

Abstract
Ehlers-Danlos syndrome (EDS) type I (the classical variety) is a dominantly inherited, genetically heterogeneous connective-tissue disorder. Mutations in the COL5A1 and COL5A2 genes, which encode type V collagen, have been identified in several individuals. Most mutations affect either the triple-helical domain of the protein or the expression of one COL5A1 allele. We identified a novel splice-acceptor mutation (IVS4-2A→G) in the N-propeptide-encoding region of COL5A1, in one patient with EDS type I. The outcome of this mutation was complex: In the major product, both exons 5 and 6 were skipped; other products included a small amount in which only exon 5 was skipped and an even smaller amount in which cryptic acceptor sites within exon 5 were used. All products were in frame. Pro-α1(V) chains with abnormal N-propeptides were secreted and were incorporated into extracellular matrix, and the mutation resulted in dramatic alterations in collagen fibril structure. The two-exon skip occurred in transcripts in which intron 5 was removed rapidly relative to introns 4 and 6, leaving a large (270 nt) composite exon that can be skipped in its entirety. The transcripts in which only exon 5 was skipped were derived from those in which intron 6 was removed prior to intron 5. The use of cryptic acceptor sites in exon 5 occurred in transcripts in which intron 4 was removed subsequent to introns 5 and 6. These findings suggest that the order of intron removal plays an important role in the outcome of splice-site mutations and provide a model that explains why multiple products derive from a mutation at a single splice site.
Introduction
Type V collagen, a minor component of many connective tissues, plays a role in the regulation of the size and configuration of fibrils of the much more abundant component type I collagen (van der Rest and Garrone 1991). The major variant of type V collagen in most tissues is a heterotrimer that is composed of two pro-α1(V) chains and a single pro-α2(V) chain, encoded by the COL5A1 and COL5A2 genes, respectively. The two genes are part of the fibrillar-collagen gene family and encode chains with uninterrupted triple helices. COL5A1 differs somewhat from the major fibrillar-collagen genes in intron-exon structure and encodes a very large amino-terminal (N-)propeptide (Greenspan et al. 1991a; Takahara et al. 1991, 1995) that is similar to those encoded by the COL11A1 and COL11A2 genes (Zhidkova et al. 1993; Tsumaki and Kimura 1995). This propeptide (extending from the signal-peptide cleavage site to the beginning of the major triple helix), which consists of 522 amino acids, is composed of an amino-terminal PARP (proline-arginine–rich protein) globular subdomain, which bears some homology to similar domains in thrombospondin and von Willebrand factor (Bork 1992); a globular subdomain, known as the “variable region”; and a small collagenous subdomain. A site for proteolytic maturation is encoded in exon 5, and cleavage at this site removes the PARP subdomain (Imamura et al. 1998; Unsöld et al. 2002). The retained variable region projects beyond the surface of large heterotypic fibrils composed of both type I and type V collagen and has been suggested to regulate fibril structure (Linsenmayer et al. 1993). These heterotypic fibrils are found in skin and other tissues.
Fibril architecture is altered in skin and other tissues, in individuals with Ehlers-Danlos syndrome (EDS) types I (MIM 130000) and II (MIM 130010) (the classical form of the disorder), which are clinically characterized by very soft hyperextensible skin, large- and small-joint hypermobility, abnormal scar formation, and easy bruising (Beighton 1993; Steinmann et al. 1993). There is increasing evidence that mutations in either of the two type V collagen genes are the usual causes of the phenotypes of EDS types I and II, but mutations have also been identified, in a small number of individuals, in the COL1A1 (Nuytinck et al. 2000), COL1A2 (Sasaki et al. 1987; Hata et al. 1988; Nicholls et al. 2001), and tenascin X (TNX) (Burch et al. 1997; Schalkwijk et al. 2001) genes. Exon-skipping mutations in the triple-helix–encoding region of COL5A1 (Nicholls et al. 1996); alterations, in the C-propeptide of pro-α1(V), that interfere with chain assembly (Wenstrup et al. 1996; De Paepe et al. 1997); a translocation that deletes the entire carboxyl-terminal half of the coding sequence (Toriello et al. 1996); and mutations that alter the triple helix of pro-α2(V) (Michalickova et al. 1998) have all been found. Collectively, these seem to account for less than one-fourth of all mutations. Recently, it has become clear that COL5A1 mutations that result in the lack, usually as a result of very unstable mRNA, of protein synthesized from one null allele account for a significant proportion of the remainder (Schwarze et al. 2000; Wenstrup et al. 2000).
Here we characterize (1) the first mutation reported to alter the N-propeptide of the pro-α1(V) chain and (2) a novel response to an acceptor-site mutation. Such N-propeptide mutations are unknown in the other fibrillar-collagen genes. The patient whom we studied has an acceptor-site mutation (IVS4-2A→G), in COL5A1 intron 4, that leads to multiple mRNA products: the major product lacks the sequences of exons 5 and 6, another product lacks the sequence of exon 5 alone, and two additional products are missing 12 or 15 nt at the 5′ end of exon 5. All mRNA products are in frame and should produce protein products. The two products missing the sequence of exon 5 lack the PARP-domain cleavage site and also lack two of the four cysteines that are normally found in the N-propeptide. The patient’s cells incorporate mutant pro-α1(V)–derived chains, with abnormal-sized N-propeptides, into extracellular matrix in culture and synthesize collagen fibrils with a bizarre architecture, providing evidence that pro-α1(V) N-propeptide sequences play an important role in fibrillogenesis. The unusual occurrence of a two-exon skip is best explained by the order of intron removal in this region such that intron 5 is removed rapidly, in most transcripts, with respect to introns 4 and 6, so that a large exon (resulting from the fusion of exons 5 and 6) can then be skipped. This novel mechanism suggests that intron splice order may be an important factor in the understanding of some outcomes of splice-site mutations and that multiple products are derived from different splicing pathways.
Patient, Material, and Methods
Case Report
When seen in 1995, the patient was a 5-year-old boy. He was adopted, and no information was available about his biological parents. He was 37 inches tall (<5th percentile) and weighed 32 pounds (5th percentile). He had very soft, hyperextensible skin and had scars, from previous injuries, on his forehead, as well as scars and hyperpigmentation on his shins. He could touch his fingers to the dorsum of each arm, with passive flexion. His large joints had mild hypermobility, and all small joints appeared to be hypermobile. The physical findings for the patient were compatible with the diagnosis of Ehlers-Danlos syndrome type I (Beighton et al. 1998). A dermal-punch-biopsy sample was taken after assent from the patient and consent from the parents were obtained. Cells were grown from the biopsy sample and were then maintained in culture as described elsewhere (Bonadio et al. 1985).
Preparation of cDNA and Genomic DNA
Total cellular RNA and genomic DNA were extracted from cultured dermal fibroblasts from the patient and from a control individual, by use of the RNeasy Mini Kit (Qiagen) and QIAamp DNA Mini Kit (Qiagen), respectively. In addition, nuclear and cytoplasmic RNA were prepared from cultured fibroblasts from the patient and a control cell strain, as described elsewhere (Schwarze et al. 1999). cDNA was synthesized by the priming of total RNA, with random hexamers, in the presence of Superscript II reverse transcriptase (Gibco BRL) or the priming of nuclear and cytoplasmic RNA separately after treatment with DNase I (Boehringer Mannheim) for 20 min at 37°C, with the incubation buffer recommended by the manufacturer. To check for residual genomic DNA in the RNA preparation, we performed a control PCR without prior reverse transcription.
Amplification of the COL5A1 Coding Sequence and Sequence Determination of Aberrant Products
COL5A1 coding sequences were amplified in six overlapping fragments, with primer sets that have been published elsewhere (Schwarze et al. 2000), and the fragments were separated on 6% polyacrylamide gels. Extra PCR bands that were derived from the patient’s cDNA were excised from the gel and reamplified. Sequences of the reamplification products were determined, by the dideoxy-chain–termination method, either directly or after cloning into the PCR II vector, according to instructions provided in the TA cloning kit (Invitrogen).
