

Abstract
Children with autism spectrum disorder (ASD) and age-matched typically-developing (TD) peers were tested on two forms of eyeblink conditioning (EBC), a Pavlovian associative learning paradigm where subjects learn to execute an appropriately-timed eyeblink in response to a previously neutral conditioning stimulus (CS). One version of the task, trace EBC, interposes a stimulus-free interval between the presentation of the CS and the unconditioned stimulus (US), a puff of air to the eye which causes subjects to blink. In delay EBC, the CS overlaps in time with the delivery of the US, usually with both stimuli terminating simultaneously. ASD children performed normally during trace EBC, exhibiting no differences from typically-developing (TD) subjects with regard to learning rate or the timing of the CR. However, when subsequently tested on delay EBC, subjects with ASD displayed abnormally-timed conditioned eye blinks that began earlier and peaked sooner than those of TD subjects, consistent with previous findings. The results suggest an impaired ability of children with ASD to properly time conditioned eye blinks which appears to be specific to delay EBC. We suggest that this deficit may reflect a dysfunction of cerebellar cortex in which increases in the intensity or duration of sensory input can temporarily disrupt the accuracy of motor timing over short temporal intervals.
Keywords: autism, eyeblink conditioning, timing, cerebellum
1. Introduction
Autism spectrum disorders (ASD) are early-onset childhood disorders characterized by core impairments in social interaction, language, and repetitive and stereotyped movements (American Psychiatric Association 2000). The severity of the core impairments varies widely across the autism spectrum, and this extreme heterogeneity can impede the ability to apply objective measures of brain function uniformly across subjects and as they pass through different stages of development (Siegel et al. 1988). The establishment of biomarkers and techniques to non-invasively probe functional brain abnormalities related to core symptoms are key challenges.
Classical conditioning of the eyeblink (Gormezano et al. 1983) – also known as eyeblink conditioning (EBC) - is an objective method to measure brain function that is attractive for the study of children with ASD. EBC does not depend upon verbal or social interaction and simple modifications of the paradigm can probe the functioning of different levels of the brain. Furthermore, EBC performance has been shown to be sensitive to functional impairments in an array of disorders including attention deficit hyperactivity disorder (Frings et al., 2010), fetal alcohol syndrome (Coffin et al., 2005; Jacobson et al., 2008, 2011), schizophrenia (Sears et al., 2000; Edwards et al., 2008), Fragile-X (Koekkoek et al., 2005), depression (Greer et al., 2005), post-traumatic stress disorder (Burriss et al., 2007), dyslexia (Coffin et al., 2005; Nicolson et al., 2002), and neurodegenerative diseases such as Huntington’s (Woodruff-Pak and Papka, 1996), and Alzheimer’s (Woodruff-Pak, 2001). There are two general forms of EBC: trace and delay. The difference between trace and delay EBC is the presence or absence of a stimulus-free (or “trace”) period between the onset of a conditioned stimulus (CS) and an unconditioned stimulus (US; Fig. 1). In trace EBC, there is a stimulus free period between the CS and US; in delay EBC, there is not. For both types of EBC, the US is usually a brief air puff directed to the eye that reliably elicits an eyeblink, called the unconditioned response (UR). The CS is often a tone, but can be any other stimulus that does not elicit an eyeblink before conditioning. “Conditioning” consists of repeated pairings of the CS followed by the US at a fixed interval, typically ranging between 250 and 1000 ms. Over time, subjects begin to blink in response to the CS in advance of the US. This adaptive behavior is termed the conditioned response (CR) and its frequency of occurrence provides a measure of associative learning. The timing of the CR provides an estimate of the precision with which the brain can encode a temporal interval in the sub-second range.
Fig. 1.
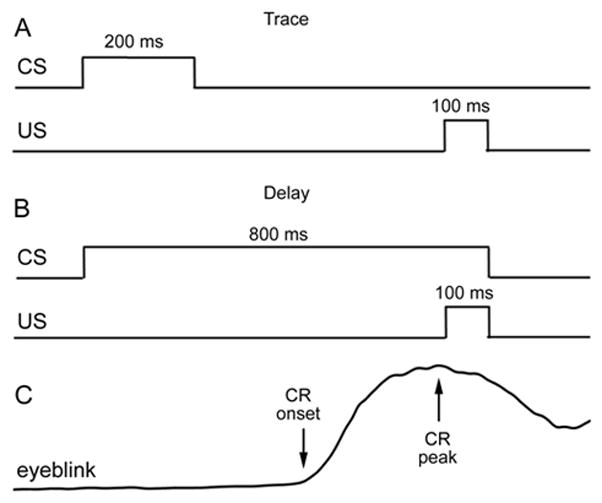
EBC protocols. Square wave plots depict stimulus timing during (a) trace EBC and (b) delay EBC. The CS-US interval (US onset - CS onset) was held constant at 700 ms in the two testing paradigms. The duration of the trace period (US onset-CS offset) was 500 ms. In delay EBC the tone CS and air puff US co-terminated. Subjects performed two sessions of trace EBC followed by one session of delay EBC. Sessions were conducted on separate days. (c) Schematic of the eyeblink CR as measured during testing. Upward deflection of the plot denotes closure of the eyelid. Eye blinks of sufficient amplitude (see Methods) occurring after the CS and prior to the US were classified as CRs and their onset and peak latencies recorded (arrows).
A wealth of fundamental neurobiology indicates that EBC paradigms can be used to probe forebrain and brainstem/cerebellar systems whose dysfunction has been implicated in ASD. Trace EBC is impaired by damage in the medial prefrontal cortex (Kronforst-Collins et al. 1998; Weible et al. 2000), a region whose cytology and connectivity is altered in ASD (Courschesne & Pierce 2005; Amaral et al. 2008; Sundaram et al. 2008). The medial prefrontal cortex demonstrates persistent activity during trace EBC that may maintain a representation of the CS during the trace period that is subsequently relayed to the hippocampus (Siegel et al. 2011). In humans, trace EBC is impaired by bilateral lesions to the medial temporal lobe that include the hippocampus (McGlinchey-Berroth et al. 1997; Clark & Squire 1998), which is functionally activated during trace EBC (Cheng et al. 2008) and necessary in non-human animals (Solomon et al. 1986; Moyer et al. 1990; McEchron et al. 1998). The combination of prefrontal and hippocampal involvement in trace EBC implicates long-range connections between the frontal and temporal lobes, which may be altered in ASD (Bode et al. 2011). In contrast, delay EBC does not require the forebrain (Mauk & Thompson 1987) and its minimal neural circuitry is believed to reside primarily in the brainstem and cerebellum (Thompson, 2005). Brainstem/cerebellar circuitry permitting the performance of CRs during delay EBC was established by lesion and recording experiments in experimental animals (McCormick & Thompson 1984; Yeo et al. 1985; Welsh & Harvey 1989, 1991; Harvey et al. 1993; Perret et al. 1993; Welsh & Harvey 1998) which have been confirmed in humans with cerebellar stroke or degeneration (Gerwig et al. 2005, 2008) and in normal humans undergoing functional imaging (Molchan et al. 1994; Cheng et al. 2008; Parker et al. 2012).
Alterations in cerebellar anatomy have been implicated in ASD, although there is considerable heterogeneity. The most often reported finding is hypoplasia of the cerebellar posterior lobe vermal lobules VI and VII (Courschesne et al. 1988; Kates et al. 1998; Kaufmann et al. 2003). However, involvement of other cerebellar lobules, such as the anterior lobe vermis and vermal lobules VIII-X has also sometimes been observed (Levitt et al. 1999; Webb et al. 2009). Not all studies have found vermal hypoplasia (Piven et al. 1997) and one report found hypoplasia only in high-functioning ASD and not low-functioning ASD or Asperger’s disorder (Scott et al. 2009). Other reports have found cerebellar enlargement (Piven et al. 1997; Harden et al. 2001; Palmen et al. 2005). Numerous histopathological studies have reported a loss of Purkinje cells in ASD, especially in the vermis and hemispheral lobules of the posterior lobe (Ritvo et al. 1986; Bauman & Kemper 1994; Palmen et al. 2004). However a recent quantitative study (Whitney et al. 2008) reported that only half of ASD cerebella (3 of 6) showed convincing Purkinje cell loss. Overall, the literature points to cerebellar involvement in ASD, with the caveat of a high degree of heterogeneity whose relevance to symptom expression is not understood.
