

Abstract
Bronchopulmonary dysplasia of the premature newborn is characterized by lung injury, resulting in alveolar simplification and reduced pulmonary function. Exposure of neonatal mice to hyperoxia enhanced sphingosine-1-phosphate (S1P) levels in lung tissues; however, the role of increased S1P in the pathobiological characteristics of bronchopulmonary dysplasia has not been investigated. We hypothesized that an altered S1P signaling axis, in part, is responsible for neonatal lung injury leading to bronchopulmonary dysplasia. To validate this hypothesis, newborn wild-type, sphingosine kinase1−/− (Sphk1−/−), sphingosine kinase 2−/− (Sphk2−/−), and S1P lyase+/− (Sgpl1+/−) mice were exposed to hyperoxia (75%) from postnatal day 1 to 7. Sphk1−/−, but not Sphk2−/− or Sgpl1+/−, mice offered protection against hyperoxia-induced lung injury, with improved alveolarization and alveolar integrity compared with wild type. Furthermore, SphK1 deficiency attenuated hyperoxia-induced accumulation of IL-6 in bronchoalveolar lavage fluids and NADPH oxidase (NOX) 2 and NOX4 protein expression in lung tissue. In vitro experiments using human lung microvascular endothelial cells showed that exogenous S1P stimulated intracellular reactive oxygen species (ROS) generation, whereas SphK1 siRNA, or inhibitor against SphK1, attenuated hyperoxia-induced S1P generation. Knockdown of NOX2 and NOX4, using specific siRNA, reduced both basal and S1P-induced ROS formation. These results suggest an important role for SphK1-mediated S1P signaling–regulated ROS in the development of hyperoxia-induced lung injury in a murine neonatal model of bronchopulmonary dysplasia.
Bronchopulmonary dysplasia (BPD) is a chronic lung disease occurring as a consequence of injury to the rapidly developing premature lungs of a preterm newborn infant.1 Preterm neonates receive ventilator care and inhaled oxygen supplementation for variable periods after delivery; prolonged exposure of preterm lungs to hyperoxia results in inflammation, pulmonary edema, lung injury, and, ultimately, death.2,3 BPD is characterized by decreased secondary septation of alveoli, resulting in the formation of enlarged simplified alveoli and reduced area for gas exchange.4,5 More than 25% of premature infants with birth weights <1500 g develop BPD.5,6 Infants with BPD have higher rehospitalization rates because of asthma, infection, pulmonary hypertension, and other respiratory tract ailments.7,8 Many surviving neonatal BPD patients reaching adulthood show a sharp decline in lung capacity, indicating that the adverse effects of insult in the neonatal stage can be long lasting.9,10 There is no effective treatment for BPD, and strategies to prevent BPD by administering gentler ventilation and other therapeutic approaches have not been effective.11 The identification of novel signaling pathways linking hyperoxia-induced lung injury in neonatal BPD is necessary for new therapeutic approaches.
Sphingolipids and their metabolites, such as ceramide, sphingosine, and sphingosine-1-phosphate (S1P), are important bioregulators, capable of modulating acute lung injury in a variety of lung disorders.12–14 S1P plays an important role in vascular development and endothelial barrier function.14,15 It is generated by the phosphorylation of sphingosine catalyzed by sphingosine kinases (SphKs) 1 and 2 and metabolized by S1P phosphatases and lipid phosphatases to yield sphingosine or by S1P lyase (S1PL; Sgpl1) that generates Δ2-hexadecenal and ethanolamine phosphate in mammalian cells.16 In addition to the previously mentioned enzymes, serine palmitoyltransferase (SPT) initiates the biosynthesis of sphingolipids by catalyzing condensation of serine and palmitoyl-CoA to form 3-ketosphinganine.17 S1P acts extracellularly and intracellularly, and most effects of extracellular S1P are mediated via a family of five highly specific G-protein–coupled S1P1-5 receptors.18,19 Significantly lower levels of S1P in plasma and lung tissues were reported in a murine model of lipopolysaccharide (LPS)–induced lung injury, most likely because of elevated expression of S1PL,20 and infusion of S1P ameliorated LPS-induced acute lung injury in murine and canine models.21,22 Taken together, these results suggest a protective role for S1P in LPS-mediated lung injury. Hyperoxia is also known to cause lung injury; however, the underlying pathological characteristics are not similar to those observed in the LPS-treated mouse model.20,23
The goal of the present study was, therefore, to elucidate the role of S1P in the development of lung injury and BPD in the murine neonatal model. Our results showed that hyperoxia-induced accumulation of S1P is detrimental and linked to BPD because SphK1-, but not SphK2-, deficient mice exhibited significantly less hyperoxia-induced reactive oxygen species (ROS) formation, lung injury, and BPD, such as morphological characteristics, whereas S1P lyase–deficient heterozygous mice showed the opposite. Furthermore, by using human lung microvascular endothelial cells (HLMVECs), we observed that exogenous S1P stimulated ROS production, and down-regulation of SphK1 with siRNA blocked hyperoxia-induced ROS generation. We also present herein evidence in support of an inflammatory role for S1P in BPD as it relates to increased expression of NADPH oxidase (NOX) proteins, such as NOX2 and NOX4, and the proinflammatory cytokine, IL-6.
Materials and Methods
Materials
HLMVECs, Eagle’s basal medium (EBM)-2, and the Bullet kit were obtained from Lonza (San Diego, CA). PBS was from Biofluids Inc. (Rockville, MD). Ampicillin, fetal bovine serum (FBS), trypsin, MgCl2, EGTA, Tris-HCl, Triton X-100, sodium orthovanadate, aprotinin, and Tween 20 were obtained from Sigma-Aldrich Inc. (St. Louis, MO). Dihydroethidium (hydroethidine) and 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate-diacetoxymethyl ester (DCFDA) were purchased from Life Technologies (Eugene, OR). The electrochemiluminescence kit was from Amersham Biosciences (Piscataway, NJ). SMART Pool small-interfering RNA duplex oligonucleotides targeting SphK1 antibody were purchased from Exalpha (Shirley, MA). SphK2 and Nox2 antibodies were from Abcam (Cambridge, MA). SGPL, SPT2, Nox1, and Nox4 antibodies and siRNA for SphK1, Nox2, and Nox4 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). SphK-I2 [2-(p-hydroxyanilino)-4-(p-chlorophenyl)] thiazole was purchased from Cayman Chemical (Ann Arbor, MI), and S1P was obtained from Avanti Polar Lipids (Alabaster, AL).
Mouse Experiments and Animal Care
All experiments using animals were previously approved by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago. We used neonatal mice to study the effect of hyperoxia in the newborn developing lungs. Lung development in the neonatal mice on the day of birth is in the late canalicular/early alveolar stage, corresponding to that of a preterm neonate at 24 to 26 weeks of gestation.24 Sphk1−/− and Sphk2−/− mice were obtained from Dr. Richard L. Proia (NIH, Bethesda, MD)25 and were backcrossed to C57BL/6 background for two generations (F2 hybrid). The resultant mixed background of C57BL/6 strain and the original background (F2 hybrid) were used as controls and are referred to hereafter as wild type (WT). Sgpl1+/− adult mice (129SV background) were purchased from Jackson Labs (Sacramento, CA). The WT, Sphk1−/−, Sphk2−/− , or Sgpl1+/− newborn (NB) mice, along with the lactating dams, were exposed to hyperoxia of 75% O2 for 1 week or normoxia from postnatal (PN) day 1 for 7 days, as previously described.26,27 The NB mice (along with their mothers) were placed in cages in an airtight Plexiglas chamber (55 × 40 × 50 cm) maintained at a hyperoxic condition. Two lactating dams were used. Mothers were alternated between hyperoxia and normoxia every 24 hours. The litter size was kept limited to six pups to control for the effects of litter size on nutrition and growth. The animals were maintained as per the University of Illinois protocol for animal use. Oxygen levels were constantly monitored by an oxygen sensor that was connected to a relay switch incorporated into the oxygen supply circuit. The inside of the chamber was kept at atmospheric pressure. The animals were sacrificed, the lung tissues were collected and homogenized, and whole cell lysates were prepared for further analysis.