An additional PCR that amplified the products from the abnormal and normal COL5A1 alleles in the region affected by the mutation was performed. This reaction used a sense primer, in exon 4 (5′-GATCGACATCAATGGCATCATCG-3′), that was end-labeled with γ[32P]-ATP (3,000 Ci/mmol; Amersham) by T4 polynucleotide kinase (New England BioLabs) and an antisense primer, in exon 7 (Schwarze et al. 2000) (1 min each at 94.5°C, 58°C, and 72°C, for 30 cycles). The products were separated on a 7% polyacrylamide gel, which was dried, and the radioactive bands were visualized by autoradiography.
Southern Blots
Genomic DNA was extracted from cultured dermal fibroblasts by use of standard phenol/chloroform methods (Ausubel et al. 1987). DNA samples were transferred from 0.8% agarose gels to Hybond-N+ membranes (Amersham), which were hybridized and washed as described elsewhere (Takahara et al. 1994). The hybridized probe was a 255-nt PCR product that was amplified, with primers 5′-ATCCAGCAGCTGCTCTTTGTCTCG-3′ and 5′-CTCTGTGGTTTCTTTGGCAGCTTCCAC-3′, from human placenta cDNA template (Clontech) and that corresponded to sequences contained in COL5A1 exons 5 and 6, comprising nucleotides 890–1144 of the published pro-α1(V) cDNA sequence (Greenspan et al. 1991a).
Synthesis of Riboprobe for RNA Protection
Total RNA was extracted from dermal fibroblasts by use of TRIzol reagent (Gibco BRL), according to the manufacturer's protocol. A 675-bp BamHI-NaeI restriction fragment, comprising nucleotides 856–1530 of the published human pro-α1(V) cDNA sequence (Greenspan et al. 1991a), was cloned between the BamHI and EcoRV sites of the vector pBluescript II KS+ (Stratagene); the resultant plasmid was linearized at the BamHI site; and a [32P]-labeled 734-base riboprobe was generated using RNA polymerase T3 in the presence of γ[32P]GTP. Conditions for the annealing of the antisense probe to 20 μg fibroblast RNA or yeast tRNA and for the incubation with RNases A and T1 were as described elsewhere (Greenspan et al. 1991b).
PCR Amplification
The following amplifications employed high-fidelity/long-distance PCR conditions (Barnes 1994; Cheng et al. 1994) and used Taq Extender PCR additive (Stratagene), according to the manufacturer's protocols. Amplifications to obtain the probe for the Southern blot (shown in fig. 2A) and the genomic region between primers P1 and P2 (in fig. 2B) were performed with denaturation at 94°C for 3 min, followed by 32 cycles of 98°C for 10 s with annealing and extension at 68°C. This two-step profile was begun at 68°C for 5 min, with autoextension adding 15 s per cycle thereafter. Final incubation was at 70°C for 8 min. Conditions for amplification, with primers P3 and P4, of the polymorphic-length repeat sequence in intron 6 were the same as above except that annealing and extension were begun at 68°C for 1.5 min, with autoextension adding 2 s per cycle. Primers P1–P4 are listed in table 1.
Figure 2.

Analysis of patient's genomic DNA. A, Southern blot analysis, with a probe for COL5A1 exons 5 and 6. Genomic DNA samples from the patient (P) and from the control individual (C) were cleaved with BamHI (B) or Pst I (P) and were probed for exon 5 and 6 sequences. B, Summary of features in the COL5A1 region affected in the patient. Exons are denoted by vertical bars or unblackened boxes. The exons are connected by horizontal lines that depict introns drawn approximately to scale. Arrowheads denote the positions of PCR primers. A circled P marks the site of a polymorphic PstI site in exon 5 of the patient's allele A. This polymorphic site contributes to the size differences between the patient’s allele A and allele B fragments and is also used as a marker in figure 5. Here and in figure 5, the control individual is also seen to be heterozygous for the PstI site. The obligatory ag dinucleotide of the intron 4 splice-acceptor site is shown for allele A. In allele B, an A→G transition (marked with an asterisk [*]) converts this dinucleotide ag to gg (IVS4-2A→G). Two cryptic splice-acceptor sites used by some allele B RNA transcripts are boxed within a 17-nt sequence shown for the allele B intron 4/exon 5 junction; intron sequences are denoted by lowercase letters, and exon sequences are denoted by uppercase letters. Restriction sites in the upper section of the panel were derived elsewhere (Takahara et al. 1995). Underlined restriction sites in the middle section of the panel were inferred from the Southern blot shown here and from another Southern blot not shown; all other restriction sites in the middle section of the panel were derived from complete sequencing of amplification products generated using primers P1 and P2. A schematic in the bottom section of the panel illustrates relationships between the restriction fragments seen in panel A and various features of the region of COL5A1 affected in the patient. B = BamHI; E = EcoRI; P = PstI; S = SacI; X = XhoI.
Table 1.
COL5A1 Primers[Note]
| Primer | Sequence(5′→3′) | Figure |
| P1 | GATCGAATTCTGAGGACAAGCTCGTCTTGAGGCTTGG | 3 |
| P2 | CAGTAAGCTTACTGGTTTCAGGCATGGGAGCAGCTTC | 3 |
| P3 | GTCCAGCCTCAGAGCCATTGTCAG | 3, 4 |
| P4 | ACTCCAGGCTCTCAGAGCAATGTG | 3, 4 |
| IN 4S | TTCAAGGCATGGGGCTGTGTCT | 5 |
| IN 4S mod | TCTGGACTTTCCCCTGCTTCAAGGC | 5 |
| E 6A | GGGTCTTCGTAGTAGGGGTATTCGTAGT | 5 |
| IN 6A | GCCCAGCCCAACCCAGTCC | 5 |
Note.— “IN” denotes intron-derived oligonucleotides; “E” denotes exon-derived oligonucleotides.
Exon Trapping
Allele A or allele B genomic sequences extending, in intron 4, from an EcoRI site 103 bp upstream from exon 5, to a BamHI site in intron 7, 395 bp downstream from exon 7, were separately inserted between the EcoRI and BamHI sites of pBluescript II KS+ and were then excised with EcoRI and XbaI. The latter sequence cut at the XbaI site within the pBluescript polylinker, 7 bp beyond the BamHI site, giving the inserts ends that were compatible for cloning between the EcoRI and NheI sites of the exon-trapping vector pSPL3 (Gibco BRL). Constructs were then transfected into COS-7 cells (American Type Culture Collection number CRL-1651), and total RNA was isolated and was reverse transcribed into first-strand cDNA, which was then amplified, according to the manufacturer's instructions, with vector-specific primers. Amplification products were sequenced after their subcloning into the pGEM-5Zf(+) vector (Promega) and after use of the Sequenase II kit (USB), as described elsewhere (Takahara et al. 1994).
Determination of Order of Intron Removal in the COL5A1 Gene
To determine the order of intron removal between exons 4 and 7 of COL5A1 pre-mRNA, we analyzed the splicing intermediates by a PCR strategy described elsewhere (Kessler et al. 1993; Schwarze et al. 1999). Nucleotide sequences of the primers described in the legend to figure 5 are listed in table 1. PCR was performed in a 50-μl volume that contained 1 μl cDNA derived from nuclear RNA after incubation of cultured control and patient fibroblasts with actinomycin D for 0, 5, 10, 20, 40, and 60 min; 5 μl 10 × PCR buffer with 20 mM MgCl2, 0.2 mM dNTPs, 1 U of Taq polymerase (Boehringer Mannheim); and 10 pmol of each primer. Amplifications were done through 30 cycles of denaturation at 94.5°C for 1 min, primer annealing at 60°C for 45 s, and primer extension at 72°C for 1 min 45 s. All PCR products (10-μl aliquots) were separated on 7% polyacrylamide gels, and the bands were visualized by ethidium bromide staining. For the estimation of the contribution of alleles A and B to splice intermediates, bands of interest were excised from gels, the DNA fragments were reamplified with the original primer sets, the reamplification products were cut with PstI (New England BioLabs), and the products were analyzed by PAGE.