There has only been one study of EBC in ASD subjects (Sears et al. 1994). That study examined 11 ASD subjects, diagnosed using DSM-III-R criteria, and 11 non-ASD subjects. The study used a single session of delay EBC and found that learning was more rapid in ASD subjects and that ASD subjects showed CRs that occurred abnormally early and which failed to adapt to the CS-US interval. Drawing analogy to EBC experiments performed on brain-lesioned rabbits from that time period, Sears et al. (1994) concluded that the pathophysiology underlying ASD involved disruptions in both hippocampal and cerebellar memory storage.
Here, we undertook a much needed follow-up study to re-examine the performance of ASD subjects on EBC. Our experimental design extended the earlier study by Sears et al. (1994) in a number of ways. First, we employed more recent clinical evaluation methods to diagnose ASD (American Psychiatric Association, 2000) than those used by Sears and colleagues. Second, we tested the same subjects over multiple sessions holding crucial parameters, such as the interstimulus interval, constant, thus allowing us to assess the performance of ASD subjects on retention of the CS-US association and CR timing. Third, we tested subject performance on trace EBC. In contrast to delay EBC, trace EBC performance is dependent on both cortical and subcortical forebrain regions, and had not been previously investigated in children with autism. Finally, the results from our trace EBC experiment prompted us to subsequently test our subjects on one session of delay EBC, allowing for a comparison of each subject’s performance on a forebrain- and brainstem/cerebellar-dependent task (trace EBC) with that on an exclusively brainstem/cerebellar-dependent task (delay EBC). Our results suggest that the previously-reported enhancement of learning and early-onset CRs for ASD subjects trained on delay EBC (Sears et al., 1994) fail to generalize to trace EBC. We then show that ASD subjects initially trained on trace EBC display early-onset CRs when switched to delay EBC with the same interstimulus interval. Our results suggest that early-onset conditioned eyeblinks are endemic to delay conditioning, and that this deficit may have its origins in cerebellar cortical dysfunction that fails to fully compensate for abrupt increases in the duration and/or intensity of sensory input.
2. Experimental Procedures
2.1 Subjects
The subjects were 30 children ranging in age from 6 to 15 years. Fourteen were diagnosed with ASD (13 male, 1 female) and 16 were typically-developing (TD; 7 male, 9 female). ASD subjects included children diagnosed with autistic disorder (n = 7), Asperger’s Disorder (Asp, n = 5), and pervasive developmental disorder-not otherwise specified (PDD-NOS, n = 2) based on the content-area scores on the revised Autism Diagnostic Interview (ADI-R; Lord et al. 1994) and the Childhood Autism Rating Scale (Schopler et al. 1980). By convention, Asp and PDD-NOS children met criteria on 2 of the 3 domains assessed on the ADI-R and showed developmental delays prior to 3 years of age based on retrospective report. Exclusion factors for the ASD group were the presence of psychiatric diagnosis including Rett’s disorder or childhood disintegrative disorder. TD subjects had no psychiatric diagnoses other than one subject being diagnosed with oppositional defiant disorder and obsessive-compulsive disorder (ODD/OCD). The ASD and TD groups were matched for age (ASD, 8.6 ± 2.7 years; TD, 9.6 ± 2.5 years, p = 0.30, t-test) and IQ (mean WASI scores: 106 ± 11 for ASD and 111 ± 13 for TD (p = 0.27). Two TD subjects (one male, one female) had a male sibling in the ASD group. Three groups of TD subjects (n = 2, 4, and 2) were siblings. However, exclusion of these individuals from the analysis did not change the conclusions of the study and statistically strengthened the group difference in onset latency measured across the first several blocks of delay conditioning. Because of this we included all subjects in our analysis. All subjects had normal or corrected-normal vision and were sufficiently cooperative to undergo the experiment.
2.2 Eyeblink Conditioning
EBC was carried out using the San Diego Instruments (San Diego, CA) portable system. Subjects sat in a quiet room, seated at a desk watching a silent movie of their choice on a video monitor. The experimenter and the experimental hardware were housed in an adjacent, acoustically-isolated room from which the subject could be observed through a one-way mirror. The subjects wore custom-designed laboratory safety goggles and headphones (Sennheiser, model eh350). Eye blinks were detected by an infrared emitter-sensor assembly (Honeywell, HOA1405) mounted on the goggles positioned approximately 1 cm from the pupil. Eye blinks were defined as a change in the sensor output that exceeded 15 standard deviations above the mean baseline output during the 750 ms prior to stimulus delivery. Sensor output was digitized at 1 kHz, recorded to a hard drive, and analyzed off-line using custom-written Matlab routines. The CS was a 1-kHz, 61-dB pure tone delivered binaurally through the headphones and calibrated with a sound level meter (RadioShack) fitted to the headphone earpad with an acoustic coupler (model DR1-R, Digital Recordings, Halifax, Nova Scotia). The US was a 100 ms puff of air (5 psi source pressure) delivered to the right cornea through a stainless steel tube (1 mm i.d.) attached to the infrared sensor. The behavioral control system was calibrated to account for the delay in the travel time of the air puff from the compressor unit to the air ejection tube. Prior to a session of EBC, subjects were given 5 presentations of the US alone to ensure that the sensor and air tube assembly were properly positioned and that the US elicited eye blinks.
The subjects underwent 3 sessions of EBC in a 2-phase procedure. Each EBC session consisted of 90 trials, divided into 9 blocks of 10 trials. The first 9 trials in each block consisted of paired CS-US trials and the 10th trial was a CS-alone trial. The intertrial interval was 20 sec, on average (range 15–25 sec). EBC sessions occurred on separate visits and the parents were paid $25 per visit. Effort was made to limit the time between sessions to no greater than 3 weeks. The mean time between trace EBC sessions was 16 ± 3 days for TD and 10 ± 4 days for the ASD subjects (mean ± 1 SEM, p = 0.12). The mean time between the second trace and delay EBC session was 39 ± 16 days for TD subjects. The large variation was due to 3 subjects who had 3 months or longer between sessions. Excluding those subjects, the average time between sessions 2 and 3 for TD subjects was 12 ± 3 days while that for the ASD subjects was 14 ± 4 days. No significant correlation was found between the intersession interval and EBC performance measures, and covariance analysis showed that variation in intersession interval could not explain the observed differences in CR latency measures. Because of this, all subjects were included in the analyses.
2.3 Phase 1: Trace EBC
Phase 1 consisted of 2 sessions of trace EBC. Trace EBC was carried out using a 200-ms CS followed by a 500-ms trace interval and then the US, defining a 700 ms CS-US interval (Fig. 1A). The 700 ms CS-US interval was chosen on the basis of a functional imaging study that demonstrated it robustly activated the hippocampus in humans during trace EBC (Cheng et al. 2008). CRs were defined as eye blinks that occurred at least 80 ms after CS onset and prior to US onset (Sears et al. 1994).
2.4 Phase 2: Delay EBC
Phase 2 consisted of 1 session of delay EBC. As shown in Fig. 1B, delay EBC used the identical CS-US interval as was used in Phase 1 (700 ms) and was accomplished simply by extending the duration of the CS by 600 ms so that the CS co-terminated with the US. All other conditioning parameters remained the same as in Phase 1. Thus, the only difference between Phases 1 and 2 was replacement of the stimulus-free portion of the CS-US interval with a lengthened tone CS.
2.5 Analysis and Statistics
A combination of parametric and non-parametric statistical tests was used to appropriately analyze the data. Differences in the mean values of CR frequency, onset latency, and peak latency were analyzed using a mixed-design analysis of variance (ANOVA) with blocks of 10 trials being the repeated measure and experimental group being the between variable, and with Scheffe post-hoc tests. In order to minimize Type I error, the Greenhouse-Geisser correction was applied in instances where the sphericity assumption (Mauchly’s test) was violated. Data for CR parameters were averaged for each 10-trial block (9 blocks/session). For ANOVAs of CR latency, in trial blocks in which a subject failed to perform a CR the missing value was replaced with the corresponding group mean for that block. However, those values were not included in calculations of mean latencies in the data plots. Paired t-tests were used to compare individual values of 10-trial blocks between EBC sessions. No replacement values were used for t-tests and any subjects not contributing latency values were excluded from the analysis. One TD subject who exhibited only one CR during the final two blocks of Phase 1 was excluded from the latency analysis. Averaged data are presented as the mean ± 1 standard error of the mean (SEM). Analyses were performed using SPSS (ver. 19) and OpenStat (Miller 2012) software. Statistical significance was set at p < 0.05.
