

Abstract
The current antiretroviral therapy (ART) can effectively reduce plasma HIV loads to undetectable levels, but cannot eliminate latently infected resting memory CD4 T cells that persist for the lifetime of infected patients. Therefore, designing new therapeutic approaches to eliminate these latently infected cells or the cells that produce HIV upon reactivation from latency is a priority in the ART era in order to progress to a cure of HIV. Here, we show that “designer” T cells expressing chimeric antigen receptor (CAR), CD4-CD28-CD3ζ, can target and kill HIV Env-expressing cells. Further, they secrete effector cytokines upon contact with HIV Env+ target cells that can reactivate latent HIV in a cell line model, thereby exposing those cells to recognition and killing by anti-HIV CAR+ T cells. Taken to the limit, this process could form the basis for an eventual functional or sterilizing cure for HIV in patients.
Keywords: HIV-1, gene-modified T cells, designer T cells, cytotoxicity, CD4-CD28-CD3ζ, chimeric antigen receptor, reactivation of latent HIV
Introduction
The currently available ART used for the treatment of HIV patients is highly effective in lowering plasma viral loads even to undetectable levels (Gulick et al., 1997; Hammer et al., 1997; Luzuriaga et al., 1997). However, apart from its toxicity during long-term use (Hawkins, 2010) and high cost (Hill, Cho, and Mrus, 2011), it possesses a limitation in that it cannot target or eliminate latent HIV residing in resting CD4 T cells in patients (Chun et al., 1997; Finzi et al., 1999; Finzi et al., 1997; Wong et al., 1997). These latently infected cells are extremely stable and therefore persist in vivo, even after prolonged suppressive therapy (Siliciano et al., 2003), posing a major obstacle to HIV's cure by using ART. Soon after therapy is withdrawn, viral loads return to pretreatment levels in most patients (Davey et al., 1999; Garcia et al., 1999; Mata et al., 2005). To achieve the complete eradication of HIV from patients, it is essential to eliminate all HIV reservoirs (Bagasra et al., 1996; Lambotte et al., 2000; Sonza et al., 2001; Zalar et al., 2010; Zhu et al., 2002), including latently infected resting CD4 T cells, from the body. Since it became clear that ART is unable to achieve this (Siliciano et al., 2003), new therapeutic approaches are required to eliminate or control the pool of these HIV reservoirs in patients.
In the field of T cell immunotherapy, chimeric antigen receptors (CARs) are created by directly linking viral or tumor antigen binding domains of antibodies or ligands with the activating signaling domains of CD3ζ or other receptors (Jena, Dotti, and Cooper, 2010; Ma, Gonzalo-Daganzo, and Junghans, 2002; Sadelain, 2009; Yang et al., 2007). The CAR-expressing T cells are often referred as “designer T cells" (dTc). Previously, HIV Env-specific, chimeric CD4-CD3ζ receptor-expressing dTc were found to efficiently target and kill HIV-infected or HIV-Env expressing cells in vitro (Roberts et al., 1994; Yang et al., 1997). In vivo, however, such a “1st generation” construct was later tested in phase I and II clinical trials without success (Deeks et al., 2002; Mitsuyasu et al., 2000; Walker et al., 2000).
Over the recent decade, there have been significant advances in our understanding of signaling requirements for effective T cell function and an appreciation of the role of costimulation, e.g., through CD28 that provides a co-stimulatory signal important for cell proliferation, cytokine production and cell survival (Beecham et al., 2000; Boise et al., 1995; Emtage et al., 2008; Green et al., 1995). Recently, we sought to apply these lessons in creating a chimeric receptor of advanced design (2nd generation) that consists of an extracellular CD4 domain attached with CD28 region, followed by the cytoplasmic effector domain of CD3ζ (namely, CD4-CD28-CD3ζ), adding CD28 signaling to the 1st generation format.
During the course of characterizing our newly designed CD4-dTc product, we examined its effects on latently infected cells in the ACH-2 cell line model of HIV latency. As expected, the CD4-dTc could target and kill control HIV Env+ cells and HIV-infected H9 or primary T cells. However, to our surprise, with ACH-2 targets, CD4-dTc cleared not only the small fraction (~5–10%) of cells making HIV in the total population of ACH-2 cells, but also nearly eliminated the entire latent cell population. This clearance of latently infected cells appeared to proceed in three steps: (i) CD4-dTc were stimulated initially by small numbers of HIV-expressing ACH-2 cells to release TNFα into the coculture supernatants, (ii) locally released TNFα induced reactivation of latent HIV in neighboring ACH-2 cells, and finally (iii) the newly virus-expressing ACH-2 cells were targeted by the same CD4-dTc population for elimination.
Results
Chimeric CD4-CD28-CD3ζ receptors
The designer T cells applied unsuccessfully in the prior clinical trials mentioned above (Deeks et al., 2002; Walker et al., 2000) possessed a CAR of the form CD4-CD3ζ, with CD4 extracellular domain to engage gp120 on the surface of virus-infected cells and CD3ζ cytoplasmically to activate T cell “Signal 1” for killing the infected targets. The construct of our design possesses the transmembrane and partial extracellular domain of CD28 (Figure 1A) that mediates dimerization of chimeric molecules (Figure 1B) and the cytoplasmic domain that adds “Signal 2” co-stimulation to transmit Signal 1+2 on gp120 engagement. We refer to the modified T cells with this new construct as 2nd generation dTc (i.e., CD4-dTc).
Figure 1.
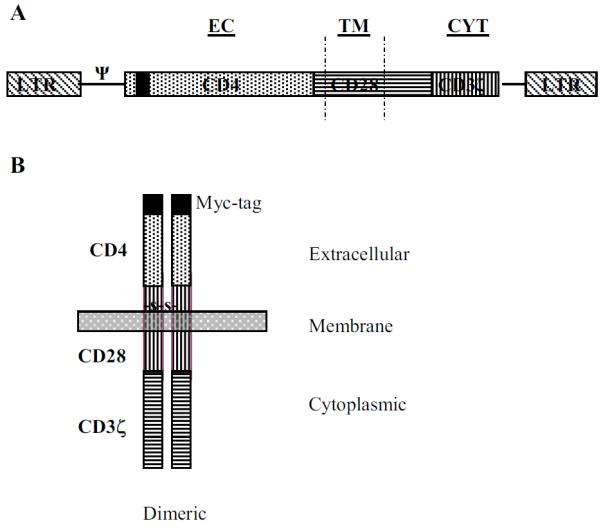
Schematic representations. (A) Retroviral vector and (B) cell-surface expression pattern of CD4-CD28-CD3ζ chimeric receptor. ψ represents retroviral packaging signal. EC, TM and CYT denote extracellular, transmembrane, and cytoplasmic portions of the construct. A 10 amino acid Myc-tag is located at the N-terminus.
