

ABSTRACT
Shiga toxins (Stx) are the main agent responsible for the development of hemolytic-uremic syndrome (HUS), the most severe and life-threatening systemic complication of infection with enterohemorrhagic Escherichia coli (EHEC) strains. We previously described Stx2 expression by eukaryotic cells after they were transfected in vitro with the stx2 gene cloned into a prokaryotic plasmid (pStx2). The aim of this study was to evaluate whether mammalian cells were also able to express Stx2 in vivo after pStx2 injection. Mice were inoculated by hydrodynamics-based transfection (HBT) with pStx2. We studied the survival, percentage of polymorphonuclear leukocytes in plasma, plasma urea levels, and histology of the kidneys and the brains of mice. Mice displayed a lethal dose-related response to pStx2. Stx2 mRNA was recovered from the liver, and Stx2 cytotoxic activity was observed in plasma of mice injected with pStx2. Stx2 was detected by immunofluorescence in the brains of mice inoculated with pStx2, and markers of central nervous system (CNS) damage were observed, including increased expression of glial fibrillary acidic protein (GFAP) and fragmentation of NeuN in neurons. Moreover, anti-Stx2B-immunized mice were protected against pStx2 inoculation. Our results show that Stx2 is expressed in vivo from the wild stx2 gene, reproducing pathogenic damage induced by purified Stx2 or secondary to EHEC infection.
IMPORTANCE
Enterohemorrhagic Shiga toxin (Stx)-producing Escherichia coli (EHEC) infections are a serious public health problem, and Stx is the main pathogenic agent associated with typical hemolytic-uremic syndrome (HUS). In contrast to the detailed information describing the molecular basis for EHEC adherence to epithelial cells, very little is known about how Stx is released from bacteria in the gut, reaching its target tissues, mainly the kidney and central nervous system (CNS). In order to develop an efficient treatment for EHEC infections, it is necessary to understand the mechanisms involved in Stx expression. In this regard, the present study demonstrates that mammals can synthesize biologically active Stx using the natural promoter associated with the Stx-converting bacteriophage genome. These results could impact the comprehension of EHEC HUS, since local eukaryotic cells transduced and/or infected by bacteriophage encoding Stx2 could be an alternative source of Stx production.
Introduction
Shiga-toxin-producing Escherichia coli (STEC) strains are emergent food-borne pathogens that can cause a range of clinical disease from uncomplicated diarrhea to hemorrhagic colitis and life-threatening complications, such as hemolytic-uremic syndrome (HUS) (1–4). The cardinal element of virulence of STEC/enterohemorrhagic E. coli (EHEC) is the production of Shiga toxins (Stx), which constitute an AB5 toxin (5). The genes encoding the two subunits of Stx are located in the genomes of lambdoid bacteriophages (6), which are efficient vectors for the transfer of stx and play an important role in the evolution of new pathogens (7–9). Stx phages can be induced from the lysogenic strain (10). As a result of prophage induction, bacterial host cells lyse and release free phage particles that can infect other bacteria (11–14). However, low levels of spontaneous phage induction can also occur. It has been demonstrated that most Stx2 prophages are spontaneously induced more readily than lambdoid prophages which do not encode Stx (15).
The fate of EHEC infections depends on the interplay between bacteria and the host, in which the inflammatory response has a key role. EHEC intestinal infection triggers cytokine synthesis, particularly of interleukin 8 (IL-8), in the submucosa, which induces recruitment, activation, and migration of neutrophils to the intestine. In addition, the migration and activation of neutrophils contribute to the destabilization or epithelial integrity disruption, increasing Stx absorption and thereby the pathogenicity of EHEC (16). Furthermore, these neutrophils could also act as carriers of phage particles.
We previously reported the ability of eukaryotic cells to recognize wild promoter sequences in the stx2 gene to drive Stx2 expression by cell lines (17). To analyze whether mammals were capable of transcribing, translating, and expressing Shiga toxins from the stx2 gene, we analyzed Stx2 expression in vivo after transfection with a plasmid construction carrying the stx2 gene under its own promoter (pStx2). For this purpose, mice were inoculated with pStx2 by a hydrodynamics-based transfection (HBT) procedure in which a volume of 8 to 12 ml of a plasmid-containing solution per 100 g of animal weight was rapidly injected (in less than 8 s) into the tail vein of mice (18). This gene administration procedure induces high levels of transgene expression mainly in the liver (18). For this reason, it has been widely used to evaluate a number of aspects, including the therapeutic activities of certain genes (19) and the analysis of regulatory functions of DNA sequences (20), but also to induce high levels of systemic cytokine expression (21). In mice with a body weight of 18 to 20 g, an injection volume of 1.6 ml per mouse is almost equivalent to the total blood volume of the animal (blood volume is estimated to be 7.3% of the body weight in mice), and then the injected DNA solution is likely to accumulate in the inferior vena cava when the injection rate exceeds the cardiac output (18). As a result, a high hydrostatic pressure develops in the inferior vena cava. Such hydrostatic pressure, being proportional to the volume and the speed of injection, forces the flow of the DNA solution into tissues such as the liver, kidney, and heart, which are directly linked to the inferior vena cava. Since the liver is the largest organ in the body with an expandable structure, a large portion of the DNA solution is forced into the liver in a direction that is opposite to that of the regular circulation. This results in a direct exposure of DNA molecules to liver cells before they are mixed with the blood.
After the pStx2 HBT procedure, levels of Stx2 mRNA in the liver and Stx2 biological activity were monitored in the animals. Mice injected with pStx2 died with typical signs of Stx2 intoxication, including acute kidney injury, neutrophilia, and central nervous system (CNS) damage. Noticeably, the Stx2 protein was detected in the brain by immunofluorescence, and anti-Stx2B-immunized mice receiving pStx2 HBT survived and lacked any of the hallmarks of Stx2 intoxication. These results show that Stx2 is expressed in vivo after pStx2 HBT, driven by its own wild stx2 promoter. Furthermore, active Stx2 thus produced is able to target the kidney and the brain and reproduces the lethal damage induced by purified Stx2 or secondary to EHEC infection.