Mean CR topography was calculated for each subject by averaging trials in which CRs were performed for a specific condition. Thereafter, the mean waveform for each group was calculated by averaging the mean CR waveforms from the individual subjects. Final waveforms were smoothed with a Gaussian kernel (full width at half maximum value of 15, a standard deviation of approximately 6.4).
3. Results
3.1 Phase 1: Trace EBC
Fig. 2A displays CR acquisition curves over the two trace EBC sessions for the TD and ASD groups. Both groups showed learning of the CS-US association by displaying a significant increase in percentage CRs from the first to the second session [F(1,17) = 3.2, p < 0.005]. Most of the increase in CR frequency occurred in the first block of the second session, with modest increases within each session. There was neither a significant difference in learning rate between the groups [F(1,27) = 0.01, p = 0.99] nor a significant difference in the shape of the learning curves across the two sessions [F(1,17) = 0.8, p = 0.67]. Additionally, no significant main effect of group (p > 0.8) or interaction of group by trial-blocks (p > 0.3) was detected during either of the trace EBC sessions. The data indicate that ASD subjects acquired CRs normally during trace EBC. The rate of CR acquisition was consistent with studies of TD children at this age under comparable stimulus parameters (Frings et al. 2010; Jacobson et al. 2011).
Fig. 2.
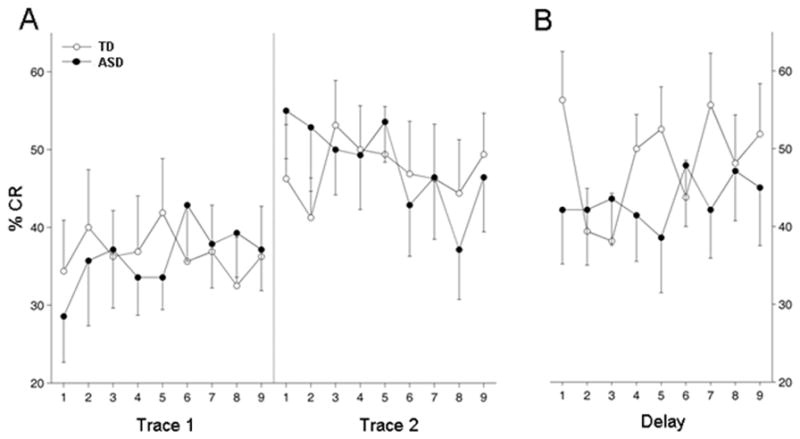
Percent CRs. Values represent percent CRs for each block of 10 trials (9 blocks per session, n = 16 for TD group and n = 14 for ASD group). (a) Results from two sessions of trace EBC. (b) Results for the single session of delay EBC. Percent CRs increased across the trace EBC sessions for both groups and did not differ between groups. Most of the increase in percent CRs occurred at the beginning of the second trace EBC session. There was no significant difference in percent CRs in the delay EBC session. Data presented as the mean ± 1 SEM.
Further analysis was performed on the timing characteristics of the CRs acquired during trace EBC. Mean values of CR onset (Fig. 3A) and peak latency (Fig. 3B) between the TD and ASD groups were not significantly different during the first session of trace EBC [F(1,28) = 0.96, p = 0.34 for onset latency; F(1,28) = 1.0, p = 0.32 for CR peak latency]. Both groups showed an increase in CR onset latency as the session progressed, from an overall average of 381 ± 25 ms, in the first block of session 1 to 469 ± 22 ms during the last block of the session, indicating that the CRs adapted to the CS-US interval by moving out in time toward US onset with conditioning [F(8,224) = 2.4, p < 0.05]. CR peak latency also showed a comparable increase, from 464 ± 23 ms in the first block of trace EBC session 1 to 537 ± 21 ms during the last block of the session [F(8,224) = 2.2, p < 0.05].
Fig. 3.
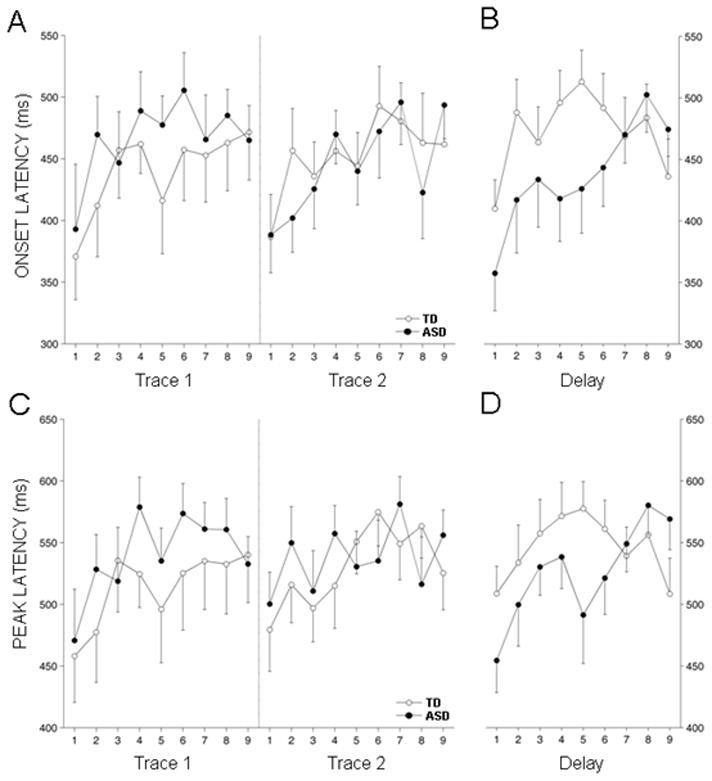
CR onset and peak latencies. (a,c) Group means for onset and peak latency, respectively, for the two sessions of trace EBC. (b,d) Mean onset and peak latency, respectively, for the session of delay EBC. In general, latencies were at their lowest values for all groups at the beginning of each session and gradually migrated toward the time of US onset as the session progressed. Onset latencies in the first 3 blocks of trace EBC were not different between the groups. In contrast, ASD group onset latencies were significantly lower at the outset of the delay EBC session (asterisks indicate p < 0.05). By the end of the session, ASD onset latencies were statistically indistinguishable from those of the TD group. Peak latency values for the groups during delay EBC displayed a similar pattern, though the decrease for the ASD group at the beginning of the session was not quite significant by ANOVA. Data presented as the mean ± 1 SEM.
At the beginning of trace session 2, the groups showed a learning enhancement, expressing a higher percentage CRs averaging 46 ± 7, 55 ± 6 for the TD and ASD groups, respectively, relative to the last block of the previous session (36 ± 7 and 37 ± 5, respectively, F(1,28) = 5.4, p < 0.05). The timing of CRs showed robust change during this period characterized by shorter onset latency (387 ± 20 ms) and shorter peak latency (489 ± 19 ms) at the beginning of session 2 than at the end of session 1. The change was evident among both groups, as was a rapid readaptation of CR timing with conditioning so that during the last block of 10 trials mean onset and peak latencies had again moved toward the US to have overall mean values of 477 ± 19 ms and 540 ± 17 ms, respectively, an effect which was highly significant [F(8,224) = 3.7, p < 0.001 for onset latency; F(8,224) = 2.6, p < 0.01 for peak latency]. There was no significant group difference in the timing parameters through session 2 [F(1,28) = 0.1, p = 0.79 for onset latency; F(1,28) = 0.1, p = 0.78 for peak latency]. It is noteworthy that despite a relatively high percentage of CRs at the beginning of trace session 2, indicating a robust CS-US association, retention of stimulus timing was weak in both groups, with the ASD group showing less retention of CR timing that the TD group.