Cell surface expression of CD4-CD28-CD3 in human T cells
Stimulated peripheral blood mononuclear cells (PBMCs)-derived from normal donors were transduced with recombinant retrovirus. To check the cell surface for CAR expression, cells were double-stained with anti-CD4 and anti-CD8 antibodies and analyzed by flow cytometry. The majority of CD8 T cells that were co-stained with these antibodies represented CAR expressing cells, estimated at 54% (Figure 2B). Because CD4 antibodies used in these stainings could not distinguish between natural versus engineered CD4 molecules expressed on transduced CD4 T cells, myc staining allowed us to judge the modification of CD4 T cells. When analyzed by staining with antibodies to myc-tag, a similar percentage (53%) of transduced CD4 T cells was separately determined (see Figure 2D). Stimulated PBMCs were also modified with irrelevant anti-carcinoembryonic antigen (αCEA) 2nd generation CAR (Emtage et al., 2008) (Figure 2F) for use as negative control dTc.
Figure 2.

Expression of CD4-CD28-CD3ζ in transduced PBMCs. Transduced and control PBMCs were stained with antibodies (A, B: CD4 vs CD8; C, D: Myc vs CD8) and analyzed by flow cytometry. CD4-CAR-modified CD8 T cells in panel B was 54%, after subtracting backgrounds observed in panel A that displays naturally present double-positive CD4+CD8+ T cells. Myc staining in Panel D shows 51% Myc+ CD8 T cells and 53% Myc+ CD4 T cells (CD8-cells), after subtracting background myc-staining derived from C and non-T cells derived from B. See Materials and Methods for calculations. Panels A, B, D pertain to a single donor PBMC activation and transduction; panel C is from a different donor PBMC activation to assess non-specific myc staining. Panel E and F represent unmodified and modified PBMCs with anti-CEAscFv-CD28-CD3ζ construct, respectively. The APC-labeled WI2 antibody reacted with the modified T cells that represent αCEA-dTc. About 25% T cells were modified (for calculation, see materials and methods).
Cytotoxic effects of CD4-dTc on cells expressing HIV Env
To determine if CD4-dTc as effectors can kill target cells expressing HIV Env, we cocultured equal numbers of CD4-dTc and CEM-Env+ cells. In conventional short-term chromium-release assays, we observed rates of 15–30% specific killing of HIV Env+ targets over 5h (not shown). To gain an impression of the extent of killing that could be obtained, we examined 24–48h time periods using manual counting of target cells when chromium-release backgrounds would be too high (Emtage et al., 2003; Junghans, 1990). As seen in Figure 3A, the number of live CEM-Env+ cells when cocultured with CD4-dTc was reduced >95% in comparison to targets in control cocultures. These data establish that CD4-dTc but not unmodified T cells preferentially target and kill HIV Env+ cells in vitro.
Figure 3.
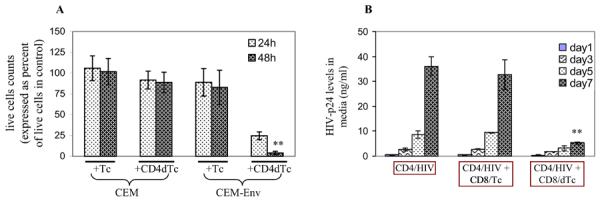
Cytotoxic effect of gene-modified T cells (dTc) on target cells. (A) Target cell killing. Mock-transduced (Tc) or CD4-CAR-transduced (CD4-dTc) PBMCs were mixed with target cells, CEM or CEM-Env, at 1:1 ratio and cultured for 2 days without exogenous IL2 in media. Live cells were counted on day 1 and day 2, and the values were normalized with the corresponding target cells cultured alone, taken as 100%. The data pooled from two independent experiments performed in duplicate are shown. Asterisks (**) indicate p<0.001. (B) Viral titer suppression. HIVJRCSF-infected primary CD4 T cells were mixed with mock-transduced CD8 T cells (CD8/Tc) or CD4-CAR-transduced CD8 T cells (CD8/dTc) at 1:1 ratio and cultured for 7 days. On days indicated, culture media were tested for HIV-p24 by ELISA (Advanced Bioscience Laboratories, Inc.). A representative data set from one of three experiments is shown. Asterisks (**) represent p<0.03.
A consequence of infected cell killing should be a reduced virus production in culture. To test CD4-dTc for their ability to control HIV replication in primary T cell cultures, we infected stimulated CD4 T cells from normal donor with a CCR5-dependent virus strain, HIVJRCSF, at moi ~0.1 and cultured for 5 days. In the meantime, stimulated CD8 T cells from the same donor were transduced with CD4-CAR vector and cultured for two days, and then incubated with the HIV-infected CD4 T cells. As a surrogate for virus production, culture supernatants were harvested and assayed for HIV-p24 levels by ELISA. As can be seen in Figure 3B, the rapid increases in p24 levels were observed to day 7 in infected CD4 T cell cultures, with or without control activated CD8 T cells (CD8/Tc) co-cultures. However, the level of p24 in the mixture with gene-modified CD8 T cells (CD8/dTc) was >6-fold lower than in control cultures. These data suggest that the gene-modified CD8 T cells can control HIV replication and spread when mixed with infected primary CD4 T cells (Figure 3B), presumably by targeting and killing specifically the infected cells in culture. The levels of p24 suppression could not be explained by any non-specific cytolytic (Grimm et al., 1982) or non-cytolytic suppressive effect (Rosok et al., 1997; Tsuchie et al., 1997) caused by CD8/dTc, because no similar effect was seen with CD8/Tc (Figure 3B, middle).