RESULTS
Mice displayed a lethal dose-related response to pStx2.
The hydrodynamics-based transfection (HBT) technique has proven to be a feasible and efficient way to deliver nonviral hepatic genes in mice (18, 22). Thus, mice were injected by HBT with different doses of pStx2 or pStx2ΔAB, a plasmid construction in which the active site of Stx2 has been deleted (23), or the pGEMT cloning vector (Promega) without any Stx2 genetic material as an additional negative control. Mice injected with the highest dose of pStx2 (3.5 µg pStx2/mouse) became agitated and restless within 6 h and died within 24 h. Lower doses of pStx2 showed lethal effects at later times, as demonstrated by the resulting dose-response curve (Fig. 1A). HBT produces mainly transfection of hepatocytes. To evaluate hepatic inflammation, the concentrations of liver-specific enzymes, including alkaline phosphatase (ALP) and aspartate aminotransferase (AST), were analyzed. All mice had hepatic enzymes in the normal concentration ranges (data not shown). Mice inoculated with pStx2ΔAB or pGEMT survived and showed no signs of toxic effects. These controls allowed us to disregard any damage caused by the plasmid construction or HBT side effects.
Analysis of gene expression in liver.
Because the liver is the main organ implicated in gene expression after the HBT technique (18), total RNA was purified from the livers of mice inoculated with pStx2 or pGEMT and analyzed through real-time PCR using primers of the Stx2 A subunit. In Fig. 1B, the real-time curves obtained for the negative control (gray; mRNA from livers of mice inoculated with pGEMT), the positive control (bold; purified pStx2), and the mRNA from the livers of mice inoculated with pStx2 (dotted line) are shown. The products of real-time PCR were loaded on an agarose gel, and the bands observed were according to the expected size (Fig. 1C).
FIG 1 .
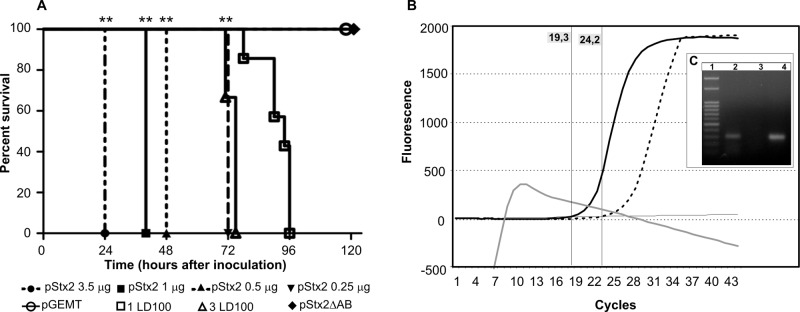
(A) Mice displayed a lethal dose-related response to pStx2. Survival rates of mice injected with different doses of pStx2 are shown. Mice injected with pStx2ΔAB or pGEMT were used as negative controls. Mice injected with 1 LD100 and 3 LD100 of purified Stx2 were used as positive controls. (B) Stx2 mRNA was detected in the liver. A real-time-PCR curve using cDNA from livers of mice inoculated with pStx2 (dotted line) or pGEMT (gray line) is shown. As a positive control, 50 pg of plasmid pStx2 (bold line) was used. (C) DNA electrophoresis. Lane 1, 50-bp ladder; lane 2. cDNA from livers of mice inoculated with pStx2; lane 3, cDNA from livers of mice inoculated with pGEMT; lane 4, pStx2 was used as positive control.
Evaluation of Stx2 biological activity in plasma.
With the aim of evaluating whether Stx2 expressed in the liver was released into circulation, plasma from mice inoculated with pStx2, pStx2ΔAB, or pGEMT was evaluated on Vero cells as the most sensitive test to detect Stx2 (24). Mice were inoculated with different doses of pStx2 (3.5 to 0.25 µg/mouse) and bled at 24 h, after HBT. We observed a dose-response effect for Stx2-cytotoxicity (Fig. 2A). To confirm the specificity in the cytotoxic assay, purified Stx2 equivalent to 1 CD50 (cytotoxic dose that kills 50% of Vero cells) (4.5 ng/ml) or plasma from mice inoculated with 3.5 µg of pStx2 was preincubated with polyclonal anti-Stx2 antibody. Significant neutralization of the cytotoxicity was observed in both samples (Fig. 2C). As expected, no cytotoxic effect on Vero cells was observed with plasma from mice inoculated with pGEMT or pStx2ΔAB.
FIG 2 .
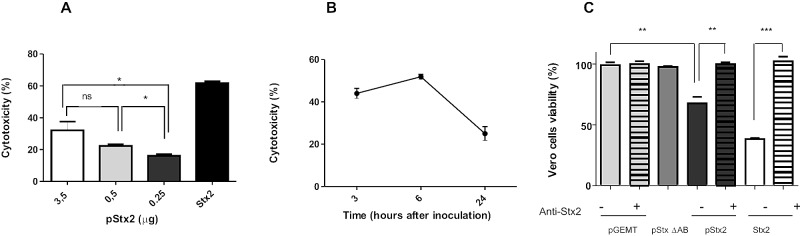
In vitro
evaluation of Stx2 activity in plasma. (A) Vero cells were incubated with plasma from mice inoculated with different doses of pStx2 obtained at 24 h postinoculation. After 48 h of incubation, cells were stained with crystal violet and the optical density at 595 nm (OD595) was measured, as detailed in Materials and Methods. Each bar represents the mean ± SEM for 4 mice. *, P < 0.03, t test. (B) Kinetic study of Stx2 activity in plasma. Vero cells were incubated with a 1:60 dilution of plasma from mice inoculated with 3.5 µg of pStx2 obtained at different times postinoculation. Each time point represents the mean ± SEM for 4 mice. (C) In vitro neutralization of Stx2 activity in plasma. Vero cells were incubated with a 1:60 dilution of plasma of mice inoculated with pStx2, pStx2ΔAB or pGEMT. The specificity of cytotoxicity was evaluated in parallel by preincubating each sample with mouse anti-Stx2 polyclonal antibody (dilution, 1/400) for 1 h at 37°C and for 1 h at 4°C (pStx2 + antibodies, pGEMT + antibodies, and Stx2 + antibodies). As positive and negative controls of cytotoxicity, Vero cells were incubated with 1 CD50 of Stx2 (Stx2 and Stx2 + antibodies) or Vero cells in medium, respectively. After 48 h, cells were stained with crystal violet and the OD595 was measured as detailed in Materials and Methods. Results are means ± SEM for 4 mice. **, P <0.005; ***, P = 0.0001.