3.2 Phase 2: Delay EBC
The absence of any difference in learning rate or onset latency between ASD and TD subjects contrasted with a previous study of delay EBC by Sears et al. (1994) in which learning occurred more rapidly in ASD subjects and CRs occurred with abnormally shorter onset latency. We asked whether the difference in the experimental outcomes might have been due to a difference between delay and trace EBC. In trace EBC, a stimulus-free (trace) period intervenes between the CS and US (Fig. 1A), while in delay EBC there is temporal overlap between the CS and US (Fig. 1B). To test this, we exposed our subjects to one additional session of EBC where the only change to the stimulus parameters was to extend the CS duration in order to fill the CS-US interval, thereby producing a delay EBC paradigm (Fig. 1B).
Analysis of CR timing parameters during delay EBC revealed significant differences between the groups (Fig. 3). The main effect was that CR onset and peak latencies of the ASD group occurred significantly earlier than those of the TD group. Both groups showed an abrupt decrease in CR onset latency (Fig. 3A) during the first block of delay EBC relative to the last block of trace EBC, but this effect was more pronounced for the ASD group (mean percent decrease of 7.4 ± 6 and 25.7 ± 6 for the TD and ASD groups, respectively; p < 0.05, t-test). CR onset latencies were significantly shorter for the ASD group through the first 6 blocks of delay EBC [F(1,28) = 4.5, p < 0.05]. Mean values for both groups increased through the session, with the ASD group changing more substantially [F(8,224) = 2.1, p < 0.05, block x group interaction], such that values for the two groups were very similar during the last three blocks of training (p = 0.54). CR peak latencies (Fig. 3B) showed a similar pattern of change from trace to delay EBC for the ASD group but a very small change for the TD group. The peak latency shift from the last block of trace EBC to the first block of delay EBC was more pronounced for the ASD group (mean percent decrease of 0.04 ± 5 and 17 ± 5 for the TD and ASD groups, respectively; p < 0.05, t-test). Peak latencies in the ASD group trended lower than those of the TD group through the first 6 blocks of delay EBC [F(1,28)=3.6, p = 0.07] but this difference disappeared during the last 3 blocks [F(1,28) = 1.4, p = 0.25]. This pattern of change resulted in a significant interaction effect through the entire session [F(8,224)=2.5, p = 0.04, Greenhouse-Geisser correction].
Importantly, the alteration in CR timing observed in the first several blocks of delay EBC occurred without a significant change in the percentage CRs, indicating that the CS-US association acquired during trace EBC was intact. Switching to delay EBC did not significantly change the CR frequency of ASD subjects from their previous trace EBC session (Fig. 2B) and there was no significant difference between the TD and ASD groups in overall mean CR frequency during delay EBC [F(1,28) = 0.41, p = 0.53 for blocks 1–6 and F(1,28) = 0.7, p = 0.41 for all 9 blocks].
An unexpected finding was that the ASD subjects were unimpaired in their ability to show normal motor adaptation of their CRs to the CS-US interval with repeated trials of delay EBC, indicating that the effect of ASD on CR timing was transient, not due to an inability of ASD subjects to adapt or perform properly timed CRs, and therefore triggered by the paradigm switch. Mean values of CR onset and peak latency for all groups increased through the session of delay EBC, with the ASD group changing more substantially [block x group interactions: F(8,224) = 2.1, p < 0.05 for onset latency, and F(8,224) = 3.08, p < 0.05 for peak latency]. During the last 3 blocks of delay EBC, neither the mean CR onset latency nor the mean CR peak latency were significantly different among the groups [F(1,28) = 0.4, p = 0.54 for onset latency and F(1,28) = 1.4, p = 0.25 for peak latency]. The experiment indicated that ASD children were not impaired in their ability to readapt the timing of their CRs to the CS-US interval.
3.3 Control Analyses for Gender and Age
While there has been no report, to our knowledge, of gender-based differences in EBC, our experimental groups were not evenly balanced with regard to gender, with 13 of 14 ASD subjects being male and only 7 of 16 TD subjects being male. It was therefore necessary to determine whether the higher percentage of females in the TD group might have contributed to the observed differences in CR latency measures. We examined the learning performance and CR metrics of males and females in the TD group during the delay EBC session and during the transition from trace to delay EBC, the two critical periods where performance differences between TD and ASD subjects were observed.
With regard to mean CR onset latency, no significant main effect of gender [F(1,14) = 1.3, p = 0.28] and no interaction effect of gender by trial-block [F(8,12) = 0.6, p = 0.81] was found during either the entire delay EBC session or through the first 6 blocks of training [group: F(1,14) = 1.0, p = 0.34; group x block interaction: F(5,70) = 0.7, p = 0.61] where the differences between TD and ASD groups were greatest. Likewise, no significant main effect of gender and no interaction effect of gender by trial blocks was found for CR peak latencies during the entire delay EBC session [group: F(1,14) = 0.8, p = 0.38; group x block interaction: F(8,112) = 0.3, p = 0.97] or during the first 6 blocks of training [group: F(1,14) = 0.8, p = 0.39; group x block interaction: F(5,70) = 0.3, p = 0.92].
Additionally, no significant difference was found between male and female TD subjects in the percent decrease in CR onset (p = 0.31) or peak (p = 0.54) latency from the final block of trace EBC to the first block of delay EBC. Finally, learning performance was not significantly different across the entire delay EBC session [F(1,14) = 1.4, p = 0.26 for group; F(8,112) = 0.2, p = 0.99 for group x block interaction] or through the first 6 blocks of training [F(1,14) = 1.1, p = 0.32 for group; F(5,70) = 0.2, p = 0.89 for group x block interaction]. The results clearly showed that there were no statistically detectable performance differences between males and females in the TD group. Therefore it is highly unlikely that gender imbalance contributed to the observed differences in CR latencies between TD and ASD subjects.
Finally, variations in subject age did not account for onset latency differences observed during the delay EBC session. Subject age was not a significant covariate in onset latency values in the first six blocks of delay EBC, where differences in CR latencies were significant [F(1,27) = 0.3, p = 0.60], nor did it correlate with the decrease in CR onset latency from the last block of trace EBC to the first block of delay EBC (r = 0.14, p = 0.94).
4. Discussion
In this experiment, we tested potential impairments in short-duration temporal processing, associative learning and motor adaptation in ASD by using a combination of trace and delay EBC. It is accepted that different, but overlapping, brain systems contribute to learning under trace and delay EBC, with the former involving a much higher engagement of forebrain areas such as the hippocampus (Clark and Squire, 1998; Solomon et al., 1986), frontal cortex (McLaughlin et al., 2002; Weiss and Disterhoft, 2011), and striatum (Flores and Disterhoft, 2009) to encode the CS-US association. It is also generally accepted that delay EBC utilizes a more restricted neural circuitry in the hindbrain in which temporal processing of the CS-US interval and the association of the CS with the US are encoded by brainstem/cerebellar systems. CS- and US-related afference from brainstem sensory areas converge within the brainstem reticular formation and cerebellum. The deep cerebellar nuclei provide a critical link between sensory input pathways and reflex motor circuitry controlling eye blinks. A known overlap in the underlying neural circuitries engaged by trace and delay EBC is the cerebellum, which is believed to be necessary for both the motor and timing aspects of EBC. Specifically, the cerebellar cortex in particular is believed to mediate the adaptive timing of the CR in both trace and delay EBC, and scales the onset and peak of the CR to match the time of US delivery (Gerwig et al. 2005, 2008; Kalmbach et al. 2010). In trained subjects, mechanical lesions or chemical inactivation of the cerebellar cortex spares CRs but disrupts their adaptive timing, resulting in short-latency conditioned blinks (Perrett et al., 1993, Kalmbach et al., 2010).