Figure 5.

Amplification of regions between introns 4 and 6 in unspliced or partially spliced COL5A1 precursor mRNA (cDNA) isolated from cultured fibroblasts after incubation with actinomycin D (Act.D) for 0, 5, 10, 20, 40, and 60 min. A, Amplification with an intron 4 (IN 4S) and exon 6 (E 6A) primer pair. The full-length product was quickly lost in the control cells but remained present for 60 min in the patient cells. In both cell strains, the shorter product, from which intron 5 had been removed, persisted. B and C, PstI restriction fragments of excised and reamplified full-length (intron 4–exon 5–intron 5–exon 6) and partially spliced (intron 4–exon 5–exon 6) products, respectively, derived from the PstI+/− control individual (C) and the PstI+/− patient (P). In the patient cells, the full-length product (B) contained mRNA derived only from the mutant (PstI−) allele, and the product from which intron 5 had been removed (C) was derived largely from the mutant allele. D, With an intron 4 (IN 4S mod) and intron 6 (IN 6A) primer pair, the amount of full-length product rapidly declined in the control cells but remained abundant in the patient cells. The partially spliced product, which removed intron 5 but contained introns 4 and 6, remained. E, PstI restriction fragments of excised and reamplified partially spliced (intron 4–exon 5–exon 6–intron 6) product derived from the PstI+/− control individual (C) and patient (P). In the patient cells, this product was derived from the mutant and normal alleles in approximately equal amounts. Molecular weight marker is λ-DNA cut with PstI. gen = genomic DNA control individual; NC = PCR-reagent control individual; +/+, +/−, and −/− = PstI-restricted cDNAs reverse transcribed from control RNA samples from individuals who were homozygous positive, heterozygous, and homozygous negative, respectively, for a polymorphic PstI site in COL5A1 exon 5.
Protein Analysis
Fibroblasts were cultured for 1 wk in the presence of 200 mM ascorbate-2-phosphate, as described elsewhere (Hazeki et al. 1998). After being washed with PBS, cell layers were scraped into extraction buffer (PBS containing 1 M NaCl, 0.5% NP-40, 5 mM EDTA, 1 mM N-ethylmaleimide, 1 mM p-aminobenzoic acid, and 0.1 mM phenylmethylsulfonyl fluoride), were freeze thawed once, and were then stirred overnight at 4°C. Insoluble material that contained matrix was collected, in this and subsequent steps, by centrifugation at 15,000 rpm at 4°C for 10 min in an Eppendorf centrifuge; was rinsed twice with extraction buffer; and either was solubilized in 2% SDS-gel sample buffer prior to electrophoresis or was subjected to bacterial collagenase treatment (as described by Imamura et al. [1998]), which degrades the major collagenous domains of collagen molecules, thus liberating noncollagenous domains from the insoluble matrix. Material liberated from matrix samples by collagenase was mixed with 4 × SDS-gel sample buffer. Samples were analyzed by SDS-PAGE with either a 5% running gel and a 4% stacking gel or a 4%–15% gradient gel and were detected by immunoblotting, with a polyclonal antibody described elsewhere (Unsöld et al. 2002), directed against sequences in the variable subdomain of the N-terminal globular portion of the human pro-α1(V) chain.
To compare efficiencies of secretion of type V procollagen, we compared the ratio of type V:type I collagen in the matrix of patient and control fibroblasts. Cells cultured for 3 wk in the presence of 200 mM ascorbate-2-phosphate were washed twice with PBS; were scraped into 200 mM sodium phosphate buffer (pH 7.2) that contained 5 mM EDTA, 1 mM N-ethylmaleimide, 0.1 mM phenylmethylsulfonyl fluoride, and 1 mM p-aminobenzoic acid; and were suspended overnight at 4°C. Insoluble material was washed twice in double-distilled water containing the same inhibitors and was then treated with pepsin (0.1 mg/ml in 0.5 M acetic acid) overnight at 4°C. Insoluble material was collected and treated with pepsin (0.1 mg/ml in 0.5 M acetic acid) overnight at room temperature. Solubilized supernatants from the two pepsin extractions were pooled, and the collagenous proteins were concentrated by precipitation with 20% NaCl. Insoluble residue that remained after the two pepsin treatments was suspended in SDS-gel sample buffer with 5% 2-mercaptoethanol, by boiling for 5 min. Proteins in both pepsin-soluble and -insoluble material were separated by SDS-PAGE and were stained with Coomassie Blue, and bands were scanned and quantified using Image software (National Institutes of Health). The ratios of type V:type I collagen were estimated by obtainment of the ratios of α1(V):α2(I) chains, which were 0.56 for the patient and 0.54 for the control individual, when values for pepsin-soluble and -insoluble materials were combined.
Electron-Microscopic Examination of Collagen Fibrils Synthesized by Cultured Cells
Dermal fibroblasts from the patient and from a control individual were grown in culture for 3 wk, with daily addition of new medium that contained ascorbic acid (50 μg/ml). Cells and matrix were fixed in culture dishes, were embedded in Epon, and were thin-sectioned, stained, and examined by transmission electron microscopy, as described elsewhere (Smith et al. 1992).
Results
Multiple Splice Products from a COL5A1 Allele
RT-PCR was performed on total RNA from the patient's dermal fibroblasts, with primers that amplified COL5A1 and COL5A2 mRNA sequences in six and five overlapping fragments, respectively. Only the fragment of COL5A1 mRNA that included the region between exons 1 and 7 was abnormal when products were analyzed on polyacrylamide gels. When the region between exons 4 and 7 was amplified, to narrow the abnormal domain, three smaller-than-expected fragments were apparent (fig. 1A). Sequence determination of the two smallest products showed that one was missing the 132 nt of exon 5 and that the other was missing both the 132 nt of exon 5 and the 138 nt of exon 6. When fragments cloned from the gel region that contained the band slightly smaller than the normal-sized band were analyzed, two additional shorter fragments were identified. In one, the first 12 nt from the 5′ end of exon 5 were deleted, and, in the other, the first 15 nt from the same exon were deleted. All four products retained the normal reading frame, and all were found in the cytoplasm (not shown).
Figure 1.

Detection of abnormalities between exons 4 and 7 sequences in RNA from one of the patient's COL5A1 alleles. A, Amplification of COL5A1 cDNA from a control individual (C) and the patient (P) with a sense primer, end-labeled with γ[32P]-ATP (see the “Patient, Material, and Methods” section), in exon 4 and with an antisense primer in exon 7. A single major band is apparent in the sample from the control individual; the sample from the patient has the normal-sized band derived from the normal COL5A1 allele, one band just below that representing fragments produced by use of two separate cryptic acceptor sites in exon 5 (removing 12 or 15 nt from exon 5), and two lower bands that result from skipping of either exon 5 or both exons 5 and 6. B, Quantitative RNase-protection analysis. Conditions for labeling and annealing of the antisense probe to yeast (Y), control (C), and patient (P) RNA are described in the “Patient, Material, and Methods” section. Nuclease-protected fragments were separated on a denaturing 6% polyacrylamide gel. Sizes (in nt) of protected fragments are given. C, Schematic depicting the relationship of the riboprobe and protected fragments to COL5A1 exon sequences. BamHI and NaeI sites used in the making of the probe—as well as a cryptic splice site used in allele B, an AT-rich region that may result in partial cleavage by RNase due to “breathing” of the RNA duplex, and an SNP at nucleotide 1318 (T/C)—are shown. A wavy line represents non-COL5A1 vector sequences on the riboprobe.