BAL Collection
Mice were euthanized, the trachea was isolated by blunt dissection, tubing was secured in the airway, and bronchoalveolar lavage (BAL) collection with PBS was undertaken, as described previously.28,29 Protein concentrations in BAL fluid were measured using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA), as previously described.19,29 Optical density readings of samples were converted to milligrams/milliliters, using values obtained from a standard curve generated with 0.1 to 1.5 mg/mL serial dilutions of bovine serum albumin. BAL samples were analyzed for levels of tumor necrosis factor-α, monocyte chemoattractant protein-1, interferon-γ, IL-6, and IL-1α, using a commercially available cytokine panel (Bio-Plex Suspended Multiplex Bead Array Assay kit; Bio-Rad), in accordance with the manufacturer’s instructions. Data from the reactions were acquired with a flow cytometry system (X Map-100; Luminex, Austin, TX) and accompanying software (Bio-Plex Manager software version 5.0; Bio-Rad). The median fluorescence intensity was used as a measure of detection of protein. The values reported represent a median reporter fluorescence intensity of at least 100 beads. All samples were read in duplicate. Samples were considered positive when the mean fluorescence intensity value was 3 SDs greater than the negative control.30–32
Analysis of Sphingoid Base-1-Phosphates
Analyses of sphingoid base-1-phosphates were performed by electrospray ionization liquid chromatography–tandem mass spectrometry, as previously described.33,34 The instrumentation used was an API4000 Q-trap hybrid triple-quadruple linear ion-trap mass spectrometer (Applied Biosystems, Foster City, CA), equipped with a turbo ion spray ionization source interfaced with an automated Agilent 1100 series liquid chromatograph and autosampler (Agilent Technologies, Wilmington, DE). S1P and DihydroS1P were analyzed as bis-acetylated derivatives, with C17-S1P as the internal standard using reverse-phase high-performance liquid chromatography separation, negative-ion electrospray ionization, and magnetic resonance mammography analysis.
Preparation of S1P for Exogenous Addition on Endothelial Cells
S1P dissolved in methanol: toluene (1:1 v/v), to a final concentration of 1 mmol/L, was stored in aliquots, in glass vials at −20°C. An aliquot of S1P solution was transferred to a glass tube, the solvent was evaporated under N2, and the thin film of S1P at the bottom of the glass tube was reconstituted by sonication with probe sonicator (3× 15 seconds) in basal EGM-2 medium containing 0.1% fatty acid–free bovine serum albumin. The S1P solution was always made fresh 10 minutes before the experiment and kept at room temperature.35,36
Histological Analysis
Animals were euthanized, a median sternotomy was performed, and perfusion of the right side of the heart was accomplished with calcium and magnesium-free PBS to clear the pulmonary intravascular space. The heart and lungs were then removed en bloc, fixed to pressure (15-cm water) with neutral-buffered 2% paraformaldehyde, fixed overnight in 2% paraformaldehyde, embedded in paraffin, divided into sections (5 μm thick), and stained with H&E at the Research Histology Laboratory (Department of Pathology, University of Illinois at Chicago). The objective assessment of the extent of alveolarization was determined by the mean linear intercept (MLI) method.37,38 Slides were examined at ×10 magnification, and the septal thickness and tissue density for a minimum of 50 alveoli for each section were measured. At least two sections from each pup were used for analysis.
TUNEL Assay
End labeling of exposed 3′-OH ends of DNA fragments was undertaken with the TUNEL in situ cell death detection kit Alkaline Phosphatase (Roche Diagnostics, Indianapolis, IN), as per the manufacturer’s instructions. The TUNEL index was calculated by randomly selecting four high-power fields in each section, counting 200 cells in each area, and expressing the number of TUNEL-positive cells as a percentage, as previously described.26,39 At least two sections were used from each pup for analyses.
Granulocyte Esterase Staining
Tissue was first fixed in citrate-acetone-formaldehyde solution and then incubated in naphthol AS-D chloroacetate solution (Fast Red Violet LB; Sigma, St. Louis), followed by counterstaining with hematoxylin solution. Enzymatic hydrolysis of ester linkages liberates free naphthol compounds. These, coupled with the diazonium salt, form highly colored deposits at sites of enzyme activity. Granulocyte esterase enzyme is considered specific for cells of granulocytic lineage. Sites of granulocyte esterase activity show bright red granulation.39,40 Activity is weak or absent in monocytes and lymphocytes. Slides were examined at ×20 magnification, cells positive for neutrophil esterase were counted in at least four high-power fields in each section, and two sections were used per pup.
Endothelial Cell Culture
HLMVECs, passages between 5 and 8, were cultured in EGM-2 complete medium (10% FBS, 100 U/mL penicillin, and streptomycin) at 37°C and 5% CO2. They were allowed to grow to approximately 90% confluence, as characterized by typical cobblestone morphological characteristics, as described previously.40,41 Cells from T-75 flasks were detached with 0.05% trypsin, resuspended in fresh complete medium, and cultured in 35- or 60-mm dishes or on glass coverslips for various studies.
Exposure of Cells to Hyperoxia
HLMVECs (approximately 90% confluence) in complete EGM-2 medium were placed in a humidity-controlled airtight modulator incubator chamber (Billups-Rothenberg, Del Mar, CA), flushed continuously with 95% O2 and 5% CO2 for 30 minutes until the oxygen level inside the chamber reached approximately 95%. HLMVECs were then placed in the cell culture incubator at 37°C for 3 hours of hyperoxia exposure. The concentration of O2 inside the chamber was monitored with a digital oxygen monitor. The buffering capacity of the cell culture medium did not change significantly during hyperoxic exposure and was maintained at a pH of approximately 7.4.
Determination of Hyperoxia-Induced Production of O2⋅− and Total ROS
To measure total ROS or O2⋅–, spectrofluorimeter- or fluorescence microscopy–based methods were used. Hyperoxia-induced O2⋅– release by HLMVECs was measured by hydroethidine fluorescence, as previously described.42 Total ROS production in HLMVECs, exposed to either normoxia or hyperoxia, was determined by the DCFDA fluorescence method.40,43 Briefly, HLMVECs (approximately 90% confluent in 35-mm dishes) were loaded with 10 μmol/L DCFDA in EGM-2 basal medium and incubated at 37°C for 30 minutes. Fluorescence of oxidized DCFDA in cell lysates, an index of formation of ROS, was measured with an Aminco Bowman series 2 Spectrofluorimeter (Thermo Fischer Scientific, West Palm Beach, FL) using excitation and emission set at 490 and 530 nm, respectively, with appropriate blanks. Hyperoxia-induced ROS formation in cells was also quantified by fluorescence microscopy. HLMVECs (approximately 90% confluent) in 35-mm dishes were loaded with 10 μmol/L DCFDA in EBM-2 basal medium for 30 minutes at 37°C in a 95% air and 5% CO2 environment. After 30 minutes of loading, the medium containing DCFDA was aspirated; cells were rinsed once with EGM-2 complete medium, and cells were pre-incubated with agents for the indicated time periods, followed by exposure to either normoxia (95% air and 5% CO2) or hyperoxia (95% O2 and 5% CO2) for 3 hours. At the end of the incubation, cells were washed twice with PBS at room temperature and were examined under a Nikon Eclipse TE 2000-S fluorescence microscope (Tokyo, Japan) with a Hamamatsu digital charge-coupled device camera (Hamamatsu, Japan) using a 20X objective lens and MetaVue software version 1.0 (Universal Imaging Corp, Downingtown, PA).
RNA Isolation and Quantitative RT-PCR
Total RNA was isolated from HLMVECs grown on 35-mm dishes or from mouse lung homogenate using TRIzol reagent (Life Technologies, Grand Island, NY), according to the manufacturer’s instructions, and purified using the RNeasy Mini Kit, according to the manufacturer’s protocol (Qiagen, Valencia, CA). iQ SYBR Green Supermix (Life Technologies) was used to perform real-time PCR using iCycler by Bio-Rad. 18S ribosomal RNA (sense, 5′-GTAACCCGTTGAACCCCATT-3′; and antisense, 5′-CCATCCAATCGGTAGTAGCG-3′) was used as an external control to normalize expression. Quantitative RT-PCR was performed as previously described.21 All primers were designed by inspection of the genes of interest and were designed using Beacon Designer software version 2.1 (Premier Biosoft, Palo Alto, CA). The sequence descriptions of mouse primers used are as follows: Sphk1, 5′-GCTGTCAGGCTGGTGTTATG-3′ (forward) and 5′-ATATGCTTGCCCTTCTGCAT-3′ (reverse); mouse Sphk2, 5′-ACTGCTCGCTTCTTCTCTGC-3′ (forward) and 5′-CCACTGACAGGAAGGAAAA-3′ (reverse); mouse Sgpl1, 5′-AACTCTGCCTGCTCAGGGTA-3′ (forward) and 5′-CTCCTGAGGCTTTCCCTTCT-3′ (reverse); Nox2, 5′-ACTCCTTGGAGCACTGG-3′ (forward) and 5′-GTTCCTGTCCAGTTGTCTTCG-3′ (reverse); and Nox4, 5′-TGAACTACAGTGAAGATTTCCTTGAAC-3′ (forward) and 5′-GACACCCGTCAGACCAGGAA-3′ (reverse). Negative controls, consisting of reaction mixtures containing all components except target RNA, were included with each of the real-time PCR runs. To verify that amplified products were derived from mRNA and did not represent genomic DNA contamination, representative PCR mixtures for each gene were run in the absence of the reverse transcriptase enzyme after first being cycled to 95°C for 15 minutes. In the absence of reverse transcription, no PCR products were observed.