Phase 1 of our experiment presented the first test, to our knowledge, of ASD subject performance on trace EBC. The results demonstrated that any impairment in cerebral cortical and lower forebrain function in our ASD subjects was not sufficient to impair trace EBC. Children with ASD were normal in the ability to temporally process the CS-US interval and to form the CS-US association during trace EBC. This was unexpected given numerous recent demonstrations of alterations in forebrain development, cytology, connectivity, and functional activation in ASD. On the other hand, while the hippocampus has been heavily implicated in trace EBC, hippocampal neuropathology is not a hallmark of ASD, and when present is subtle compared to other areas of the cerebrum (Amaral et al. 2008) and scales with ASD severity (Dager et al., 2007). Changes in columnar organization (Casanova et al. 2002, 2003), packing density, neuron numbers (Courschesne et al. 2011), and synaptic inhibition (Oblak et al. 2009; Blatt & Fatemi 2011) have been documented in ASD neocortex, and those alterations have been hypothesized to underlie changes in cortical activity patterns during the resting state and sensory processing (Wilson et al. 2007; Cornew et al. 2011). If those neocortical changes were present in our high-functioning ASD subjects, they did not affect trace EBC.
The most unexpected finding during trace EBC was that ASD subjects were normal in their ability to appropriately time their CRs to the CS-US interval, indicating intact cerebellar processing of motor adaptation. The normal CR adaptation by ASD children during EBC stands in contrast to the previous report of impaired CR timing during EBC (Sears et al. 1994). Given the current understanding that cerebellar function helps determine CR timing in both trace and delay EBC (Gerwig et al. 2005, 2008; Kalmbach et al. 2010), our results did not support the hypothesis that a significant disruption of cerebellar motor adaptation occurs in ASD. The finding of substantial motor adaptation during trace EBC adds to other demonstrations that cerebellar-dependent motor adaptation is intact in ASD as measured by vestibulo-ocular reflex adaptation (Goldberg et al. 2000), adaptive catching (Mostofsky et al. 2004), adaptive throwing during visual field shift (Gidley Larson et al. 2008), and adaptive reaching during external force perturbations (Gidley Larson et al. 2008).
The major finding of our study was that once having normally learned trace EBC, ASD subjects transiently expressed a high prevalence of early-onset CRs during subsequent exposure to delay EBC with an identical CS-US interval. This result is in concordance with the previous observations of Sears et al. (1994) of early onset CRs in ASD subjects trained exclusively on a single session of delay EBC with a considerably shorter interstimulus interval. Notably, however, unlike in the Sears et al. (1994) study, ASD children in our experiment showed robust CR adaptation with continued delay EBC, having a time-course that was not different than the de novo adaptation during trace EBC. ASD subjects, therefore, behaved as if they were naive to the CS-US interval but possessed intact cerebellar motor adaptation. The timing adaptation observed in our subjects during delay EBC, but absent from the subjects tested in Sears et al. (1994), may have been due to the extensive training our subjects received on trace EBC with the same interstimulus interval prior to delay EBC testing.
The behavioral phenotype of short-latency CRs that we observed in ASD subjects is similar to effects previously reported in ASD (Sears et al. 1994), in other neurological conditions including ADHD (Frings et al. 2010), schizophrenia (Bolbecker et al., 2009; Sears et al., 2000), and Huntington’s disease (Woodruff-Pak and Papka, 1996). Perhaps noteworthy here is the fact that ASD and ADHD exhibit substantial overlap in symptomatology, including hyperactivity and deficits in attention (Dickerson Mayes et al., 2012). Interestingly, other disorders such as fetal alcohol syndrome (Coffin et al., 2005; Jacobson et al., 2008, 2011) and dyslexia (Coffin et al., 2005) result in longer CR latencies, demonstrating that EBC performance is to some degree sensitive to different neuropathologies.
With regard to the current findings, short-latency CRs have been observed in experimental animals with destruction of cerebellar cortex (e.g., Perrett et al., 1993). However, our study was unique in showing that short-latency CRs in ASD subjects were neither permanent nor general (occurring both during delay and trace EBC). The cerebellar cortex has been implicated in imparting adaptive timing to eyeblink CRs (McCormick & Thompson 1984; Harvey et al. 1993; Perrett et al. 1993; Garcia & Mauk 1998; Garcia et al. 1999; Kalmbach et al. 2010), as large destruction of anterior lobe and paravermal cortex can shorten CR onset latency without reducing their prevalence. Genetically-altered mice with impairments in synaptic plasticity between Purkinje cells and their presynaptic neurons also exhibited shorter-latency CRs (Koekkek et al. 2003, but see Welsh et al. 2005; Schonewille et al. 2011), as did mice that completely lacked Purkinje cells (Chen et al. 1996). Likewise, rats born of dams exposed to the teratogenic, anti-convulsant drug valproic acid exhibit Purkinje neuron abnormalities and a decrease in overall cerebellar volume (Rodier et al., 2001), as well as early-onset CRs during EBC (Murawski et al., 2009; Stanton et al., 2001).
Extending from these observations, one hypothesis is that the behavioral phenotype of ASD children we observed was a cerebellar disorder due to a loss of Purkinje cells. Qualitative histology studies have often reported a loss of Purkinje cells in ASD (Ritvo et al. 1986; Bauman & Kemper 1994; Palmen et al. 2004), although the generality of that finding has been brought into question by the first quantitative analysis of Purkinje cell numbers in ASD (Whitney et al. 2008). Under the assumption that Purkinje cell loss is widespread in ASD, Sears et al. (1994) postulated that the loss of inhibitory Purkinje cells in ASD would increase deep cerebellar nuclear activity to potentiate motor neuron activation and reduce CR latency. In this respect, the increase in CS duration that our subjects experienced when they were switched from trace to delay EBC would have provided more prolonged excitatory drive throughout the brain due to the longer auditory input, including onto deep cerebellar nuclear neurons which, when disinhibited by a loss of Purkinje cells, might have reduced CR latency (Sears et al., 1994). This hypothesis has foundational support from a detailed study of the computational properties of the cerebellar Purkinje neuronal network (Steuber et al., 2007). It can also be regarded as consistent with a sensorimotor gating deficit in ASD, such as has been observed using the prepulse inhibition paradigm in which ASD subjects’ startle response was less likely to be inhibited by a preceding auditory stimulus (McAlonan et al. 2002; Perry et al. 2007). Sensorimotor gating deficits have been proposed to underlie the inability of individuals with ASD to properly gate stereotypic patterns of movement and thought (McAlonan et al. 2002). The current understanding that there is heterogeneity of Purkinje cell loss in ASD may explain the heterogeneity observed in CR timing among ASD subjects as a whole. The fact that Purkinje cell loss in ASD is sporadic may account for the ability of ASD subjects to readapt with continued EBC training.
A second hypothesis is that CR timing during trace and delay EBC is controlled by different circuitries that were differentially impaired in our ASD subjects. Although a recent study (Kalmbach et al. 2010) provided evidence for a critical role of cerebellar cortex in regulating CR timing in both trace and delay EBC, it has been hypothesized that CR timing in trace EBC is mediated by a forebrain timing circuit (Woodruff-Pak et al. 2006; Disterhoft & Woodruff-Pak 2008). While it is demonstrated that cerebellar cortical lesions tend to have less impact on CR expression in trace than in delay EBC, the effects of cerebellar damage on CR timing generalize across paradigms (Gerwig et al. 2006; Kalmbach et al. 2010). The absence of any evidence of CR timing abnormalities during trace EBC in our study was consistent with a lack of impairment in any forebrain circuits that have been hypothesized to be involved in CR timing but also reduces the probability that the transient deficits in CR timing upon the switch to delay EBC were due to a generalized impairment in motor performance.
In summary, our data suggest that the timing of conditioned eye blink responses in ASD subjects is disrupted during delay, but not trace, EBC. However, we stress that given our relatively small group sizes and constraints pertaining to subject selection (the presence of sibling controls and imprecise gender matching between experimental groups) further investigations will be required to corroborate these findings. If our results are substantiated, additional studies could examine potential correlations between EBC performance metrics and autism phenotype severity. Though additional analyses of our data revealed no significant correlations between CR timing or CR timing changes and individual ADI-R subscores in our ASD subjects, this is perhaps not surprising given our modest group sizes and the considerable heterogeneity in our cohort. Studies with larger experimental groups will allow for more effective comparisons between EBC performance measures and ASD severity. Further investigations should also seek to examine CR latency differences in younger subjects, particularly given that EBC can be applied to individuals at very early stages of development, including infants (Herbert et al., 2003; Reeb-Sutherland et al. 2012). A wealth of fundamental neurobiology implicates brainstem/cerebellar systems in EBC which might be impacted by the changes in cerebellar anatomy that have been reported in ASD. As EBC is an objective paradigm that can be applied equally to humans and non-human animals, it can facilitate bidirectional research to establish mechanisms of neurological dysfunction in ASD.