To confirm further the direct killing of HIV infected cells, CD4-dTc were used to treat H9 T cell line infected with HIV213, a CXCR4-dependent virus (Song, Li, and Cloyd, 1996), and then the cocultures were evaluated by flow cytometry. To distinguish the targets from the effectors in the assay, 7-day infected H9 cells were labeled with carboxyfluorescein succinimidyl ester (CFSE). When assayed by intracellular staining with anti-p24 antibodies prior to mixing, these H9 cultures showed >90% infection (not shown). Then infected target cells (i.e., H9/213 cells) were mixed with mock unmodified T cells (Tc) or αCEA-dTc (Emtage et al., 2008) as controls (see Figure 4A), or with CD4-dTc at 1:1 ratio and incubated for 22h at 37°C. The αCEA-dTc were T cells modified with αCEAscFv-CD28-CD3ζ (see Figure 2F) that could produce effector cytokines in response to CEA-expressing cells and kill them (Emtage et al., 2008). The exogenous IL2 was not added to media in these assays in order to reduce lymphokine activated killer (LAK) activity of stimulated primary T cells (Grimm et al., 1982). In parallel, equivalent numbers of effector and target cells were cultured separately, and 22h later they were mixed (served as 0h time point). Cell-mixtures were stained intracellularly with anti-HIV-p24 antibody and evaluated by flow cytometry. As can be seen in Figure 4A, H9/213 target cells were 96% reduced upon 22h of coculture with CD4-dTc (see right, 3rd panel from top), compared to control cocultures with Tc or αCEA-dTc (top two panels on right). These data demonstrated that the observed killing of target cells was due to the T cells modified with CD4-CAR construct (i.e., CD4-dTc), but not because of mere gene-modification of these cells with any non-specific construct by retroviral transduction. The cytotoxic effect of CD4-dTc could be specifically reversed when these effectors were preincubated with an HIV-blocking anti-human CD4 (domain I) antibody (20μg/ml) for 1h (Figure 4A, 2nd panel from bottom), demonstrating that interaction between the CAR CD4 and HIV Env is crucial for CD4-dTc's cytotoxicity towards HIV-infected target cells.
Figure 4.


dTc kill HIV-infected cells. (A) Specific CAR is required for dTc killing. CFSE-labeled H9/213 cells were mixed with Tc or αCEA-dTc or CD4-dTc at 1:1 ratio and incubated for 22h. Then cell-mixtures were stained intracellularly with anti-HIV-p24 antibody and analyzed by flow cytometry. Bottom three panels show the effect of HIV-blocking anti-human CD4 antibody or anti-human HLA-DR antibody or Concanamycin A (CMA) (see Materials and Methods). Raw counts of p24+ H9 cells are represented in upper right quadrants for control and experimental samples at 22 hr. Percent specific lysis expressed in terms of target cell loss was calculated as described in Materials and Methods. (B) Lack of killing of uninfected DR+ H9 cells by CD4-dTc. CFSE-labeled H9 cells were mixed with Tc or CD4-dTc at 1:1 and incubated for 22h and analyzed by flow cytometry as in A. Numbers shown in lower right quadrants represent raw counts of H9 cells at 22 hr. (C) Expression of HLA-DR on various cells. Cells were stained with anti-human HLA-DR antibody or matched isotype control antibody and analyzed by flow cytometry. The stainings with isotype control antibody are shown by histograms to the left (top panels) or as overlapped with HLA-DR staining (bottom panels). Stimulated PBMCs cultured for two weeks in media with 300U/ml IL-2 before staining with antibody.
CD4 receptors are known to interact with HLA-DR molecules, albeit at much lower affinity (Kd >100μM) than with HIV Env (Kd ~6nM) (Gao, Rao, and Bell, 2002; Lasky et al., 1987; Thali et al., 1991; Wang et al., 2001; Weber and Karjalainen, 1993). Because H9 cells express HLA-DR (Figure 4C), we tested whether CD4-CAR/HLA-DR interaction contributed to the CD4-dTc-mediated killing of target (H9/213) cells in Figure 4A. Our results showed no loss of cytotoxicity against infected H9/213 cells with a blocking anti-HLA-DR antibody (Fu and Karr, 1994; Wang et al., 1999) (Figure 4A, 3rd panel from bottom). Further, CD4-dTc exhibited no cytotoxicity towards HLA-DR+ uninfected H9 cells (Figure 4B, lower right). Finally, killing of CEM-Env+ cells, which are negative for HLA-DR, is highly efficient with the CD4-dTc, indicating no obligate role for HLA class II in the killing and recognition of gp120+ targets. Overall, these data rule out a role of HLA-DR/CD4-CAR interactions in the observed killing of infected target cells by CD4-dTc.
During the 22h incubation period, CD4 T cells in Tc or in αCEA-dTc became infected with HIV produced by the H9/213 targets, as expected (Figure 4A, left upper quadrants). While a fraction of CD4 T cells or CD4-CAR-modified CD8 T cells in CD4-dTc population would similarly be infected, the CD4-dTc were able to perform self-targeting and -killing for an in-vitro “self-cure” as reflected by the absence of p24+ effectors after 22h (Figure 4A, compare upper left quadrants in top two right panels with that in 3rd right panel from top).
In attempt to understand the mechanisms of CD4-dTc-mediated killing of target cells, we used Concanamycin A (CMA) in our assays to inhibit vacuolar type H+ATPase important for perforin-based cytolytic activity of CTLs (Emtage et al., 2003; Kataoka et al., 1996). CMA partially suppressed the killing ability of CD4-dTc towards HIV infected cells (from 96% to 68% killing; Figure 4A), suggesting that the cytolytic effector mechanisms of these CD4-dTc are at least partly mediated by perforin, as noted with HIV-specific CTLs present in HIV controllers (Hersperger et al., 2010; Saez-Cirion et al., 2009).
HIV-specific gene-modified T cell-mediated killing of a latently infected cell line, ACH-2
Because HIV-Env expressing cells were found to be targeted and killed by CD4-CAR modified T cells (as shown above), we sought to examine their effects on ACH-2, a latently HIV infected, transformed CEM cell line (Clouse et al., 1989; Folks et al., 1989). Approximately 5% of these cells in culture express HIV constitutively, whereas other ~95% cells remain latent. We speculated that the CD4-dTc would eliminate this positive cell fraction, while sparing p24-negative cells with latent HIV.
We cocultured ACH-2 cells with CD4-dTc or Tc in 1:1 proportion for 44h. Unexpectedly, instead of eliminating only the small 5% virus expressing fraction of ACH-2 cells that we predicted, 85% of ACH-2 cells were killed by CD4-dTc (Figure 5, compare ovals in bottom panels). By the preceding control tests, it is apparent that this killing must have occurred in an antigen-specific manner, i.e., through CD4-CAR/HIV Env interaction. To explain this CD4-dTc-specific lysis of latently infected ACH-2 cells, we speculated that CD4-dTc somehow reactivated latent HIV in these cells, making them targets for CD4-dTc-mediated killing in culture.
Figure 5.