Immunoblotting
Protein expression was detected in mouse lungs and HLMVECs by immunoblotting, as previously described.20,26 HLMVECs grown on 35-mm dishes (approximately 90% confluence) were rinsed twice with ice-cold PBS and lysed in 100 μL of modified lysis buffer (50 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 0.25% sodium deoxycholate, 1 mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L Na3VO4, 1 mmol/L NaF, 10 μg/mL aprotinin, 10 μg/mL leupeptin, and 1 μg/mL pepstatin), sonicated on ice with a probe sonicator (3× for 15 seconds), and centrifuged at 5000 × g in a microcentrifuge (4°C for 5 minutes). Protein concentrations of the supernatants were determined using the Pierce bicinchoninic acid protein assay kit (Thermo Scientific, Rockford, IL). The supernatants, adjusted to 1 mg of protein/mL (cell lysates), were denatured by boiling in SDS sample buffer for 5 minutes, and samples were separated on 10% SDS-polyacrylamide gels. Protein bands were transferred overnight (24 V at 4°C) onto a nitrocellulose membrane (0.45 μm thick; Bio-Rad), probed with primary and secondary antibodies, according to the manufacturer’s protocol, and immunodetected by using the electrochemiluminescence kit (GE Amersham Healthcare Life Sciences, Pittsburgh, PA). The blots were scanned (UMAX Power Lock II, Dallas, TX) and quantified by an automated digitizing system, UN-SCAN-IT GEL (Silk Scientific Corp, Orem, UT). On sacrifice, the neonatal mouse lungs were perfused with PBS, resulting in removal of blood from pulmonary blood vessels. Protein extraction was performed using T-PER (Thermo Scientific). A ratio of 20 mL of T-PER/1 g of tissue was added to the tissue and homogenized. Lysate was centrifuged at 10,000 × g for 5 minutes to pellet cell/tissue debris at 4°C. Supernatant was collected, equal amounts of protein (20 μg) were loaded onto 10% SDS-PAGE gels, and Western blot analysis was performed according to standard protocols.
Transient Transfection of HLMVECs
For siRNA experiments, HLMVECs were transfected with Fl-luciferase GL2 duplex siRNA (target sequence, 5′-CGTACGCGGAATACTTCGA-3′; Dharmacon, Lafayette, CO) as a positive control (scrambled RNA). HLMVECs grown to approximately 50% confluence in 6-well plates were transfected with Gene Silencer transfection agent (Genlantis, San Diego, CA) plus scrambled RNA or siRNA specific for SphK1, Nox2, or Nox4 (50 nmol/L) in serum-free EBM-2 medium, according to the manufacturer’s recommendations. After 3 hours of transfection, the serum-free medium was replaced by 1 mL of fresh complete EGM-2 medium containing 10% FBS and the growth factors, and cells were cultured for an additional 72 hours before experiments.
Treatment with Exogenous S1P
HLMVECs (approximately 90% confluence), grown on 35-mm dishes, were starved for 3 hours in EBM-2 without growth factors. Cells were treated with 1 μmol/L S1P for 5 and 30 minutes, respectively, followed by ROS measurements with DCFDA. Control cells were always treated accordingly in the presence of vehicle for the indicated times.
H2O2 Measurement
HLMVECs were exposed to normoxia and hyperoxia for 5 and 30 minutes, respectively. Cell culture media were collected and centrifuged at 3000 × g, for 10 minutes at 4°C, and H2O2 in the supernatant was measured immediately using a commercial kit and according to the manufacturer’s protocol. In brief, 50 μL of control or S1P-treated samples and standards were mixed with a working solution of 100 μmol/L Amplex Red reagent and 0.2 U/mL horseradish peroxidase (Life Technologies, Grand Island, NY). Reaction solutions were protected from light and incubated at room temperature for 30 minutes, and absorbance was read at 560 nm.
Pretreatment of Cells with SKI-II
HLMVECs grown to approximately 90% confluence were pre-incubated with an SphK inhibitor (SKI-II; 1 to 10 μmol/L) in serum-free media containing 1% FBS, as indicated for 24 hours before stimulation with hyperoxia (95% O2 and 5% CO2) for 3 hours.44
Statistical Analysis
An analysis of variance and a Student-Newman-Keul’s test were used to compare means of two or more different treatment groups. The level of significance was set to P < 0.05, unless otherwise stated. Results are expressed as means ± SEM.
Results
Expression of S1P-Metabolizing Enzymes and S1P Levels in Lung Tissues from Hyperoxia-Challenged Neonatal Mice
The relative protein expression levels of SphK 1 and 2, S1PL, and SPT in lung tissues from neonatal pups, exposed to normoxia or hyperoxia, were determined by immunoblotting (Figure 1, A–E). SphK1, SPT2, and S1PL levels were significantly elevated in neonatal lung tissues from hyperoxic mice compared with normoxic animals. SphK2 protein expression, however, remained unaltered in response to hyperoxia. There was some discrepancy between protein and mRNA levels. Hyperoxia resulted in a decreased mRNA expression of Sphk2 and Sgpl1, but not Sphk1 and SPT2 (Figure 1, F–I). Furthermore, S1P levels were significantly elevated in lung tissues from hyperoxia-exposed mice (Figure 1J). These results suggested that hyperoxia differentially modulates the expression levels of the S1P-metabolizing enzymes and S1P generation in neonatal lung tissues.
Figure 1.

Hyperoxia increases S1P level and alters expression of enzymes involved in S1P metabolism. WT (C57BL/6J) NB mice, along with the lactating dams, were exposed to normoxia (NO; white bars) or hyperoxia (HO; black bars) of 75% O2 for 1 week from PN day 1 for 7 days. After exposure, NB mice were euthanized, lungs were removed for protein, and RNA was extracted as described in Materials and Methods. A: Whole lung homogenates subjected to SDS-PAGE and Western blot analysis showed that HO enhanced expression of SphK1, SPT2, and S1PL after exposure to HO for 7 days. No significant difference was noted in the expression of SphK2. B–E: Western blots probed with anti-SphK1 (B), anti-SphK2 (C), anti-SPT2 (D), and anti-S1P lyase (E) antibodies were quantified by densitometry and normalized to total actin. F–I: Real-time RT-PCR quantification of mRNA demonstrated that HO induced an increase in mRNA expression of Sphk1 (F) and SPT2 (H), whereas a decrease in mRNA expression of Sphk2 (G) and S1P lyase (Sgpl1) (I) was noted. J: C18-S1P levels in lung tissue from WT NB mice exposed to HO were significantly elevated in the lung tissue compared with normoxia. Significantly increased from NO control: ∗P < 0.05, ∗∗∗P < 0.001 (n = 5 to 8 per group).
SphK1, But Not SphK2, Deficiency Attenuates Hyperoxia-Induced BPD in Neonatal Mice
SphK1 and SphK2 are widely expressed in most mammalian tissues, including the lung.45,46 Because the expression of SphK1, but not that of SphK2, was enhanced in neonatal lung tissues from hyperoxia-challenged mice, we investigated the effect of genetic deletion of SphK1 and SphK2 on hyperoxia-induced BPD. As anticipated, lung tissues from Sphk1−/− or Sphk2−/− mice did not express SphK1 or SphK2, respectively, and deletion of either SphK1 or SphK2 had no overt phenotypic effects under normoxic conditions (data not shown). Plasma S1P levels were significantly lower in Sphk1−/− mice (approximately 30%) compared with WT mice (plasma S1P, 658 ± 34 versus 1107 ± 218 pmol/mL; P < 0.05). However, plasma S1P levels were not significantly different between SphK2-deficient mice and the WT mice exposed to hyperoxia (data not shown). To investigate the effects of SphK1 or SphK2 deficiency on hyperoxia-induced BPD, 1-day-old WT, Sphk1−/−, or Sphk2−/− pups were exposed to normoxia or hyperoxia (75% O2) for 7 days. The rate of survival in hyperoxia-treated WT, Sphk1−/−, or Sphk2−/− groups was not significantly different in all of the genotypes, and this exposure regimen did not significantly affect the mortality of the pups. A litter size of six neonatal mice was used in the study for the normoxia and hyperoxia groups. A minimum of two mothers per group were used in the study. The survival rate was 11 to 12 of 12 pups in each of the groups.
The effect of SphK1 deficiency on hyperoxia-induced alveolarization defect and lung injury were evaluated by the MLI method and H&E staining, respectively. Exposure to hyperoxia significantly increased injury and reduced alveolarization in the lung by day 7 in the WT mice, which was ameliorated in Sphk1−/− mice exposed to hyperoxia (Figure 2A). Improved alveolarization was noted in the Sphk1−/− group, as shown by histopathological quantification for alveolarization (MLI) (Figure 2B). More important, Sphk1−/− deficiency alone had no significant effect on MLI in the absence of hyperoxia challenge. Interestingly, hyperoxia did not alter lung collagen content in SphK1−/− and WT mice (data not shown). In contrast to SphK1 deficiency, loss of SphK2 (Sphk2−/− mice) had no effect on hyperoxia-induced lung injury, BPD-like morphological characteristics (Figure 3A), and impaired alveolarization, as characterized by enlarged simplified alveoli showing a higher MLI (Figure 3B).
Figure 2.