The amount of Stx2 in plasma was calculated through the Vero cytotoxic assay using a standard dose-response curve made with purified Stx2. The toxicity observed in plasma from mice inoculated with 3.5 µg of pStx2 was equivalent to that of 172 ± 51 Stx2 ng/ml; the value was 119 ± 7 Stx2 ng/ml for mice inoculated with 0.5 µg of pStx2 and 88 ± 13 Stx2 ng/ml for mice inoculated with 0.25 µg of pStx2. These results support the high lethal effect of pStx2 shown in the survival data previously described (Fig. 1A).
Then, we evaluated Stx2 cytotoxicity in plasma at different time points after inoculation of 3.5 µg of pStx2. The kinetic study showed the maximal Stx2 activity in plasma at 6 h after HBT (Fig. 2B). As previously reported, the transfection induced by HBT is transient, and after this time point, the levels of produced protein slowly decreased.
Laboratory and histological parameters in mice injected with pStx2.
To further characterize the pathogenic effect induced by pStx2 HBT, mice were inoculated with high or low lethal doses of pStx2, and biochemical and histopathological changes in the kidneys and brains of the animals were analyzed. It has been previously demonstrated using murine models that plasma urea levels are good indicators of acute renal damage during Stx2 intoxication. Mice injected with 0.25 µg of pStx2 died at 72 h after injection, showing neutrophilia (Fig. 3A), leucopenia, and a significant increased in the plasma urea level (Fig. 3B). These results suggest that mice were able to endogenously produce Stx2, in an amount and/or timing that led mice to death by acute kidney injury. In addition, alterations in leukocyte populations were similar to those obtained after exogenous injection of purified Stx2 or oral infection with Stx2-producing E. coli (25, 26). On the other hand, mice inoculated with 3.5 µg or 1 µg of plasmid died within 24 h after inoculation with signs of neurological involvement but no alterations in the laboratory markers related to hepatic or renal function or leukocyte counts. All mice injected with pStx2ΔAB or pGEMT remained alive and showed biochemical values similar to those for nontreated mice.
FIG 3 .
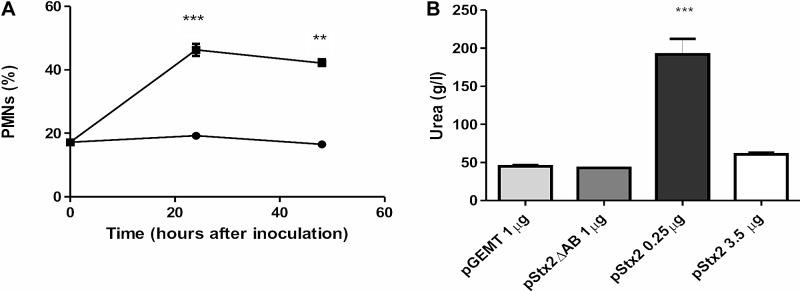
Laboratory parameters for mice injected with pStx2. (A) Percentages of polymorphonuclear leukocytes for mice inoculated with 0.25 µg of pStx2 (■) or with 0.25 µg pGEMT (●) at 24 and 48 h after inoculation. Controls versus pStx2, ***, P = 0.0004; **, P = 0.0037. Each time point represents the mean ± SEM for 4 mice. (B) Mice were injected with different doses of pStx2, and plasma urea values were evaluated before death. Each bar represents the mean ± SEM for 4 mice. ***, P = 0.0002.
Histology of kidneys and brains of mice injected with pStx2.
Mice inoculated with pStx2 or pGEMT were histologically studied. Kidneys from mice inoculated with 0.25 µg of pStx2 at 48 h after HBT showed enlargement of glomeruli due to mesangial hypercellularity with subsequent obliteration of Bowman’s space. Tubular epithelia showed features of edema with thickening cytoplasmatic alterations and ill-defined luminal edges (Fig. 4B). As expected, a normal structure of the cortex and medulla was observed in the kidneys of pGEMT-inoculated mice (Fig. 4A). On the other hand, leukocyte infiltration around necrotic areas was observed in the brains of mice inoculated with 3.5 µg of pStx2 and sacrificed at 24 h after HBT (Fig. 5B). No alterations in the brains of control mice injected with pGEMT were observed (Fig. 5A).
FIG 4 .
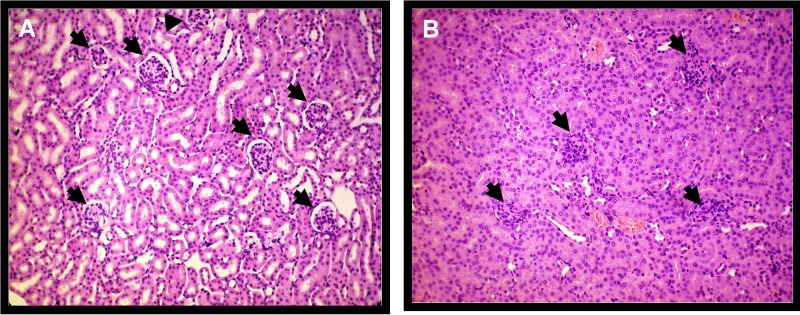
Kidney histology of cortical zone. (A) Renal glomeruli of a control mouse showed normal structure; lobular organization of the glomeruli and a flat epithelium lining the glomerular capsule can be seen (arrows). The renal tubules are lined with typical thick cubic epithelium with relatively regular distinct lumen. H&E stain; magnification, ×250. (B) Kidney of a mouse inoculated with pStx2 showed glomeruli enlarged with mesangial hypercellularity tightly filling the Bowmann’s space (arrows). The tubular epithelium showed enlarged cells with ill-defined luminal edges and clumpy cytoplasm. Capillaries are filled with blood cells; some tubules contain single desquamated cells. H&E; magnification, ×250.
