella typhimurium [122]. CRP recognizes pathogens through recognition of PC expressed on the surface of S. pneumoniae, Hemophilus influenza, and other pathogenic organisms. Mutagenesis studies have determined that the PC-binding pocket is necessary for protection in the S. pneumoniae model [123]. Weiser et al. determined that expression of PC on H. influenzae allowed for CRP binding and killing by complement [124]. Furthermore it was shown that CRP is expressed in the respiratory tract and could be found in these secretions [125]. Thus CRP may provide a barrier function, much like IgA and a direct protective effect from respiratory tract pathogens through complement activation.
Although SAP binds preferentially to ligands containing phosphoethanolamine, it has been shown to bind to S. pneumoniae, which results in classical complement activation and enhanced phagocytosis [114]. Thus CRP and SAP can both participate in protection from S. pneumoniae, a common and often fatal infection in the young and the elderly.
SAP binds to the LPS component of the Gram-negative cell wall, and the effects of SAP deficiency on Gram-negative infection as well as LPS shock have been studied with conflicting results. One group reported increased resistance to LPS shock in SAP−/− mice [126]. A second group reported that SAP−/− mice were more susceptible to LPS shock and to E. coli 0111:B4 but more resistant to lethal infection with Streptococcus pyogenes or E. coli J5, organisms to which SAP binds [40]. Human SAP binds to and neutralizes Shiga toxin 2, the main mediator of sever hemorrhagic colitis and hemolytic uremic syndrome that occurs following ingestion of enterohemorrhagic E. coli O157:H7 [127, 128].
Recently, it has been reported that SAP is an inhibitor of influenza viral infection [129, 130]. SAP was found to inhibit viral binding to hyaluronic acid in a calcium-independent manner. These results were consistent with earlier studies of SAP and viral infection [131, 132] although the mechanisms of action were deemed to be different.
SAP has also been found associated with invasive Candida albicans and amyloid associated with this fungal infection in the gut [133]. No functional consequences were examined although it was speculated that SAP might inhibit the neutrophil response.
CRP has also been shown to bind to nonbacterial pathogens. CRP was found to bind avidly to Leishmania donovani. Binding was specific for the lipophosphoglycan on the surface of metacyclic L. donovani [134]. The result of this binding was a kind of silent phagocytosis that did not induce cytokine production or protect the host from infection.
Others have examined the interaction of CRP with malarial parasites. Early studies indicated that CRP bound to Plasmodium falciparum and P. yoelii sporozoite surface membranes and that CRP could protect rats from experimental infection with P. yoelii sporozoites [135]. CRP elevation has been proposed as an excellent measure of parasitemia in falciparum malaria [136].
A correlation between genetically determined levels of CRP expression and malaria infection was carried out in a Sudanese population. This study examined an upstream polymorphism in the CRP gene, −286 (C > T > A) that is known to influence CRP levels. The A form has the highest levels of baseline CRP expression. In this study, a cohort of 192 Sudanese donors followed for malaria infection for 9 years had their CRP −286 gene locus genotyped. The prevalence of the CRP alleles A, C, and T were 21%, 52%, and 27%, respectively. The number of malaria episodes experienced by each individual over the study period was used as an index for malaria susceptibility. The A-allele, unlike the C- and T-alleles or CRP genotypes, was significantly associated with an increased number of malaria episodes, P = 0.007 and increased parasite counts. The proportion of A-allele carriers among donors not known to have had malaria during the study period was 18%, whereas it was 43% and 63% among donors who had experienced 1–4 and ≥5 malaria episodes, respectively, over the same period (P = 0.002). A second study was done in Ghana on the genetic association between FcγRIIA 131R/H polymorphisms and malaria. Using a recessive model the FcγRIIA R allele, which has higher binding to CRP and lower binding to IgG, was positively associated with severe malaria, but not with cerebral malaria [137]. Together these epidemiological and genetic associations suggest that, in addition to its utility as a prognostic marker in falciparum malaria [136], CRP may play a deleterious role in the disease.
10. CRP and SAP and the Kidney
CRP deposition in the kidney has been demonstrated in various forms of glomerular injury. Salmon's group found CRP deposited in renal glomeruli from patients with lupus nephritis [138]. They further suggested that the R form of the FcγRIIA gene was associated with more severe renal disease. The R form of FcγRIIA is the form that binds CRP more avidly than the H form. Szalai et al. also found CRP deposited in CRPtg (NZB x NZW) F1 lupus prone mice and showed by in situ hybridization that it was produced locally [139]. More recently Sjöwall et al. reported colocalization of IgG, CRP, complement (C1q and C3c), and dsDNA in glomerular basement membrane/subendothelial electron dense deposits in a small number of lupus nephritis patients.
CRP has also been detected in kidneys undergoing acute rejection, and it was shown that CRP production could be induced in renal tubular epithelial cells [140]. Nakahara et al. [141] examined a wide variety of kidneys from children with various types of glomerular diseases. They found that CRP deposition was encountered more often in patients with proliferative diseases than in those with nonproliferative diseases. CRP deposition was primarily in the peritubular capillary walls and small vessels in the interstitium.
Recently evidence has been provided that CRP may play a pathologic role in certain mouse models of renal injury. Acute renal injury was induced in CRPtg mice and controls by clamping both renal pedicles for 30 min and then allowing reperfusion for 24 h. The transgenic mice had worse outcomes in all parameters measured [142]. The same research group had previously shown increased inflammation and fibrosis in CRPtg mice 3 days after induction of unilateral ureteral obstruction [143]. However, progression of renal injury at days 7 and 14 was equivalent for CRPtg and wild-type mice in this study.
SAP was shown to be a normal constituent of the glomerular basement membrane [144, 145]. SAP has been shown to potently inhibit renal fibrosis [146] in vivo. This effect was initiated by SAP binding to cell debris, followed by suppression of inflammatory macrophages through activation FcγRI and IL-10.
11. Pentraxins in Autoimmune Disease
A role for CRP in autoimmune diseases was suggested years ago when it was found that CRP was deposited in the nuclei of cells in the synovium of rheumatoid arthritis patients [147] and localized with polymorphonuclear cells in vasculitis [148] and experimental allergic encephalomyelitis [149]. These findings led to an exploration of the nature of the nuclear ligands recognized. Once it was established that CRP bound specifically to nuclear autoantigens including snRNPs, histones, and chromatin [28, 150–153], its role in SLE was investigated further.
Our group initially hypothesized that CRP binding to nuclear autoantigens would promote their clearance and regulate the autoantibody response. We tested this in the (NZB x NZW) F1 female (NZB/W) mouse model of SLE [110]. NZB/W mice make a strong antichromatin and anti-DNA response and die of glomerulonephritis at about 9 months of age. In our study, NZB/W mice were injected with chromatin, which accelerates their autoimmune disease, in the presence or absence of CRP. The results showed a prolonged survival of mice injected with CRP and chromatin compared to chromatin alone, and a transient decrease in autoantibodies. However, the mice developed antibodies to CRP, which may have neutralized its later effectiveness. To circumvent this problem, Volanakis's group crossed a human CRPtg mouse strain [111] with NZB/W mice. They found that transgenic expression of CRP even at low levels (<5 μg/mL) prolonged the survival of these mice by about 8 weeks. The CRPtg mice had decreased IgM anti-dsDNA, but increased IgG anti-dsDNA decreased renal disease. These results supported a protective role for CRP in SLE, but suggested that the mechanism probably was not suppression of autoantigen presentation or autoantibody responses.
Two additional papers showed that CRP given as a single injection of 200 μg per mouse had a rapid and long-lasting protective effect on renal disease in both NZB/W and MRL/lpr mice [154, 155]. This work clearly established a predominant effect of CRP on ongoing renal pathology and showed a similar protective effect of CRP in accelerated nephrotoxic nephritis (NTN), an immune complex-mediated glomerulonephritis that is not autoimmune in nature [154]. The establishment of the NTN model allowed further analysis of the mechanism of CRP suppression of renal disease. The effect of CRP in NTN required macrophages, FcγRI, and IL-10, consistent with the induction of a regulatory macrophage phenotype [156]. In subsequent studies of experimental autoimmune thrombocytopenia, spleen cells or bone marrow macrophages treated with CRP in vitro transferred suppression of platelet clearance to recipient mice further supporting an FcγRI-dependent regulatory macrophage mechanism [157]. Further studies are needed to determine the steps subsequent to the induction of regulatory macrophages that result in long-term suppression of disease in SLE models. In the MRL/lpr mouse, mAb depletion experiments implicated regulatory T cells in the long-term suppression of renal disease [155].
Szalai's group examined the effect of both CRP deficiency (CRP−/− mice) and overexpression (CRPtg) mice on the course of collagen-induced arthritis a model for human rheumatoid arthritis. CRP−/− mice were more susceptible to induction of collagen-induced arthritis and developed more severe disease, whereas CRPtg mice were more resistant to disease induction and had a milder disease course [112]. These authors also showed a protective effect of CRP in the mouse experimental allergic encephalomyelitis model of human multiple sclerosis [158]. Similar to other inflammatory models, CRP increased the production IL-10. In addition CRP inhibited proliferation of encephalitogenic T cells and decreased production of inflammatory chemokines in vitro. They went on to demonstrate that CRP-mediated protection required the presence of the inhibitory FcγR, FcγRIIb [159]. The effect of transgenic rabbit CRP was also examined in mice generated by Jiang et al. [160]. Rabbit CRP was expressed under the PEPCK promoter, which is upregulated by diet manipulation. Induction of CRP expression led to a very marked inhibition of monoarticular antigen-induced arthritis.
SAP has also been implicated in the pathogenesis of SLE largely because of studies that demonstrated that SAP bound to DNA and chromatin. It was postulated that SAP was responsible for clearance and degradation of these autoantigens from the blood [116]. Studies on SAP-deficient mice showed spontaneous antinuclear antibodies and severe glomerulonephritis, which supported this hypothesis [116]. However, this concept was challenged when an SAP “knock in” failed to correct the defect. It was determined that the background of the SAP−/− mice was influenced by the process of gene knockout and that genes from the 129 strain contributed to the autoimmune manifestations [161]. SAP−/− mice generated by a different group also spontaneously produced anti-nuclear antibodies but did not develop glomerulonephritis [126]. Recently, mouse SAP was reported to inhibit renal disease and autoantibody production in a model of SLE initiated by immunization of BALB/c mice with activated lymphocyte DNA in complete Freund's adjuvant [162]. Consistent with findings in CRP suppression of immune-mediated diseases, the mechanism of disease suppression by SAP involved the induction of regulatory macrophages producing IL-10 [163]. There is no indication of an involvement of SAP in SLE in man.
Another mechanism by which CRP is proposed to influence B cell activity is through shedding of membrane BLyS/BAFF by immune complex binding to Fcγ receptors. These investigators reported that Fcγ receptor cross-linking by either CRP or IgG IC induced the release of BLyS/BAFF from myeloid cells [164]. They further found that CRP, like IC, induced release of BLyS through the high affinity receptor for IgG, FcγRI.
CRP may display neoepitopes when bound to the surface of ELISA wells and some patients may develop antibodies that only react with these altered molecules. Bell et al. described autoantibodies directed towards CRP in patients who developed a type of illness resembling graft versus host disease following ingestion of contaminated cooking oil [165]. Surprisingly these antibodies reacted with cryptic epitopes of CRP but not to native CRP. Subsequently the presence of similar autoantibodies was reported in patients with SLE as well [166, 167]. The specificity for SLE is not complete as similar autoantibodies were seen in patients with chronic hepatitis C infection [168]. Although the clinical significance of these antibodies remains unknown, it has been proposed recently that complexes of CRP and anti-CRP along with anti-DNA antibodies may exacerbate inflammation by binding to necrotic remnants of apoptotic cells [169].
12. CRP in Cardiovascular Disease
Several years ago the identification of elevated baseline serum CRP as a predictor for cardiovascular events led to multiple studies by several groups to examine the role of CRP in mouse models of atherogenesis. Early studies suggested that CRP could facilitate the uptake of LDL by macrophages through opsonization. The interaction was reported to be dependent on micropinocytosis through FcγRIIa [26]. However, it remains controversial as to whether CRP binds to oxidized or otherwise modified LDL [25, 170, 171].