Deficiency of sphingosine kinase 1 protects alveolarization of murine neonatal lungs under hyperoxia. WT (C57BL/6J) or Sphk1−/− NB mice, along with the lactating dams, were exposed to normoxia (NO; white bars) or hyperoxia (HO; black bars; 75% O2) from PN day 1 for 7 days. On completion of exposure, NB mice were euthanized, and lungs were removed, embedded in paraffin, and cut into sections (5 μm thick) for staining. A: Representative H&E photomicrographs of lung sections obtained from WT and Sphk1−/− NB mice exposed to NO or HO. Sphk1−/− NB mice showed significantly improved alveolarization under HO compared with WT NB. Original magnification, ×10. B: The objective assessment of alveolarization of neonatal lungs was determined by the MLI method. After exposure to HO, lung MLI in Sphk1−/− NB is significantly lower compared with WT. ∗P < 0.05 (n = 5 to 8 per group).
Figure 3.

Deficiency of sphingosine kinase 2 does not protect murine neonatal lungs from hyperoxia. WT (C57BL/6J) or Sphk2−/− NB mice, along with the lactating dams, were exposed to normoxia (NO; white bars) or hyperoxia (HO; black bars; 75% O2) for 1 week from PN day 1 for 7 days. On completion of exposure, NB mice were euthanized, and lungs were removed, embedded in paraffin, and cut into sections (5 μm thick) for staining. A: Representative H&E photomicrographs of lung sections obtained from Sphk2−/− NB mice exposed to NO or HO. Sphk2−/− NB mice, similar to WT NB, showed disruption of normal alveolarization under HO. Original magnification, ×10. B: The objective assessment of alveolarization of neonatal lungs was determined by the MLI method. MLI in both Sphk2−/− NB and WT NB mice lungs is significantly higher compared with NB mice exposed to NO. ∗P < 0.05 versus normoxia control. No significant difference was found between WT and Sphk2−/− exposed to hyperoxia (n = 5 to 8 per group).
Next, the effect of SphK1 deficiency on hyperoxia-induced lung inflammation was assessed. The total protein, cell number, and IL-6 levels were significantly elevated in BAL fluids from WT pups exposed to hyperoxia compared with those from Sphk1−/− pups (Figure 4, A–C). There was an increase in the level of monocyte chemoattractant protein-1 in both groups exposed to hyperoxia, whereas no significant elevation was noted with tumor necrosis factor-α, interferon-γ, and IL-1α. The BAL fluids from WT and Sphk1−/− mice showed macrophage dominance (mean, 91.2% ± 2.3% for WT and 90.4% ± 2.7% for Sphk1−/−) under normoxia and hyperoxia. The total neutrophil count was decreased in the BAL of Sphk1−/− pups compared with WT exposed to hyperoxia (mean, 144.1 ± 58.8 for WT and 74.9 ± 30.6 for Sphk1−/−), and specific neutrophil esterase staining of lung tissue also showed a significant decrease in Sphk1−/− pups compared with WT exposed to hyperoxia (mean, 4.05 ± 0.3 for WT and 1.92 ± 0.31 for Sphk1−/−) (Figure 4, D and E). For a better understanding of the pathways involved in the protection against alveolar simplification, we determined apoptosis using TUNEL staining on lung tissues. A significant decrease of TUNEL-positive cells was noted in the lungs of Sphk1−/− pups compared with WT exposed to hyperoxia (Figure 4F). These results suggest a detrimental role for SphK1, but not SphK2, in hyperoxia-induced BPD.
Figure 4.

Deficiency of sphingosine kinase 1 attenuates hyperoxia-induced inflammatory changes in neonatal mouse lungs. WT (C57BL/6J) or Sphk1−/− NB mice were exposed to normoxia (NO; white bars) or hyperoxia (HO; black bars) (75% O2) from PN day 1 for 7 days. On completion of exposure, the mice were euthanized, lungs were lavaged by PBS solution, and BAL fluids were analyzed as described in Materials and Methods. Deficiency of SphK1 reduced pulmonary vascular leak after HO (A), as shown by a significant decrease in BAL fluid total protein levels, decreased total cell count (B), lower levels of IL-6 (C), and decreased neutrophils in Sphk1−/− mice compared with WT mice exposed to HO (D). Lung tissue showed reduced granulocyte esterase staining (E) and reduced TUNEL-positive cells (F) in Sphk1−/− mice compared with WT mice exposed to HO. Data are expressed as means ± SEM. Significantly different from NO control: ∗P < 0.05, significant decrease in Sphk1−/− compared with WT under HO (n = 5 to 8 per group), ∗∗∗P < 0.001.
S1P Lyase Deficiency Potentiates Hyperoxia-Induced BPD in Neonatal Mice
Having established that SphK1 deficiency confers protection against hyperoxia-induced BPD, we further determined the role of S1PL, the S1P-metabolizing enzyme, in BPD using Sgpl1+/− heterozygous mice. Sgpl1−/− mice do not survive beyond 3 to 4 weeks after birth and exhibit vascular defects.47 The Sgpl1+/− mice that have partial deficiency of S1PL enzyme exhibited elevated S1P levels in lung tissue, but not in plasma, under normoxia (data not shown). Hyperoxia challenge of 1-day-old pups significantly increased injury and alveolar simplification in WT (Sgpl1+/+) mice after 7 days of hyperoxia exposure, which was further aggravated in Sgpl1+/− mice (Figure 5A). Histopathological quantification for alveolar simplification (MLI) showed a significant negative effect of partial S1PL deficiency in the form of enlarged simplified alveoli (Figure 5B). More important, S1PL deficiency alone had no significant effect on alveolar development in the absence of hyperoxia challenge. Furthermore, hyperoxia-induced protein leakage into BAL fluids was markedly higher in Sgpl1+/− mice compared with WT mice (Figure 5C).
Figure 5.

Partial deficiency of S1P lyase aggravates hyperoxia-induced neonatal lung injury. WT or Sgpl1+/− heterozygous NB mice in a 129SV background, along with the lactating dams, were exposed to normoxia (NO; white bars) or hyperoxia (HO; black bars; 75% O2) for 1 week from PN day 1 for 7 days. On completion of exposure, NB mice were euthanized, and lungs were removed, embedded in paraffin, and cut into sections (5 μm thick) for staining. A: Representative H&E photomicrographs of lung sections obtained from WT and Sgpl1+/− NB mice exposed to NO or HO. Sgpl1+/− NB mice exposed to HO showed significant lung injury with impaired alveolarization compared with WT NB exposed HO. Original magnification, ×10. B: The objective assessment of alveolarization of neonatal lungs was determined by the MLI method. MLI in Sgpl1+/− NB mouse lungs exposed to HO was significantly higher compared with WT NB exposed to HO. C: Lungs were lavaged by PBS solutions, and BAL fluids were analyzed as described in Materials and Methods. Partial deficiency of S1P lyase increased pulmonary vascular leak after HO, as shown by a significant increase in BAL total protein levels. Data are expressed as means ± SEM. Significantly different from control: ∗P < 0.05, ∗∗∗P < 0.001 (n = 5 to 8 per group).
SphK1, But Not SphK2, Deficiency Modulates Hyperoxia-Induced NOX Expression and ROS Generation in Neonatal Lung Tissue
We have shown earlier that hyperoxia enhances the expression of NOX2 and NOX4, which, in turn, mediate increased ROS generation in lung endothelium.41,42 Because SphK1 deficiency ameliorated hyperoxia-induced lung injury and BPD in neonatal mice, we investigated the potential link between SphK1, NOX expression, and ROS generation. Hyperoxia challenge of WT neonatal 1-day-old mice increased mRNA levels and protein expression of NOX2 and NOX4 in lung tissues compared with normoxic controls at day 7 after the challenge (Figure 6, A–F). Interestingly, in Sphk1−/− mice, the hyperoxia-induced up-regulation of NOX mRNA and protein levels was less pronounced (Figure 6, E and F). In contrast to SphK1 deficiency, loss of SphK2 had no significant effect on hyperoxia-induced protein expression of NOX2 and NOX4 (Figure 7, A–C). The previously described data suggested that SphK1, but not SphK2, modulates hyperoxia-induced expression of NOX proteins in neonatal lung.
Figure 6.

Effect of hyperoxia on expression of NOX2 and NOX4 in neonatal lungs of WT and Sphk1−/− mice. WT NB or Sphk1−/− NB mice, along with the lactating dams, were exposed to normoxia (NO; white bars) or hyperoxia (HO; black bars; 75% O2) for 1 week from PN day 1 for 7 days. After exposure, NB mice were euthanized, lungs were removed for protein, and RNA was extracted as described in Materials and Methods. A: Whole lung homogenates were subjected to SDS-PAGE and Western blot analysis. Immunoblot showed increased expression of NOX2 and NOX4 in lungs from WT after exposure to HO for 7 days. NOX2 and NOX4 protein expressions were significantly decreased in Sphk1−/− exposed to HO, compared with WT, mice. Western blots probed with anti-NOX1 (B), anti-NOX2 (C), and anti-NOX4 (D) antibodies were quantified by densitometry and normalized to the corresponding total actin. E and F: Total RNA was isolated and subjected to real-time RT-PCR. Quantification of mRNA demonstrated that HO induced an increase in mRNA expression of NOX2 (E) and NOX4 (F) in WT mice, which was significantly less in the lungs of Sphk1−/− NB exposed to HO. Significantly increased from NO control: ∗P < 0.05, ∗∗∗P < 0.001 (n = 5 to 8 per group).