FIG 5 .
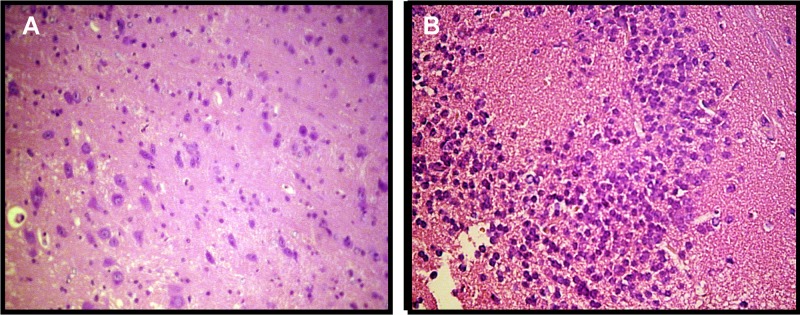
Brain histology. (A) Brain of a control mouse inoculated with pGEMT; no alterations were observed. H&E; magnification, ×400. (B) Brain of a mouse inoculated with 0.25 µg pStx2 at 24 h after inoculation. Lymphocyte infiltration was observed. H&E; magnification, ×400.
Immunofluorescence analysis of Stx2 expression in brains of mice inoculated with pStx2.
To elucidate whether the histological alterations in the brain were the consequence of a direct effect of Stx2, brains of mice inoculated with pStx2 or pGEMT were evaluated by an immunofluorescence assay using a polyclonal anti-Stx2 antibody (27). As a positive control, we tested mice inoculated with purified Stx2. We observed Stx2-immunopositive cells in the CA2 hippocampus region of mice inoculated with 3 µg of pStx2 24 h after HBT (Fig. 6B). Stx2 was also detected in the brains of mice injected with 0.25 µg of pStx2 at 48 h after HBT (Fig. 6C). Control mice inoculated with purified Stx2 (Fig. 6D) showed numbers of cells immunopositive for Stx2 similar to those of pStx2 HBT mice (Fig. 6F). The Cortex and hypothalamus also showed Stx2-positive cells (data not shown).
FIG 6 .
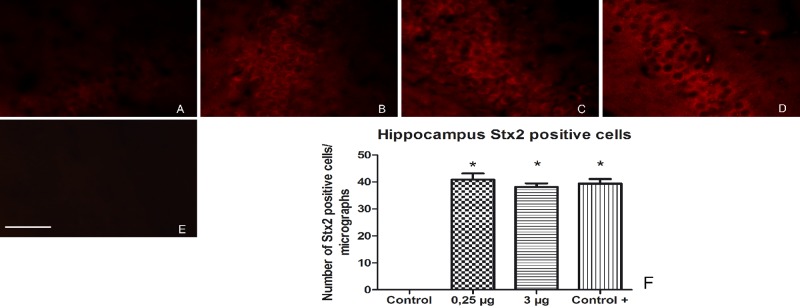
Localization of Stx2 in brains of mice inoculated with pStx2. The area observed in this study is located in the mouse CA2 hippocampus. (A) Brain from control mouse inoculated with pGEMT. (B) Brain from mouse inoculated with 3 µg of pStx2 24 h after inoculation. (C) Brain from mouse inoculated with 0.25 µg of pStx2 48 h after inoculation. (D) Brain from mouse injected with 0.5 ng of Stx2 4 days after injection. (E) Brain from mouse inoculated with 0.25 µg of pStx2 48 h after inoculation, without primary antibody. (F) Number of Stx2-immunopositive cells in hippocampus per micrograph. Scale bar = 50 µm.
pStx2 increased expression of GFAP in astrocytes.
The hallmark of astrocyte (AST) activation is the enhanced expression of the major intermediate filament protein, glial fibrillary acidic protein (GFAP) (28). Previously, it has been demonstrated that Stx increases GFAP expression in AST, both in vitro (29) and in vivo (30). As depicted in Fig. 7, pStx2 HBT induced the upregulation of GFAP in the CA2 hippocampus zone at each time point evaluated: 24 h post-3-µg dose of pStx2 or 48 h post-0.25-µg dose of pStx2. GFAP has an important role in repairing CNS after injury (31). We did not observe differences in GFAP expression between the two different pStx2 doses used (Fig. 7B and C). Vehicle/saline injection did not induce upregulation of GFAP in comparison with the treatment.
FIG 7 .
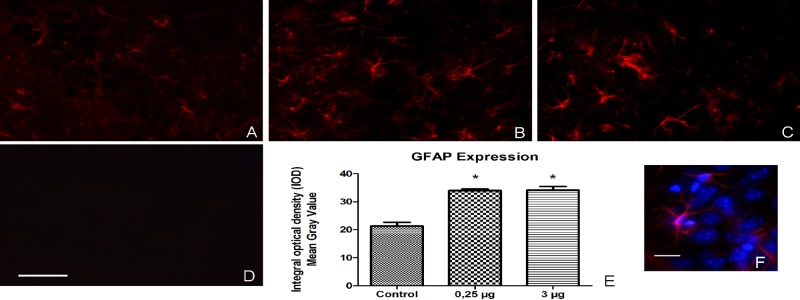
GFAP expression was increased in astrocytes after pStx2 injection. The area observed in this study is located in the mouse CA2 hippocampus. (A) Brain from control mouse inoculated with pGEMT. (B) Brain from mouse inoculated with 0.25 µg of pStx2 48 h after inoculation. (C) Brain from mouse inoculated with 3 µg of pStx2 24 h after inoculation. (D) Brain from mouse inoculated with 0.25 µg of pStx2 48 h after inoculation without primary antibody. Scale bar = 25 µm. (E) GFAP expression measure by Integral optical density in hippocampus. (F) Colocalization of GFAP and Hoescht 33342 stain. Scale bar = 20 µm.
pStx2 caused neuronal damage, evidenced by NeuN immunoreactivity.