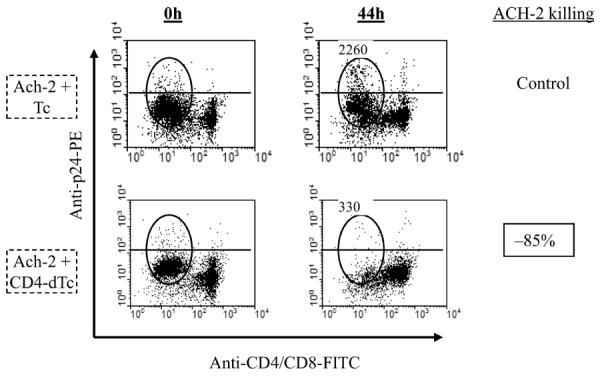
CD4-dTc kill latently infected cells. Equal numbers of ACH-2 cells were mixed with at least two wks grown Tc or dTc at 1:1 ratio and incubated for 44h. The cell mixtures were stained at 0h and 44h with antibodies to CD4, CD8 and intracellularly expressed HIV-p24 antigens, followed by flow cytometry. The stained CD4 and CD8 T cells clustered together in flow diagrams, whereas ACH-2 cells clustered separately (oval) as they are negative for both CD4 and CD8 expression. The horizontal line indicates the cut-off for p24 expression. Raw counts of total ACH-2 cells for control and experimental cultures at 44 hr are shown above ovals. Percent specific killing of ACH-2 cells by CD4-dTc was calculated as in Materials and Methods.
To investigate whether a soluble factor was mediating reactivation, fresh ACH-2 cells were incubated with coculture supernatants and analyzed for p24 expression (Fig.6A). When incubated with fresh media or control culture supernatants, ACH-2 cells were p24+ in the range of 7.5–11%. With supernatant from ACH-2/CD4-dTc cocultures, ACH-2 was induced to 67% being positive for p24-expression.
Figure 6.
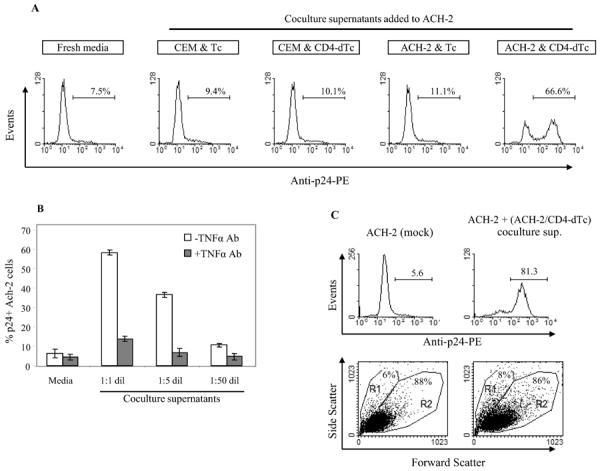
Reactivation of latent HIV during CD4-dTc stimulation by target cells. (A) Supernatants from cocultures reactivate latent HIV in ACH-2 cells. Supernatants were collected from 24 hour co-cultures of effector and various target cells mixed at 1:1 ratio. ACH-2 cells were incubated with the coculture supernatants for 24h and assayed for HIV-p24 expression by intracellular staining and flow cytometry. (B) TNFα neutralization inhibits latent HIV reactivation in ACH-2 cells. Supernatants harvested from ACH-2 and CD4-dTc coculture were incubated at different dilutions with or without TNF-α antibody (5μg/ml) at 37°C for 1h, and then added to ACH-2 cells for 18h, followed by detection of p24+ cells in flow cytometry as in A. (C) ACH-2 cells' viability after latent HIV reactivation. Coculture supernatant from ACH-2 and CD4-dTc were added to fresh ACH-2 cells and incubated for 44h at 37°C. Upper panels: Potent reactivation of latent HIV occurred by coculture supernatant (right) compared to untreated control (mock) (left). Lower panels: Scatter plots were analyzed for viable (R2) versus dead (R1) cells before (left) and after (right) treatment.
These data suggested that the interaction between low percent virus-expressing ACH-2 cells (7.5%) and CD4-dTc might have induced a factor in coculture supernatant that could reactivate latent HIV in ACH-2 cells. We examined this hypothesis first by testing if CD4-dTc could produce various effector cytokines in response to HIV-Env+ targets and controls (Table 1). We found only CD4-dTc incubated with CEM-Env+ cells could produce IL2, IFNγ and TNFα in culture, with all other cell combinations negative for these cytokines.
Table I.
Production of effector cytokines in gene-modified T cells upon engagement with HIV-gpl60.
| Mixtures of effector and target cells | Levels of cytokines or effector molecules (in pg/ml) |
||
|---|---|---|---|
| IL2 | TNFα | IFNγ | |
|
|
|
||
| CEM + Tc | <4 | <4 | <4 |
| CEM + CD4-dTc | <4 | <4 | <4 |
| CEM-Env + Tc | <4 | <4 | <4 |
| CEM-Env + CD4-dTc | 318 6 | 249 46 | 838 169 |
, SD. These data are representative of 3 independent experiments.
It was previously reported that TNFα could reactivate latent HIV in ACH-2 cells (Folks et al., 1989). To assay for a role for TNFα as soluble inducer under our conditions, we preincubated varying dilutions of coculture supernatant with TNFα neutralizing antibody prior to adding to ACH-2 cells. After overnight incubation, we analyzed the percentage of HIVp24+ cells in culture by flow cytometry, and found that anti-TNFα antibody could suppress the stimulatory effect of coculture supernatant on latent HIV in ACH-2 cells (Figure 6B). This demonstrated that TNFα released from CD4-dTc in coculture supernatants caused the reactivation of latent HIV in ACH-2 cells, leading to their killing by CD4-dTc.
As a final control, the ACH-2 cells' viability was not affected by the secreted TNFα or other factors in these supernatants or by the re-expressed HIV, even after 44h of incubation (Fig. 6C). Hence, the extensive killing of ACH-2 cells shown in Figure 5 required the presence of CD4-dTc cell fraction in cocultures. That is, the two functions of reactivating latent HIV in ACH-2 cells followed by the killing of virus expressing cells were each separately mediated activities of CD4-dTc.
Discussion
The HIV-specific, cytotoxic designer T cells of this study can recognize and kill HIV envelope expressing cells in an MHC-independent fashion, whereas natural CTLs in vivo require MHC-I dependent antigen presentation on the surface of infected cells for recognition and killing. The cytotoxicity of HIV-specific CD4-dTc requires CD3ζ linked to CD4 molecules C-terminally, either directly or through a domain of other molecules, in this case, costimulatory domain of CD28. The modification of CTLs by CD4 alone without attached signaling domains would not be expected to mediate cytotoxic effects against HIV+ cells, as previously reported with various constructs (Finney, Akbar, and Lawson, 2004; Haynes et al., 2002). Further, the cytolytic activity of CD4-CAR expressing dTc towards HIV infected cells requires the high affinity interaction between CD4-CAR and HIV Env (Lasky et al., 1987; Thali et al., 1991) that is not duplicated with a low affinity interaction between CD4-CAR and MHC-II (Gao, Rao, and Bell, 2002; Wang et al., 2001; Weber and Karjalainen, 1993).