Figure 7.
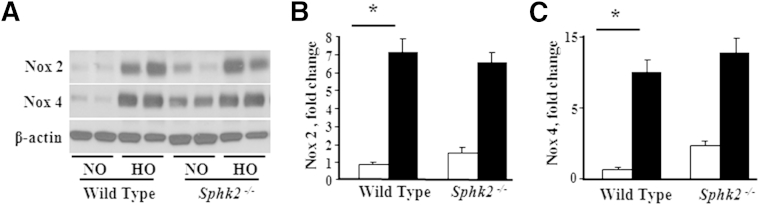
Effect of hyperoxia on expression of NOX2 and NOX4 in neonatal lungs of Sphk2−/− NB mice compared with WT NB. WT NB or Sphk2−/− NB mice were exposed to normoxia (NO; white bars) or hyperoxia (HO; black bars) (75% O2) from PN day 1 for 7 days. After exposure, NB mice were euthanized and the lungs were removed for protein extraction, as described in Materials and Methods. A: Whole lung homogenates were subjected to SDS-PAGE and Western blot analysis. A representative immunoblot showed increased expression of NOX2 and NOX4 in the lungs of WT and Sphk2−/− NB mice after exposure to HO for 7 days. Western blots probed with anti-NOX2 (B) and anti-NOX4 (C) antibodies were quantified by densitometry and normalized to the corresponding total actin. ∗P < 0.05, NO control. No significant difference was found compared with WT HO (n = 5 to 8 per group).
Role of Endogenous and Exogenous S1P on Hyperoxia-Induced ROS Generation in HLMVECs
To further characterize the potential link between hyperoxia-induced SphK1/S1P signaling axis and ROS generation, SphK1 was knocked down using siRNA in HLMVECs and exposed to hyperoxia (95% O2) for 3 hours. Hyperoxia stimulated ROS generation, as evidenced by DCFDA oxidation, and hydrogen peroxide accumulation in conditioned media was attenuated in SphK1 siRNA-treated HLMVECs (Figure 8, A and B). SphK1 siRNA transfection effectively knocked down >80% of SphK1 protein in HLMVECs (Figure 8C). In addition to SphK1 siRNA, blocking SphK1/SphK2 activity with SKI-II, an inhibitor of both the isoforms of SphK, attenuated hyperoxia-induced S1P generation and ROS formation in HLMVECs (Figure 8, D and E). Consistent with the previously described data, exposure of HLMVECs to 1 μmol/L exogenous S1P for 30 minutes also stimulated intracellular ROS generation (Figure 8, F and G). These results further strengthened the role of SphK1/S1P signaling in hyperoxia-induced ROS generation in human lung ECs.
Figure 8.

S1P enhances ROS generation in human lung endothelial cells. A: HLMVECs were transfected with 50 nmol/L scrambled RNA or SphK1 siRNA for 72 hours and exposed to normoxia (NO) or hyperoxia (HO) for 3 hours, and total ROS production was measured by DCFDA fluorescence. Original magnification, ×20. B: Hyperoxia-induced increase in ROS production in HLMVECs was significantly decreased by SphK1 knockdown in cells. Images were quantified by ImageJ (NIH, Bethesda, MD). Values for ROS production are means ± SD from three independent experiments and normalized to percentage control. C: Immunoblot showing effective knockdown of expression of SphK1 by SphK1siRNA. D: HLMVECs grown to approximately 90% confluence were pre-incubated with 1 to 10 μmol/L SKI-II (SphK1/SphK2 inhibitor) in serum-free or media containing 1% FBS, as indicated for 24 hours before stimulation with hyperoxia (95% O2 and 5%CO2) for 3 hours. After incubation, cells were washed twice with PBS at room temperature, and total ROS production was measured by DCFDA fluorescence. SKI-II blocked ROS production in HLMVECs under hyperoxia. Original magnification, ×20. E: Data were quantified based on the number of DCFDA pixels. Values for ROS production are means ± SD from three independent experiments and normalized to percentage control. F: HLMVECs (approximately 90% confluence), grown on 35-mm dishes, were starved for 3 hours in EBM-2 containing 0.1% FBS, without growth factors, and treated with 1 μmol/L S1P for 5 and 30 minutes, respectively. Cells were loaded with DCFDA and exposed to 1 μmol/L S1P for 5 and 30 minutes, respectively, followed by washing. Intracellular ROS generation in HLMVECs was quantified by DCFDA measurement. G: H2O2 in medium from normoxia and hyperoxia cells. ∗P < 0.05, versus control; †P < 0.05, significant decrease of ROS formation under HO by SphK1 inhibition.
Down-Regulation of NOX2 and NOX4 Expression Attenuates S1P-Induced ROS Generation in HLMVECs
Our earlier studies showed that hyperoxia-induced ROS formation in human lung ECs is NOX2 and NOX4 dependent.42 However, the contribution of S1P signaling and the role of activated NOX proteins in the generation of superoxide/ROS in human lung ECs are unclear. Therefore, we investigated the link between S1P and NOX proteins by selectively down-regulating NOX2 and NOX4 with specific siRNAs. We also suppressed Rac1, an essential component of NOX2 activation, using the inhibitor, NSC23766.48,49 Down-regulation of NOX2 or NOX4 with 50 nmol/L siRNA for 48 hours reduced both basal (approximately 70%) and S1P-induced (approximately 80%) ROS formation (Figure 9, A–D). Effective knockdown of expression of NOX2 and NOX4 by siRNA is demonstrated by immunoblot (Figure 9E). Similarly, pretreatment of HLMVECs with NSC23766 significantly attenuated S1P-induced ROS production (Figure 9, F and G). Furthermore, it also blocked S1P-mediated translocation of p47phox to the cell periphery (Figure 9G), a prerequisite for NOX2 activation. These results showed that S1P-induced ROS production is, in part, dependent on NOX2 and NOX4 in HLMVECs.
Figure 9.

Role of NOX2, NOX4, and Rac1 on S1P-induced ROS production in HLMVECs. A: HLMVECs were transfected with 50 nmol/L scrambled (sc) or Nox2 siRNA for 72 hours, washed with ice-cold PBS, loaded with DCFDA, and then exposed to 1 μmol/L S1P for 30 minutes. ROS production was measured by DCFDA fluorescence. Nox2 siRNA inhibits S1P-stimulated formation of ROS. Original magnification, ×20. B: Quantified data show that Nox2 siRNA inhibits S1P-stimulated formation of ROS. Values for ROS production are means ± SD from three independent experiments. C: HLMVECs were transfected with 50 nmol/L scrambled (sc) or Nox4 siRNA for 72 hours, loaded with DCFDA, and exposed to 1 μmol/L S1P for 30 minutes, as previously described. Nox4 siRNA inhibits S1P-stimulated formation of ROS. D: Quantified data show that Nox4 siRNA inhibits S1P-stimulated formation of ROS. Values for ROS production are means ± SD from three independent experiments. E: Immunoblot demonstrating effective knockdown of expression of NOX2 and NOX4 by siRNA. F: Inhibition of Rac1 with NSC23766 blunts S1P-induced translocation of p47phox to the cell periphery and ROS production. HLMVECs grown on slide chambers were pretreated with 50 μmol/L NSC23766 for 30 minutes, exposed to 1 μmol/L S1P for 30 minutes, washed, fixed, permeabilized, probed with anti-p47phox antibody, and examined by immunofluorescence microscopy. Original magnification, ×60 (oil objective). Exposure of cells to S1P resulted in redistribution of p47phox to the cell periphery, whereas NSC23766 blunted p47phox redistribution. A representative image from one of the three independent experiments is shown. G: HLMVECs were pretreated with 50 μmol/L NSC23766 for 30 minutes and exposed to 1 μmol/L S1P for 30 minutes, and total ROS production was measured by DCFDA fluorescence. Original magnification, ×20. Values for ROS production are means ± SD from three independent experiments. Significantly increased from control untreated cells (sc). ∗P < 0.05, significant decrease of ROS formation compared with control. NO, normoxia; Veh, vehicle.