Human CRPtg or rabbit CRPtg mice were crossed onto mouse strains deficient in apolipoprotein E (apoE−/−) or low-density lipoprotein receptor (LDLR−/−) or CRP was infused into APOE*-Leiden mice. Although one study reported accelerated atherosclerosis in CRPtg/apoE−/− mice [172], 5 subsequent studies found no effect [173–177]. A more recent study noted that the apoE−/− mouse models have more severe hypercholesterolemia than humans as well as continuous low-grade inflammation and used a model of LDLR−/− mice expressing apolipoprotein B100, crossed onto human CRPtg [178]. In this study the presence of human CRP slowed lesion progression and was thus atheroprotective. Recently CRP-deficient mice were developed by gene targeting. Studies done comparing CRP-deficient and -sufficient mice in ApoE−/− and LDLR−/− atherogenesis models produced results consistent with an atheroprotective role for CRP [113]. The combined findings of these studies indicate that CRP is either neutral or protective in atherosclerosis given the limitations of the mouse models.
Extensive epidemiological studies of human CRP polymorphisms do not support the hypothesis that genetically determined elevated baseline levels of CRP contribute to human cardiovascular disease [179]. However, this does not preclude participation of CRP-induced cellular responses within the atherosclerotic plaque or in reperfusion injury.
13. Pentraxins and Monocytes and Macrophages
CRP and SAP bind preferentially to monocytes and neutrophils among human peripheral blood cells and opsonize targets for phagocytosis both directly through FcγR and FcαRI [14, 96, 180, 181] and indirectly through the activation of complement [182]. Activation of peripheral blood mononuclear cells (PBMC) by CRP with production of inflammatory cytokines was originally reported by Ballou and Lozanski [183]. Subsequent studies identified a strong synergy between CRP and LPS as well as differential proinflammatory or anti-inflammatory cytokine release depending on whether PBMC or macrophages were used [184–186]. Interpretation of studies of pentraxins and cytokine induction is further complicated by the lack of receptor crosslinking by pentameric CRP and SAP. A recent analysis that addressed these issues identified IL-6, IL-10, and IL-8 release by monocytes activated by aggregated SAP [14]. These responses were inhibited by mAb to FcγRI and FcγRIII, and by Syk inhibitors.
During infection macrophages may be exposed to both CRP and TLR ligands in the form of pathogen-associated or damage-associated molecular patterns (PAMPs or DAMPs). In this regard CRP acting through FcγRI and FcγRIIA enhanced PBMC production of proinflammatory cytokines, TNF-α and IL-1β in response to S. pneumoniae [91].
CRP has been injected into human volunteers to measure in vivo cytokine responses although the findings remain controversial. Bisoendial et al. injected healthy volunteers with 1.25 mg/kg recombinant human CRP. Cytokine profiles were generated by RT-PCR. He found upregulation of MMP9, MCP-1 (CCL2), uPA, MIP-1α, and IκBα mRNAs in peripheral leukocytes [187]. However, these findings were disputed by Pepys who maintained that the injected CRP must contain contaminants [188]. It appears unlikely that uncomplexed CRP will induce proinflammatory events. However, CRP is frequently found at sites of tissue injury along with complement where it likely participates in the clearance of complexes and activation of cells through complement and FcγR.
14. Pentraxins and Dendritic Cells
As CRP binds to pathogenic organisms and enhances their uptake by macrophages and dendritic cells (DCs), it was predicted to enhance antigen presentation and immunization. A model in which DCs pulsed with S. pneumoniae are used to immunize mice was used to test this hypothesis [189]. It was found that opsonization of S. pneumoniae with CRP prior to incubation with DC enhanced antibody responses compared to DC pulsed with unopsonized bacteria [190]. CRP had the greatest effect on the IgG secondary and memory responses to both protein (pneumococcal surface protein A) and polysaccharide (PC) antigens. CRP opsonization also increased the effectiveness of pulsed DC vaccination in protecting mice from intranasal challenge. The effects of CRP on S. pneumoniae uptake, antibody responses, and protection all required the FcR γ-chain.
CRP interactions with human DC have also been studied. A study by Zhang et al. [191] reported that CRP at low concentrations (10 μg/mL) inhibited the differentiation of CD14+ monocytes into DC in the presence of GM-CSF and IL-4 as well as the maturation of immature DC by LPS. The inhibitory effect of CRP on DC differentiation was blocked by antibody to FcγRII. A second group [192] isolated myeloid DC from blood and showed that CRP at 10 μg/mL or higher decreased the expression of the chemokine receptor CCR5 as well as the migration of these cells in response to the CCR5 ligand, MIP-1β. In contrast a third study [193] reported that CRP at very low concentrations (2 μg/mL) induced further maturation of immature monocyte-derived DC, and that this also was inhibited by antibody to FcγRII. These conflicting results may in part be due to the use of commercial CRP preparations, which may contain denatured CRP as well as preservatives and contaminating LPS.
We have recently examined the effect of CRP on a distinct DC type, the plasmacytoid DC (pDC). Like myeloid DC, pDCs are found in low numbers in the blood. The pDCs play an important role in innate defense against viral infection by producing large quantities of type I interferon (IFN) [194]. More recently, pDCs have been implicated in the increased levels of IFN and the IFN-inducible gene expression pattern in the peripheral blood of patients with SLE [195]. In this case, autoantibody immune complexes containing nucleoprotein autoantigens induce IFN production. Immune complexes are taken up by pDC through FcγRIIa and activate intracellular TLR for RNA or DNA in the endosomal compartment to stimulate IFN synthesis. CRP binds to nucleoprotein autoantigens, snRNPs, and chromatin, as well as to FcγRIIa. However, we found that CRP-snRNP complexes did not induce IFN synthesis by pDC, and CRP inhibited the IFN response to autoantibody-snRNP complexes [196]. This inhibitory effect of CRP was associated with increased pDC maturation and with more rapid processing of IC into late endosome/lysosomes. IFN produced by pDC contributes to pathogenesis of SLE and other autoimmune diseases, so these results are consistent with the protective effect of CRP in mouse models of SLE [110, 139, 155, 156].
SAP binds to DNA, which is a TLR9 agonist. Recent studies showed that SAP binding to DNA blocks innate immune responses to DNA-based vaccines [197]. The authors found that, in mice tg for human SAP, T cell and antibody responses to DNA vaccines were decreased. The defective responses were shown to be the result of SAP binding to DNA, which facilitated uptake through FcγRI and FcγRIII. SAP prevented DNA binding to other DNA-binding molecules and inhibited activation of NFκB and type I interferon responses in a human macrophage cell line.
15. CRP and Neutrophil Activation Chemotaxis and Phagocytosis
The interaction of CRP with neutrophils has been studied over many years. The first defined activity of CRP on neutrophils was its ability to opsonize both Gram-positive and Gram-negative pathogens [118, 198]. Mortensen et al. went on to show that CRP and complement acting in concert could induce phagocytosis of erythrocytes coated with the C-polysaccharide of S. pneumoniae (PnC) [182]. In 1985, Kilpatrick and Volanakis demonstrated that phagocytosis of PnC-coated RBC required activation of neutrophils by phorbol myristate acetate, a treatment that downregulates CD32A-dependent phagocytosis and increases FcαRI-dependent binding and phagocytosis [181]. More recently, it has been shown that CRP-mediated phagocytosis by neutrophils may proceed through FcαRI or FcγRIIA [96].
CRP has also been demonstrated to have inhibitory effects on neutrophils, particularly on neutrophil chemotaxis. For example, Webster's group found that CRP was able to inhibit neutrophil chemotaxis in vitro and in vivo [200, 201]. These effects were thought to be mediated through inhibition of p38 MAP kinase [202]. Zhong et al. also examined the effect of CRP on neutrophil chemotaxis with similar findings. He found that CRP inhibited neutrophil chemotaxis to IL-8 and fMLP (formyl-methionyl-leucyl-pheylalanine) chemotactic stimuli [203].
Zeller and Sullivan found that aggregated CRP could enhance chemoluminescence induced by IgG. This activity was strongly inhibited by antibodies to FcγRII/III [204, 205]. Unfortunately, many of these studies were performed before the characterization of the mouse and human FcγR and before blocking antibodies specific for each receptor were available.
More recently, the effects of CRP on phagocytosis have been reexamined with the finding that CRP also binds to CD89 [96]. Regulation of FcR on human neutrophils is rather complex and depends on several factors. Although human neutrophils normally express CD32A and GPI-anchored CD16B, they may express CD64 in response to signals like IFN-γ. It has also been reported that CD32A is maintained in a low affinity state unless stimulated by fMLP [199, 206]. In contrast CD89, the receptor for IgA, is upregulated by PMA resulting in preferential binding. CRP is capable of enhancing bacterial uptake through CD89 and upregulating its surface expression [96].
16. CRP Effects on the Vascular Endothelium
CRP has been extensively studied in cardiovascular disease and much of this work has revolved around its effect on the endothelium. One of the first studies suggested that CRP has a direct inflammatory effect on endothelial cells [207]. The investigators found that low concentrations of CRP in the presence of serum, acting through unknown mechanisms, would increase levels of adhesive molecules 10-fold. Unfortunately, the role of complement was not explored. These findings suggested that CRP might contribute to vascular injury and cardiovascular disease.
A pathogenic role for CRP interaction was also supported by the finding that CRP could decrease eNOS expression in human aortic endothelial cells leading to attraction of monocytes to endothelial cells [208]. These activities were found to be due to CRP engagement of FcγR on the endothelial cells [209]. However, the finding of eNOS inhibition by CRP was challenged by others who suggested that CRP actually increased NO production in vitro and in vivo leading to a decreased response of phenylephrine-induced vasoconstriction [210]. CRP effects on the endothelium after experimental induced injury were also studied [211–213]. The prothrombotic effects of CRP in this model required FcγRI [214]. It was later shown by the same group that vascular damage induced by CRP required complement [215].
It has also been reported that CRP can induce apoptosis of vascular smooth muscle cells through stabilization of GADD153 mRNA [216]. These effects were seen at very low levels of CRP and it is unclear whether these in vitro findings are relevant in vivo although colocalization of CRP and GADD153 was found in atherosclerotic lesions.
Investigations of whether CRP contributes mechanistically to cardiovascular disease have been extensive and controversial. The absence of a clear effect in mouse models as described above has further impeded progress in this area. For a thorough discussion of the findings both supporting and disputing a role for CRP in atherogenesis, the reader is referred to three recent review articles [217–219].
17. CRP Genetics
The genes for the classical pentraxins lie on chromosome 1q23.2 in man. This is an immunological hot spot with genes for the FcγR lying close by at 1q23.3. This region is also associated with the risk for SLE in man and in mouse models of SLE. As discussed above, many studies have focused on the CRP gene due to its perceived involvement in cardiovascular disease. More than 100 SNPs in and around the CRP gene have now been identified. None of these polymorphisms is associated with the coding region of CRP and no variations in the protein sequence of CRP have been identified. However, polymorphisms in the noncoding regions in the promoter and the untranslated region have a substantial effect on baseline CRP levels. Polymorphisms in genes that stimulate CRP production like IL-6, IL-1, and several others also contribute to baseline CRP levels. Groups of CRP SNPs inherited together have been identified with five common major haplotypes in Northern European subjects. Two of these haplotypes are associated with high baseline CRP levels and two are associated with lower CRP levels [220]. Differences in acute phase levels of CRP are also influenced by these haplotypes [221]. Moreover CRP haplotypes have been linked to several disease states. In the case of cardiovascular disease, there is now at least some degree of consensus that genetically determined that baseline levels of CRP do not influence disease risk in a causative relationship despite their strong association reviewed in [222]. However, associations between CRP and individuals with genetically determined lower baseline levels of CRP are at increased risk of SLE and lupus nephritis [223, 224]. See the genetic association database at NCBI.
Since many of the effects of CRP in inflammatory states are related to FcγR and these receptors display polymorphisms association between these polymorphisms and disease risk efforts have been made to determine the importance of these FcγRs in relation to CRP. Jönsen et al. studied FcγRIIA, FcγRIIIA, and CRP polymorphisms in relation to multiple SLE disease manifestations including glomerulonephritis [224]. They found associations between a low expressing CRP allele and more severe glomerulonephritis and an interactive effect between this CRP allele and the low IgG-binding FcγRIIIA allele (F/F).
18. Clinical Use of CRP Levels
CRP levels are used clinically in two different ways. The initial assays used to measure CRP in the circulation were relatively insensitive and for many years a positive value was used as an indication of inflammation or infection. CRP is an excellent marker of the acute inflammatory response and is used extensively for diagnosis and prognosis of rheumatologic and other diseases. CRP levels of 10 μg/mL up to 500 μg/mL can be seen in the acute phase response. CRP is routinely used to measure disease activity in rheumatoid arthritis and is part of the Disease Activity Score 28. Similarly measuring CRP levels is helpful in monitoring disease activity of various forms of vasculitis. However, CRP monitoring is of little value in measuring disease activity in SLE, scleroderma, polymyositis, or dermatomyositis where CRP levels do not correlate well with disease activity [225].