Since Stx-induced toxicity for neurons has been previously demonstrated (32, 33), we analyzed neurons by using the specific neuronal marker NeuN. This marker protein is localized in the nucleus and the cytoplasm of neurons. Under injury conditions, NeuN in the nucleus becomes immunonegative or fragmented but cytoplasmic NeuN keeps stable (34). According to previous data, while neurons showing intense nuclear reactivity are considered normal, neurons with NeuN fragmentation in the nucleus are considered abnormal (34). We observed abnormal neurons in the brains of mice injected with pStx2 (Fig. 8B and C). The number of abnormal neurons in mice injected with pStx2 was significantly higher than that in control mice injected with pGEMT (Fig. 8G). No significant differences were observed between pStx2 doses (Fig. 8G).
FIG 8 .
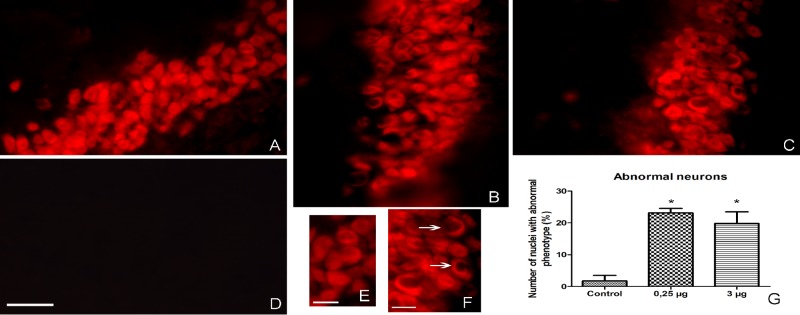
NeuN immunoreactivity as neuronal damage marker. A significant increase in the number of abnormal nuclei was observed by immunofluorescence with anti-NeuN antibodies. (A) Brain from control mouse inoculated with pGEMT. (B) Brain from mouse inoculated with 0.25 µg of pStx2 48 h after inoculation. (C) Brain from mouse inoculated with 3 µg of pStx2 24 h after inoculation. (D) Brain from mouse inoculated with 0.25 µg of pStx2 48 h after inoculation with no primary antibody. Scale bar = 50 µm. (E) Zoomed-in view of picture A. Scale bar = 15 µm. (F) Zoomed-in view of picture C. Scale bar = 25 µm. (G) Numbers of nuclei with abnormal phenotype.
In vivo neutralization of pStx2 toxicity.
We recently reported that the chimeric protein BLS-Stx2B induces high anti-Stx2B neutralizing antibody titers in mice (35). Then, to confirm that pStx2 toxicity and mortality were dependent on Stx2 production in vivo, anti-Stx2B-immunized and nonimmunized mice were inoculated with 0.25 µg of pStx2. As shown in Fig. 9A, BLS-Stx2B-immunized mice showed a significantly higher survival rate (80%) than nonimmunized control mice (0%). As biochemical parameters that correlate with systemic Stx2 toxicity, the polymorphonuclear leukocyte (PMN) percentage and urea level in plasma were assayed in both groups. All survivors in the immunized group presented normal plasma urea values (urea g/liter, 36.6 ± 3.6; n = 8). Although immunized mice showed an increase in the percentage of circulating PMN, it was significantly lower than that in the nonimmunized mice (Fig. 9B). These results indicate that Stx2 immunized mice were protected against pStx2 toxicity, confirming that lethal effects of pStx2 are dependent on Stx2.
FIG 9 .
In vivo
neutralization of Stx2-like biological activity. (A) Offspring from BLS-Stx2B-immunized mice were inoculated with 0.25 µg of pStx2 (fill line) (n = 10). Offspring from nonimmunized mice also were inoculated with 0.25 µg of pStx2 (broken line) (n = 6). Log rank (Mantel-Cox) test, P = 0.0229. (B) Polymorphonuclear leukocyte percentages for mice inoculated with 0.25 µg of pStx2 after 48 h of inoculation. *, P = 0.0231; **, P = 0.0024; ***, P < 0.0001. Each bar represents the mean ± SEM for live mice at 48 h.
DISCUSSION
The major finding of this study is the demonstration that mammals are able to express functional Stx2 from the wild stx2 gene sequence. Furthermore, mice transfected with pStx2 in vivo developed the main pathogenic signs associated with Stx2 intoxication, including CNS and renal alterations.
Nowadays there is no doubt about the importance of Stx2 and Stx2 phage biology to the understanding of HUS. The current knowledge states that Stx2 expression is under control of phage promoters controlled by the phage replication cycle. Therefore, induction of Stx-converting prophages triggers increased Stx production (7, 36, 37). At present, the linkage between the phage growth cycle and Stx expression is well established on a molecular level. Besides, following induction, Stx phages can infect other bacteria present in the gut in vivo and in vitro, and they could also infect eukaryotic cells, such as epithelial cells, macrophages, or neutrophils, present in the local inflamed zone. We used a hydrodynamics-based transfection (HBT) procedure in mice to test our hypothesis that the gene for Stx2 can be expressed in the tissues of a mammal, with pathological consequences.