A quantitative RNase-protection assay was performed, to measure relative amounts of the various spliced RNA forms produced by the patient's COL5A1 alleles and to ascertain whether, as appeared to be the case on the basis of the RT-PCR results (above), the product of the two-exon skip was the major species derived from the mutant allele. The patient's RNA was found to protect approximately one-half the amount, as did control RNA, of a 675-nt fragment corresponding to correctly spliced mRNA (fig. 1B)—consistent with the interpretation that only one of the patient's COL5A1 alleles produces correctly spliced mRNA. The patient's RNA also protected an ∼638-nt fragment, not protected by control RNA, that corresponded to mRNA derived by use of the exon 5 cryptic splice sites. However, an even greater amount of a 380-nt fragment, corresponding to RNA from which both exons 5 and 6 were spliced out, was protected by the patient's RNA, demonstrating that the patient's fibroblasts produce more of this form of RNA than of RNA that has used exon 5 cryptic splice sites. In fact, since the riboprobe was uniformly labeled along its length, the RNase-protection autoradiogram underrepresents the abundance of the mRNA that corresponds to the relatively small 380-nt fragment. A 518-nt fragment, corresponding in size to RNA from which exon 5 but not exon 6 was spliced out (figs. 1B and 1C), was protected by the patient's RNA, with lesser amounts protected by control RNA. The lesser amounts protected by control RNA may correspond to partially processed nuclear RNA, since total RNA was used. However, this fragment was not seen after amplification of the cDNA (fig. 1A), suggesting that it is an artifact of the RNase-protection assay, perhaps resulting from the AU-rich nature of sequences at the juncture of exons 5 and 6 (AAUAUUA [nucleotides 1011–1017]), which may result in limited cleavage by RNase at this site because of “breathing” of the RNA duplex (Ausubel et al. 1987). A 462-nt fragment protected by both patient and control RNA (figs. 1B and 1C) probably resulted from limited cleavage at an SNP at nucleotide 1321 (in exon 7; see fig. 1B), which is T both in the published sequence (Greenspan et al. 1991a) and in the cDNA used to prepare the riboprobe but which is C in both COL5A1 alleles of both the patient and the control individual.
Identification of a Mutation (IVS4-2A→G) in the Splice-Acceptor Site of Intron 4
To identify the genomic defect(s) leading to the aberrant forms of COL5A1 mRNAs described above (see the “Multiple Splice Products from a COL5A1 Allele” subsection), we performed Southern blot analysis of the patient's genomic DNA, with a cDNA probe that contained the sequences of exons 5 and 6. As can be seen, the blot detected restriction fragments that correspond in size to those of the control individual, as well as a larger fragment not seen in the control individual, after digestion of DNA with BamHI or PstI (fig. 2A). PCR amplification of the patient's genomic DNA, with primers located just upstream from exon 5 (P1) and within exon 7 (P2) (see fig. 2B), yielded two different-sized products of ∼3.5 and ∼3.8 kb (not shown), which were designated alleles “A” and “B,” respectively. The ∼300-bp size difference reflected different numbers of a 56-bp repeat sequence in intron 6. This genomic segment, with nine different alleles detected, is highly polymorphic in the general population (table 2). Alleles 1 and 2, the two smallest alleles, contain five and six of the 56-bp repeats, respectively. The patient's alleles A and B, respectively, corresponded in size with alleles 4 and 6 in the general population. Since the ∼300-bp size difference between these two normally occurring repeat-sequence alleles is sufficient to account for the size differences between the normal- and abnormal-sized restriction fragments in the genomic Southern blot (fig. 2A), it is unlikely that these size differences are related to the patient’s EDS phenotype.
Table 2.
Allele Sizes and Frequencies of a Polymorphic Repeat in COL5A1 Intron 6[Note]
| Allele | Size(bp) | Frequencya |
| 1 | 481 | .03 |
| 2 | 537 | .31 |
| 3 | ∼950 | .03 |
| 4 | ∼1,000 | .39 |
| 5 | ∼1,050 | .11 |
| 6 | ∼1,250 | .05 |
| 7 | ∼1,300 | .02 |
| 8 | ∼1,350 | .05 |
| 9 | ∼1,400 | .01 |
Note.— The patient had alleles 4 (normal) and 6 (mutant). The repeat sequence is AGCG(TG)3CATGCGGGCACGGG(TG)2CATGAGCC(TG)5AGCG(TG)3. (An SphI site that was present in some repeats, but absent in others, is underlined.)
Frequencies were estimated on the basis of 96 chromosomes from unrelated individuals. Observed heterozygosity was 0.73. The polymorphism-information-content value was 0.69.
In addition to the difference in the length of the polymorphic repeat sequence in intron 6, two additional DNA sequence differences were observed between alleles A and B: (1) an A→G transition at the −2 position of the obligatory AG dinucleotide of the intron 4 splice-acceptor site (i.e., IVS4-2A→G) in allele B (figs. 2B and 3); and (2) substitution of a T for the C at nucleotide 738 (i.e., 738C→T, with the A of the initiator methionine being reference position 1 in cDNA) in allele A, in the coding region of exon 5 (fig. 2B). The latter substitution introduced a PstI site into exon 5 of allele A (fig. 2B) that did not affect the codon. In fact, the control individual was also found to be heterozygous for this PstI site. PstI restriction and direct sequencing of RT-PCR products that correspond to this region showed that, as expected on the basis of the splice-site mutation in allele B, the majority of expressed sequences that contained exon 5 were derived from allele A.
Figure 3.

DNA sequence through the acceptor splice junction of intron 4 in the two alleles (A and B) from the patient. In the normal allele, the intron ends with CAG, whereas, in the mutant allele, there is a substitution of G for the A so that the intron ends with CGG. Intron sequences are denoted by lowercase letters, and exon sequences are denoted by uppercase letters.
Splice-Junction Sequences That Flank the Two Spliced Exons 5 and 6 in the Patient's Mutant Allele Are Not Sufficient to Produce the Two-Exon Skip
The skipping of two exons as a consequence of a single splice-acceptor mutation is unexpected. One factor that could contribute to the skipping of both exons 5 and 6 is the poor fit of the intron 6 splice-donor site—which contains pyrimidines, rather than purines, at positions 3 and 4 (i.e., AGgtctgg, instead of AGgtaagt) (Takahara et al. 1995) and the donor-site consensus sequence (Shapiro and Senapathy 1987). To determine whether the splice-junction sequences that flank exons 5 and 6 in the patient's mutant allele were sufficient to produce the skipping of the two exons, the portions of alleles A and B comprising 103 bp of intron 4, all of exons 5–7, all of introns 5 and 6, and 395 bp of intron 7 were separately placed in an exon-trapping vector and transfected into COS-7 cells. Genomic sequences that included exon 4 and the remainder of intron 4 were not included in these constructs because of the large size (∼26 kb) of intron 4 (Takahara et al. 1995). As expected, the construct that contained sequences from normal allele A produced only correctly spliced mRNA that contained exons 5–7 (fig. 4). Surprisingly, the construct that contained the mutant allele B sequence gave rise to only one major mRNA species, somewhat smaller than that produced by the normal allele (fig. 4). This product, identical to one of the low-abundance splice products identified in the patient’s fibroblast mRNA, resulted from use of the cryptic splice site, within exon 5, constituted by an AG dinucleotide pair 11 and 12 nt from the normal intron 4/exon 5 junction (see fig. 2B). The absence of additional splice products in this system demonstrated that exons 5 and 6 (and 7) splice-junction sequences and other sequences contained within the exon-trapping vector were insufficient for the replication, in the patient’s allele, of the effects of the mutation, suggesting that additional COL5A1 sequences were involved in the production of the full range of splice products found in the patient’s fibroblast mRNA.
Figure 4.