About 20 years ago highly sensitive assays were developed that could detect baseline CRP levels in apparently healthy individuals. These assays were said to measure high sensitivity (hs)-CRP although the only difference is in the ability to detect lower levels of CRP. There is little consistent evidence for the presence of CRP that is glycosylated or otherwise modified from native pentameric CRP in the circulation. The advent of the hs-CRP assay led to numerous studies that showed utility in individuals at risk for cardiovascular disease, metabolic syndrome, periodontal disease, and other chronic diseases associated with a low level of inflammation. The American Heart Association established ranges of CRP levels that were associated with risk of cardiovascular events [226]. These values probably reflect both genetic differences in CRP production and stimuli for its synthesis as well as underlying inflammation due to factors like periodontal disease and the metabolic syndrome. Whether there is a direct contribution of mildly elevated CRP levels to cardiovascular disease has been extensively debated [227]. Regardless of its role in pathogenesis it remains a strong marker of cardiovascular disease risk with equivalence to the widely measured risk factor, cholesterol [228].
19. CRP in Sepsis and Shock
As noted above CRP levels are highly elevated in patients with sepsis. The levels of CRP in sepsis have been shown to be related to mortality and organ failure [229]. In sepsis CRP was shown to participate in complement activation [230]. Recently it was reported that CRP strikingly downregulates the C5aR on PMN in patients with sepsis [231]. These finding are reminiscent of earlier findings that suggested that CRP could induce shedding of the IL-6 receptor on neutrophils [232] and suggest a regulatory role for CRP in the inflammatory response during sepsis.
These findings in human neutrophils are consistent with earlier findings in mouse models. An anti-inflammatory role for CRP was first shown in mouse models of lethal inflammation induced by LPS, platelet-activating factor (PAF), or TNF-α plus IL-1β [233]. These investigators developed a CRPtg strain of mice in which rabbit CRP was expressed under the diet-inducible phosphoenolpyruvate carboxykinase (PEPCK) promoter. They found that mice expressing acute phase levels of CRP were protected from lethal endotoxin shock as well as shock induced by PAF and TNF-α plus IL-1β. Subsequent studies indicated that CRP protection from PAF required an intact PC binding site and might be mediated by direct binding of CRP to the PC group on PAF [234].
Several other groups reported protection of mice from LPS shock by injection of human CRP as well [40, 121, 235]. These studies also showed that SAP, although it binds to LPS, was not protective [40]. CRP was protective and the mechanism required both activating and inhibitory FcγRs [121]. This study demonstrated induction of a regulatory macrophage phenotype by CRP and LPS, similar to the regulatory macrophages induced by LPS and IC [236]. A similar anti-inflammatory pathway is induced by CRP in immune-mediated diseases as discussed above.
Although these studies are consistent with a regulatory role for CRP in the acute phase response, it is more difficult to test this in humans. One study injected endotoxin into healthy volunteers with genetically different baseline CRP levels and measured the proinflammatory cytokines response (TNF-α and IL-6). Consistent with the results in the mouse models, individuals with higher CRP levels had lower TNF-α and IL-6 responses to LPS injection [237].
Traumatic injury induces dramatic changes in both pro-inflammatory mediators that can result in shock as well as anti-inflammatory mediators that can suppress the immune system [238]. Monocytes and macrophages are key initiators and regulators of innate immune responses following trauma. In a study of 50 trauma patients, we observed an increase in an activated monocyte/macrophage population (CD14highCD16+CD163+) in the blood that was highly correlated with CRP levels, as well as M-CSF and TGF-β [239], M-CSF and TGF-β found in trauma plasma could induce this phenotype in normal monocytes. Although it was not essential for inducing the phenotype, CRP activated M-CSF differentiated monocytes to produce anti-inflammatory cytokines, IL-10 and IL-1RA. These findings are consistent with a role for CRP in the anti-inflammatory response following trauma that helps prevent shock.
20. SAP in Disease
The other major member of the pentraxin family is SAP. In this section I focus on properties of SAP related to human disease. Unlike CRP, SAP is a constitutively expressed protein in man that is normally present at about 40 μg/mL in blood. SAP was named for its physical association with amyloid deposits associated with various forms of amyloidosis, which is associated with a variety of inflammatory hereditary, malignant and infectious conditions. These amyloid deposits are normally detected by biopsy of the affected organ and fluorescent staining. Amyloid deposits progressively affect organ function by massively infiltrating the parenchyma.
The function of SAP in the amyloid plaque is not completely understood. It has been proposed that SAP serves to stabilize the amyloid fibrils against degradative enzymes [240]. Like CRP, SAP is very resistant to enzymatic attack due to its tightly packed structure. In mice targeted deletion of the SAP gene leads to delayed amyloidogenesis in a reactive model of systemic amyloidosis [115]. This finding has led to several approaches designed to block this stabilization by depleting SAP systemically. It has been shown that the extent and localization of amyloid deposits may be determined by imaging studies that use injected, labeled SAP as a marker [43]. Treatment of amyloidosis with agents that clear SAP from circulation along with anti-SAP antibodies has been tried and found to be effective in an animal model [241]. More recently, similar studies were done with patients suffering from amyloidosis [242]. However, the results of follow-up studies are unavailable so far. Similar studies have been reported in Alzheimer's disease [243]. However, the clinical utility of this agent remains unknown.
There is a growing body of studies suggesting unique properties of SAP in wound healing and regulation of fibrosis. Pilling et al. first demonstrated that SAP could prevent the generation of fibrocytes in vitro and suggested that this might contribute to delayed wound healing and facilitated connective tissue disease [244]. They went on to show that this effect was mediated by binding FcγR and specifically to FcγRI [245]. These studies have led to a series of human clinical studies related to treatment of chronic fibrosing conditions such as interstitial pulmonary fibrosis, macular degeneration and myelofibrosis. These are severe irreversible conditions, and any new therapeutic agents would be welcomed. An in vivo correlate of this function was shown as well. SAP was found to downregulate the conversion of monocytes to fibroblasts in a mouse model of fibrotic cardiomyopathy [246].
21. Summary
The pentraxins are a family of evolutionarily conserved proteins that trace their evolutionary roots back to the early invertebrates. They have evolved along with the innate and adaptive immune system interacting with the ancient complement system and the Fc receptors. Their best described role is in host defense although they are important pattern recognition molecules for altered self-antigens as well.
The pentraxins have been studied for over 80 years now and we have learned a great deal about their structure, function and evolution. Despite this intensive study we have only recently begun to understand their role in disease and host defense (Figure 4). Figure 4 provides a cartoon representation of the major known properties of the pentraxins. CRP production is stimulated inflammatory events whereas SAP is constitutively expressed. Both pentraxins activate complement and play a role in host defense although this is much better studied for CRP. Both CRP and SAP also bind to damaged cells and nuclear components and facilitate their safe removal in a nonimmunogenic manner. When binding of pentraxins to multivalent ligands leads to FcR crosslinking, macrophage polarization is seen, which in the case of CRP suppresses inflammation and in the case of SAP prevents fibrosis. Overall the pentraxins provide a regulatory pathway to control the inflammatory response to tissue injury.
Figure 4.
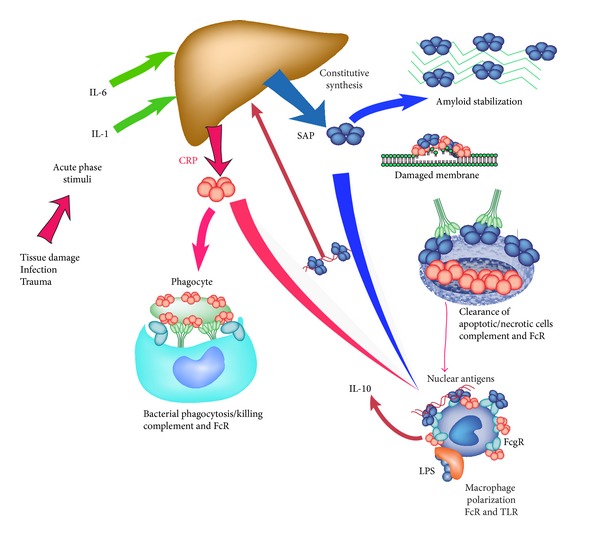
Overview of the major activities of the pentraxins. Both CRP and SAP are predominantly serum proteins, produced in the liver. In man SAP is constitutive and CRP is a major acute phase reactant. Both contribute to host defense as direct opsonins and through complement activation. Both bind to ligands exposed during cell death and tissue damage leading to opsonization and removal. In addition to these activities many studies support a role for pentraxins in regulating the inflammatory response to immune complexes and TLR agonists. This regulation is initiated by pentraxin interactions with FcγR and mediated by polarized macrophages.
Abbreviations
- CRP:
C-reactive protein
- DC:
Dendritic cells
- FcγR:
Fcγ receptors
- fH:
Factor H
- FcαRI (CD89):
The IgA receptor
- ITAM:
Immunoreceptor tyrosine-based activation motif
- LPS:
Lipopolysaccharide
- PC:
Phosphocholine
- NPTXI and NPTXII:
Pentraxin I and II
- PTX3:
Long pentraxin 3
- snRNPs:
Small nuclear ribonucleoproteins
- SAP:
Serum amyloid P component
- SPR:
Surface plasmon resonance
- SLE:
Systemic lupus erythematosus
- TLR:
Toll-like receptor
- tg:
Transgenic
- NZB/W (NZB x NZW):
F1 female mouse model of SLE
- apoE:
Apolipoprotein E
- LDLR:
Low density lipoprotein receptor
- PBMC:
Peripheral blood mononuclear cells
- pDC:
Plasmacytoid DC
- PEPCK:
Phosphoenolpyruvate carboxykinase promoter.