Discussion
BPD is a severe debilitating disease affecting the preterm newborn, with no effective treatment. Identification of new therapeutic targets for drug development is critical to improve the prognosis of this increasingly prevalent condition. By using a neonatal mouse model, our study provides the first direct in vivo evidence that SphK1 is a novel therapeutic target for BPD in the newborn. The major findings of this study are as follows: i) increased expression of SphK1 and elevated S1P levels, along with increased expression of NOX2 and NOX4 in the neonatal lung tissue after exposure to hyperoxia; ii) Sphk1−/− mice exposed to hyperoxia showed improved alveolarization, and decreased ROS accumulation, neutrophil influx into the lungs, apoptosis, and protein expression of NOX2, NOX4, and IL-6 levels; iii) in vitro, SphK1siRNA attenuated hyperoxia-induced S1P generation and ROS formation in HLMVECs; and iv) down-regulation of NOX2 or NOX4 with siRNA reduced both basal and S1P-induced ROS formation. This study suggests a novel link between the hyperoxia-induced SphK/S1P signaling axis, NOX proteins, and ROS; and raises the possibility that these are potential therapeutic targets against BPD.
The pathogenesis of BPD is well described, and its development is associated with lung inflammation, epithelial/endothelial injury, and impaired postnatal lung growth.4,50,51 Several factors, including angiogenesis proteins, proinflammatory cytokines, and oxidative stress, have been described to protect against or contribute to the pathogenesis of BPD.52 However, molecular mechanisms contributing to impaired alveolar formation and development of BPD are incompletely understood. ROS accumulation and an imbalance in cellular reduction-oxidation status have been implicated in hyperoxia-induced lung injury and BPD.53,54 Earlier, we have demonstrated that exposure of adult mice to hyperoxia increased ROS production in the lung, which was NADPH oxidase dependent, with minimal or no contribution of mitochondrial electron transport.55 Furthermore, the hyperoxia-induced ROS production in the mouse lung and human lung ECs was mediated by enhanced expression of NOX2 and NOX4, because blocking NOX2 or NOX4 attenuated hyperoxia-induced ROS generation, lung injury, and inflammation (Figure 6A).42 Expression levels of NOX2 and NOX4 were elevated in the neonatal BPD mouse lung, confirming that the physiological role of NOX proteins is the key regulator of lung inflammation and injury.
The first interesting and novel finding of the present study is the potential involvement of SphK1/S1P signaling in impaired alveolarization and lung injury in neonatal mice exposed to hyperoxia. S1P is a naturally occurring bioactive sphingophospholipid, which is present in plasma and tissues at concentrations ranging from nM to μM.56 In tissues, S1P levels are maintained by its synthesis and catabolism, and changes in the tissue environment can alter S1P homeostasis. In the present study, S1P accumulation in the lungs of neonatal pups exposed to hyperoxia correlated with increased SphK1 and SPT expression, the key enzymes of sphingolipid homeostasis in mammalian cells. In contrast to the mRNA levels, the protein expression of S1PL was higher in WT mice exposed to hyperoxia. Protein expression is dictated by several factors, including level of mRNA, its half-life, translational efficiency, and turnover rate of the protein of interest. Also, it is evident that there is no direct correlation between mRNA expression levels and protein expression, and in many instances, an increase in mRNA expression does not necessarily translate to a similar level of protein expression.57,58 Thus, our current observation of a lack of correlation between mRNA and protein expression of S1PL is in accordance with reports in the literature.44
Our data indicate that enhanced S1P accumulation in lungs is linked to BPD because Sphk1−/−, but not Sphk2−/−, mice exposed to hyperoxia showed better alveolar development and had a reduced vascular leak. This beneficial effect of SphK1 deficiency against BPD is probably because of a direct consequence of reduced S1P levels in circulation and lung tissues in SphK1-deficient mice. Additional support for this contention comes from experiments performed with Sgpl1+/− mice, in which alveolar formation and vascular leak were significantly impaired after exposure to hyperoxia, and earlier studies have shown elevated S1P levels in plasma, lungs, and other tissues of Sgpl1+/− mice.20,59 The role of S1P in pulmonary diseases is complex. In an ovalbumin-challenged murine model of asthma, increased S1P levels in lung tissue aggravated airway inflammation and hyperresponsiveness.60 Earlier studies showed that the administration of SphK inhibitors, N,N-dimethylsphingosine or SKI-II, significantly reduced eosinophilia, pulmonary inflammatory cell infiltrate, IL-4 and IL-5 levels, and peroxidase activity61 in BAL fluid in response to inhaled ovalbumin challenge.61,62 Similarly, we noted reduced markers of inflammation in Sphk1+/− neonatal mice exposed to hyperoxia. In contrast, in the LPS-induced murine model of acute lung injury, a decrease in S1P in the lungs, as evident in SphK1 deficiency, potentiated the lung injury,63 whereas S1PL suppression in the same model ameliorated pulmonary inflammatory response and barrier disruption, both in vivo and in vitro.20,21 Recently, knocking down of SphK1 or treatment with SphK inhibitor, SKI-II, attenuated S1P generation and development of bleomycin-induced pulmonary fibrosis in mice.64 Thus, S1P is a double-edged sword with both beneficial and detrimental effects in different pathological conditions.65,66 A role for SPT in the development of BPD is unclear. It is reasonable to assume that enhanced expression of SPT2 in hyperoxia can also contribute to altered sphingolipid metabolism and S1P accumulation in lungs. Further studies are necessary to delineate the potential role of SPT in BPD.
The second interesting aspect of this study is the potential cross talk between the SphK1/S1P signaling and NOX proteins in ROS generation in response to hyperoxia. Our results show, for the first time to our knowledge, that SphK1, but not SphK2, modulates NOX expression in the neonatal lung. Knockdown of SphK1 blunted hyperoxia-induced NOX2 and NOX4 expression in mouse lung. In vitro, exogenous addition of S1P to HLMVECs stimulated redistribution of NADPH oxidase components, such as Rac1 and p47phox to cell periphery and ROS production, which was blocked by SphK1 siRNA (Figure 9). The exact mechanism of up-regulation of NOX2/NOX4 by S1P is yet to be defined; however, in adult murine lungs, Pseudomonas aeruginosa–mediated NOX2 expression is regulated by NF-κB67 and hyperoxia-induced NOX4 by Nrf2.68 Interestingly, S1P activates Nrf2 in HLMVECs, and inhibition of SphK1 attenuated S1P-induced translocation of Nrf2 to the cell nucleus (data not shown). Thus, the potential link between SphK1/S1P→Nrf2→ROS in the development of lung injury needs to be explored in BPD.
In conclusion, our findings provide correlative evidence for the SphK1/S1P signaling pathway as an essential mediator of hyperoxia-induced lung injury and development of BPD-like morphological characteristics in mice. In addition, we have identified a potential link between SphK1/S1P signaling in NOX2/NOX4 activation, ROS generation, and subsequent development of lung injury and BPD (Figure 10). These findings suggest that targeting SphK1/S1P signaling with small-molecule inhibitors may represent a novel therapeutic approach against human BPD.
Figure 10.

Scheme showing the summary of role of sphingolipids in hyperoxia-induced neonatal lung injury. Hyperoxia causes an increase in sphingosine kinase 1, which stimulates formation of S1P. Stimulation by S1P, in turn, increases levels of NOX2 and NOX4, leading to increased formation of ROS. The process of secondary septation, leading to an increase in lung surface area in the developing neonatal lung, is affected by ROS, leading to BPD, such as morphological characteristics. SphK1 knockout (KO) mice demonstrated protection against hyperoxia because alveoli formation was better preserved compared with WT exposed to hyperoxia.
Acknowledgment
We thank Dr. Richard L. Proia (NIH/National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD) for providing breeding pairs of Sphk1−/− and Sphk2−/− mice.
Footnotes
Supported by University of Illinois at Chicago Center for Clinical and Translational Science grant UL1TR000050 (A.H.) and NIH grants HL P01 98050 and P01 58064 (V.N.), HL66109 (S.P.R.), and ES11869 (S.P.R.).