Following this experimental approach, we showed that pStx2 could be efficiently transfected. The RNA analysis of the liver revealed the presence of Stx2-mRNA at 3 h post-pStx2 HBT but not in mice injected with pGEMT. The totality of previous studies using HBT to study gene function and regulation, gene therapy, and DNA vaccination or gene therapy relied on in vivo delivery of genes under control of eukaryotic promoters, and it is known that promoter strength is one of the most important parameters determining the level of gene expression (18, 38). In this regard, among the many promoters that have been evaluated in mice, cytomegalovirus immediate-early promoter (CMV) and human α1-antitrypsin promoter are generally selected. Both promoters are similarly effective in expressing genes and also exhibit almost identical time-response curves. In both cases, the level of gene expression peaks at 8 h after DNA administration and drops quickly thereafter.
In our experimental design, a bacterial gene was translated and expressed in vivo under its own wild bacterial promoter sequences by mammalian cells. Although the translation efficiency of pStx2 is probably low, the importance of this finding relies mainly on two facts. First, this protein has a strong impact in human health. Infections with STEC/EHEC strains cause HUS, acute renal failure (1, 4, 39), and less commonly CNS impairment (40). In particular, it has been reported that the mortality rate from HUS increases from 0 to 5% to 7 to 40% when the CNS is involved (41–43). This issue became prominent in Germany in 2011, when the consumption of sprouts containing Stx2 from the unusual enteroaggregative E. coli O104:H4 resulted in 3,816 cases of gastroenteritis, 845 of which evolved to HUS and 54 to death (44). Second, Stx2 presents one of the highest biological activities among bacterial toxins. In fact, concentrations lower than the pg/ml range in mice (ng/mouse) have been shown to be lethal. It has been recently demonstrated that a concentration of Stx2 as low as 10 fM is able to induce ribosome damage and to modulate selected cell signaling pathways that change cellular functions (45). However, it should be noted that the Stx2 concentration in blood of HUS patients has never been determined. Therefore, although the efficiency of protein synthesis by eukaryotic cells under bacterial promoter sequences must be significantly lower than that under the control of a eukaryotic promoter, the protein concentration systemically raised was enough to trigger a pathogenic cascade and death in mice. We were not able to directly detect free Stx2 in the plasma obtained from mice by Western blotting (WB) after pStx2 HBT. However, plasma from mice inoculated with pStx2 showed Stx2-specific cytotoxicity in the Vero cell assay. We observed a peak of cytotoxicity 6 h after inoculation of mice with 3 µg of pStx2. No cytotoxicity was observed in plasma from control mice injected with pGEMT or pStx2ΔAB, which had lost the active site of the toxin. Moreover, in vitro neutralization of plasma cytotoxicity by anti-Stx2 polyclonal antibodies strongly supports the specificity of the reaction and the similarity between wild bacterial Stx2 and that produced in vivo by eukaryotic cells.
Once Stx reaches the tissue, the initial step in the pathogenesis of STEC HUS is binding of the B subunit to Gb3 on the cell membrane. This step is followed by retrograde transport of the A subunit to the Golgi apparatus and inhibition of protein synthesis in the ribosome (45). In fact, we not only demonstrated that eukaryotic cells expressed active and functional Stx2 in vivo but also that it was able to reproduce all signs and symptoms of Stx2 intoxication in mice. It is noteworthy that we observed that the death of the mice was dependent on the dose of pStx2 injected. When pStx2 was injected at a concentration of 0.25 µg/mouse, mice died at 72 h with acute renal damage (biochemically and histologically demonstrated) and with typical neutrophilia (25, 26). These pathological signs are similarly observed in mice injected with purified Stx2 or orally infected with EHEC. In addition, they partially resembled pathological parameters observed in HUS patients. In particular, the increase in the number of peripheral blood neutrophils has been correlated with a poor prognosis (46, 47).
However, mice inoculated with the highest pStx2 dose (3.5 µg) died within 24 h post-HBT, with no increase in plasma urea. Because longer times are necessary to induce lethality secondary to acute renal damage, death was associated with CNS damage. Supporting this hypothesis, histological brain damage and Stx2 presence in brains of mice were demonstrated. Those mice showed CNS injury, evidenced by leukocyte infiltration and activation of AST, demonstrated by upregulation of GFAP expression. In this regard, it has been previously reported that intracerebroventricular administration of Stx2 increases the expression of GFAP, leading to astrogliosis (30), and that Stx2 causes neuronal death and glial cell damage in rat brains (33). We also found an increase in abnormal neuronal nuclei in the brains of mice injected with pStx2 evaluated by NeuN staining. No data for NeuN and Stx2 are published, but it is known that normal neurons express a high signal for NeuN in the nucleus. A decrease in nuclear NeuN expression was reported for several pathological conditions (48). All these results led us to conclude that when high doses of pStx2 were used, mice died of CNS damage as a consequence of direct Stx2 effect. Moreover, we found strong evidence supporting the immunolocalization of Stx2 in the brains of mice. This result would also indicate that the Stx2 protein is expressed retaining the quaternary conformation necessary to bind to its specific receptor, Gb3.
A definitive demonstration that toxicity upon pStx2 HBT was mediated by Stx2 is that anti-Stx2B-immunized mice were protected against lethality of pStx2 HBT. In addition, the fact that high titters of circulating anti-Stx2B antibodies blocked pStx2 HBT toxicity indicates that Stx2 is being expressed in the liver and probably, after cell death, the toxin is released into systemic circulation to reach its target tissues. In line with this reasoning, Stx2 toxicity was directly detected in the serum from pStx2 HBT mice.
In conclusion, we demonstrated that biologically active Stx2 is being expressed by mammals cells using the natural promoter associated with the Stx-converting bacteriophage genome. The protein thus synthesized by the host retains the natural conformation that allows it to interact with its target tissues and induces the typical pathological features associated with bacterial Stx2. These results could impact STEC HUS comprehension, since local eukaryotic cells transduced with and/or infected by bacteriophage encoding Stx2 could be an alternative source of Stx2 production.