References
- 1.Robey FA, Liu TY. Limulin: a C-reactive protein from Limulus polyphemus. Journal of Biological Chemistry. 1981;256(2):969–975. [PubMed] [Google Scholar]
- 2.Tillett WS, Francis T. Serological reactions in pneumonia with a non-protein somatic fraction of pneumococcus. Journal of Experimental Medicine. 1930;52(4):561–571. doi: 10.1084/jem.52.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osmand AP, Friedenson B, Gewurz H. Characterization of C reactive protein and the complement subcomponent C1t at homologous proteins displaying cyclic pentameric symmetry (pentraxins) Proceedings of the National Academy of Sciences of the United States of America. 1977;74(2):739–743. doi: 10.1073/pnas.74.2.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hutchinson WL, Hohenester E, Pepys MB. Human serum amyloid P component is a single uncomplexed pentamer in whole serum. Molecular Medicine. 2000;6(6):482–493. [PMC free article] [PubMed] [Google Scholar]
- 5.Kinoshita CM, Ying SC, Hugli TE, et al. Elucidation of a protease-sensitive site involved in the binding of calcium to C-reactive protein. Biochemistry. 1989;28(25):9840–9848. doi: 10.1021/bi00451a044. [DOI] [PubMed] [Google Scholar]
- 6.Srinivasan N, White HE, Emsley J, Wood SP, Pepys MB, Blundell TL. Comparative analyses of pentraxins: implications for protomer assembly and ligand binding. Structure. 1994;2(11):1017–1027. doi: 10.1016/S0969-2126(94)00105-7. [DOI] [PubMed] [Google Scholar]
- 7.Pepys MB, Rademacher TW, Amatayakul-Chantler S, et al. Human serum amyloid P component is an invariant constituent of amyloid deposits and has a uniquely homogeneous glycostructure. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(12):5602–5606. doi: 10.1073/pnas.91.12.5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emsley J, White HE, O’Hara BP, et al. Structure of pentameric human serum amyloid P component. Nature. 1994;367(6461):338–345. doi: 10.1038/367338a0. [DOI] [PubMed] [Google Scholar]
- 9.Shrive AK, Cheetham GMT, Holden D, et al. Three dimensional structure of human C-reactive protein. Nature Structural Biology. 1996;3(4):346–354. doi: 10.1038/nsb0496-346. [DOI] [PubMed] [Google Scholar]
- 10.Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999;7(2):169–177. doi: 10.1016/S0969-2126(99)80023-9. [DOI] [PubMed] [Google Scholar]
- 11.Agrawal A, Shrive AK, Greenhough TJ, Volanakis JE. Topology and structure of the C1q-binding site on C-reactive protein. Journal of Immunology. 2001;166(6):3998–4004. doi: 10.4049/jimmunol.166.6.3998. [DOI] [PubMed] [Google Scholar]
- 12.Agrawal A, Volanakis JE. Probing the C1q-binding site on human C-reactive protein by site-directed mutagenesis. Journal of Immunology. 1994;152(11):5404–5410. [PubMed] [Google Scholar]
- 13.Bang R, Marnell L, Mold C, et al. Analysis of binding sites in human C-reactive protein for FcγRI, FcγRIIA, and C1q by site-directed mutagenesis. Journal of Biological Chemistry. 2005;280(26):25095–25102. doi: 10.1074/jbc.M504782200. [DOI] [PubMed] [Google Scholar]
- 14.Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. Structural recognition and functional activation of FcγR by innate pentraxins. Nature. 2008;456(7224):989–992. doi: 10.1038/nature07468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garianda C, Hirsch E, Bozza S, et al. Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature. 2002;420(6912):182–186. doi: 10.1038/nature01195. [DOI] [PubMed] [Google Scholar]
- 16.Moalli F, Doni A, Deban L, et al. Role of complement and Fcγ receptors in the protective activity of the long pentraxin PTX3 against Aspergillus fumigatus. Blood. 2010;116(24):5170–5180. doi: 10.1182/blood-2009-12-258376. [DOI] [PubMed] [Google Scholar]
- 17.Mantovani A, Valentino S, Gentile S, Inforzato A, Bottazzi B, Garlanda C. The long pentraxin PTX3: a paradigm for humoral pattern recognition molecules. Annals of the New York Academy of Sciences. 2013;1285:1–14. doi: 10.1111/nyas.12043. [DOI] [PubMed] [Google Scholar]
- 18.Inforzato A, Bottazzi B, Garlanda C, Valentino S, Mantovani A. Pentraxins in humoral innate immunity. Advances in Experimental Medicine and Biology. 2012;946:1–20. doi: 10.1007/978-1-4614-0106-3_1. [DOI] [PubMed] [Google Scholar]
- 19.Abernethy TJ, Avery OT. The occurrence during acute infections of a protein not normally present in the blood. I: distribution of the reactive protein in patients; sera and the effect of calcium on the flocculation reaction with C polysaccharide of pneumococcus. Journal of Experimental Medicine. 1941;73:173–182. doi: 10.1084/jem.73.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Volanakis JE, Kaplan MH. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proceedings of the Society for Experimental Biology and Medicine. 1971;136(2):612–614. doi: 10.3181/00379727-136-35323. [DOI] [PubMed] [Google Scholar]
- 21.Narkates AJ, Volanakis JE. C-reactive protein binding specificities: artificial and natural phospholipid bilayers. Annals of the New York Academy of Sciences. 1982;389:172–182. doi: 10.1111/j.1749-6632.1982.tb22135.x. [DOI] [PubMed] [Google Scholar]
- 22.Li YP, Mold C, Du Clos TW. Sublytic complement attack exposes C-reactive protein binding sites on cell membranes. Journal of Immunology. 1994;152(6):2995–3005. [PubMed] [Google Scholar]
- 23.Kushner I, Kaplan MH. Studies of acute phase protein. I. An immunohistochemical method for the localization of Cx-reactive protein in rabbits. Association with necrosis in local inflammatory lesions. Journal of Experimental Medicine. 1961;114:961–974. doi: 10.1084/jem.114.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kushner I, Rakita L, Kaplan MH. Studies of acute-phase protein. II. Localization of Cx-reactive protein in heart in induced myocardial infarction in rabbits. The Journal of Clinical Investigation. 1963;42:286–292. doi: 10.1172/JCI104715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang MK, Binder CJ, Torzewski M, Witztum JL. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: phosphorylcholine of oxidized phospholipids. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(20):13043–13048. doi: 10.1073/pnas.192399699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zwaka TP, Hombach V, Torzewski J. C-reactive protein-mediated low density lipoprotein uptake by macrophages: implications for atherosclerosis. Circulation. 2001;103(9):1194–1197. doi: 10.1161/01.cir.103.9.1194. [DOI] [PubMed] [Google Scholar]
- 27.Tan EM. Autoantibodies to Nuclear Antigens (ANA): their Immunobiology and Medicine. Advances in Immunology. 1982;33:167–240. doi: 10.1016/s0065-2776(08)60836-6. [DOI] [PubMed] [Google Scholar]
- 28.Du Clos TW. C-reactive protein reacts with the U1 small nuclear ribonucleoprotein. Journal of Immunology. 1989;143(8):2553–2559. [PubMed] [Google Scholar]
- 29.Du Clos TW. The interaction of C-reactive protein and serum amyloid P component with nuclear antigens. Molecular Biology Reports. 1996;23(3-4):253–260. doi: 10.1007/BF00351177. [DOI] [PubMed] [Google Scholar]
- 30.Pepys MB, Butler PJG. Serum amyloid P component is the major calcium-dependent specific DNA binding protein of the serum. Biochemical and Biophysical Research Communications. 1987;148(1):308–313. doi: 10.1016/0006-291x(87)91111-9. [DOI] [PubMed] [Google Scholar]
- 31.DiCamelli R, Potempa LA, Siegel J. Binding reactivity of C-reactive protein for polycations. Journal of Immunology. 1980;125(5):1933–1938. [PubMed] [Google Scholar]
- 32.Potempa LA, Siegel JN, Gewurz H. Binding reactivity of C-reactive protein for polycations. II. Medulatory effects of calcium and phosphocholine. Journal of Immunology. 1981;127(4):1509–1514. [PubMed] [Google Scholar]
- 33.Black S, Agrawal A, Samols D. The phosphocholine and the polycation-binding sites on rabbit C-reactive protein are structurally and functionally distinct. Molecular Immunology. 2003;39(16):1045–1054. doi: 10.1016/s0161-5890(03)00031-2. [DOI] [PubMed] [Google Scholar]
- 34.Lee RT, Takagahara I, Lee YC. Mapping the binding areas of human C-reactive protein for phosphorylcholine and polycationic compounds. Relationship between the two types of binding sites. Journal of Biological Chemistry. 2002;277(1):225–232. doi: 10.1074/jbc.M106039200. [DOI] [PubMed] [Google Scholar]
- 35.Siegel J, Osmand AP, Wilson MF, Gewurz H. Interactions of C reactive protein with the complement system. II. C reactive protein mediated consumption of complement by poly L lysine polymers and other polycations. Journal of Experimental Medicine. 1975;142(3):709–721. doi: 10.1084/jem.142.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaplan MH, Volanakis JE. Interaction of C reactive protein complexes with the complement system. I. Consumption of human complement associated with the reaction of C reactive protein with pneumococcal C polysaccharide and with the choline phosphatides, lecithin and sphingomyelin. Journal of Immunology. 1974;112(6):2135–2147. [PubMed] [Google Scholar]
- 37.Schwalbe RA, Dahlback B, Coe JE, Nelsestuen GL. Pentraxin family of proteins interact specifically with phosphorylcholine and/or phosphorylethanolamine. Biochemistry. 1992;31(20):4907–4915. doi: 10.1021/bi00135a023. [DOI] [PubMed] [Google Scholar]
- 38.Gillmore JD, Hutchinson WL, Herbert J, et al. Autoimmunity and glomerulonephritis in mice with targeted deletion of the serum amyloid P component gene: SAP deficiency or strain combination? Immunology. 2004;112(2):255–264. doi: 10.1111/j.1365-2567.2004.01860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hind CRK, Collins PM, Baltz ML, Pepys MB. Human serum amyloid P component, a circulating lectin with specificity for the cyclic 4,6-pyruvate acetal of galactose. Interactions with various bacteria. Biochemical Journal. 1985;225(1):107–111. doi: 10.1042/bj2250107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noursadeghi M, Bickerstaff MCM, Gallimore JR, Herbert J, Cohen J, Pepys MB. Role of serum amyloid P component in bacterial infection: protection of the host or protection of the pathogen. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(26):14584–14589. doi: 10.1073/pnas.97.26.14584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hind CRK, Collins PM, Renn D. Binding specificity of serum amyloid P component for the pyruvate acetal of galactose. Journal of Experimental Medicine. 1984;159(4):1058–1069. doi: 10.1084/jem.159.4.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pepys MB, Dyck RF, de Beer FC. Binding of serum amyloid P-component (SAP) by amyloid fibrils. Clinical and Experimental Immunology. 1979;38(2):284–293. [PMC free article] [PubMed] [Google Scholar]
- 43.Hawkins PN, Lavender JP, Pepys MB. Evaluation of systemic amyloidosis by scintigraphy with123I-labeled serum amyloid P component. New England Journal of Medicine. 1990;323(8):508–513. doi: 10.1056/NEJM199008233230803. [DOI] [PubMed] [Google Scholar]
- 44.Siegel J, Rent R, Gewurz H. Interactions of C reactive protein with the complement system. I. Protamine induced consumption of complement in acute phase sera. Journal of Experimental Medicine. 1974;140(3):631–646. doi: 10.1084/jem.140.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du Clos TW, Mold C. Complement and complement deficiencies. In: Rich RR, Fleisher HW Jr., Shearer WT, Frew AJ, Weyand CM, editors. Clinical Immunology Principles and Practice. 4th edition. Elsevier; 2013. pp. 252–269. [Google Scholar]
- 46.Claus DR, Siegel J, Petras K. Interactions of C reactive protein with the first component of human complement. Journal of Immunology. 1977;119(1):187–192. [PubMed] [Google Scholar]
- 47.Jiang H, Siegel JN, Gewurz H. Binding and complement activation by C-reactive protein via the collagen-like region of C1q and inhibition of these reactions by monoclonal antibodies to C-reactive protein and C1q. Journal of Immunology. 1991;146(7):2324–2330. [PubMed] [Google Scholar]
- 48.Jiang H, Robey FA, Gewurz H. Localization of sites through which C-reactive protein binds and activates complement to residues 14-26 and 76-92 of the human C1q A chain. Journal of Experimental Medicine. 1992;175(5):1373–1379. doi: 10.1084/jem.175.5.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roumenina LT, Ruseva MM, Zlatarova A, et al. Interaction of C1q with IgG1, C-reactive protein and pentraxin 3: mutational studies using recombinant globular head modules of human C1q A, B, and C chains. Biochemistry. 2006;45(13):4093–4104. doi: 10.1021/bi052646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaboriaud C, Juanhuix J, Gruez A, et al. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. Journal of Biological Chemistry. 2003;278(47):46974–46982. doi: 10.1074/jbc.M307764200. [DOI] [PubMed] [Google Scholar]