HIV can down-modulate MHC-I molecules in infected cells (Peterlin and Trono, 2003), which serves as one of the immune evasive mechanisms for HIV (Yewdell and Hill, 2002). However, such down-modulation would not abrogate the recognition of infected cells by HIV-specific designer T cells. Thus, there could be a potential utility of these gene-modified T cells in vivo in the post-ART era, for example, infusion of CD4-dTc into patients on ART could result in targeting of persistently active HIV reservoirs (Chun et al., 2008; Poles et al., 2006), reducing or eliminating their pool in the body.
The ACH-2 cell line has been applied as a model for HIV latency, with 90–98% of the cells expressing no virus although harboring HIV provirus. Unexpectedly, most ACH-2 cells were found to be attacked and killed in vitro by our HIV-specific CD4-dTc rather than just the minor HIV-expressing fraction. This outcome was attributed to induction of HIV expression in latent cells by locally secreted TNFα from CD4-dTc responding to the small percent of HIV-expressing ACH-2 cells. Although it was previously reported that TNFα can reactivate these latent cells (Folks et al., 1989), it was uncertain prior to this study whether TNFα could be produced at sufficient levels by CD4-dTc engaging with small numbers of reactive targets, so that it can reactivate nearby latent targets.
Yet it is unclear whether such scenario will also apply to in vivo where patients are infused with these designer T cells. In in-vitro models of HIV latency that employ primary CD4 T cells, TNFα by itself is unable to reactivate latent HIV (Bosque and Planelles, 2009; Tyagi, Pearson, and Karn, 2010). TNFα has the property of inducing NF-kB activation and nuclear translocation, which is sufficient for reactivating latent virus in ACH-2 cells. In contrast, the latently infected quiescent CD4 T cells in the primary cell model additionally require activated pTEF-b complex for viral emergence (Tyagi, Pearson, and Karn, 2010), because this complex is nearly absent in primary cells (Herrmann et al., 1998). Thus, the CD4-dTc are able to deliver the one of two important “signals” for latent HIV reactivation; if means are discovered to selectively activate pTEF-b as the other “signal,” by pharmacological means or as a component of dTc engineering, then an opportunity for a progressive spreading of latent HIV reactivation and elimination can be envisioned that could deplete latent reservoirs in patients.
Alternatively, the HIV-specific designer T cells, if remaining active and engaging in immune surveillance in infused patients, could target and kill latently infected cells that re-express HIV, as shown in this report. Such a scenario could lead to sustained control of viremia at clinically undetectable levels, even after ART withdrawal, and suggests the possibility of a `functional cure' for HIV.
Materials and methods
Retroviral vectors, Cell lines, Plasmids, and Antibodies
A retroviral vector with MFG vector backbone expressing anti-CEA-scFV-CD28-CD3ζ was used (Emtage et al., 2008) to generate recombinant plasmids for the study. Phoenix amphotropic and ecotropic cells and murine helper cell line, PG13, were purchased from American Type Culture Collection (ATCC), USA. A clonal T cell line (CEM-Env) expressing HIV Env at the cell surface and a virus strain, HIV213 were the gifts from Dr. Miles Cloyd, University of Texas Medical Branch at Galveston.
A DNA fragment for the expression of extracellular portion of human CD4 molecule with myc epitope tag at the N-terminus was generated and ligated into the vector as shown in Figure 1A to express chimeric CD4-CD28-CD3ζ protein in transduced cells (Figure 1B). The pseudotyped retroviral vector was produced and used to transduce stimulated T cells (see below).
The following antibodies reactive to human target molecules were used: Anti-CD4, −CD8 antibodies conjugated to PE (phycoerythrin), APC (allophycocyanin) and FITC (fluorescein isothiocyanate) antibodies were purchased from eBioscience. The anti-HIVp24-PE, anti-Myc-FITC, HIV-blocking anti-CD4 antibody (clone QS4120) and anti-human HLA-DR antibody (clone G46-6[L243]) were purchased from Coulter, Sigma, Ancell Corporation, and BD Biosciences, respectively. The APC-labeled WI2, an anti-idiotype antibody to anti-CEAscFv (αCEA) were obtained from Immunomedics (Emtage et al., 2008).
Preparation of a stable cell line expressing recombinant retrovirus with CAR-cassette
The retroviral construct (plasmid) encoding 2nd generation chimeric CD4-CD28-zeta molecule (see Figure 1A) was used to transfect a mixture of Phoenix ampho- and eco-tropic cells (ATCC, USA) as previously described (Beaudoin, Bais, and Junghans, 2008). Twenty-four hours later, the transduced Phoenix-mix cells expressing chimeric protein were sorted once. The culture supernatant from the sorted virus-producing cells was used to transduce a PG13 (empty) murine helper cell line for stable integration and virus particle production. The PG13 cells express gibbon ape leukemia virus (GaLV) envelope, in addition to murine leukemia virus gag protein, for pseudotyping the recombinant retroviral vector. The human cells, in contrast to mouse cells, can be infected readily with these pseudotyped retroviruses (Miller et al., 1991). The transduced cells that express 2nd generation chimera at the cell surface were identified by staining with antihuman CD4 antibodies (eBioscience, USA), followed by flow cytometry. The antibody stained CD4+ PG13 cells were sorted by fluorescence activated cell sorter (FACS) and expanded in culture. This stable line was used as the source of retrovirus particles that are cable of expressing CD4-CAR (i.e., CD4-CD28-CD3ζ chimera) upon transduction into human T cells.
Transduction of human PBMCs with recombinant retrovirus and culture
PBMCs were isolated from normal donor's blood by ficoll-hypaque method, and stimulated with anti-CD3ε antibody (clone OKT3) at 100ng/ml concentration in RPMI media, 10% fetal bovine serum for two days. Then stimulated PBMCs were cultured in presence of 300IU/ml IL2 for another 2 days. For transduction, ~8×106 cells were mixed with 3 ml of culture supernatant harvested from vector producing cells in 6-well plate coated with retronectin (10μg/ml, Takara) and centrifuged for 2h at 32°C. After overnight incubation at 37°C in CO2 incubator, culture supernatants were removed and cells were transduced for the second time to increase transduction efficiency. Next day, supernatants were replaced with fresh media containing 300U/ml IL2 and cells were maintained in culture for 2–3 weeks in presence of IL2 (100IU/ml). Within this time period, portions of cells were harvested and used in various experiments.