Highlights.
Children with autism spectrum disorders (ASD) underwent eyeblink conditioning.
Children with ASD performed normally during trace eyeblink conditioning.
Children with ASD showed short-latency conditioned eyeblinks on delay conditioning.
Short-latency conditioned eyeblinks may reflect cerebellar cortical dysfunction.
ASD may be associated with a deficit in sensorimotor timing.
Acknowledgments
This study was supported by National Institute of Mental Health grant R21 MH084219 to RPM. JPW was supported by National Institute of Neurological Disorders and Stroke grant R01 NS31224-19. We sadly acknowledge the passing of Professors Harvey and Malone during the course of this study. We thank Alicia Fuscellaro for assistance with data tabulation and experimental protocols, and Mark Shiber at the Drexel Machine shop for technical assistance with experimental hardware.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends in Neuroscience. 2008;31:137–142. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. 2000. [Google Scholar]
- Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism. In: Bauman ML, Kemper TL, editors. The Neurobiology of Autism. Baltimore, MD: Johns Hopkins University Press; 1994. pp. 119–145. [Google Scholar]
- Bode MK, Mattila ML, Kiviniemi V, Rahko J, Moilanen I, Ebeling H, et al. White matter in autism spectrum disorders – evidence of impaired fiber formation. Acta Radiology. 2011;52:1169–1174. doi: 10.1258/ar.2011.110197. [DOI] [PubMed] [Google Scholar]
- Blatt GJ, Fatemi SH. Alterations in GABAergic biomarkers in the autism brain: Research findings and clinical implications. Anatomical Record (Hoboken) 2011;294:1646–1652. doi: 10.1002/ar.21252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolbecker AR, Mehta CS, Edwards CR, Steinmetz JE, O’Donnell BF, Hetrick WP. Eye-blink conditioning deficits indicate temporal processing abnormalities in schizophrenia. Schizophrenia Research. 2009;111:182–191. doi: 10.1016/j.schres.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burriss L, Ayers E, Powell DA. Combat veterans show normal discrimination during differential trace eyeblink conditioning, but increased responsivity to the conditioned and unconditioned stimulus. Journal of Psychiatric Research. 2007;41:785–794. doi: 10.1016/j.jpsychires.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Casanova MF, Buxhoeveden D, Gomez J. Disruption in the inhibitory architecture of the cell minicolumn: implications for autism. Neuroscientist. 2003;9:496–507. doi: 10.1177/1073858403253552. [DOI] [PubMed] [Google Scholar]
- Casanova MF, Buxhoeveden DP, Switala AE, Roy I. Minicolumnar pathology in autism. Neurology. 2002;58:428–432. doi: 10.1212/wnl.58.3.428. [DOI] [PubMed] [Google Scholar]
- Chen L, Bao S, Lockard JM, Kim JJ, Thompson RF. Impaired classical eyeblink conditioning in cerebellar-lesioned and purkinje cell degeneration (pcd) mutant mice. Journal of Neuroscience. 1996;16:2829–2838. doi: 10.1523/JNEUROSCI.16-08-02829.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng DT, Disterhoft JF, Power JM, Ellis DA, Desmond JE. Neural substrates underlying human delay and trace eyeblink conditioning. Proceedings of the National Academy of Sciences (USA) 2008;105:8108–8113. doi: 10.1073/pnas.0800374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RE, Squire LR. Classical conditioning and brain systems: the role of awareness. Science. 1998;280:77–81. doi: 10.1126/science.280.5360.77. [DOI] [PubMed] [Google Scholar]
- Coffin JM, Baroody S, Schneider K, O’Neill J. Impaired cerebellar learning in children with prenatal alcohol exposure: a comparative study of eyeblink conditioning in children with ADHD and dyslexia. Cortex. 2005;41:389–398. doi: 10.1016/s0010-9452(08)70275-2. [DOI] [PubMed] [Google Scholar]
- Cornew L, Roberts TP, Blaskey L, Edgar JC. Resting-state oscillatory activity in autism spectrum disorders. Journal of Autism and Developmental Disorders. 2011 doi: 10.1007/s10803-011-1431-6. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, et al. Neuron number and size in prefrontal cortex of children with autism. Journal of the American Medical Association. 2011;306:2001–2010. doi: 10.1001/jama.2011.1638. [DOI] [PubMed] [Google Scholar]
- Courschesne E, Pierce K. Why the frontal cortex in autism might be talking only to itself: local over-connectivity but long-distance disconnection. Current Opinion in Neurobiology. 2005;15:225–230. doi: 10.1016/j.conb.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Yeung-Courchesne R, Press G, Hesselink J, Jernigan T. Hypoplasia of cerebellar vermal lobules VI and VII in autism. New England Journal of Medicine. 1988;318:1349–1354. doi: 10.1056/NEJM198805263182102. [DOI] [PubMed] [Google Scholar]
- Dager SR, Wang L, Friedman SD, Shaw DW, Constantino JN, Artru AA, Dawson G, Csernansky JG. Shape mapping of the hippocampus in young children with autism spectrum disorder. American Journal of Radiology. 2007;28:672–677. [PMC free article] [PubMed] [Google Scholar]
- Dickerson Mayes S, Calhoun SL, Mayes RD, Molitoris S. Autism and ADHD: Overlapping and discriminating symptoms. Research in Autism Spectrum Disorders. 2012;6:277–285. [Google Scholar]
- Edwards CR, Newman S, Bismark A, Skosnik PD, O’Donnell BF, Shekhar A, Steinmetz JE, Hetrick WP. Cerebellum volume and eyeblink conditioning in schizophrenia. Psychiatry Research: Neuroimaging. 2008;162:185–194. doi: 10.1016/j.pscychresns.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores LC, Disterhoft JF. Caudate nucleus is critically involved in trace eyeblink conditioning. Journal of Neuroscience. 2009;29:14511–14520. doi: 10.1523/JNEUROSCI.3119-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frings M, Gaertner K, Buderath P, Gerwig M, Christiansen H, Schoch B, et al. Timing of conditioned eyeblink responses is impaired in children with attention-deficit/hyperactivity disorder. Experimental Brain Research. 2010;201:167–176. doi: 10.1007/s00221-009-2020-1. [DOI] [PubMed] [Google Scholar]
- Garcia KS, Mauk MD. Pharmacological analysis of cerebellar contributions to the timing and expression of conditioned eyelid responses. Neuropharmacology. 1998;37:471–480. doi: 10.1016/s0028-3908(98)00055-0. [DOI] [PubMed] [Google Scholar]