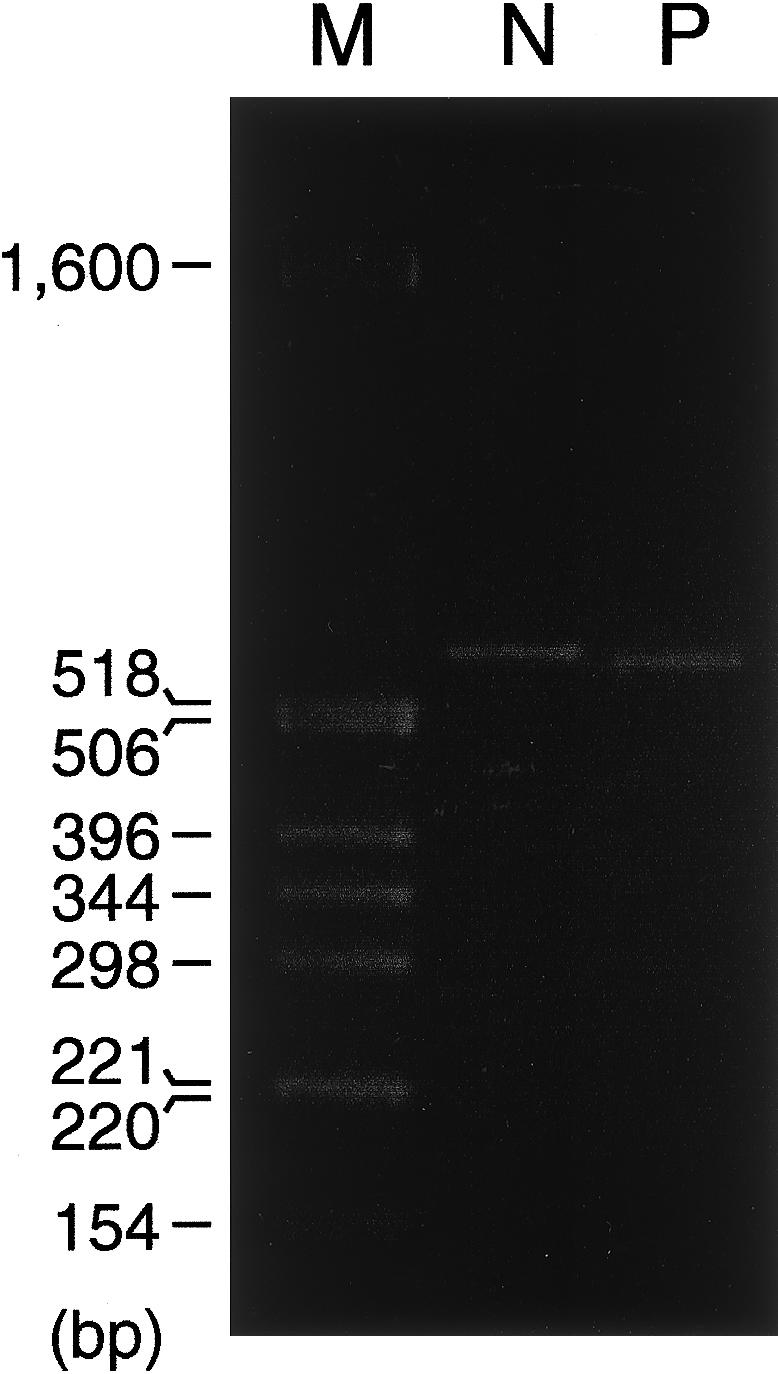
Spliced products after transfection of COS-7 cells with constructs that contain the normal and mutant sequences. Transfection with a construct containing wild-type normal (N) sequences produces a single band that corresponds to correctly spliced mRNA containing exons 5–7, whereas transfection with a construct containing the patient’s mutation (P) produces a single major band that corresponds to mRNA resulting from use of a cryptic splice site constituted by an AG dinucleotide pair 11 and 12 nt from the normal intron 4/exon 5 junction. M = size markers.
Order of Intron Removal in COL5A1 Precursor mRNA Indicates That Rapid Removal of Intron 5 Could Account for the Two-Exon Skip
Cells from the patient and from a control individual were incubated with actinomycin D, to stop transcription, and nuclear RNA was harvested after 0, 5, 10, 20, 40, and 60 min and was reverse transcribed to cDNA. Paired intron/exon or intron/intron primers were used to amplify different regions between intron 4 and exon 7 in the unspliced or partially spliced precursor mRNA, and the products were separated by PAGE. When an intron 4/exon 6 primer pair was used to determine the relative order of intron 4 and intron 5 removal, the full-length product was quickly lost in the control cells but remained present for 60 min in the patient cells (fig. 5A). This is consistent with a pathway in which intron 4 can be removed prior to removal of intron 5 in control cells. In the patient cells, this product contained only mRNA derived from the mutant allele (fig. 5B), consistent with the delayed removal of intron 4, presumably as a consequence of the acceptor-site mutation. A shorter product, from which intron 5 had been removed, persisted in both cell strains, although, in the patient cells, this product was derived largely from the mutant allele (fig. 5C). In both cell strains, this indicates that there is a separate pathway in which intron 5 is removed prior to excision of intron 4.
When the intron 4/intron 6 primer pair was used to determine whether there was a pathway in which intron 5 was removed prior to the two flanking introns, the amount of full-length product rapidly declined in the control cells but remained abundant in the patient cells (fig. 5D). A partially spliced product, from which intron 5 was removed but still containing introns 4 and 6, remained apparent in both cells. In the patient cells, this product was derived from the mutant and normal alleles in an approximately equal ratio, indicating that, in these cells, the pathway in which intron 5 was removed prior to the flanking introns was equally active in both alleles (fig. 5E). In additional studies, with primers in intron 5 and exon 7, very little of the full-length product was apparent, but the product that contained intron 5 but lacked intron 6 was present, consistent with a pathway in which intron 6 was removed prior to intron 5 in both patient and control cells (data not shown).
These data are consistent with a model in which there are multiple pathways of ordered intron removal in the region between exons 4 and 7 (fig. 6). In the control cells, there are pathways in which intron 4 is removed prior to intron 5, in which intron 5 is removed prior to intron 4, in which intron 5 is removed prior to intron 6, and in which intron 6 is removed prior to intron 5. In the cells from the patient, the removal of intron 4 appears to be delayed as a consequence of the acceptor-site mutation, but the removal of neither intron 5 nor intron 6 is appreciably altered. As a result, in most instances, intron 5 is spliced first (fig. 6) in the mutant allele. For the mutant allele, the next step in this splicing pathway can be either the skipping of the fused “5-6” exon or the removal of intron 6. The latter product, which contains fused exons 5–7 sequences, is then a substrate for use of the cryptic exon 5 acceptor sites. The product of the single exon 5 skip is generated from the pathway in which intron 6 is removed prior to intron 5 (fig. 6).
Figure 6.

Predicted pathways of intron removal in the mutant COL5A1 allele (the asterisk [*] indicates IVS4-2A→G), in which either intron 5 or intron 6 is removed first. If intron 5 is removed first, then the next step in this pathway can be either skipping of both exons 5 and 6 or removal of intron 6. The latter product is then a substrate for use in the cryptic exon 5 acceptor sites. The other pathway in which intron 6 is removed prior to intron 5 leads to the product of a single exon 5 skip.