References
- 1.Northway W.H., Jr. An introduction to bronchopulmonary dysplasia. Clin Perinatol. 1992;19:489–495. [PubMed] [Google Scholar]
- 2.Bourbon J., Boucherat O., Chailley-Heu B., Delacourt C. Control mechanisms of lung alveolar development and their disorders in bronchopulmonary dysplasia. Pediatr Res. 2005;57:38R–46R. doi: 10.1203/01.PDR.0000159630.35883.BE. [DOI] [PubMed] [Google Scholar]
- 3.Bhandari V. Hyperoxia-derived lung damage in preterm infants. Semin Fetal Neonatal Med. 2010;15:223–229. doi: 10.1016/j.siny.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Husain A.N., Siddiqui N.H., Stocker J.T. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol. 1998;29:710–717. doi: 10.1016/s0046-8177(98)90280-5. [DOI] [PubMed] [Google Scholar]
- 5.Jobe A.H., Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;163:1723–1729. doi: 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 6.Bland R.D. Neonatal chronic lung disease in the post-surfactant era. Biol Neonate. 2005;88:181–191. doi: 10.1159/000087581. [DOI] [PubMed] [Google Scholar]
- 7.Luu T.M., Lefebvre F., Riley P., Infante-Rivard C. Continuing utilisation of specialised health services in extremely preterm infants. Arch Dis Child Fetal Neonatal Ed. 2010;95:F320–F325. doi: 10.1136/adc.2009.173138. [DOI] [PubMed] [Google Scholar]
- 8.Smith V.C., Zupancic J.A., McCormick M.C., Croen L.A., Greene J., Escobar G.J., Richardson D.K. Rehospitalization in the first year of life among infants with bronchopulmonary dysplasia. J Pediatr. 2004;144:799–803. doi: 10.1016/j.jpeds.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 9.Doyle L.W., Anderson P.J. Long-term outcomes of bronchopulmonary dysplasia. Semin Fetal Neonatal Med. 2009;14:391–395. doi: 10.1016/j.siny.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 10.Doyle L.W., Faber B., Callanan C., Freezer N., Ford G.W., Davis N.M. Bronchopulmonary dysplasia in very low birth weight subjects and lung function in late adolescence. Pediatrics. 2006;118:108–113. doi: 10.1542/peds.2005-2522. [DOI] [PubMed] [Google Scholar]
- 11.Kugelman A., Durand M. A comprehensive approach to the prevention of bronchopulmonary dysplasia. Pediatr Pulmonol. 2011;46:1153–1165. doi: 10.1002/ppul.21508. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y., Uhlig S. The role of sphingolipids in respiratory disease. Ther Adv Respir Dis. 2011;5:325–344. doi: 10.1177/1753465811406772. [DOI] [PubMed] [Google Scholar]
- 13.Takuwa Y., Okamoto Y., Yoshioka K., Takuwa N. Sphingosine-1-phosphate signaling in physiology and diseases. Biofactors. 2010;38:329–337. doi: 10.1002/biof.1030. [DOI] [PubMed] [Google Scholar]
- 14.Wang L., Dudek S.M. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res. 2009;77:39–45. doi: 10.1016/j.mvr.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swan D.J., Kirby J.A., Ali S. Vascular biology: the role of sphingosine 1-phosphate in both the resting state and inflammation. J Cell Mol Med. 2010;14:2211–2222. doi: 10.1111/j.1582-4934.2010.01136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gault C.R., Obeid L.M., Hannun Y.A. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol. 2010;688:1–23. doi: 10.1007/978-1-4419-6741-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Natarajan V., Dudek S.M., Jacobson J.R., Moreno-Vinasco L., Huang L.S., Abassi T., Mathew B., Zhao Y., Wang L., Bittman R., Weichselbaum R., Berdyshev E., Garcia J.G. Sphingosine-1-phosphate, FTY720 and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am J Respir Cell Mol Biol. 2013;49:6–17. doi: 10.1165/rcmb.2012-0411TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bode C., Graler M.H. Immune regulation by sphingosine 1-phosphate and its receptors. Arch Immunol Ther Exp (Warsz) 2012;60:3–12. doi: 10.1007/s00005-011-0159-5. [DOI] [PubMed] [Google Scholar]
- 19.Sammani S., Moreno-Vinasco L., Mirzapoiazova T., Singleton P.A., Chiang E.T., Evenoski C.L., Wang T., Mathew B., Husain A., Moitra J., Sun X., Nunez L., Jacobson J.R., Dudek S.M., Natarajan V., Garcia J.G. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol. 2010;43:394–402. doi: 10.1165/rcmb.2009-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y., Gorshkova I.A., Berdyshev E., He D., Fu P., Ma W., Su Y., Usatyuk P.V., Pendyala S., Oskouian B., Saba J.D., Garcia J.G., Natarajan V. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am J Respir Cell Mol Biol. 2011;45:426–435. doi: 10.1165/rcmb.2010-0422OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szczepaniak W.S., Zhang Y., Hagerty S., Crow M.T., Kesari P., Garcia J.G., Choi A.M., Simon B.A., McVerry B.J. Sphingosine 1-phosphate rescues canine LPS-induced acute lung injury and alters systemic inflammatory cytokine production in vivo. Transl Res. 2008;152:213–224. doi: 10.1016/j.trsl.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng X., Hassoun P.M., Sammani S., McVerry B.J., Burne M.J., Rabb H., Pearse D., Tuder R.M., Garcia J.G. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–1251. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- 23.Warner B.B., Stuart L.A., Papes R.A., Wispe J.R. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol. 1998;275:L110–L117. doi: 10.1152/ajplung.1998.275.1.L110. [DOI] [PubMed] [Google Scholar]
- 24.Copland I., Post M. Lung development and fetal lung growth. Paediatr Respir Rev. 2004;5(Suppl A):S259–S264. doi: 10.1016/s1526-0542(04)90049-8. [DOI] [PubMed] [Google Scholar]
- 25.Allende M.L., Sasaki T., Kawai H., Olivera A., Mi Y., van Echten-Deckert G., Hajdu R., Rosenbach M., Keohane C.A., Mandala S., Spiegel S., Proia R.L. Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. J Biol Chem. 2004;279:52487–52492. doi: 10.1074/jbc.M406512200. [DOI] [PubMed] [Google Scholar]
- 26.Harijith A., Choo-Wing R., Cataltepe S., Yasumatsu R., Aghai Z.H., Janer J., Andersson S., Homer R.J., Bhandari V. A role for matrix metalloproteinase 9 in IFNgamma-mediated injury in developing lungs: relevance to bronchopulmonary dysplasia. Am J Respir Cell Mol Biol. 2011;44:621–630. doi: 10.1165/rcmb.2010-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reddy N.M., Potteti H.R., Mariani T.J., Biswal S., Reddy S.P. Conditional deletion of Nrf2 in airway epithelium exacerbates acute lung injury and impairs the resolution of inflammation. Am J Respir Cell Mol Biol. 2011;45:1161–1168. doi: 10.1165/rcmb.2011-0144OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhandari V., Choo-Wing R., Lee C.G., Yusuf K., Nedrelow J.H., Ambalavanan N., Malkus H., Homer R.J., Elias J.A. Developmental regulation of NO-mediated VEGF-induced effects in the lung. Am J Respir Cell Mol Biol. 2008;39:420–430. doi: 10.1165/rcmb.2007-0024OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choo-Wing R., Nedrelow J.H., Homer R.J., Elias J.A., Bhandari V. Developmental differences in the responses of IL-6 and IL-13 transgenic mice exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2007;293:L142–L150. doi: 10.1152/ajplung.00434.2006. [DOI] [PubMed] [Google Scholar]
- 30.Djoba Siawaya J.F., Roberts T., Babb C., Black G., Golakai H.J., Stanley K., Bapela N.B., Hoal E., Parida S., van Helden P., Walzl G. An evaluation of commercial fluorescent bead-based luminex cytokine assays. PLoS One. 2008;3:e2535. doi: 10.1371/journal.pone.0002535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Houser B. Bio-Rad’s Bio-Plex(R) suspension array system, xMAP technology overview. Arch Physiol Biochem. 2012;118:192–196. doi: 10.3109/13813455.2012.705301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moncunill G., Aponte J.J., Nhabomba A.J., Dobano C. Performance of multiplex commercial kits to quantify cytokine and chemokine responses in culture supernatants from Plasmodium falciparum stimulations. PLoS One. 2013;8:e52587. doi: 10.1371/journal.pone.0052587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berdyshev E.V., Gorshkova I.A., Garcia J.G., Natarajan V., Hubbard W.C. Quantitative analysis of sphingoid base-1-phosphates as bisacetylated derivatives by liquid chromatography-tandem mass spectrometry. Anal Biochem. 2005;339:129–136. doi: 10.1016/j.ab.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Mathew B., Jacobson J.R., Berdyshev E., Huang Y., Sun X., Zhao Y., Gerhold L.M., Siegler J., Evenoski C., Wang T., Zhou T., Zaidi R., Moreno-Vinasco L., Bittman R., Chen C.T., LaRiviere P.J., Sammani S., Lussier Y.A., Dudek S.M., Natarajan V., Weichselbaum R.R., Garcia J.G. Role of sphingolipids in murine radiation-induced lung injury: protection by sphingosine 1-phosphate analogs. FASEB J. 2011;25:3388–3400. doi: 10.1096/fj.11-183970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaphorst K.L., Chiang E., Jacobs K.N., Zaiman A., Natarajan V., Wigley F., Garcia J.G. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol. 2003;285:L258–L267. doi: 10.1152/ajplung.00311.2002. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y., Kalari S.K., Usatyuk P.V., Gorshkova I., He D., Watkins T., Brindley D.N., Sun C., Bittman R., Garcia J.G., Berdyshev E.V., Natarajan V. Intracellular generation of sphingosine 1-phosphate in human lung endothelial cells: role of lipid phosphate phosphatase-1 and sphingosine kinase 1. J Biol Chem. 2007;282:14165–14177. doi: 10.1074/jbc.M701279200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hasleton P.S. The internal surface area of the lung in emphysema. Pathol Eur. 1976;11:211–218. [PubMed] [Google Scholar]