- Garcia KS, Steele PM, Mauk MD. Cerebellar cortex lesions prevent acquisition of conditioned eyelid responses. Journal of Neuroscience. 1999;19:10940–10947. doi: 10.1523/JNEUROSCI.19-24-10940.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerwig M, Eber AC, Guberina H, Frings M, Kolb FP, Forsting M, et al. Trace eyeblink conditioning in patients with cerebellar degeneration: comparison of short and long trace intervals. Experimental Brain Research. 2008;187:85–96. doi: 10.1007/s00221-008-1283-2. [DOI] [PubMed] [Google Scholar]
- Gerwig M, Haijar K, Dimitrova A, Maschke M, Kolb FP, Frings M, et al. Timing of conditioned eyeblink responses is impaired in cerebellar patients. Journal of Neuroscience. 2005;25:3919–3931. doi: 10.1523/JNEUROSCI.0266-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidley Larson JC, Bastian AJ, Donchin O, Shadmehr R, Mostofsky SH. Acquisition of internal models of motor tasks in children with autism. Brain. 2008;131:2894–2903. doi: 10.1093/brain/awn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MC, Landa R, Lasker A, Cooper L, Zee DS. Evidence of normal cerebellar control of the vestibulo-ocular reflex (VOR) in children with high-functioning autism. Journal of Autism and Developmental Disorders. 2000;30:519–524. doi: 10.1023/a:1005631225367. [DOI] [PubMed] [Google Scholar]
- Gormezano I, Kehoe EJ, Marshall BS. Twenty years of classical conditioning research with the rabbit. Progress in Psychobiology and Physiological Psychology. 1983;10:197–275. [Google Scholar]
- Greer TL, Trivedi MH, Thompson LT. Impaired delay and trace eyeblink conditioning performance in major depressive disorder. Journal of Affective Disorders. 2005;86:235–245. doi: 10.1016/j.jad.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Hardan AY, Minshew N, Harenski K, Keshavan M. Posterior fossa magnetic resonance imaging in autism. Journal of the American Academy of Child and Adolescent Psychiatry. 2001;40:666–672. doi: 10.1097/00004583-200106000-00011. [DOI] [PubMed] [Google Scholar]
- Harvey JA, Welsh JP, Yeo CH, Romano AG. Recoverable and nonrecoverable deficits in conditioned responses after cerebellar cortical lesions. Journal of Neuroscience. 1993;13:1624–1635. doi: 10.1523/JNEUROSCI.13-04-01624.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert JS, Eckerman CO, Stanton ME. The ontogeny of human learning in delay, long-delay and trace eyeblink conditioning. Behavioral Neuroscience. 2003;117:1196–1210. doi: 10.1037/0735-7044.117.6.1196. [DOI] [PubMed] [Google Scholar]
- Highnam CL, Bleile KM. Language in the cerebellum. American Journal of Speech and Language Pathology. 2011;20:337–347. doi: 10.1044/1058-0360(2011/10-0096). [DOI] [PubMed] [Google Scholar]
- Hoffland BS, Bologna M, Kassavetis P, Teo JTH, Rothwell JC, Yeo CH, et al. Cerebellar theta burst stimulation impairs eyeblink classical conditioning. Journal of Physiology (London) 2012;590.4:887–897. doi: 10.1113/jphysiol.2011.218537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SW, Stanton ME, Dodge NC, Pienaar M, Fuller DS, Molteno CD, et al. Impaired delay and trace eyeblink conditioning in school-age children with fetal alcohol syndrome. Alcoholism: Clinical and Experimental Research. 2011;35:250–264. doi: 10.1111/j.1530-0277.2010.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SW, Stanton ME, Molteno CD, Burden MJ, Fuller DS, Hoyme E, Robinson LK, Khaole N, Jacobson JL. Impaired eyeblink conditioning in children with fetal alcohol syndrome. Alcoholism: Clinical and Experimental Research. 2008;32:365–372. doi: 10.1111/j.1530-0277.2007.00585.x. [DOI] [PubMed] [Google Scholar]
- Kalmbach BE, Davis T, Ohyama T, Riuesch F, Nores WL, Mauk MD. Cerebellar cortex contributions to the expression of timing of conditioned eyelid responses. Journal of Neurophysiology. 2010;103:2039–2049. doi: 10.1152/jn.00033.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates WR, Mostofsky SH, Zimmerman AW, Mazzocco MM, Landa R, Warsofsky IS, et al. Neuroanatomical and neurocognitive differences in a pair of monozygous twins discordant for strictly defined autism. Annals of Neurology. 1998;43:782–791. doi: 10.1002/ana.410430613. [DOI] [PubMed] [Google Scholar]
- Koekkoek SKE, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJH, Maex R, De Graaf W, Smit AE, Vanderwerf F, Bakker CE, Willemsen R, Ikeda T, Kakizawa S, Onodera K, Nelson DL, Mientjes E, Joosten M, De Schutter E, Oostra BA, Ito M, De Zeeuw CI. Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X Syndrome. Neuron. 2005;47:339–352. doi: 10.1016/j.neuron.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Kronforst-Collins MA, Disterhoft JF. Lesions of the caudal area of rabbit medial prefrontal cortex impair trace eyeblink conditioning. Neurobiology of Learning and Memory. 1998;69:147–162. doi: 10.1006/nlme.1997.3818. [DOI] [PubMed] [Google Scholar]
- Levitt JG, Blanton R, Capetillo-Cunliffe L, Guthrie D, Toga A, Cracken JT. Cerebellar vermis lobules VIII–X in autism. Progress in Neuropsychopharmacology and Biological Psychiatry. 1999;23:625–633. doi: 10.1016/s0278-5846(99)00021-4. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, LeCouteur A. Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of Autism and Developmental Disorders. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Mauk MD, Thompson RF. Retention of classically conditioned eyelid responses following acute decerebration. Brain Research. 1987;403:89–95. doi: 10.1016/0006-8993(87)90126-0. [DOI] [PubMed] [Google Scholar]
- McAlonan GM, Daly E, Kumari V, Critchley HD, van Amelsvoort T, Suckling J, et al. Brain anatomy and sensorimotor gating in Asperger’s syndrome. Brain. 2002;127:1594–1606. doi: 10.1093/brain/awf150. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Thompson RF. Cerebellum: Essential involvement in the classically conditioned eyelid response. Science. 1984;223:296–299. doi: 10.1126/science.6701513. [DOI] [PubMed] [Google Scholar]
- McEchron MD, Bouwmeester H, Tseng W, Weiss C, Disterhoft JF. Hippocampectomy disrupts auditory trace fear conditioning and contextual fear conditioning in the rat. Hippocampus. 1998;8:638–646. doi: 10.1002/(SICI)1098-1063(1998)8:6<638::AID-HIPO6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- McGlinchey-Berroth R, Carrillo MC, Gabrieli JD, Brawn CM, Disterhoft JF. Impaired trace eyeblink conditioning in bilateral, medial-temporal lobe amnesia. Behavioral Neuroscience. 1997;111:873–882. doi: 10.1037//0735-7044.111.5.873. [DOI] [PubMed] [Google Scholar]
- McLaughlin J, Skaggs H, Churchwell J, Powell DA. Medial prefrontal cortex and pavlovian conditioning: trace versus delay conditioning. Behavioral Neuroscience. 2002;116:37–47. [PubMed] [Google Scholar]
- Miller WG. OpenStat. 2012 http://www.statprograms4U.com.