Cultured Fibroblasts Incorporate Abnormal Pro-α1(V) Chains into Extracellular Matrix and Assemble Collagen Fibrils with Very Abnormal Structures
To determine the effects of the patient’s mutation at the protein level, we examined dermal fibroblasts cultured from the patient and from a control individual for pro-α1(V)–derived chains that were incorporated into the extracellular matrix associated with cell layers. The patient’s mutation did not appear to result in a secretion defect for type V procollagen, as ratios of type V:type I collagen were essentially the same in patient and control matrices (see the “Patient, Material, and Methods” section). Patient and control fibroblast matrix each contained multimeric forms of pro-α1(V)–derived chains (fig. 7A), probably from intact type V collagen molecules covalently cross-linked to each other and to chains of type I collagen, as occurs in heterotypic fibrils (Niyibizi and Eyre 1994). In addition, the matrix from each contained monomeric pro-α1(V)–derived forms that corresponded in size to pNα1(V) (from which the C-propeptide was removed, but in which the full-length N-propeptide was intact) and mature α1(V) chains (from which the C-propeptide and the PARP subdomain of the N-propeptide were removed, but which retained the N-propeptide variable region subdomain) (see figs. 7A, 7B, and 7D). Molecules that retained C-propeptides were not observed in either sample, consistent with the probability that retained C-propeptides prevent the incorporation of type V procollagen–derived forms into matrix (Fessler and Fessler 1987). Notably, matrix made by the patient’s cells contained two novel pro-α1(V)–derived chains not seen in control matrix, with mobilities intermediate between those of pNα1(V) and α1(V) forms (figs. 7A and 7B). The mobility differences between the novel chains and the normal α1(V) and pNα1(V) forms appear to reflect the different molecular weights of the chains translated from the different abnormal mRNA species. This conclusion is supported by analysis of matrix samples treated with bacterial collagenase. Such treatment of control matrix liberated fragments of 85 and 50 kD (fig. 7C) that corresponded, respectively, to the full-length N-propeptide and to that portion of the N-propeptide that remained in mature α1(V) chains subsequent to excision of the PARP subdomain by bone morphogenetic protein–1 (BMP-1) (fig. 7D). In contrast, similar treatment of matrix samples from the patient’s cells produced, in addition to the 85-kD form, two novel bands of 76 and 63 kD, which probably represent mutant N-propeptides missing sequences encoded by exon 5 (76 kD) and by both exons 5 and 6 (63 kD) (figs. 7C and 7D). The very small amount of the 50-kD band in the patient sample reflects the relatively small amount of total protein produced by the patient’s cells plus the decreased access that collagenase appears to have to mature α1(V) chains, compared to pNα1(V) forms, in matrix samples—for example, compare the ratio of pNα1(V):α1(V) chains to that of 85 kD:50 kD forms in the control lanes in figs. 7A and 7C, respectively.
Figure 7.

Secreted pro-α1(V)–derived chains incorporated into the extracellular matrix of control and patient cells. Bands on immunoblots were detected with polyclonal antibodies that were directed against sequences in the variable subdomain of the pro-α1(V) N-propeptide. A, Matrix showing multimeric and monomeric forms of pro-α1(V)–derived chains. Braces ({) indicate the multimeric forms. B, Close-up of a portion of a longer exposure of the immunoblot seen in panel A. C, Peptides liberated from matrix by treatment with bacterial collagenase. D, Representation of the 5′ end of COL5A1, the intron-exon structure, the predicted protein domains encoded by each exon, and the peptides derived from the matrix after cleavage with bacterial collagenase, which degrades the major collagenous domain (COL). The antibody-recognition domain (*), the site of BMP-1 cleavage, the PARP and variable domains, and the signal peptide (SP) are shown. Although the peptides have predicted molecular masses that are below the apparent molecular weights, these differences are probably due to N-linked glycosylation at sites in the PARP and variable subdomains.
When dermal fibroblasts from the patient and from a control individual were grown in culture for 3 wk, the fibrils formed by the control cells were small and compact with normal periodic staining. The fibrils from the patient cells had a complex architecture with a longitudinal twist (fig. 8), reminiscent of fibrils seen in skin from individuals with EDS type I (Vogel et al. 1979; Schwarze et al. 2000).
Figure 8.

Electron micrograph of collagen fibrils synthesized in culture by cells from a control individual (left) and from the patient (right). Fibrils formed by the control cells are small and compact with smooth contours and normal periodic staining. Fibrils from the patient cells have a complex architecture with irregular, jagged contours and longitudinal twists.
Discussion
We think that the findings in this study are important for two reasons: First, they show that the complex outcome of splice-site mutations can be explained by analysis of the order of intron removal. Second, they demonstrate that the amino-terminal end of the pro-α1(V) chain plays a significant role in fibril formation in the extracellular matrix.
In most previous reports of mutations at single splice junctions that result in the skipping of two or more exons (Schneider et al. 1993; Hayashida et al. 1994; Haire et al. 1997; Minami et al. 1999; Fang et al. 2001), the product of the multiexon skip is a low-abundance species. Usually, these are seen in the context of an alternatively spliced adjacent exon (Schneider et al. 1993; Hayashida et al. 1994; Minami et al. 1999), so that the effects of the mutations add to and are obscured by the unusual splicing features of the domain. An exception is a splice-donor mutation (IVS12a+1G→A), in the NF1 gene, in which exons 11 and 12a are skipped (Fang et al. 2001). In that instance, Fang et al. (2001) propose that there is excision, by use of the intron 10c donor site and intron 12a acceptor site, of the entire region. However, no demonstration of this pathway is provided, and the in vitro and transfection studies that were done use truncated intron extensions, which, as we found, may distort the outcome. In contrast to previous reports, the mutation that we describe is not in the context of an alternatively spliced region, and the two-exon skip is the major outcome.
The mechanisms whereby mutations at a single splice junction result in the skipping of multiple exons are not apparent from current exon- and intron-definition models of splicing (Berget 1995). We think that, by adding a mechanistic structure to the usual exon- and intron-definition models, analysis of the order of intron removal provides a way to explain the two-exon skip. The cells from the patient whom we studied derive multiple in-frame products from the mutant allele, which harbors the IVS4-2A→G sequence alteration. These products include one in which both exons 5 and 6 are skipped (the major product), one in which only exon 5 is skipped, and two other minor products in which cryptic acceptor sites in the first 15 nt of exon 5 are used. After transfection of a fragment of the mutant allele into COS-7 cells, when we attempted to replicate the effects of the mutation, we found, to our surprise, that only one product was produced—one of the minor products seen in cells from the affected individual, in which a cryptic site in exon 5 is used. These findings suggest that additional mRNA sequences are an important factor in the determination of the outcome of the mutation, since the sequences of introns 4 and 7 had been truncated for the purposes of the COS-7 cell transfection.
In cells from the patient, in some transcripts, intron 5 was removed rapidly, but, in other transcripts, intron 6 was removed prior to intron 5. Transcripts of the mutant allele from which intron 5 but not introns 4 and 6 had been removed accumulated in the nucleus, providing a substrate for the skipping of the fused 5-6 exon in one splicing event. Additional products accumulated in which introns 5 and 6 were removed such that neither exon 5 nor the combination of exons 5 and 6 could be removed. These instead provided the substrate in which the cryptic splice-acceptor sites in exon 5 could be used (see fig. 6). Because the internal intron (intron 5, in this case) was removed rapidly, the fused 5-6 exon could be removed rapidly, which could explain why the product of the two-exon skip was the major outcome of the mutation. As generally described, exon- and intron-definition models do not predict the proportion of different outcomes that result from a single mutation. When the order of intron removal is considered, it is apparent that the different products derive from different pathways, in proportion to the molecules that traverse each pathway.
Our studies suggest that, if splice-site mutations lead to two-exon skipping, then the exons chosen will differ depending on whether the mutation is at an acceptor site or a donor site. In the example here, the acceptor-site mutation leads to the removal of the two downstream exons, because those exons can be fused to a “single” exon while the next downstream intron and the mutation-bearing intron remain in the transcript. If, however, the mutation is at a donor site, then the removal of the upstream intron creates a “single” exon that, if the next upstream intron is retained, allows the skipping of the two exons upstream from the mutation site. This outcome is seen with a splice-donor site in the NF1 gene (Fang et al. 2001). Other studies of naturally occurring splice-site mutations in the human HPRT gene (O'Neill et al. 1998) and of induced mutations in the dhfr gene in Chinese hamster ovary cells (Carothers et al. 1993) have identified examples of two-exon skips that often conform to this model. In instances that do not, the loss of the second exon of the pair re-creates an in-frame product that is then stable. In these instances, it is often difficult to determine the extent to which the product of the two-exon skip represents a major splicing pathway or is simply the only product that is in-frame and thus is not subject to nonsense-mediated decay. Thus, whereas pathways in which the intervening intron is removed first may be the explanation for those events in which the skipping of either exon would provide a stable product, other mechanisms may be involved in other situations.