- 51.Ying SC, Jiang H, Gewurz A, Gewurz H. Human serum amyloid P component (SAP) binds and activates the classical complement pathjway via collagen-like region of C1q. The FASEB Journal. 1992;(article A1451) [PubMed] [Google Scholar]
- 52.Hicks PS, Saunero-Nava L, Du Clos TW, Mold C. Serum amyloid P component binds to histones and activates the classical complement pathway. Journal of Immunology. 1992;149(11):3689–3694. [PubMed] [Google Scholar]
- 53.Berman S, Gewurz H, Mold C. Binding of c-reactive protein to nucleated cells leads to complement activation without cytolysis. Journal of Immunology. 1986;136(4):1354–1359. [PubMed] [Google Scholar]
- 54.Gershov D, Kim S, Brot N, Elkon KB. C-reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. Journal of Experimental Medicine. 2000;192(9):1353–1363. doi: 10.1084/jem.192.9.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mold C, Kingzette M, Gewurz H. C-reactive protein inhibits pneumococcal activation of the alternative pathway by increasing the interaction between factor H and C3b. Journal of Immunology. 1984;133(2):882–885. [PubMed] [Google Scholar]
- 56.Suankratay C, Mold C, Zhang Y, Potempa LA, Lint TF, Gewurz H. Complement regulation in innate immunity and the acute-phase response: inhibition of mannan-binding lectin-initiated complement cytolysis by C-reactive protein (CRP) Clinical and Experimental Immunology. 1998;113(3):353–359. doi: 10.1046/j.1365-2249.1998.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunological Reviews. 2008;223(1):300–316. doi: 10.1111/j.1600-065X.2008.00641.x. [DOI] [PubMed] [Google Scholar]
- 58.Sjöberg AP, Trouw LA, McGrath FDG, Hack CE, Blom AM. Regulation of complement activation by C-reactive protein: targeting of the inhibitory activity of C4b-binding protein. Journal of Immunology. 2006;176(12):7612–7620. doi: 10.4049/jimmunol.176.12.7612. [DOI] [PubMed] [Google Scholar]
- 59.Jarva H, Jokiranta TS, Hellwage J, Zipfel PF, Meri S. Regulation of complement activation by C-reactive protein: targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8-11. Journal of Immunology. 1999;163(7):3957–3962. [PubMed] [Google Scholar]
- 60.Giannakis E, Jokiranta TS, Male DA, et al. A common site within factor H SCR 7 responsible for binding heparin, C-reactive protein and streptococcal M protein. European Journal of Immunology. 2003;33(4):962–969. doi: 10.1002/eji.200323541. [DOI] [PubMed] [Google Scholar]
- 61.Okemefuna AI, Nan R, Miller A, Gor J, Perkins SJ. Complement factor H binds at two independent sites to C-reactive protein in acute phase concentrations. Journal of Biological Chemistry. 2010;285(2):1053–1065. doi: 10.1074/jbc.M109.044529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edwards AO, Ritter R, III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 63.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 64.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(20):7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sjöberg AP, Trouw LA, Clark SJ, et al. The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. Journal of Biological Chemistry. 2007;282(15):10894–10900. doi: 10.1074/jbc.M610256200. [DOI] [PubMed] [Google Scholar]
- 67.Yu J, Wiita P, Kawaguchi R, et al. Biochemical analysis of a common human polymorphism associated with age-related macular degeneration. Biochemistry. 2007;46(28):8451–8461. doi: 10.1021/bi700459a. [DOI] [PubMed] [Google Scholar]
- 68.Boekhoorn SS, Vingerling JR, Witteman JCM, Hofman A, de Jong PTVM. C-reactive protein level and risk of aging macula disorder: the Rotterdam study. Archives of Ophthalmology. 2007;125(10):1396–1401. doi: 10.1001/archopht.125.10.1396. [DOI] [PubMed] [Google Scholar]
- 69.Nakayama S, Gewurz H, Holzer T. The role of the spleen in the protective effect of C-reactive protein in Streptococcus pneumoniae infection. Clinical and Experimental Immunology. 1983;54(2):319–326. [PMC free article] [PubMed] [Google Scholar]
- 70.Horowitz J, Volanakis JE, Briles DE. Blood clearance of Streptococcus pneumoniae by C-reactive protein. Journal of Immunology. 1987;138(8):2598–2603. [PubMed] [Google Scholar]
- 71.Szalai AJ, Briles DE, Volanakis JE. Role of complement in C-reactive-protein-mediated protection of mice from Streptococcus pneumoniae . Infection and Immunity. 1996;64(11):4850–4853. doi: 10.1128/iai.64.11.4850-4853.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mold C, Rodic-Polic B, Du Clos TW. Protection from Streptococcus pneumoniae infection by C-reactive protein and natural antibody requires complement but not Fcγ, receptors. Journal of Immunology. 2002;168(12):6375–6381. doi: 10.4049/jimmunol.168.12.6375. [DOI] [PubMed] [Google Scholar]
- 73.Nijmeijer R, Lagrand WK, Lubbers YTP, et al. C-reactive protein activates complement in infarcted human myocardium. American Journal of Pathology. 2003;163(1):269–275. doi: 10.1016/S0002-9440(10)63650-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Griselli M, Herbert J, Hutchinson WL, et al. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. Journal of Experimental Medicine. 1999;190(12):1733–1739. doi: 10.1084/jem.190.12.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nimmerjahn F, Ravetch JV. FcgammaRs in health and disease. Current Topics in Microbiology and Immunology. 2011;350:105–125. doi: 10.1007/82_2010_86. [DOI] [PubMed] [Google Scholar]
- 76.Zahedi K, Tebo JM, Siripont J, Klimo GF, Mortensen RF. Binding of human C-reactive protein to mouse macrophages is mediated by distinct receptors. Journal of Immunology. 1989;142(7):2384–2392. [PubMed] [Google Scholar]
- 77.Marnell LL, Mold C, Volzer MA, Burlingame RW, Du Clos TW. C-reactive protein binds to FcγRI in transfected COS cells. Journal of Immunology. 1995;155(4):2185–2193. [PubMed] [Google Scholar]
- 78.Tron K, Manolov DE, Röcker C, Kächele M, Torzewski J, Nienhaus GU. C-reactive protein specifically binds to Fcγ receptor type I on a macrophage-like cell line. European Journal of Immunology. 2008;38(5):1414–1422. doi: 10.1002/eji.200738002. [DOI] [PubMed] [Google Scholar]
- 79.Bodman-Smith KB, Melendez AJ, Campbell I, Harrison PT, Allen JM, Raynes JG. C-reactive protein-mediated phagocytosis and phospholipase D signalling through the high-affinity receptor for immunoglobulin G (FcγRI) Immunology. 2002;107(2):252–260. doi: 10.1046/j.1365-2567.2002.01481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Röcker C, Manolov DE, Kuzmenkina EV, et al. Affinity of C-reactive protein toward FcγRI is strongly enhanced by the γ-chain. American Journal of Pathology. 2007;170(2):755–763. doi: 10.2353/ajpath.2007.060734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bharadwaj D, Stein MP, Volzer M, Mold C, Du Clos TW. The major receptor for C-reactive protein on leukocytes is Fcγ receptor II. Journal of Experimental Medicine. 1999;190(4):585–590. doi: 10.1084/jem.190.4.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Manolov DE, Röcker C, Hombach V, Nienhaus GU, Torzewski J. Ultrasensitive confocal fluorescence microscopy of C-reactive protein interacting with FcγRIIa. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(12):2372–2377. doi: 10.1161/01.ATV.0000147407.17137.02. [DOI] [PubMed] [Google Scholar]
- 83.Warmerdam PAM, van de Winkel JGJ, Gosselin EJ, Capel PJA. Molecular basis for a polymorphism of human Fcγ receptor II (CD32) Journal of Experimental Medicine. 1990;172(1):19–25. doi: 10.1084/jem.172.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Warmerdam PAM, van de Winkel JGJ, Vlug A, Westerdaal NAC, Capel PJA. A single amino acid in the second Ig-like domain of the human Fcγ receptor II is critical for human IgG2 binding. Journal of Immunology. 1991;147(4):1338–1343. [PubMed] [Google Scholar]
- 85.Cooke GS, Aucan C, Walley AJ, et al. Association of Fcγ receptor IIa (CD32) polymorphism with severe malaria in West Africa. American Journal of Tropical Medicine and Hygiene. 2003;69(6):565–568. [PubMed] [Google Scholar]
- 86.Solé-Violán J, García-Laorden MI, Marcos-Ramos JA, et al. The Fcγ receptor IIA-H/H131 genotype is associated with bacteremia in pneumococcal community-acquired pneumonia. Critical Care Medicine. 2011;39(6):1388–1393. doi: 10.1097/CCM.0b013e31820eda74. [DOI] [PubMed] [Google Scholar]
- 87.Bredius RGM, Derkx BHF, Fijen CAP, et al. Fcγ receptor IIa (CD32) polymorphism in fulminant meningococcal septic shock in children. Journal of Infectious Diseases. 1994;170(4):848–853. doi: 10.1093/infdis/170.4.848. [DOI] [PubMed] [Google Scholar]
- 88.Salmon JE, Millard S, Schachter LA, et al. FcγRIIA alleles are heritable risk factors for lupus nephritis in African Americans. Journal of Clinical Investigation. 1996;97(5):1348–1354. doi: 10.1172/JCI118552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stein MP, Edberg JC, Kimberly RP, et al. C-reactive protein binding to FcγRIIa on human monocytes and neutrophils is allele-specific. Journal of Clinical Investigation. 2000;105(3):369–376. doi: 10.1172/JCI7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bodman-Smith KB, Gregory RE, Harrison PT, Raynes JG. FcgammaRIIa expression with FcgammaRI results in C-reactive protein- and IgG-mediated phagocytosis. Journal of Leukocyte Biology. 2004;75:1029–1035. doi: 10.1189/jlb.0703306. [DOI] [PubMed] [Google Scholar]
- 91.Mold C, Du Clos TW. C-reactive protein increases cytokine responses to Streptococcus pneumoniae through interactions with Fγy receptors. Journal of Immunology. 2006;176(12):7598–7604. doi: 10.4049/jimmunol.176.12.7598. [DOI] [PubMed] [Google Scholar]
- 92.Aas V, Sand KL, Asheim HC, Benestad HB, Iversen JG. C-reactive protein triggers calcium signalling in human neutrophilic granulocytes via FcgammaRIIa in an allele-specific way. Scandinavian Journal of Immunology. 2013;77(6):442–451. doi: 10.1111/sji.12049. [DOI] [PubMed] [Google Scholar]
- 93.Pepys MB, Gallimore JR, Lloyd J, et al. Isolation and characterization of pharmaceutical grade human pentraxins, serum amyloid P component and C-reactive protein, for clinical use. Journal of Immunological Methods. 2012;384:92–102. doi: 10.1016/j.jim.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Segal DM, Taurog JD, Metzger H. Dimeric immunoglobulin E serves as a unit signal for mast cell degranulation. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(7):2993–2997. doi: 10.1073/pnas.74.7.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gallo P, Gonçalves R, Mosser DM. The influence of IgG density and macrophage Fc (gamma) receptor cross-linking on phagocytosis and IL-10 production. Immunology Letters. 2010;133(2):70–77. doi: 10.1016/j.imlet.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lu J, Marjon KD, Marnell LL, et al. Recognition and functional activation of the human IgA receptor (FcαRI) by C-reactive protein. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(12):4974–4979. doi: 10.1073/pnas.1018369108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chi M, Tridandapani S, Zhong W, Coggeshall KM, Mortensen RF. C-reactive protein induces signaling through FcγRIIa on HL-60 granulocytes. Journal of Immunology. 2002;168(3):1413–1418. doi: 10.4049/jimmunol.168.3.1413. [DOI] [PubMed] [Google Scholar]
- 98.Yang J, Wezeman M, Zhang X, et al. Human C-reactive protein binds activating Fcγ receptors and protects Myeloma Tumor Cells from Apoptosis. Cancer Cell. 2007;12(3):252–265. doi: 10.1016/j.ccr.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 99.Lu J, Marjon KD, Mold C, du Clos TW, Sun PD. Pentraxins and Fc receptors. Immunological Reviews. 2012;250:230–238. doi: 10.1111/j.1600-065X.2012.01162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hurlimann J, Thorbecke GJ, Hochwald GM. The liver as the site of C-reactive protein formation. Journal of Experimental Medicine. 1966;123(2):365–378. doi: 10.1084/jem.123.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Toniatti C, Arcone R, Majello B, Ganter U, Arpaia G, Ciliberto G. Regulation of the human C-reactive protein gene, a major marker of inflammation and cancer. Molecular Biology and Medicine. 1990;7(3):199–212. [PubMed] [Google Scholar]