To determine the percentage of gene-modified T cells in culture in Figure 2, (A) mock and (B) transduced cells were stained using anti-CD8-APC and anti-CD4-PE antibodies or, (C) mock and (D) transduced cells were stained with anti-CD8-APC and anti-Myc-FITC antibody and analyzed by flow cytometry.
In Figure 2B, the formula used to calculate the modified CD8 cells was: %Ttransduced = 100×Q2/(Q2+Q4). The fraction of double-positive cells (CD4+CD8+) naturally present at baseline in Figure 2A (Q2: 2.8%) was subtracted from Q2 in numerator and denominator because these cells could not be judged for modification.
In Figure 2D, the formula used to calculate the modified CD8 T cells was: %Ttransduced = 100×Q2/(Q2+Q4), in which background myc staining from (C) (Q2: 0.9%) was subtracted from Q2 in numerator and denominator. We examine the CD8- population to infer the fraction of CD4 cells. The formula to calculate modified CD4 T cells was: %Ttransduced = 100×Q1/(Q1+Q3) in which background myc staining from (C) (Q1: 0.7%) was subtracted from Q1 in numerator and denominator. In addition, the fraction of non-T cells (CD4−CD8−) from PBMC that are present in the transduction from (B) (Q3: 9.1%) is subtracted from Q3 in D in estimating the unmodified CD4+ T cells in the denominator. This fraction is seen as 9.7% in Figure 2A and 9.1% in Figure 2B, essentially unchanged by the transduction. Non-dividing non-T cells would not be expected to be modified.
dTc killing assay by flow cytometry
Specific killing of various HIV-infected target cells by dTc was also determined by flow cytometric analysis. Effector and target cells were mixed at 1:1 ratio in equal volumes in 48-well plate and incubated for specified times. Using changes in cell percents in the mixtures per usual practice cannot be applied if the total cells in the analysis change due to target cell killing. Instead, the same proportion of the sample is counted and the numbers of targets in that portion is representative of the surviving fraction. To achieve this, total samples were thoroughly collected, processed equivalently and resuspended in equal volumes for flow cytometric analyses. To allow for different total counts due to target killing in some samples, each sample was analyzed for an identical fixed time (e.g., 20 seconds). Because the flow setting is in microliters per second, this ensures the same sample volume is processed and counted. To determine the specific killing due to dTc, the target cell counts in control cultures with activated non-transduced T cells (Tc) were taken as 100% and then target cell counts in dTc coculture were normalized accordingly. The percent specific lysis (%SL) of HIV-infected target cells due to dTc was calculated by using a formula: %SL= 100×(1 – (surviving targets with dTc/surviving targets with Tc). Killing is expressed as a negative number in figures to emphasize the loss of cells as opposed to a positive measure of lysis such as chromium-release.
Carboxyfluorescein succinimidyl ester (CFSE) labeling and HIV-p24 antibody staining
These were done by following the previously published methods (Lyons and Parish, 1994; Sahu et al., 2006).
Treatment of effectors (CD4-dTc) with various antibodies or Concanamycin A prior to mixing with target cells, whenever required
To examine the blocking effects of various reagents in our killing assays, effectors (CD4-dTc) were incubated with HIV-blocking anti-human CD4 (domain I) antibody (20μg/ml) or anti-human HLA-DR antibody (10μg/ml) or Concanamycin A (160nM, Sigma) at 37°C for 1h prior to mixing and incubation with infected (H9/213) cells for 22h, followed by intracellular staining and flow cytometry.
Highlights
The anti-HIV activities of CD4 CAR+ designer T cells (dTc) are tested in vitro.
Killing of HIV-infected cells by CD4 CAR+ T cells requires CAR/HIV-Env interaction.
Target cell lysis by CD4 CAR+ T cells was partly perforin-mediated.
dTc could reactivate and then eradicate latently infected cells in the ACH-2 model.
Acknowledgments
This work was supported by the NIH grant, 5R21AI076145 to RPJ and the research funds to GK Sahu from Roger Williams Medical Center, Providence, RI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bagasra O, Lavi E, Bobroski L, Khalili K, Pestaner JP, Tawadros R, Pomerantz RJ. Cellular reservoirs of HIV-1 in the central nervous system of infected individuals: identification by the combination of in situ polymerase chain reaction and immunohistochemistry. AIDS. 1996;10(6):573–85. doi: 10.1097/00002030-199606000-00002. [DOI] [PubMed] [Google Scholar]
- Beaudoin EL, Bais AJ, Junghans RP. Sorting vector producer cells for high transgene expression increases retroviral titer. J Virol Methods. 2008;148(1–2):253–9. doi: 10.1016/j.jviromet.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Beecham EJ, Ma Q, Ripley R, Junghans RP. Coupling CD28 co-stimulation to immunoglobulin T-cell receptor molecules: the dynamics of T-cell proliferation and death. J Immunother. 2000;23(6):631–42. doi: 10.1097/00002371-200011000-00004. [DOI] [PubMed] [Google Scholar]
- Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3(1):87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2009;113(1):58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387(6629):183–8. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, Kottilil S, Moir S, Mican JM, Mullins JI, Ward DJ, Kovacs JA, Mannon PJ, Fauci AS. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis. 2008;197(5):714–20. doi: 10.1086/527324. [DOI] [PubMed] [Google Scholar]
- Clouse KA, Powell D, Washington I, Poli G, Strebel K, Farrar W, Barstad P, Kovacs J, Fauci AS, Folks TM. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J Immunol. 1989;142(2):431–8. [PubMed] [Google Scholar]
- Davey RT, Jr., Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, Kovacs JA, Polis MA, Walker RE, Falloon J, Masur H, Gee D, Baseler M, Dimitrov DS, Fauci AS, Lane HC. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(26):15109–14. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, Macken C, Richman DD, Christopherson C, June CH, Lazar R, Broad DF, Jalali S, Hege KM. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther. 2002;5(6):788–97. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- Emtage PC, Clarke D, Gonzalo-Daganzo R, Junghans RP. Generating potent Th1/Tc1 T cell adoptive immunotherapy doses using human IL-12: Harnessing the immunomodulatory potential of IL-12 without the in vivo-associated toxicity. J Immunother. 2003;26(2):97–106. doi: 10.1097/00002371-200303000-00002. [DOI] [PubMed] [Google Scholar]