Relationships between COL5A1 mutations and the EDS phenotype are not entirely clear. In the present instance, unlike a number of other reported cases of classical EDS, the phenotype does not result from haploinsufficiency, as pro-α1(V)–derived chains encoded by the mutant allele are efficiently incorporated into the extracellular matrix. Rather, the mutation results in pro-α1(V) chains with an altered N-propeptide (most of which are missing the BMP-1 cleavage site encoded in exon 5) that appears to interfere with normal processing.
Dermal collagen fibrils are abnormal in several forms of EDS (Holbrook and Byers 1989). The most dramatic alterations occur in EDS type VIIC, or dermatosparaxis—in which deficiency of the enzyme procollagen N-proteinase leaves the N-propeptides of type I procollagen intact and sheets, rather than cylindrical fibrils, form (Smith et al. 1992). In EDS types VIIA and VIIB—in which part or all of the telopeptide sequences encoded by exon 6 of COL1A1 or COL1A2, respectively, are deleted from the protein—fibril formation is disrupted, but not as dramatically (Cole et al. 1987; Byers et al. 1997). Most individuals with EDS types I and II also have abnormal fibrils in skin (Holbrook and Byers 1989; Hausser and Anton-Lamprecht 1994). Fibril diameter is generally larger than normal, fibril borders are irregular, and composite fibrils are common. These aberrations occur in individuals who have alterations in the triple-helical domains of the type V collagen genes (Nicholls et al. 1996) and also in individuals in whom the mRNA from one allele of the COL5A1 gene is extremely unstable (Schwarze et al. 2000). In culture, cells from the individual studied here produced strikingly abnormal fibrils. Presumably, retention of the additional amino-terminal sequences on some pro-α1(V)–derived molecules—and/or derangement, due to loss of cysteines involved in intramolecular disulfide bonding, in the tertiary configuration of the N-propeptides—interferes with normal fibrillogenesis. Retained N-terminal variable subdomain sequences of mature α1(V) chains protrude beyond the surface of type I/type V heterotypic collagen fibrils and are hypothesized to play a role in the regulation of their diameters (Linsenmayer et al. 1993). Our results present in vivo evidence that the pro-α1(V) globular N-terminal sequences play an important role in the regulation of collagen fibril architecture. Moreover, the bizarre architecture of collagen fibrils that result from the N-propeptide mutation in the present report suggests roles for pro-α1(V) N-terminal sequences that lie beyond their postulated role in the prevention of addition, to heterotypic fibril surfaces via steric hindrance, of type I collagen monomers (Linsenmayer et al. 1993). The extent of derangement of the patient’s collagen fibrils here suggests the presence of additional mechanisms that have yet to be understood. Rare individuals such as this patient who have mutations that affect coding sequences and that have either unknown or only postulated roles in fibril formation provide those occasional glimpses into the biological prerequisites for collagen fibrillogenesis.
Acknowledgments
This work was supported by National Institutes of Health grants GM46846 and AR43621 (both to D.S.G.) and AR21557 (to P.H.B.).
Footnotes
The first two authors contributed equally to this work.
Electronic-Database Information
Accession numbers and the URL for data presented herein are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for EDS types I [MIM 130000] and II [MIM 130010])
References
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman SD, Smith JA, Struhl K (1987) Current protocols in molecular biology. Green Publishing and Wiley Interscience, New York [Google Scholar]
- Barnes WM (1994) PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc Natl Acad Sci USA 91:2216–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beighton P (1993) The Ehlers-Danlos syndromes. In: Beighton P (ed) McKusick's heritable disorders of connective tissue. Mosby-Year Book, St Louis [Google Scholar]
- Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ (1998) Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Am J Med Genet 77:31–37 [DOI] [PubMed] [Google Scholar]
- Berget SM (1995) Exon recognition in vertebrate splicing. J Biol Chem 270:2411–2414 [DOI] [PubMed] [Google Scholar]
- Bonadio J, Holbrook KA, Gelinas RE, Jacob J, Byers PH (1985) Altered triple helical structure of type I procollagen in lethal perinatal osteogenesis imperfecta. J Biol Chem 260:1734–1742 [PubMed] [Google Scholar]
- Bork P (1992) The modular architecture of vertebrate collagens. FEBS Lett 307:49–54 [DOI] [PubMed] [Google Scholar]
- Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Miller WL, Bristow J (1997) Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet 17:104–108 [DOI] [PubMed] [Google Scholar]
- Byers PH, Duvic M, Atkinson M, Robinow M, Smith LT, Krane SM, Greally MT, Ludman M, Matalon R, Pauker S, Quanbeck D, Schwarze U (1997) Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am J Med Genet 72:94–105 [DOI] [PubMed] [Google Scholar]
- Carothers AM, Urlaub G, Grunberger D, Chasin LA (1993) Splicing mutants and their second-site suppressors at the dihydrofolate reductase locus in Chinese hamster ovary cells. Mol Cell Biol 13:5085–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Fockler C, Barnes WM, Higuchi R (1994) Effective amplification of long targets from cloned inserts and human genomic DNA. Proc Natl Acad Sci USA 91:5695–5699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole WG, Evans R, Sillence DO (1987) The clinical features of Ehlers-Danlos syndrome type VII due to a deletion of 24 amino acids from the proα1(I) chain of type I procollagen. J Med Genet 24:698–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paepe A, Nuytinck L, Hausser I, Anton-Lamprecht I, Naeyaert JM (1997) Mutations in the COL5A1 gene are causal in the Ehlers-Danlos syndromes I and II. Am J Hum Genet 60:547–554 [PMC free article] [PubMed] [Google Scholar]
- Fang LJ, Simard MJ, Vidaud D, Assouline B, Lemieux B, Vidaud M, Chabot B, Thirion JP (2001) A novel mutation in the neurofibromatosis type 1 (NF1) gene promotes skipping of two exons by preventing exon definition. J Mol Biol 307:1261–1270 [DOI] [PubMed] [Google Scholar]
- Fessler JH, Fessler LI (1987) Type V collagen. In: Mayne R, Burgeson RE (eds) Structure and function of collagen types. Academic Press, Orlando, pp 81–103 [Google Scholar]
- Greenspan DS, Cheng W, Hoffman GG (1991a) The pro-α1(V) collagen chain: complete primary structure, distribution of expression, and comparison with the pro-α1(XI) collagen chain. J Biol Chem 266:24727–24733 [PubMed] [Google Scholar]
- Greenspan DS, Lee ST, Lee BS, Hoffman GG (1991b) Homology between α2(V) and α1(III) collagen promoters and evidence for negatively acting elements in the α2(V) first intron and 5′ flanking sequences. Gene Expr 1:29–39 [PMC free article] [PubMed] [Google Scholar]
- Haire RN, Ohta Y, Strong SJ, Litman RT, Liu Y, Prchal JT, Cooper MD, Litman GW (1997) Unusual patterns of exon skipping in Bruton tyrosine kinase are associated with mutations involving the intron 17 3′ splice site. Am J Hum Genet 60:798–807 [PMC free article] [PubMed] [Google Scholar]
- Hata R, Kurata S, Shinkai H (1988) Existence of malfunctioning proα2(I) collagen genes in a patient with a proα2(I)-chain-defective variant of Ehlers-Danlos syndrome. Eur J Biochem 174:231–237 [DOI] [PubMed] [Google Scholar]
- Hausser I, Anton-Lamprecht I (1994) Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum Genet 93:394–407 [DOI] [PubMed] [Google Scholar]