- 102.Zhang D, Sun M, Samols D, Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. Journal of Biological Chemistry. 1996;271(16):9503–9509. doi: 10.1074/jbc.271.16.9503. [DOI] [PubMed] [Google Scholar]
- 103.Agrawal A, Cha-Molstad H, Samols D, Kushner I. Transactivation of C-reactive protein by IL-6 requires synergistic interaction of CCAAT/enhancer binding protein β (C/EBPβ) and Rel p50. Journal of Immunology. 2001;166(4):2378–2384. doi: 10.4049/jimmunol.166.4.2378. [DOI] [PubMed] [Google Scholar]
- 104.Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. 2008;112(10):3959–3964. doi: 10.1182/blood-2008-05-155846. [DOI] [PubMed] [Google Scholar]
- 105.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. New England Journal of Medicine. 1999;340(6):448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 106.Pepys MB, Dash AC, Markham RE. Comparative clinical study of protein SAP (amyloid P component) and C-reactive protein in serum. Clinical and Experimental Immunology. 1978;32(1):119–124. [PMC free article] [PubMed] [Google Scholar]
- 107.Pepys MB, Baltz M, Gomer K. Serum amyloid P-component is an acute-phase reactant in the mouse. Nature. 1979;278(5701):259–261. doi: 10.1038/278259a0. [DOI] [PubMed] [Google Scholar]
- 108.Mold C, Gresham HD, Du Clos TW. Serum amyloid P component and C-reactive protein mediate phagocytosis through murine FcγRs. Journal of Immunology. 2001;166(2):1200–1205. doi: 10.4049/jimmunol.166.2.1200. [DOI] [PubMed] [Google Scholar]
- 109.Lin CS, Xia D, Yun JS, et al. Expression of rabbit C-reactive protein in transgenic mice. Immunology and Cell Biology. 1995;73(6):521–531. doi: 10.1038/icb.1995.82. [DOI] [PubMed] [Google Scholar]
- 110.Du Clos TW, Zlock LT, Hicks PS, Mold C. Decreased autoantibody levels and enhance survival of (NZB x NZW) F1 mice treated with C-reactive protein. Clinical Immunology and Immunopathology. 1994;70(1):22–27. doi: 10.1006/clin.1994.1005. [DOI] [PubMed] [Google Scholar]
- 111.Ciliberto G, Arcone R, Wagner EF, Rüther U. Inducible and tissue-specific expression of human C-reactive protein in transgenic mice. EMBO Journal. 1987;6(13):4017–4022. doi: 10.1002/j.1460-2075.1987.tb02745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jones NR, Pegues MA, McCrory MA, et al. Collagen-induced arthritis is exacerbated in C-reactive protein-deficient mice. Arthritis and Rheumatism. 2011;63(9):2641–2650. doi: 10.1002/art.30444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Teupser D, Weber O, Rao TN, Sass K, Thiery J, Jörg Fehling H. No reduction of atherosclerosis in C-reactive protein (CRP)-deficient mice. Journal of Biological Chemistry. 2011;286(8):6272–6279. doi: 10.1074/jbc.M110.161414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yuste J, Botto M, Bottoms SE, Brown JS. Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae . PLoS Pathogens. 2007;3(9):1208–1219. doi: 10.1371/journal.ppat.0030120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Botto M, Hawkins PN, Bickerstaff MCM, et al. Amyloid deposition is delayed in mice with targeted deletion of the serum amyloid P component gene. Nature Medicine. 1997;3(8):855–859. doi: 10.1038/nm0897-855. [DOI] [PubMed] [Google Scholar]
- 116.Bickerstaff MCM, Botto M, Hutchinson WL, et al. Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nature Medicine. 1999;5(6):694–697. doi: 10.1038/9544. [DOI] [PubMed] [Google Scholar]
- 117.Kindmark CO. In vitro binding of human C-reactive protein by some pathogenic bacteria and zymosan. Clinical and Experimental Immunology. 1972;11(2):283–289. [PMC free article] [PubMed] [Google Scholar]
- 118.Kindmark CO. Stimulating effect of C-reactive protein on phagocytosis of various species of pathogenic bacteria. Clinical and Experimental Immunology. 1971;8(6):941–948. [PMC free article] [PubMed] [Google Scholar]
- 119.Ganrot PO, Kindmark CO. C-reactive protein—a phagocytosis-promoting factor. Scandinavian Journal of Clinical and Laboratory Investigation. 1969;24(3):215–219. doi: 10.3109/00365516909080155. [DOI] [PubMed] [Google Scholar]
- 120.Mold C, Nakayama S, Holzer TJ. C-reactive protein is protective against Streptococcus pneumoniae in mice. Journal of Experimental Medicine. 1981;154(5):1703–1708. doi: 10.1084/jem.154.5.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mold C, Rodriguez W, Rodic-Polic B, Du Clos TW. C-reactive protein mediates protection from lipopolysaccharide through interactions with FcγR. Journal of Immunology. 2002;169(12):7019–7025. doi: 10.4049/jimmunol.169.12.7019. [DOI] [PubMed] [Google Scholar]
- 122.Szalai AJ, VanCott JL, McGhee JR, Volanakis JE, Benjamin WH., Jr. Human C-reactive protein is protective against fatal Salmonella enterica serovar Typhimurium infection in transgenic mice. Infection and Immunity. 2000;68(10):5652–5656. doi: 10.1128/iai.68.10.5652-5656.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gang TB, Hammond DJ, Jr., Singh SK, Ferguson DA, Jr., Mishra VK, Agrawal A. The phosphocholine-binding pocket on C-reactive protein is necessary for initial protection of mice against pneumococcal infection. Journal of Biological Chemistry. 2012;287(51):43116–43125. doi: 10.1074/jbc.M112.427310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Weiser JN, Pan N, McGowan KL, Musher D, Martin A, Richards J. Phosphorylcholine on the lipopolysaccharide of Haemophilus influenzae contributes to persistence in the respiratory tract and sensitivity to serum killing mediated by C-reactive protein. Journal of Experimental Medicine. 1998;187(4):631–640. doi: 10.1084/jem.187.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gould JM, Weiser JN. Expression of C-reactive protein in the human respiratory tract. Infection and Immunity. 2001;69(3):1747–1754. doi: 10.1128/IAI.69.3.1747-1754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Soma M, Tamaoki T, Kawano H, et al. Mice lacking serum amyloid p component do not necessarily develop severe autoimmune disease. Biochemical and Biophysical Research Communications. 2001;286(1):200–205. doi: 10.1006/bbrc.2001.5364. [DOI] [PubMed] [Google Scholar]
- 127.Kimura T, Tani S, Matsumoto YI, Takeda T. Serum amyloid P component is the shiga toxin 2-neutralizing factor in human blood. Journal of Biological Chemistry. 2001;276(45):41576–41579. doi: 10.1074/jbc.M107819200. [DOI] [PubMed] [Google Scholar]
- 128.Armstrong GD, Mulvey GL, Marcato P, et al. Human serum amyloid P component protects against Escherichia coli O157:H7 shiga toxin 2 in vivo: therapeutic implications for hemolytic-uremic syndrome. Journal of Infectious Diseases. 2006;193(8):1120–1124. doi: 10.1086/501472. [DOI] [PubMed] [Google Scholar]
- 129.Job ER, Bottazzi B, Gilbertson B, et al. Serum amyloid P is a sialylated glycoprotein inhibitor of influenza a viruses. PLoS One. 2013;8(3) doi: 10.1371/journal.pone.0059623.e59623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reading PC, Bozza S, Gilbertson B, et al. Antiviral activity of the long chain pentraxin PTX3 against influenza viruses. Journal of Immunology. 2008;180(5):3391–3398. doi: 10.4049/jimmunol.180.5.3391. [DOI] [PubMed] [Google Scholar]
- 131.Andersen O, Ravn KV, Sørensen IJ, Jonson G, Holm Nielsen E, Svehag SE. Serum amyloid P component binds to influenza A virus haemagglutinin and inhibits the virus infection in vitro. Scandinavian Journal of Immunology. 1997;46(4):331–337. doi: 10.1046/j.1365-3083.1997.d01-147.x. [DOI] [PubMed] [Google Scholar]
- 132.Horváth A, Andersen I, Junker K, et al. Serum amyloid P component inhibits influenza A virus infections: in vitro and in vivo studies. Antiviral Research. 2001;52(1):43–53. doi: 10.1016/s0166-3542(01)00158-9. [DOI] [PubMed] [Google Scholar]
- 133.Gilchrist KB, Garcia MC, Sobonya R, Lipke PN, Klotz SA. New features of invasive candidiasis in humans: amyloid formation by fungi and deposition of serum amyloid P component by the host. Journal of Infectious Diseases. 2012;206:1473–1478. doi: 10.1093/infdis/jis464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Culley FJ, Harris RA, Kaye PM, McAdam KPWJ, Raynes JG. C-reactive protein binds to a novel ligand on Leishmania donovani and increases uptake into human macrophages. Journal of Immunology. 1996;156(12):4691–4696. [PubMed] [Google Scholar]
- 135.Pied S, Nussler A, Pontet M, et al. C-reactive protein protects against preerythrocytic stages of malaria. Infection and Immunity. 1989;57(1):278–282. doi: 10.1128/iai.57.1.278-282.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Naik P, Voller A. Serum C-reactive protein levels and falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1984;78(6):812–813. doi: 10.1016/0035-9203(84)90027-0. [DOI] [PubMed] [Google Scholar]
- 137.Schuldt K, Esser C, Evans J, et al. FCGR2A functional genetic variant associated with susceptibility to severe malarial anaemia in Ghanaian children. Journal of Medical Genetics. 2010;47(7):471–475. doi: 10.1136/jmg.2009.073643. [DOI] [PubMed] [Google Scholar]
- 138.Zuniga R, Markowitz GS, Arkachaisri T, Imperatore EA, D’Agati VD, Salmon JE. Identification of IgG subclasses and C-reactive protein in lupus nephritis: the relationship between the composition of immune deposits and Fcγ receptor type IIA alleles. Arthritis and Rheumatism. 2003;48(2):460–470. doi: 10.1002/art.10930. [DOI] [PubMed] [Google Scholar]
- 139.Szalai AJ, Weaver CT, McCrory MA, et al. Delayed lupus onset in (NZB × NZW)F1 mice expressing a human C-reactive protein transgene. Arthritis and Rheumatism. 2003;48(6):1602–1611. doi: 10.1002/art.11026. [DOI] [PubMed] [Google Scholar]
- 140.Jabs WJ, Lögering BA, Gerke P, et al. The kidney as a second site of human C-reactive protein formation in vivo. European Journal of Immunology. 2003;33(1):152–161. doi: 10.1002/immu.200390018. [DOI] [PubMed] [Google Scholar]
- 141.Nakahara C, Kanemoto K, Saito N, et al. C-reactive protein frequently localizes in the kidney in glomerular diseases. Clinical Nephrology. 2001;55(5):365–370. [PubMed] [Google Scholar]
- 142.Pegues MA, McCrory MA, Zarjou A, Szalai AJ. C-reactive protein exacerbates renal ischemia-reperfusion injury. American Journal of Physiology. 2013;304(11):F1358–F1365. doi: 10.1152/ajprenal.00476.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li Z, Chung ACK, Zhou L, et al. C-reactive protein promotes acute renal inflammation and fibrosis in unilateral ureteral obstructive nephropathy in mice. Laboratory Investigation. 2011;91(6):837–851. doi: 10.1038/labinvest.2011.42. [DOI] [PubMed] [Google Scholar]
- 144.Dyck RF, Evans DJ, Lockwood CM. Amyloid P-component in human glomerular basement membrane. Abnormal patterns of immunofluorescent staining in glomerular disease. Lancet. 1980;2(8195):606–609. doi: 10.1016/s0140-6736(80)90281-0. [DOI] [PubMed] [Google Scholar]
- 145.Dyck RF, Lockwood CM, Kershaw M. Amyloid P-component is a constituent of normal human glomerular basement membrane. Journal of Experimental Medicine. 1980;152(5):1162–1174. doi: 10.1084/jem.152.5.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Castaño AP, Lin SL, Surowy T, et al. Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Science Translational Medicine. 2009;1(5, article ra13) doi: 10.1126/scitranslmed.3000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gitlin JD, Gitlin JI, Gitlin D. Localizing of C-reactive protein in synovium of patients with rheumatoid arthritis. Arthritis and Rheumatism. 1977;20(8):1491–1499. doi: 10.1002/art.1780200808. [DOI] [PubMed] [Google Scholar]
- 148.Parish WE. Studies on vasculitis. I. Immunoglobulins, 1C, C-reactive protein, and bacterial antigens in cutaneous vasculitis lesions. Clinical Allergy. 1971;1(1):97–109. doi: 10.1111/j.1365-2222.1971.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 149.Du Clos TW, Mold C, Paterson PY. Localization of C-reactive protein in inflammatory lesions of experimental allergic encephalomyelitis. Clinical and Experimental Immunology. 1981;43(3):565–573. [PMC free article] [PubMed] [Google Scholar]
- 150.Jewell WS, Marnell LL, Rokeach LA, Du Clos TW. C-reactive protein (CRP) binding to the Sm-D protein of snRNPS. Identification of a short polypeptide binding region. Molecular Immunology. 1993;30(8):701–708. doi: 10.1016/0161-5890(93)90141-w. [DOI] [PubMed] [Google Scholar]