- Molchan SE, Sunderland T, McIntosh AR, Herscovitch P, Schreurs BG. A functional anatomical study of associative learning in humans. Proceedings of the National Academy of Sciences (USA) 1994;91:8122–8126. doi: 10.1073/pnas.91.17.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostofsky SH, Bunoski R, Morton SM, Goldberg MC, Bastian AJ. Children with autism adapt normally during a catching task requiring the cerebellum. Neurocase. 2004;10:60–64. doi: 10.1080/13554790490960503. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Devo RA, Disterhoft JF. Hippocampectomy disrupts trace eye-blink conditioning in rabbits. Behavioral Neuroscience. 1990;104:243–252. doi: 10.1037//0735-7044.104.2.243. [DOI] [PubMed] [Google Scholar]
- Murawski NJ, Brown KL, Stanton ME. Interstimulus interval (ISI) discrimination of the conditioned eyeblink response in a rodent model of autism. Behavioural Brain Research. 2009;196:297–303. doi: 10.1016/j.bbr.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Nicolson RI, Daum I, Schugens MM, Fawcett AJ, Schulz A. Eyeblink conditioning indicates cerebellar abnormality in dyslexia. Experimental Brain Research. 2002;143:42–50. doi: 10.1007/s00221-001-0969-5. [DOI] [PubMed] [Google Scholar]
- Oblak A, Gibbs TT, Blatt GJ. Decreased GABAA receptors and benzodiazelpine binding sites in the anterior cingulated cortex in autism. Autism Research. 2009;2:205–219. doi: 10.1002/aur.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmen SJ, Engeland H, Hof P, Schmitz C. Neuropathological findings in autism. Brain. 2004;127:2572–2583. doi: 10.1093/brain/awh287. [DOI] [PubMed] [Google Scholar]
- Palmen SJ, Hulshoff Pol HE, Kemner C, Schnack HG, Durston S, Lahuis BE, et al. Increased gray-matter volume in medication-naïve high-functioning children with autism spectrum disorder. Psychological Medicine. 2005;35:561–570. doi: 10.1017/s0033291704003496. [DOI] [PubMed] [Google Scholar]
- Parker KL, Andreasen NC, Liu D, Freeman JH, Ponto LL, O’Leary DS. Eyeblink conditioning in healthy adults: A positron emission tomotraphic study. Cerebellum. 2012 doi: 10.1007/s12311-012-0377-3. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrett SP, Mauk MD. Extinction of conditioned eyelid responses requires the anterior lobe of cerebellar cortex. Journal of Neuroscience. 1995;15:2074–2080. doi: 10.1523/JNEUROSCI.15-03-02074.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrett SP, Ruiz BP, Mauk MD. Cerebellar cortex lesions disrupt learning-dependent timing of conditioned eyelid responses. Journal of Neuroscience. 1993;13:1708–1718. doi: 10.1523/JNEUROSCI.13-04-01708.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biological Psychiatry. 2007;61:482–486. doi: 10.1016/j.biopsych.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Piven J, Saliba K, Bailey J, Arndt S. An MRI study of autism: the cerebellum revisited. Neurology. 1997;49:546–551. doi: 10.1212/wnl.49.2.546. [DOI] [PubMed] [Google Scholar]
- Reeb-Sutherland BC, Levitt P, Fox NA. The predictive nature of individual differences in early associative learning and emerging social behavior. PLoS One. 2012;7:e30511. doi: 10.1371/journal.pone.0030511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritvo ER, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, et al. Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA–NSAC autopsy research project. American Journal of Psychiatry. 1986;143:862–866. doi: 10.1176/ajp.143.7.862. [DOI] [PubMed] [Google Scholar]
- Rodier PM, Ingram JL, Tisdale B. Morphology and function in an animal model of autism. Neurotoxicology and Teratology. 2001;23:284. [Google Scholar]
- Schonewille M, Gao Z, Boele HJ, Veloz MF, Amerika WE, Simek AA, et al. Reevaluating the role of LTD in cerebellar motor learning. Neuron. 2011;70:43–50. doi: 10.1016/j.neuron.2011.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopler E, Reichler R, DeVellis R. Toward objective classi cation of childhood autism: childhood autism rating scale (CARS) Journal of Autism and Developmental Disorders. 1980;10:91–103. doi: 10.1007/BF02408436. [DOI] [PubMed] [Google Scholar]
- Scott JA, Schumann CM, Goodlin-Jones BL, Amaral DG. A comprehensive volumetric analysis of the cerebellum in children and adolescents with autism spectrum disorder. Autism Research. 2009;2:246–257. doi: 10.1002/aur.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears LL, Andreasen NC, O’Leary DS. Cerebellar functional abnormalities in schizophrenia are suggested by classical eyeblink conditioning. Biological Psychiatry. 2000;48:204–209. doi: 10.1016/s0006-3223(00)00247-x. [DOI] [PubMed] [Google Scholar]
- Sears LL, Finn PR, Steinmetz JE. Abnormal classical eye-blink conditioning in autism. Journal of Autism and Developmental Disorders. 1994;24:737–751. doi: 10.1007/BF02172283. [DOI] [PubMed] [Google Scholar]
- Siegel B, Pliner C, Eschler J, Elliott G. How children with autism are diagnosed: Difficulties in identification of children with multiple developmental delays. Journal of Developmental and Behavioral Pediatrics. 1988;9:199–204. [PubMed] [Google Scholar]
- Siegel JJ, Kalmbach B, Chitwood RA, Mauk MD. Persistent activity in a cortical-to-subcortical circuit: bridging the temporal gap in trace eyelid conditioning. Journal of Neurophysiology. 2012;107:50–64. doi: 10.1152/jn.00689.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon PR, Vander Schaff ER, Thompson RF, Weisz DJ. Hippocampus and trace conditioning of the rabbit’s classically conditioned nictitating membrane response. Behavioral Neuroscience. 1986;100:729–744. doi: 10.1037//0735-7044.100.5.729. [DOI] [PubMed] [Google Scholar]
- Stanton ME, Erwin RJ, Rush AN, Robinette BL, Rodier PM. Eyeblink conditioning in autism and a developmental rodent model. Neurotoxicology and Teratology. 2001;23:297. [Google Scholar]
- Steuber V, Mittmann W, Hoebeek FE, Silver RA, DeZeeuw CI, Hausser M, De Schutter E. Cerebellar LTD and pattern recognition in Purkinje cells. Neuron. 2007;54:121–136. doi: 10.1016/j.neuron.2007.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram SK, Kumar A, Makki MI, Behen ME, Chugani HT, Chugani DC. Diffusion tensor imaging of frontal lobe in autism spectrum disorder. Cerebral Cortex. 2008;18:2659–2665. doi: 10.1093/cercor/bhn031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RF. In search of memory traces. Annual Review of Psychology. 2005;56:1–23. doi: 10.1146/annurev.psych.56.091103.070239. [DOI] [PubMed] [Google Scholar]
- Webb SJ, Sparks BF, Friedman SD, Shaw DW, Giedd J, Dawson G, Dager SR. Cerebellar vermal volumes in children with autism spectrum disorder. Psychiatry Research. 2009;172:61–67. doi: 10.1016/j.pscychresns.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weible AP, McEchron MD, Disterhoft JF. Cortical involvement in acquisition and extinction of trace eyeblink conditioning. Behavioral Neuroscience. 2000;114:1058–1067. doi: 10.1037//0735-7044.114.6.1058. [DOI] [PubMed] [Google Scholar]
- Weiss C, Disterhoft JF. Exploring prefrontal cortical memory mechanisms with eyeblink conditioning. Behavioral Neuroscience. 2011;125:318–326. doi: 10.1037/a0023520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Estes AM, Dager SR. Establishing links between cerebellar imaging findings and symptom expression in autistic disorder. In: Fatemi SH, et al., editors. Consensus paper: Pathological role of the cerebellum in autism. Cerebellum. 2012. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Harvey JA. Cerebellar lesions and the nictitating membrane reflex: Performance deficits of the conditioned and unconditioned response. Journal of Neuroscience. 1989;9:299–311. doi: 10.1523/JNEUROSCI.09-01-00299.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Harvey JA. Pavlovian conditioning in the rabbit during inactivation of the interpositus nucleus. Journal of Physiology (London) 1991;444:459–480. doi: 10.1113/jphysiol.1991.sp018888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Harvey JA. Acute inactivation of the inferior olive blocks associative learning. European Journal of Neuroscience. 1998;10:3321–3332. doi: 10.1046/j.1460-9568.1998.00400.x. [DOI] [PubMed] [Google Scholar]
- Welsh JP, Yamaguchi H, Zeng XH, Kojo M, Nakada Y, Takagi A, et al. Normal motor learning during pharmacological prevention of Purkinje cell long-term depression. Proceedings of the National Academy of Sciences (USA) 2005;102:17166–17171. doi: 10.1073/pnas.0508191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney ER, Kemper TL, Bauman ML, Rosene DL, Blatt GJ. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum. 2008;7:406–416. doi: 10.1007/s12311-008-0043-y. [DOI] [PubMed] [Google Scholar]
- Wilson TW, Rojas DC, Reite ML, Teale PD, Rogers SJ. Children and adolescents with autism exhibit reduced MEG steady-state gamma responses. Biological Psychiatry. 2007;62:192–197. doi: 10.1016/j.biopsych.2006.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff-Pak DS. Eyeblink classical conditioning differentiates normal aging from Alzheimer’s disease. Integrative Physiological and Behavioral Science. 2001;36:87–108. doi: 10.1007/BF02734044. [DOI] [PubMed] [Google Scholar]
- Woodruff-Pak DS, Papka M. Huntington’s disease and eyeblink classical conditioning: normal learning but abnormal timing. Journal of the International Neuropsychological Society. 1996;2:323–334. doi: 10.1017/s135561770000134x. [DOI] [PubMed] [Google Scholar]
- Yeo CH, Hardiman MH, Glickstein M. Classical conditioning of the nictitating membrane response of the rabbit. II Lesions of the cerebellar cortex. Experimental Brain Research. 1985;60:99–113. doi: 10.1007/BF00237023. [DOI] [PubMed] [Google Scholar]