- 38.Shaffer S.G., O’Neill D., Bradt S.K., Thibeault D.W. Chronic vascular pulmonary dysplasia associated with neonatal hyperoxia exposure in the rat. Pediatr Res. 1987;21:14–20. doi: 10.1203/00006450-198701000-00005. [DOI] [PubMed] [Google Scholar]
- 39.Funke M., Zhao Z., Xu Y., Chun J., Tager A.M. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am J Respir Cell Mol Biol. 2012;46:355–364. doi: 10.1165/rcmb.2010-0155OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Usatyuk P.V., Romer L.H., He D., Parinandi N.L., Kleinberg M.E., Zhan S., Jacobson J.R., Dudek S.M., Pendyala S., Garcia J.G., Natarajan V. Regulation of hyperoxia-induced NADPH oxidase activation in human lung endothelial cells by the actin cytoskeleton and cortactin. J Biol Chem. 2007;282:23284–23295. doi: 10.1074/jbc.M700535200. [DOI] [PubMed] [Google Scholar]
- 41.Pendyala S., Usatyuk P.V., Gorshkova I.A., Garcia J.G., Natarajan V. Regulation of NADPH oxidase in vascular endothelium: the role of phospholipases, protein kinases, and cytoskeletal proteins. Antioxid Redox Signal. 2009;11:841–860. doi: 10.1089/ars.2008.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pendyala S., Gorshkova I.A., Usatyuk P.V., He D., Pennathur A., Lambeth J.D., Thannickal V.J., Natarajan V. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal. 2009;11:747–764. doi: 10.1089/ars.2008.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chowdhury A.K., Watkins T., Parinandi N.L., Saatian B., Kleinberg M.E., Usatyuk P.V., Natarajan V. Src-mediated tyrosine phosphorylation of p47phox in hyperoxia-induced activation of NADPH oxidase and generation of reactive oxygen species in lung endothelial cells. J Biol Chem. 2005;280:20700–20711. doi: 10.1074/jbc.M411722200. [DOI] [PubMed] [Google Scholar]
- 44.Kalari S., Moolky N., Pendyala S., Berdyshev E.V., Rolle C., Kanteti R., Kanteti A., Ma W., He D., Husain A.N., Kindler H.L., Kanteti P., Salgia R., Natarajan V. Sphingosine kinase 1 is required for mesothelioma cell proliferation: role of histone acetylation. PLoS One. 2012;7:e45330. doi: 10.1371/journal.pone.0045330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olivera A., Kohama T., Edsall L., Nava V., Cuvillier O., Poulton S., Spiegel S. Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J Cell Biol. 1999;147:545–558. doi: 10.1083/jcb.147.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pyne S., Pyne N.J. Sphingosine 1-phosphate signalling in mammalian cells. Biochem J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishii I., Ye X., Friedman B., Kawamura S., Contos J.J., Kingsbury M.A., Yang A.H., Zhang G., Brown J.H., Chun J. Marked perinatal lethality and cellular signaling deficits in mice null for the two sphingosine 1-phosphate (S1P) receptors, S1P(2)/LP(B2)/EDG-5 and S1P(3)/LP(B3)/EDG-3. J Biol Chem. 2002;277:25152–25159. doi: 10.1074/jbc.M200137200. [DOI] [PubMed] [Google Scholar]
- 48.Kawakami-Mori F., Shimosawa T., Mu S., Wang H., Ogura S., Yatomi Y., Fujita T. NADPH oxidase-mediated Rac1 GTP activity is necessary for nongenomic actions of the mineralocorticoid receptor in the CA1 region of the rat hippocampus. Am J Physiol Endocrinol Metab. 2012;302:E425–E432. doi: 10.1152/ajpendo.00227.2011. [DOI] [PubMed] [Google Scholar]
- 49.Hamalukic M., Huelsenbeck J., Schad A., Wirtz S., Kaina B., Fritz G. Rac1-regulated endothelial radiation response stimulates extravasation and metastasis that can be blocked by HMG-CoA reductase inhibitors. PLoS One. 2011;6:e26413. doi: 10.1371/journal.pone.0026413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stenmark K.R., Abman S.H. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Annu Rev Physiol. 2005;67:623–661. doi: 10.1146/annurev.physiol.67.040403.102229. [DOI] [PubMed] [Google Scholar]
- 51.Thebaud B., Abman S.H. Bronchopulmonary dysplasia: where have all the vessels gone? roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med. 2007;175:978–985. doi: 10.1164/rccm.200611-1660PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bry K., Hogmalm A., Backstrom E. Mechanisms of inflammatory lung injury in the neonate: lessons from a transgenic mouse model of bronchopulmonary dysplasia. Semin Perinatol. 2010;34:211–221. doi: 10.1053/j.semperi.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 53.Chess P.R., D’Angio C.T., Pryhuber G.S., Maniscalco W.M. Pathogenesis of bronchopulmonary dysplasia. Semin Perinatol. 2006;30:171–178. doi: 10.1053/j.semperi.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 54.Deulofeut R., Dudell G., Sola A. Treatment-by-gender effect when aiming to avoid hyperoxia in preterm infants in the NICU. Acta Paediatr. 2007;96:990–994. doi: 10.1111/j.1651-2227.2007.00365.x. [DOI] [PubMed] [Google Scholar]
- 55.Parinandi N.L., Kleinberg M.A., Usatyuk P.V., Cummings R.J., Pennathur A., Cardounel A.J., Zweier J.L., Garcia J.G., Natarajan V. Hyperoxia-induced NAD(P)H oxidase activation and regulation by MAP kinases in human lung endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L26–L38. doi: 10.1152/ajplung.00123.2002. [DOI] [PubMed] [Google Scholar]
- 56.Edsall L.C., Spiegel S. Enzymatic measurement of sphingosine 1-phosphate. Anal Biochem. 1999;272:80–86. doi: 10.1006/abio.1999.4157. [DOI] [PubMed] [Google Scholar]
- 57.Gygi S.P., Rochon Y., Franza B.R., Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 59.Bandhuvula P., Honbo N., Wang G.Y., Jin Z.Q., Fyrst H., Zhang M., Borowsky A.D., Dillard L., Karliner J.S., Saba J.D. S1P lyase: a novel therapeutic target for ischemia-reperfusion injury of the heart. Am J Physiol Heart Circ Physiol. 2011;300:H1753–H1761. doi: 10.1152/ajpheart.00946.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roviezzo F., Di Lorenzo A., Bucci M., Brancaleone V., Vellecco V., De Nardo M., Orlotti D., De Palma R., Rossi F., D’Agostino B., Cirino G. Sphingosine-1-phosphate/sphingosine kinase pathway is involved in mouse airway hyperresponsiveness. Am J Respir Cell Mol Biol. 2007;36:757–762. doi: 10.1165/rcmb.2006-0383OC. [DOI] [PubMed] [Google Scholar]
- 61.Nishiuma T., Nishimura Y., Okada T., Kuramoto E., Kotani Y., Jahangeer S., Nakamura S. Inhalation of sphingosine kinase inhibitor attenuates airway inflammation in asthmatic mouse model. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1085–L1093. doi: 10.1152/ajplung.00445.2007. [DOI] [PubMed] [Google Scholar]
- 62.Lai W.Q., Goh H.H., Bao Z., Wong W.S., Melendez A.J., Leung B.P. The role of sphingosine kinase in a murine model of allergic asthma. J Immunol. 2008;180:4323–4329. doi: 10.4049/jimmunol.180.6.4323. [DOI] [PubMed] [Google Scholar]
- 63.Wadgaonkar R., Patel V., Grinkina N., Romano C., Liu J., Zhao Y., Sammani S., Garcia J.G., Natarajan V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;296:L603–L613. doi: 10.1152/ajplung.90357.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang L.S., Berdyshev E., Mathew B., Fu P., Gorshkova I.A., He D., Ma W., Noth I., Ma S.F., Pendyala S., Reddy S.P., Zhou T., Zhang W., Garzon S.A., Garcia J.G., Natarajan V. Targeting sphingosine kinase 1 attenuates bleomycin-induced pulmonary fibrosis. FASEB J. 2013;27:1749–1760. doi: 10.1096/fj.12-219634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwalm S., Pfeilschifter J., Huwiler A. Sphingosine-1-phosphate: a Janus-faced mediator of fibrotic diseases. Biochim Biophys Acta. 2013;1831:239–250. doi: 10.1016/j.bbalip.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 66.Limaye V. The role of sphingosine kinase and sphingosine-1-phosphate in the regulation of endothelial cell biology. Endothelium. 2008;15:101–112. doi: 10.1080/10623320802125342. [DOI] [PubMed] [Google Scholar]
- 67.Fu P., Mohan V., Mansoor S., Tiruppathi C., Sadikot R.T., Natarajan V. Role of NOX proteins in Pseudomonas aeruginosa induced lung inflammation and permeability. Am J Respir Cell Mol Biol. 2013;48:477–488. doi: 10.1165/rcmb.2012-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pendyala S., Moitra J., Kalari S., Kleeberger S.R., Zhao Y., Reddy S.P., Garcia J.G., Natarajan V. Nrf2 regulates hyperoxia-induced Nox4 expression in human lung endothelium: identification of functional antioxidant response elements on the Nox4 promoter. Free Radic Biol Med. 2011;50:1749–1759. doi: 10.1016/j.freeradbiomed.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]