- 151.Du Clos TW, Marnell L, Zlock LR, Burlingame RW. Analysis of the binding of C-reactive protein to chromatin subunits. Journal of Immunology. 1991;146(4):1220–1225. [PubMed] [Google Scholar]
- 152.Du Clos TW, Zlock LT, Marnell L. Definition of a C-reactive protein binding determinant on histones. Journal of Biological Chemistry. 1991;266(4):2167–2171. [PubMed] [Google Scholar]
- 153.Du Clos TW, Zlock LT, Rubin RL. Analysis of the binding of c-reactive protein to histones and chromatin. Journal of Immunology. 1988;141(12):4266–4270. [PubMed] [Google Scholar]
- 154.Rodriguez W, Mold C, Kataranovski M, Hutt J, Marnell LL, Du Clos TW. Reversal of ongoing proteinuria in autoimmune mice by treatment with C-reactive protein. Arthritis and Rheumatism. 2005;52(2):642–650. doi: 10.1002/art.20846. [DOI] [PubMed] [Google Scholar]
- 155.Rodriguez W, Mold C, Marnell LL, et al. Prevention and reversal of nephritis in MRL/lpr mice with a single injection of C-reactive protein. Arthritis and Rheumatism. 2006;54(1):325–335. doi: 10.1002/art.21556. [DOI] [PubMed] [Google Scholar]
- 156.Rodriguez W, Mold C, Kataranovski M, et al. C-reactive protein-mediated suppression of nephrotoxic nephritis: role of macrophages, complement, and Fcγ receptors. Journal of Immunology. 2007;178(1):530–538. doi: 10.4049/jimmunol.178.1.530. [DOI] [PubMed] [Google Scholar]
- 157.Marjon KD, Marnell LL, Mold C, Du Clos TW. Macrophages activated by C-reactive protein through FcγRI transfer suppression of immune thrombocytopenia. Journal of Immunology. 2009;182(3):1397–1403. doi: 10.4049/jimmunol.182.3.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Szalai AJ, Nataf S, Hu XZ, Barnum SR. Experimental allergic encephalomyelitis is inhibited in transgenic mice expressing human C-reactive protein. Journal of Immunology. 2002;168(11):5792–5797. doi: 10.4049/jimmunol.168.11.5792. [DOI] [PubMed] [Google Scholar]
- 159.Hu XZ, Wright TT, Jones NR, et al. Inhibition of experimental autoimmune encephalomyelitis in human C-reactive protein transgenic mice is FcγRIIB dependent. Autoimmune Diseases. 2011;2011:6 pages. doi: 10.4061/2011/484936.484936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jiang S, Xia D, Samols D. Expression of rabbit C-reactive protein in transgenic mice inhibits development of antigen-induced arthritis. Scandinavian Journal of Rheumatology. 2006;35(5):351–355. doi: 10.1080/03009740600757963. [DOI] [PubMed] [Google Scholar]
- 161.Bygrave AE, Rose KL, Cortes-Hernandez J, et al. Spontaneous autoimmunity in 129 and C57BL/6 mice-implications for autoimmunity described in gene-targeted mice. PLoS Biology. 2004;2(8, article E243) doi: 10.1371/journal.pbio.0020243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang W, Wu J, Qiao B, Xu W, Xiong S. Amelioration of lupus nephritis by serum amyloid P component gene therapy with distinct mechanisms varied from different stage of the disease. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0022659.e22659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang W, Xu W, Xiong S. Macrophage differentiation and polarization via phosphatidylinositol 3-kinase/Akt-ERK signaling pathway conferred by serum amyloid P component. Journal of Immunology. 2011;187(4):1764–1777. doi: 10.4049/jimmunol.1002315. [DOI] [PubMed] [Google Scholar]
- 164.Li X, Su K, Ji C, et al. Immune opsonins modulate BLyS/BAFF release in a receptor-specific fashion. Journal of Immunology. 2008;181(2):1012–1018. doi: 10.4049/jimmunol.181.2.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bell SA, Du Clos TW, Khursigara G, Picazo JJ, Rubin RL. Autoantibodies to cryptic epitopes of C-reactive protein and other acute phase proteins in the toxic oil syndrome. Journal of Autoimmunity. 1995;8(2):293–303. doi: 10.1006/jaut.1995.0022. [DOI] [PubMed] [Google Scholar]
- 166.Sjöwall C, Eriksson P, Almer S, Skogh T. Autoantibodies to C-reactive protein is a common finding in SLE, but not in primary Sjögren’s syndrome, rheumatoid arthritis or inflammatory bowel disease. Journal of Autoimmunity. 2002;19(3):155–160. doi: 10.1006/jaut.2002.0608. [DOI] [PubMed] [Google Scholar]
- 167.Sjöwall C, Bengtsson AA, Sturfelt G, Skogh T. Serum levels of autoantibodies against monomeric C-reactive protein are correlated with disease activity in systemic lupus erythematosus. Arthritis Research & Therapy. 2004;6(2):R87–R94. doi: 10.1186/ar1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sjöwall C, Cardell K, Boström EA, et al. High prevalence of autoantibodies to C-reactive protein in patients with chronic hepatitis C infection: association with liver fibrosis and portal inflammation. Human Immunology. 2012;73(4):382–388. doi: 10.1016/j.humimm.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 169.Janko C, Franz S, Munoz LE, et al. CRP/anti-CRP antibodies assembly on the surfaces of cell remnants switches their phagocytic clearance toward inflammation. Front Immunol. 2011;2(article 70) doi: 10.3389/fimmu.2011.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bhakdi S, Torzewski M, Klouche M, Hemmes M. Complement and atherogenesis: binding of CRP to degraded, nonoxidized LDL enhances complement activation. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(10):2348–2354. doi: 10.1161/01.atv.19.10.2348. [DOI] [PubMed] [Google Scholar]
- 171.Singh SK, Thirumalai A, Hammond DJ, Jr., et al. Exposing a hidden functional site of C-reactive protein by site-directed mutagenesis. Journal of Biological Chemistry. 2012;287(5):3550–3558. doi: 10.1074/jbc.M111.310011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Paul A, Ko KWS, Li L, et al. C-Reactive protein accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109(5):647–655. doi: 10.1161/01.CIR.0000114526.50618.24. [DOI] [PubMed] [Google Scholar]
- 173.Hirschfield GM, Gallimore JR, Kahan MC, et al. Transgenic human C-reactive protein is not proatherogenic in apolipoprotein E-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(23):8309–8314. doi: 10.1073/pnas.0503202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Trion A, de Maat MPM, Jukema JW, et al. No effect of C-reactive protein on early atherosclerosis development in apolipoprotein E∗3-Leiden/human C-reactive protein transgenic mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(8):1635–1640. doi: 10.1161/01.ATV.0000171992.36710.1e. [DOI] [PubMed] [Google Scholar]
- 175.Tennent GA, Hutchinson WL, Kahan MC, et al. Transgenic human CRP is not pro-atherogenic, pro-atherothrombotic or pro-inflammatory in apoE-/- mice. Atherosclerosis. 2008;196(1):248–255. doi: 10.1016/j.atherosclerosis.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 176.Torzewski M, Reifenberg K, Cheng F, et al. No effect of C-reactive protein on early atherosclerosis in LDLR−/−/human C-reactive protein transgenic mice. Thrombosis and Haemostasis. 2008;99(1):196–201. doi: 10.1160/TH07-10-0595. [DOI] [PubMed] [Google Scholar]
- 177.Ortiz MA, Campana GL, Woods JR, et al. Continuously-infused human C-reactive protein is neither proatherosclerotic nor proinflammatory in apolipoprotein E-deficient mice. Experimental Biology and Medicine. 2009;234(6):624–631. doi: 10.3181/0812-RM-347. [DOI] [PubMed] [Google Scholar]
- 178.Kovacs A, Tornvall P, Nilsson R, Tegnér J, Hamsten A, Björkegren J. Human C-reactive protein slows atherosclerosis development in a mouse model with human-like hypercholesterolemia. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(34):13768–13773. doi: 10.1073/pnas.0706027104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zacho J, Tybjærg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. New England Journal of Medicine. 2008;359(18):1897–1908. doi: 10.1056/NEJMoa0707402. [DOI] [PubMed] [Google Scholar]
- 180.Bharadwaj D, Mold C, Markham E, Du Clos TW. Serum amyloid P component binds to Fcγ receptors and opsonizes particles for phagocytosis. Journal of Immunology. 2001;166(11):6735–6741. doi: 10.4049/jimmunol.166.11.6735. [DOI] [PubMed] [Google Scholar]
- 181.Kilpatrick JM, Volanakis JE. Opsonic properties of C-reactive protein. Stimulation by phorbol myristate acetate enables human neutrophils to phagocytize C-reactive protein-coated cells. Journal of Immunology. 1985;134(5):3364–3370. [PubMed] [Google Scholar]
- 182.Mortensen RF, Osmand AP, Lint TF, Gewurz H. Interaction of C reactive protein with lymphocytes and monocytes: complement dependent adherence and phagocytosis. Journal of Immunology. 1976;117(3):774–781. [PubMed] [Google Scholar]
- 183.Ballou SP, Lozanski G. Induction of inflammatory cytokine release from cultured human monocytes by C-reactive protein. Cytokine. 1992;4(5):361–368. doi: 10.1016/1043-4666(92)90079-7. [DOI] [PubMed] [Google Scholar]
- 184.Tilg H, Vannier E, Vachino G, Dinarello CA, Mier JW. Antiinflammatory properties of hepatic acute phase proteins: preferential induction of interleukin 1 (IL-1) receptor antagonist over IL-1β synthesis by human peripheral blood mononuclear cells. Journal of Experimental Medicine. 1993;178(5):1629–1636. doi: 10.1084/jem.178.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pue CA, Mortensen RF, Marsh CB, Pope HA, Wewers MD. Acute phase levels of C-reactive protein enhance IL-1β and IL-1ra production by human blood monocytes but inhibit IL-1β and IL-1ra production by alveolar macrophages. Journal of Immunology. 1996;156(4):1594–1600. [PubMed] [Google Scholar]
- 186.Galve-de Rochemonteix B, Wiktorowicz K, Kushner I, Dayer JM. C-reactive protein increases production of IL-1α, IL-1β, and TNF-α, and expression of mRNA by human alveolar macrophages. Journal of Leukocyte Biology. 1993;53(4):439–445. doi: 10.1002/jlb.53.4.439. [DOI] [PubMed] [Google Scholar]
- 187.Bisoendial RJ, Kastelein JJP, Levels JHM, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circulation Research. 2005;96(7):714–716. doi: 10.1161/01.RES.0000163015.67711.AB. [DOI] [PubMed] [Google Scholar]
- 188.Pepys MB. CRP or not CRP? That is the question. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(6):1091–1094. doi: 10.1161/01.ATV.0000169644.88847.28. [DOI] [PubMed] [Google Scholar]
- 189.Colino J, Shen Y, Snapper CM. Dendritic cells pulsed with intact Streptococcus pneumoniae elicit both protein- and polysaccharide-specific immunoglobulin isotype responses in vivo through distinct mechanisms. Journal of Experimental Medicine. 2002;195(1):1–13. doi: 10.1084/jem.20011432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Thomas-Rudolph D, Du Clos TW, Snapper CM, Mold C. C-reactive protein enhances immunity to Streptococcus pneumoniae by targeting uptake to FcγR on dendritic cells. Journal of Immunology. 2007;178(11):7283–7291. doi: 10.4049/jimmunol.178.11.7283. [DOI] [PubMed] [Google Scholar]
- 191.Zhang R, Becnel L, Li M, Chen C, Yao Q. C-reactive protein impairs human CD14+ monocyte-derived dendritic cell differentiation, maturation and function. European Journal of Immunology. 2006;36(11):2993–3006. doi: 10.1002/eji.200635207. [DOI] [PubMed] [Google Scholar]
- 192.Frenzel H, Pries R, Brocks CP, Jabs WJ, Wittkopf N, Wollenberg B. Decreased migration of myeloid dendritic cells through increased levels of C-reactive protein. Anticancer Research B. 2007;27(6):4111–4115. [PubMed] [Google Scholar]
- 193.van Vré EA, Bult H, Hoymans VY, van Tendeloo VFI, Vrints CJ, Bosmans JM. Human C-reactive protein activates monocyte-derived dendritic cells and induces dendritic cell-mediated T-cell activation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(3):511–518. doi: 10.1161/ATVBAHA.107.157016. [DOI] [PubMed] [Google Scholar]
- 194.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nature Reviews Immunology. 2008;8(8):594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 195.Rönnblom L, Eloranta ML, Alm GV. The type I interferon system in systemic lupus erythematosus. Arthritis and Rheumatism. 2006;54(2):408–420. doi: 10.1002/art.21571. [DOI] [PubMed] [Google Scholar]
- 196.Mold C, Du Clos TW. C-reactive protein inhibits plasmacytoid dendritic cell interferon responses to autoantibody immune complexes. Arthritis & Rheumatism. 2013;65(7):1891–1901. doi: 10.1002/art.37968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang Y, Guo Y, Wang X, Huang J, Shang J, Sun S. Human serum amyloid P functions as a negative regulator of the innate and adaptive immune responses to DNA vaccines. Journal of Immunology. 2011;186(5):2860–2870. doi: 10.4049/jimmunol.1003641. [DOI] [PubMed] [Google Scholar]