- Hayashida Y, Mitsubuchi H, Indo Y, Ohta K, Endo F, Wada Y, Matsuda I (1994) Deficiency of the E1β subunit in the branched-chain α-keto acid dehydrogenase complex due to a single base substitution of the intron 5, resulting in two alternatively spliced mRNAs in a patient with maple syrup urine disease. Biochim Biophys Acta 1225:317–325 [DOI] [PubMed] [Google Scholar]
- Hazeki N, Yamato M, Imamura Y, Sasaki T, Nakazato K, Yamamoto K, Konomi H, Hayashi T (1998) Analysis of matrix protein components of the dermis-like structure formed in a long-term culture of human fibroblasts: type VI collagen is a major component. J Biochem (Tokyo) 123:587–595 [DOI] [PubMed] [Google Scholar]
- Holbrook KA, Byers PH (1989) Skin is a window on heritable disorders of connective tissue. Am J Med Genet 34:105–121 [DOI] [PubMed] [Google Scholar]
- Imamura Y, Steiglitz BM, Greenspan DS (1998) Bone morphogenetic protein-1 processes the NH2-terminal propeptide, and a furin-like proprotein convertase processes the COOH-terminal propeptide of pro-α1(V) collagen. J Biol Chem 273:27511–27517 [DOI] [PubMed] [Google Scholar]
- Kessler O, Jiang Y, Chasin LA (1993) Order of intron removal during splicing of endogenous adenine phosphoribosyltransferase and dihydrofolate reductase pre-mRNA. Mol Cell Biol 13:6211–6222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsenmayer TF, Gibney E, Igoe F, Gordon MK, Fitch JM, Fessler LI, Birk DE (1993) Type V collagen: molecular structure and fibrillar organization of the chicken α1(V) NH2-terminal domain, a putative regulator of corneal fibrillogenesis. J Cell Biol 121:1181–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalickova K, Susic M, Willing MC, Wenstrup RJ, Cole WG (1998) Mutations of the α2(V) chain of type V collagen impair matrix assembly and produce Ehlers-Danlos syndrome type I. Hum Mol Genet 7:249–255 [DOI] [PubMed] [Google Scholar]
- Minami N, Nishino I, Kobayashi O, Ikezoe K, Goto Y, Nonaka I (1999) Mutations of calpain 3 gene in patients with sporadic limb-girdle muscular dystrophy in Japan. J Neurol Sci 171:31–37 [DOI] [PubMed] [Google Scholar]
- Nicholls AC, Oliver JE, McCarron S, Harrison JB, Greenspan DS, Pope FM (1996) An exon skipping mutation of a type V collagen gene (COL5A1) in Ehlers-Danlos syndrome. J Med Genet 33:940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls AC, Valler D, Wallis S, Pope FM (2001) Homozygosity for a splice site mutation of the COL1A2 gene yields a non-functional proα2(I) chain and an EDS/OI clinical phenotype. J Med Genet 38:132–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niyibizi C, Eyre DR (1994) Structural characteristics of cross-linking sites in type V collagen of bone: chain specificities and heterotypic links to type I collagen. Eur J Biochem 224:943–950 [DOI] [PubMed] [Google Scholar]
- Nuytinck L, Freund M, Lagae L, Pierard GE, Hermanns-Le T, De Paepe A (2000) Classical Ehlers-Danlos syndrome caused by a mutation in type I collagen. Am J Hum Genet 66:1398–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill JP, Rogan PK, Cariello N, Nicklas JA (1998) Mutations that alter RNA splicing of the human HPRT gene: a review of the spectrum. Mutat Res 411:179–214 [DOI] [PubMed] [Google Scholar]
- Sasaki T, Arai K, Ono M, Yamaguchi T, Furuta S, Nagai Y (1987) Ehlers-Danlos syndrome: a variant characterized by the deficiency of proα2 chain of type I procollagen. Arch Dermatol 123:76–79 [DOI] [PubMed] [Google Scholar]
- Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL, Bristow J (2001) A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med 345:1167–1175 [DOI] [PubMed] [Google Scholar]
- Schneider S, Wildhardt G, Ludwig R, Royer-Pokora B (1993) Exon skipping due to a mutation in a donor splice site in the WT-1 gene is associated with Wilms' tumor and severe genital malformations. Hum Genet 91:599–604 [DOI] [PubMed] [Google Scholar]
- Schwarze U, Atkinson M, Hoffman GG, Greenspan DS, Byers PH (2000) Null alleles of the COL5A1 gene of type V collagen are a cause of the classical forms of Ehlers-Danlos syndrome (types I and II). Am J Hum Genet 66:1757–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze U, Starman BJ, Byers PH (1999) Redefinition of exon 7 in the COL1A1 gene of type I collagen by an intron 8 splice-donor–site mutation in a form of osteogenesis imperfecta: influence of intron splice order on outcome of splice-site mutation. Am J Hum Genet 65:336–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MB, Senapathy P (1987) RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15:7155–7174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LT, Wertelecki W, Milstone LM, Petty EM, Seashore MR, Braverman IM, Jenkins TG, Byers PH (1992) Human dermatosparaxis: a form of Ehlers-Danlos syndrome that results from failure to remove the amino-terminal propeptide of type I procollagen. Am J Hum Genet 51:235–244 [PMC free article] [PubMed] [Google Scholar]
- Steinmann B, Royce PM, Superti-Furga A (1993) The Ehlers-Danlos syndrome. In: Royce PM, Steinmann B (eds) Connective tissue and its heritable disorders: molecular, genetic and medical aspects. Wiley-Liss, New York, pp 351–407 [Google Scholar]
- Takahara K, Hoffman GG, Greenspan DS (1995) Complete structural organization of the human α1(V) collagen gene (COL5A1): divergence from the conserved organization of other characterized fibrillar collagen genes. Genomics 29:588–597 [DOI] [PubMed] [Google Scholar]
- Takahara K, Lyons GE, Greenspan DS (1994) Bone morphogenetic protein-1 and a mammalian tolloid homologue (mTld) are encoded by alternatively spliced transcripts which are differentially expressed in some tissues. J Biol Chem 269:32572–32578 [PubMed] [Google Scholar]
- Takahara K, Sato Y, Okazawa K, Okamoto N, Noda A, Yaoi Y, Kato I (1991) Complete primary structure of human collagen α1(V) chain. J Biol Chem 266:13124–13129 [PubMed] [Google Scholar]
- Toriello HV, Glover TW, Takahara K, Byers PH, Miller DE, Higgins JV, Greenspan DS (1996) A translocation interrupts the COL5A1 gene in a patient with Ehlers-Danlos syndrome and hypomelanosis of Ito. Nat Genet 13:361–365 [DOI] [PubMed] [Google Scholar]
- Tsumaki N, Kimura T (1995) Differential expression of an acidic domain in the amino-terminal propeptide of mouse pro-α2(XI) collagen by complex alternative splicing. J Biol Chem 270:2372–2378 [DOI] [PubMed] [Google Scholar]
- Unsöld C, Pappano WN, Imamura Y, Steiglitz BM, Greenspan DS (2002) Biosynthetic processing of the pro-α1(V)2pro-α2(V) collagen heterotrimer by bone morphogenetic protein-1 and furin-like proprotein convertases. J Biol Chem 277:5596–5602 [DOI] [PubMed] [Google Scholar]
- van der Rest M, Garrone R (1991) Collagen family of proteins. FASEB J 5:2814–2823 [PubMed] [Google Scholar]
- Vogel A, Holbrook KA, Steinmann B, Gitzelmann R, Byers PH (1979) Abnormal collagen fibril structure in the gravis form (type I) of Ehlers-Danlos syndrome. Lab Invest 40:201–206 [PubMed] [Google Scholar]
- Wenstrup RJ, Florer JB, Willing MC, Giunta C, Steinmann B, Young F, Susic M, Cole WG (2000) COL5A1 haploinsufficiency is a common molecular mechanism underlying the classical form of EDS. Am J Hum Genet 66:1766–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenstrup RJ, Langland GT, Willing MC, D'Souza VN, Cole WG (1996) A splice-junction mutation in the region of COL5A1 that codes for the carboxyl propeptide of proα1(V) chains results in the gravis form of the Ehlers-Danlos syndrome (type I). Hum Mol Genet 5:1733–1736 [DOI] [PubMed] [Google Scholar]
- Zhidkova NI, Brewton RG, Mayne R (1993) Molecular cloning of PARP (proline/arginine-rich protein) from human cartilage and subsequent demonstration that PARP is a fragment of the NH2-terminal domain of the collagen α2(XI) chain. FEBS Lett 326:25–28 [DOI] [PubMed] [Google Scholar]