MATERIALS AND METHODS
Plasmid construction. (i) pStx2.
The plasmids were constructed by standard cloning techniques, according to the NIH policy manual on Working Safely with Hazardous Biological Materials. The complete stx2 sequence was amplified by PCR from total DNA from E. coli O157:H7 C600 (933W), using the primers Stx2Fw (5′ GAATTCATTATGCGTTGTTAG 3′) and Stx2R (5′ GAATTCTCAGTCATTATTAAACTG 3′), both containing an EcoRI restriction site (49). The resulting fragment (1,413 bp in length) was cloned in a pGEMT Easy vector (Promega Inc.), generating the plasmid pStx2. This plasmid was replicated in E. coli DH5α competent cells and purified using the Wizard Plus Minipreps DNA purification system following standardized instructions.
(ii) Control plasmids.
The pGEMT Easy vector (Promega Inc.) was used as a negative control. The pGEMT Easy vector carrying the sequence of Stx2 was digested and religated in order to delete the active site of Stx2, generating the plasmid pStx2ΔAB (23) as a Stx2-specific biological activity control.
In vivo gene expression.
Mice were inoculated with 3.5 µg of pStx2 or pGEMT using HBT and were sacrificed 3 h after inoculation. Total RNA was isolated from livers of mice inoculated with pStx2 or pGEMT as a control. The isolation was made using a single-step phenol/chloroform extraction procedure (TRIzol; Invitrogen Life Technologies). DNase treatment was made using RQ1 RNase-free DNase (Promega) following standardized instructions. cDNA was obtained using Fermentas avian myeloblastosis virus (AMV) reverse transcriptase (RT).
Real-time PCR was performed on a Smart Cycler real-time machine (Cepheid, Sunnyvale, CA) using Smart Cycler tubes from Fisher (Sunnyvale, CA). The reaction was run in the Smart Cycler at an initial 92°C for 120 s and then at 92°C for 10 s and 55°C for 15 s and 72°C for 15 s for 45 cycles using the primers giving a fragment of 147 bp on the stx2 A subunit (F2A-3, TGGCGTTAATGGAGTTCAGTGG; R2A-3, ACACAGGAGCAGTTTCAGACAG).
Mice.
BALB/c mice were bred in the animal facility of the Institute of Experimental Medicine, (IMEX), Academia Nacional de Medicina, Buenos Aires. Male mice aged 6 weeks (18 to 20 g) were used throughout the experiments. They were maintained under a 12-h light-dark cycle at 22 ± 2°C and fed with standard diet and water ad libitum.
HBT of pStx2.
The hydrodynamic gene transfer procedure was conducted as described previously (18). In brief, animals were separated into different groups and injected in the tail vein with different doses of pStx2 (3.5 µg, 1 µg, 0.5 µg, and 0.25 µg). As controls, we used 3.5 µg of pGEMT or 1 µg and 0.5 µg of pStx2ΔAB. Injections were performed in 1.6 ml of phosphate-buffered saline (PBS) by hydrodynamic inoculation via the tail vein. Plasmid injection was completed in less than 8 s.
Hematological studies.
Heparinized blood samples were obtained by puncture of the retroorbital plexus 3 h after pStx2 HBT injection for laboratory analyses that included total and differential blood cell count in the Neubauer chamber, blood smears, plasma urea determination, and liver enzymes. Biochemical determinations of urea and liver enzymes in plasma were performed using commercial kits (urea color 2R kit and alkaline phosphatase [ALP] and aspartate aminotransferase [AST] kits; Wiener Lab, Rosario, Argentina) following standardized instructions
Histological studies.
For histological analysis, mice were sacrificed 24 h or 48 h after pStx2 HBT injection (depending of the doses used) and subjected to necropsy. Mice were transcardially perfused with PBS in order to remove the blood completely, followed by 5% buffer-formaldehyde. Kidneys and brains were removed, sectioned, fixed in 5% buffer-formaldehyde, and paraffin embedded. Kidney and brain sections of paraffin-embedded tissues were stained with hematoxylin and eosin (H&E) and examined by light microscopy. Tubular injury was evaluated by assessing the presence of alterations in tubular epithelium, basement membrane integrity, and necrosis. Vascular interstitial congestion was also assessed. Another group of mice was sacrificed for immunofluorescence. Mice were anesthetized with chloral hydrate (350 mg/kg of body weight) and perfused transcardially with 0.9% NaCl solution followed by 4% paraformaldehyde in 0.1 M phosphate buffer solution (fixative per animal weight [ml/g]). Brains were removed from skulls and postfixed in the same fixative solution for 2 h. Brain sections were cut on a cryostat. Serial 20-µm-thick coronal sections were obtained and collected in 0.1 M phosphate buffer solution. Brain floating sections obtained were processed either for GFAP or NeuN immunofluorescence.
Stx2 preparation.
The Stx2 toxin was purified as reported previously (23). Briefly, E. coli JM109 transformed with plasmid pGEMT-Stx2 was used to express the Stx2 protein. Supernatants and bacterial lysates were precipitated with ammonium sulfate at 70% saturation, suspended in PBS, and dialyzed against the same buffer for 24 h. The total protein concentration was determined by standard methods. The Stx2 concentration was determined using a Ridascreen verotoxin kit (R-biopharm AG, Darmstadt, Germany). The CD50 (cytotoxic dose that kills 50% of Vero cells) was equivalent to 670 pg of Stx2. One 100% lethal dose (LD100) of Stx2 is 2.2 ng/mouse, and 3 LD100 is 6.6 ng/mouse.
Anti-Stx2 polyclonal antibodies.
BALB/c mice were immunized with Stx2 toxoid. Stx2 toxoid was prepared by formalin treatment of Stx2 (50). Briefly, 100 µg of Stx2 (Phoenix Lab, Tufts University, Boston, MA) was incubated overnight in 2% formalin and then dialyzed extensively against PBS. Mice were immunized biweekly with three injections of 5 µg of Stx2 toxoid emulsified in Freund’s complete or incomplete adjuvant.