- Emtage PC, Lo AS, Gomes EM, Liu DL, Gonzalo-Daganzo RM, Junghans RP. Second-generation anti-carcinoembryonic antigen designer T cells resist activation-induced cell death, proliferate on tumor contact, secrete cytokines, and exhibit superior antitumor activity in vivo: a preclinical evaluation. Clin Cancer Res. 2008;14(24):8112–22. doi: 10.1158/1078-0432.CCR-07-4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004;172(1):104–13. doi: 10.4049/jimmunol.172.1.104. [DOI] [PubMed] [Google Scholar]
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5(5):512–7. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278(5341):1295–300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- Folks TM, Clouse KA, Justement J, Rabson A, Duh E, Kehrl JH, Fauci AS. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc Natl Acad Sci U S A. 1989;86(7):2365–8. doi: 10.1073/pnas.86.7.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XT, Karr RW. HLA-DR alpha chain residues located on the outer loops are involved in nonpolymorphic and polymorphic antibody-binding epitopes. Hum Immunol. 1994;39(4):253–60. doi: 10.1016/0198-8859(94)90268-2. [DOI] [PubMed] [Google Scholar]
- Gao GF, Rao Z, Bell JI. Molecular coordination of alphabeta T-cell receptors and coreceptors CD8 and CD4 in their recognition of peptide-MHC ligands. Trends Immunol. 2002;23(8):408–13. doi: 10.1016/s1471-4906(02)02282-2. [DOI] [PubMed] [Google Scholar]
- Garcia F, Plana M, Vidal C, Cruceta A, O'Brien WA, Pantaleo G, Pumarola T, Gallart T, Miro JM, Gatell JM. Dynamics of viral load rebound and immunological changes after stopping effective antiretroviral therapy. AIDS. 1999;13(11):F79–86. doi: 10.1097/00002030-199907300-00002. [DOI] [PubMed] [Google Scholar]
- Green JM, Noel PJ, Sperling AI, Walunas TL, Lenschow DJ, Stack R, Gray GS, Bluestone JA, Thompson CB. T cell costimulation through the CD28 receptor. Proc Assoc Am Physicians. 1995;107(1):41–6. [PubMed] [Google Scholar]
- Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med. 1982;155(6):1823–41. doi: 10.1084/jem.155.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, Emini EA, Chodakewitz JA. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy.[see comment] New England Journal of Medicine. 1997;337(11):734–9. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, Eron JJ, Jr., Feinberg JE, Balfour HH, Jr., Deyton LR, Chodakewitz JA, Fischl MA. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team.[see comment] New England Journal of Medicine. 1997;337(11):725–33. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- Hawkins T. Understanding and managing the adverse effects of antiretroviral therapy. Antiviral Res. 2010;85(1):201–9. doi: 10.1016/j.antiviral.2009.10.016. [DOI] [PubMed] [Google Scholar]
- Haynes NM, Trapani JA, Teng MW, Jackson JT, Cerruti L, Jane SM, Kershaw MH, Smyth MJ, Darcy PK. Rejection of syngeneic colon carcinoma by CTLs expressing single-chain antibody receptors codelivering CD28 costimulation. J Immunol. 2002;169(10):5780–6. doi: 10.4049/jimmunol.169.10.5780. [DOI] [PubMed] [Google Scholar]
- Herrmann CH, Carroll RG, Wei P, Jones KA, Rice AP. Tat-associated kinase, TAK, activity is regulated by distinct mechanisms in peripheral blood lymphocytes and promonocytic cell lines. J Virol. 1998;72(12):9881–8. doi: 10.1128/jvi.72.12.9881-9888.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, Kovacs CM, Rodriguez B, Sieg SF, Teixeira-Johnson L, Gudonis D, Goepfert PA, Lederman MM, Frank I, Makedonas G, Kaul R, Walker BD, Betts MR. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog. 2010;6(5):e1000917. doi: 10.1371/journal.ppat.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AM, Cho M, Mrus JM. The costs of full suppression of plasma HIV RNA in highly antiretroviral-experienced patients. AIDS Rev. 2011;13(1):41–8. [PubMed] [Google Scholar]
- Jena B, Dotti G, Cooper LJ. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 2010;116(7):1035–44. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junghans RP. A strategy for evaluating lymphokine activation and novel monoclonal antibodies in antibody-dependent cell-mediated cytotoxicity and effector cell retargeting assays. Cancer Immunol Immunother. 1990;31(4):207–12. doi: 10.1007/BF01789170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka T, Shinohara N, Takayama H, Takaku K, Kondo S, Yonehara S, Nagai K. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin- and Fas-based lytic pathways in cell-mediated cytotoxicity. J Immunol. 1996;156(10):3678–86. [PubMed] [Google Scholar]
- Lambotte O, Taoufik Y, de Goer MG, Wallon C, Goujard C, Delfraissy JF. Detection of infectious HIV in circulating monocytes from patients on prolonged highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2000;23(2):114–9. doi: 10.1097/00126334-200002010-00002. [DOI] [PubMed] [Google Scholar]
- Lasky LA, Nakamura G, Smith DH, Fennie C, Shimasaki C, Patzer E, Berman P, Gregory T, Capon DJ. Delineation of a region of the human immunodeficiency virus type 1 gp120 glycoprotein critical for interaction with the CD4 receptor. Cell. 1987;50(6):975–85. doi: 10.1016/0092-8674(87)90524-1. [DOI] [PubMed] [Google Scholar]
- Luzuriaga K, Bryson Y, Krogstad P, Robinson J, Stechenberg B, Lamson M, Cort S, Sullivan JL. Combination treatment with zidovudine, didanosine, and nevirapine in infants with human immunodeficiency virus type 1 infection. New England Journal of Medicine. 1997;336(19):1343–9. doi: 10.1056/NEJM199705083361902. [DOI] [PubMed] [Google Scholar]
- Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171(1):131–7. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- Ma Q, Gonzalo-Daganzo RM, Junghans RP. Genetically engineered T cells as adoptive immunotherapy of cancer. Cancer Chemother Biol Response Modif. 2002;20:315–41. [PubMed] [Google Scholar]