- 198.Hokama Y, Coleman MK, Riley RF. In vitro effects of C-reactive protein on phagocytosis. Journal of bacteriology. 1962;83:1017–1024. doi: 10.1128/jb.83.5.1017-1024.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nagarajan S, Fifadara NH, Selvaraj P. Signal-specific activation and regulation of human neutrophil Fcγ receptors. Journal of Immunology. 2005;174(9):5423–5432. doi: 10.4049/jimmunol.174.9.5423. [DOI] [PubMed] [Google Scholar]
- 200.Kew RR, Hyers TM, Webster RO. Human C-reactive protein inhibits neutrophil chemotaxis in vitro: possible implications for the adult respiratory distress syndrome. Journal of Laboratory and Clinical Medicine. 1990;115(3):339–345. [PubMed] [Google Scholar]
- 201.Heuertz RM, Piquette CA, Webster RO. Rabbits with elevated serum C-reactive protein exhibit diminished neutrophil infiltration and vascular permeability in C5a-induced alveolitis. American Journal of Pathology. 1993;142(1):319–328. [PMC free article] [PubMed] [Google Scholar]
- 202.Heuertz RM, Tricomi SM, Ezekiel UR, Webster RO. C-reactive protein inhibits chemotactic peptide-induced p38 mitogen-activated protein kinase activity and human neutrophil movement. Journal of Biological Chemistry. 1999;274(25):17968–17974. doi: 10.1074/jbc.274.25.17968. [DOI] [PubMed] [Google Scholar]
- 203.Zhong W, Zen Q, Tebo J, Schlottmann K, Coggeshall M, Mortensen RF. Effect of human C-reactive protein on chemokine and chemotactic factor-induced neutrophil chemotaxis and signaling. Journal of Immunology. 1998;161(5):2533–2540. [PubMed] [Google Scholar]
- 204.Zeller JM, Sullivan BL. Monoclonal antibody to the type II Fc receptor for human IgG blocks potentiation of monocyte and neutrophil IgG-induced respiratory burst activation by aggregated C-reactive protein. Cellular Immunology. 1993;149(1):144–154. doi: 10.1006/cimm.1993.1143. [DOI] [PubMed] [Google Scholar]
- 205.Zeller JM, Sullivan BL. C-reactive protein selectively enhances the intracellular generation of reactive oxygen products by IgG-stimulated monocytes and neutrophils. Journal of Leukocyte Biology. 1992;52(4):449–455. doi: 10.1002/jlb.52.4.449. [DOI] [PubMed] [Google Scholar]
- 206.Nagarajan S, Venkiteswaran K, Anderson M, Sayed U, Zhu C, Selvaraj P. Cell-specific, activation-dependent regulation of neutrophil CD32A ligand-binding function. Blood. 2000;95(3):1069–1077. [PubMed] [Google Scholar]
- 207.Pasceri V, Willerson JT, Yeh ETH. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102(18):2165–2168. doi: 10.1161/01.cir.102.18.2165. [DOI] [PubMed] [Google Scholar]
- 208.Venugopal SK, Devaraj S, Yuhanna I, Shaul P, Jialal I. Demonstration that C-reactive protein decreases eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106(12):1439–1441. doi: 10.1161/01.cir.0000033116.22237.f9. [DOI] [PubMed] [Google Scholar]
- 209.Devaraj S, Du Clos TW, Jialal I. Binding and internalization of C-reactive protein by Fcgamma receptors on human aortic endothelial cells mediates biological effects. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(7):1359–1363. doi: 10.1161/01.ATV.0000168573.10844.ae. [DOI] [PubMed] [Google Scholar]
- 210.Clapp BR, Hirschfield GM, Storry C, et al. Inflammation and endothelial function: direct vascular effects of human C-reactive protein on nitric oxide bioavailability. Circulation. 2005;111(12):1530–1536. doi: 10.1161/01.CIR.0000159336.31613.31. [DOI] [PubMed] [Google Scholar]
- 211.Danenberg HD, Szalai AJ, Swaminathan RV, et al. Increased thrombosis after arterial injury in human C-reactive protein-transgenic mice. Circulation. 2003;108(5):512–515. doi: 10.1161/01.CIR.0000085568.13915.1E. [DOI] [PubMed] [Google Scholar]
- 212.Teoh H, Quan A, Lovren F, et al. Impaired endothelial function in C-reactive protein overexpressing mice. Atherosclerosis. 2008;201(2):318–325. doi: 10.1016/j.atherosclerosis.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 213.Danenberg HD, Grad E, Swaminathan RV, et al. Neointimal formation is reduced after arterial injury in human crp transgenic mice. Atherosclerosis. 2008;201(1):85–91. doi: 10.1016/j.atherosclerosis.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Xing D, Hage FG, Chen YF, et al. Exaggerated neointima formation in human C-reactive protein transgenic mice is IgG Fc receptor type I (FcγRI)-dependent. American Journal of Pathology. 2008;172(1):22–30. doi: 10.2353/ajpath.2008.070154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hage FG, Oparil S, Xing D, Chen YF, McCrory MA, Szalai AJ. C-reactive protein-mediated vascular injury requires complement. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(6):1189–1195. doi: 10.1161/ATVBAHA.110.205377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Blaschke F, Bruemmer D, Yin F, et al. C-reactive protein induces apoptosis in human coronary vascular smooth muscle cells. Circulation. 2004;110(5):579–587. doi: 10.1161/01.CIR.0000136999.77584.A2. [DOI] [PubMed] [Google Scholar]
- 217.Bisoendial RJ, Kastelein JJP, Stroes ESG. C-reactive protein and atherogenesis: from fatty streak to clinical event. Atherosclerosis. 2007;195(2):e10–e18. doi: 10.1016/j.atherosclerosis.2007.04.053. [DOI] [PubMed] [Google Scholar]
- 218.Devaraj S, Singh U, Jialal I. The evolving role of C-reactive protein in atherothrombosis. Clinical Chemistry. 2009;55(2):229–238. doi: 10.1373/clinchem.2008.108886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Casas JP, Shah T, Hingorani AD, Danesh J, Pepys MB. C-reactive protein and coronary heart disease: a critical review. Journal of Internal Medicine. 2008;264(4):295–314. doi: 10.1111/j.1365-2796.2008.02015.x. [DOI] [PubMed] [Google Scholar]
- 220.Dehghan A, Dupuis J, Barbalic M, et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation. 2011;123(7):731–738. doi: 10.1161/CIRCULATIONAHA.110.948570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Rhodes B, Fürnrohr BG, Vyse TJ. C-reactive protein in rheumatology: biology and genetics. Nature Reviews Rheumatology. 2011;7(5):282–289. doi: 10.1038/nrrheum.2011.37. [DOI] [PubMed] [Google Scholar]
- 222.Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell. 2012;148(6):1242–1257. doi: 10.1016/j.cell.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Russell AI, Cunninghame Graham DS, Shepherd C, et al. Polymorphism at the C-reactive protein locus influences gene expression and predisposes to systemic lupus erythematosus. Human Molecular Genetics. 2004;13(1):137–147. doi: 10.1093/hmg/ddh021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jönsen A, Gunnarsson I, Gullstrand B, et al. Association between SLE nephritis and polymorphic variants of the CRP and FcγRIIIa genes. Rheumatology. 2007;46(9):1417–1421. doi: 10.1093/rheumatology/kem167. [DOI] [PubMed] [Google Scholar]
- 225.Pepys MB, Hirschfield GM. C-reactive protein: a critical update. Journal of Clinical Investigation. 2003;111(12):1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sabatine MS, Morrow DA, Jablonski KA, et al. Prognostic significance of the Centers for Disease Control/American Heart Association high-sensitivity C-reactive protein cut points for cardiovascular and other outcomes in patients with stable coronary artery disease. Circulation. 2007;115(12):1528–1536. doi: 10.1161/CIRCULATIONAHA.106.649939. [DOI] [PubMed] [Google Scholar]
- 227.Kaptoge S, Di Angelantonio E, Pennells L, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. New England Journal of Medicine. 2012;367:1310–1320. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ridker PM, Kastelein JJ, Genest J, Koenig W. C-reactive protein and cholesterol are equally strong predictors of cardiovascular risk and both are important for quality clinical care. European Heart Journal. 2013;34:1258–1261. doi: 10.1093/eurheartj/eht022. [DOI] [PubMed] [Google Scholar]
- 229.Lobo SMA, Lobo FRM, Peres Bota D, et al. C-reactive protein levels correlate with mortality and organ failure in critically III patients. Chest. 2003;123(6):2043–2049. doi: 10.1378/chest.123.6.2043. [DOI] [PubMed] [Google Scholar]
- 230.Wolbink GJ, Bossink AWJ, Groeneveld ABJ, de Groot MCM, Thijs LG, Hack CE. Complement activation in patients with sepsis is in part mediated by C-reactive protein. Journal of Infectious Diseases. 1998;177(1):81–87. doi: 10.1086/513803. [DOI] [PubMed] [Google Scholar]
- 231.Unnewehr H, Rittirsch D, Sarma JV, et al. Changes and regulation of the C5a receptor on neutrophils during septic shock in humans. Journal of Immunology. 2013;190(8):4215–4225. doi: 10.4049/jimmunol.1200534. [DOI] [PubMed] [Google Scholar]
- 232.Jones SA, Novick D, Horiuchi S, Yamamoto N, Szalai AJ, Fuller GM. C-reactive protein: a physiological activator of interleukin 6 receptor shedding. Journal of Experimental Medicine. 1999;189(3):599–604. doi: 10.1084/jem.189.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Xia D, Samols D. Transgenic mice expressing rabbit C-reactive protein are resistant to endotoxemia. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(6):2575–2580. doi: 10.1073/pnas.94.6.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Black S, Wilson A, Samols D. An intact phosphocholine binding site is necessary for transgenic rabbit C-reactive protein to protect mice against challenge with platelet-activating factor. Journal of Immunology. 2005;175(2):1192–1196. doi: 10.4049/jimmunol.175.2.1192. [DOI] [PubMed] [Google Scholar]
- 235.Chae MR, Park BH, Kim JS, Rho HW, Park JW, Kim HR. Protective effect of C-reactive protein against the lethality induced by Vibrio vulnificus lipopolysaccharide. Microbiology and Immunology. 2000;44(5):335–340. doi: 10.1111/j.1348-0421.2000.tb02503.x. [DOI] [PubMed] [Google Scholar]
- 236.Gerber JS, Mosser DM. Reversing lipopolysaccharide toxicity by ligating the macrophage Fcγ receptors. Journal of Immunology. 2001;166(11):6861–6868. doi: 10.4049/jimmunol.166.11.6861. [DOI] [PubMed] [Google Scholar]
- 237.Marsik C, Sunder-Plassmann R, Jilma B, et al. The C-reactive protein +1444C/T alteration modulates the inflammation and coagulation response in human endotoxemia. Clinical Chemistry. 2006;52(10):1952–1957. doi: 10.1373/clinchem.2006.069823. [DOI] [PubMed] [Google Scholar]
- 238.Xiao W, Mindrinos MN, Seok J, et al. A genomic storm in critically injured humans. Journal of Experimental Medicine. 2011;208(13):2581–2590. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.West SD, Goldberg D, Ziegler A, Krencicki M, Du Clos TW, Mold C. Transforming growth factor-beta, macrophage colony-stimulating factor and C-reactive protein levels correlate with CD14(high)CD16+ monocyte induction and activation in trauma patients. PLoS One. 2012;7 doi: 10.1371/journal.pone.0052406.e52406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Tennent GA, Lovat LB, Pepys MB. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(10):4299–4303. doi: 10.1073/pnas.92.10.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bodin K, Ellmerich S, Kahan MC, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468(7320):93–97. doi: 10.1038/nature09494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Gillmore JD, Tennent GA, Hutchinson WL, et al. Sustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosis. British Journal of Haematology. 2010;148(5):760–767. doi: 10.1111/j.1365-2141.2009.08036.x. [DOI] [PubMed] [Google Scholar]
- 243.Koistoe SE, Ridha BH, Bellotti V, et al. Molecular dissection of Alzheimer’s disease neuropathology by depletion of serum amyloid P component. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(18):7619–7623. doi: 10.1073/pnas.0902640106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. Journal of Immunology. 2003;171(10):5537–5546. doi: 10.4049/jimmunol.171.10.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Crawford JR, Pilling D, Gomer RH. FcgammaRI mediates serum amyloid P inhibition of fibrocyte differentiation. Journal of Leukocyte Biology. 2012;92:699–711. doi: 10.1189/jlb.0112033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Haudek SB, Trial J, Xia Y, Gupta D, Pilling D, Entman ML. Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(29):10179–10184. doi: 10.1073/pnas.0804910105. [DOI] [PMC free article] [PubMed] [Google Scholar]