- Mata RC, Viciana P, de Alarcon A, Lopez-Cortes LF, Gomez-Vera J, Trastoy M, Cisneros JM. Discontinuation of antiretroviral therapy in patients with chronic HIV infection: clinical, virologic, and immunologic consequences. AIDS Patient Care & Stds. 2005;19(9):550–62. doi: 10.1089/apc.2005.19.550. [DOI] [PubMed] [Google Scholar]
- Miller AD, Garcia JV, von Suhr N, Lynch CM, Wilson C, Eiden MV. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol. 1991;65(5):2220–4. doi: 10.1128/jvi.65.5.2220-2224.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, Bakker A, Roberts MR, June CH, Jalali S, Lin AA, Pennathur-Das R, Hege KM. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96(3):785–93. [PubMed] [Google Scholar]
- Peterlin BM, Trono D. Hide, shield and strike back: how HIV-infected cells avoid immune eradication. Nat Rev Immunol. 2003;3(2):97–107. doi: 10.1038/nri998. [DOI] [PubMed] [Google Scholar]
- Poles MA, Boscardin WJ, Elliott J, Taing P, Fuerst MM, McGowan I, Brown S, Anton PA. Lack of decay of HIV-1 in gut-associated lymphoid tissue reservoirs in maximally suppressed individuals. J Acquir Immune Defic Syndr. 2006;43(1):65–8. doi: 10.1097/01.qai.0000230524.71717.14. [DOI] [PubMed] [Google Scholar]
- Roberts MR, Qin L, Zhang D, Smith DH, Tran AC, Dull TJ, Groopman JE, Capon DJ, Byrn RA, Finer MH. Targeting of human immunodeficiency virus-infected cells by CD8+ T lymphocytes armed with universal T-cell receptors. Blood. 1994;84(9):2878–89. [PubMed] [Google Scholar]
- Rosok B, Voltersvik P, Larsson BM, Albert J, Brinchmann JE, Asjo B. CD8+ T cells from HIV type 1-seronegative individuals suppress virus replication in acutely infected cells. AIDS Res Hum Retroviruses. 1997;13(1):79–85. doi: 10.1089/aid.1997.13.79. [DOI] [PubMed] [Google Scholar]
- Sadelain M. T-cell engineering for cancer immunotherapy. Cancer J. 2009;15(6):451–5. doi: 10.1097/PPO.0b013e3181c51f37. [DOI] [PubMed] [Google Scholar]
- Saez-Cirion A, Sinet M, Shin SY, Urrutia A, Versmisse P, Lacabaratz C, Boufassa F, Avettand-Fenoel V, Rouzioux C, Delfraissy JF, Barre-Sinoussi F, Lambotte O, Venet A, Pancino G. Heterogeneity in HIV suppression by CD8 T cells from HIV controllers: association with Gag-specific CD8 T cell responses. J Immunol. 2009;182(12):7828–37. doi: 10.4049/jimmunol.0803928. [DOI] [PubMed] [Google Scholar]
- Sahu GK, Lee K, Ji J, Braciale V, Baron S, Cloyd MW. A novel in vitro system to generate and study latently HIV-infected long-lived normal CD4+ T-lymphocytes. Virology. 2006;355(2):127–37. doi: 10.1016/j.virol.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nature Medicine. 2003;9(6):727–8. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- Song SK, Li H, Cloyd MW. Rates of shutdown of HIV-1 into latency: roles of the LTR and tat/rev/vpu gene region. Virology. 1996;225(2):377–86. doi: 10.1006/viro.1996.0612. [DOI] [PubMed] [Google Scholar]
- Sonza S, Mutimer HP, Oelrichs R, Jardine D, Harvey K, Dunne A, Purcell DF, Birch C, Crowe SM. Monocytes harbour replication-competent, non-latent HIV-1 in patients on highly active antiretroviral therapy. AIDS. 2001;15(1):17–22. doi: 10.1097/00002030-200101050-00005. [DOI] [PubMed] [Google Scholar]
- Thali M, Olshevsky U, Furman C, Gabuzda D, Li J, Sodroski J. Effects of changes in gp120-CD4 binding affinity on human immunodeficiency virus type 1 envelope glycoprotein function and soluble CD4 sensitivity. J Virol. 1991;65(9):5007–12. doi: 10.1128/jvi.65.9.5007-5012.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchie H, Detorio MA, Hossain MM, Tesfamariam N, Trickett A, Lam-Po-Tang PR, Yamada O, Ichimura H, Dwyer JM, Kurimura T. Suppression of HIV replication in vitro by CD8+ T-cells from HIV-infected and HIV-seronegative individuals. Int J STD AIDS. 1997;8(5):307–10. doi: 10.1258/0956462971920145. [DOI] [PubMed] [Google Scholar]
- Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 2010;84(13):6425–37. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RE, Bechtel CM, Natarajan V, Baseler M, Hege KM, Metcalf JA, Stevens R, Hazen A, Blaese RM, Chen CC, Leitman SF, Palensky J, Wittes J, Davey RT, Jr., Falloon J, Polis MA, Kovacs JA, Broad DF, Levine BL, Roberts MR, Masur H, Lane HC. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96(2):467–74. [PubMed] [Google Scholar]
- Wang JH, Meijers R, Xiong Y, Liu JH, Sakihama T, Zhang R, Joachimiak A, Reinherz EL. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc Natl Acad Sci USA. 2001;98(19):10799–804. doi: 10.1073/pnas.191124098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RF, Wang X, Atwood AC, Topalian SL, Rosenberg SA. Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science. 1999;284(5418):1351–4. doi: 10.1126/science.284.5418.1351. [DOI] [PubMed] [Google Scholar]
- Weber S, Karjalainen K. Mouse CD4 binds MHC class II with extremely low affinity. Int Immunol. 1993;5(6):695–8. doi: 10.1093/intimm/5.6.695. [DOI] [PubMed] [Google Scholar]
- Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278(5341):1291–5. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- Yang OO, Tran AC, Kalams SA, Johnson RP, Roberts MR, Walker BD. Lysis of HIV-1-infected cells and inhibition of viral replication by universal receptor T cells. Proc Natl Acad Sci USA. 1997;94(21):11478–83. doi: 10.1073/pnas.94.21.11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Beaudoin EL, Lu L, Du Pasquier RA, Kuroda MJ, Willemsen RA, Koralnik IJ, Junghans RP. Chimeric immune receptors (CIRs) specific to JC virus for immunotherapy in progressive multifocal leukoencephalopathy (PML) Int Immunol. 2007;19(9):1083–93. doi: 10.1093/intimm/dxm076. [DOI] [PubMed] [Google Scholar]
- Yewdell JW, Hill AB. Viral interference with antigen presentation. Nat Immunol. 2002;3(11):1019–25. doi: 10.1038/ni1102-1019. [DOI] [PubMed] [Google Scholar]
- Zalar A, Figueroa MI, Ruibal-Ares B, Bare P, Cahn P, de Bracco MM, Belmonte L. Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Antiviral Res. 2010;87(2):269–71. doi: 10.1016/j.antiviral.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Zhu T, Muthui D, Holte S, Nickle D, Feng F, Brodie S, Hwangbo Y, Mullins JI, Corey L. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. Journal of Virology. 2002;76(2):707–16. doi: 10.1128/JVI.76.2.707-716.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]