Expression and purification of Stx2-based immunogen.
We recently developed a novel immunogen based on the B subunit of Shiga toxin 2 (Stx2B) and the enzyme lumazine synthase from Brucella spp. (BLS) (BLS-Stx2B). This chimera was expressed and purified as previously described (35). Briefly, inclusion bodies containing BLS-Stx2B were solubilized by overnight incubation in 8 M urea–50 mM Tris-HCl–5 mM EDTA, pH 8, buffer and dialyzed against 1 M urea–50 mM Tris-HCl–5 mM EDTA, pH 8.5, buffer. The solubilized proteins were purified by anion-exchange chromatography in a Q-Sepharose (Pharmacia, GE Healthcare Life Sciences) column using a high-performance liquid chromatography (HPLC) apparatus (Gilson model 320). Elution was performed using a linear gradient between 0 and 1 M NaCl in a 1 M urea–50 mM Tris-HCl, pH 8.5, buffer. Protein was dialyzed against PBS previous to each immunization.
BLS-Stx2B immunization protocol.
Adult BALB/c female mice were immunized with 3 doses of BLS-Stx2B, with aluminum hydroxide (subcutaneous), on days 0, 15, and 30. The dose of BLS-Stx2B was equivalent to 20 µg of Stx2B. Females were mated 10 days after the last dose, their pups were bled every 15 days, and Stx2B-specific antibody titers were monitored by ELISA. Stx2B-immunized pups were HBT-pStx2 challenged at 6 weeks postpartum.
Immunofluorescence of Stx2 and GFAP expression.
Immunofluorescence analysis of Stx2 expression localized in the brain was performed according to a previously published procedure (27). Briefly, brain cryostat sections (20 µm) were kept at −20° C in cryopreservant solution (50% of PBS plus 30% ethylene glycol plus 20% glycerol). Sections were incubated in Triton X-100 (1%) in PBS. Sections were first incubated with 10% of fetal goat serum in PBS and then were incubated with polyclonal anti-Stx2 antibody (1/50; 1.35 mg/ml) (51) at 4°C for 48 h. Three washes with Triton X-100 (0.025% in PBS) were done, and then sections were incubated for 1 h with goat IgG anti-rabbit Alexa 555 (1:100) (Sigma, St. Louis, MO, USA) in PBS. After 3 washes with PBS, sections were mounted on glass slides and coverslipped in fluorescence mounting solution. Controls were carried out using the same procedure but omitting the primary anti-Stx2 antibody.
NeuN, GFAP, and Hoescht stain detection.
In parallel, other brain sections were incubated with polyclonal anti-GFAP polyclonal rabbit (Z0334; Dako, Glostrup, Denmark) antibody (or anti-NeuN monoclonal antibody MAB377; Millipore, Temecula, CA, USA) diluted in PBS (10 mM) plus 0.3% of Triton X-100 (1/500) at 4°C for 4 h at room temperature. Three washes with Triton X-100 (0.025% in PBS) were made, and then sections were incubated for 90 min with goat IgG anti-mouse (1:200) (Sigma, St. Louis, MO, USA) in PBS. After 3 washes with PBS, sections were incubated with Hoescht 33342 stain (1 µg/µl) for 30 min. After three washes with PBS, sections were mounted on glass slides and coverslipped in fluorescence mounting solution. Controls were carried out using the same procedure but omitting the primary anti-Stx2 antibody.
Eight sections per treatment were analyzed, and 16 micrographs per section were used to take images. Images were captured from a Zeiss Axiophot microscope (Germany) with the Fluo View application software. Serial optical sections were obtained using a Simple 32 C-imaging computer. Images were taken with a scan zoom of ×1. Adobe Photoshop software was used to assemble the images and obtain merged images.
Statistical analysis.
The significance of the difference between treatments was analyzed using Prism 5.0 software (GraphPad Software), and the P value is indicated by asterisks in the figures.
Survival and frequency data were analyzed for significance using the log rank test and χ2 test. All other data correspond to the means ± standard errors of the means (SEM) for individual mice. Statistical differences were determined using the one-way analysis of variance (ANOVA). Comparisons a posteriori between two groups were performed using Student-Newman-Keuls analysis.
Ethics statement.
Experiments performed herein were approved by the local Institutional Animal Care and Use Committee of Instituto de Medicina Experimental (IMEX), in accordance with the principles set forth in the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, 1985).
ACKNOWLEDGMENTS
This work was supported by PICT 08/417 from the Agencia Nacional de Promoción Científica y Tecnológica, Argentina (to M.S.P.). L.V.B., M.F.B., M.C.R.-G., P.D.G., J.G., and M.S.P. are members of the Research Career of CONICET (Consejo Nacional de Ciência y Tecnología).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank Victoria Campodónico for grammatical correction of the manuscript.
Footnotes
Citation Bentancor LV, Mejías MP, Pinto A, Bilen MF, Meiss R, Rodriguez-Galán MC, Baez N, Pedrotti LP, Goldstein J, Ghiringhelli PD, Palermo MS. 2013. Promoter sequence of Shiga toxin 2 (Stx2) is recognized in vivo, leading to production of biologically active Stx2. mBio 4(5):e00501-13. doi:10.1128/mBio.00501-13.
REFERENCES
- 1. Tarr PI, Gordon CA, Chandler WL. 2005. Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome. Lancet 365:1073–1086 [DOI] [PubMed] [Google Scholar]
- 2. Karmali MA. 1989. Infection by verocytotoxin-producing Escherichia coli. Clin. Microbiol. Rev. 2:15–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karmali MA, Steele BT, Petric M, Lim C. 1983. Sporadic cases of haemolytic-uraemic syndrome associated with faecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet i:619–620 [DOI] [PubMed] [Google Scholar]
- 4. Palermo MS, Exeni RA, Fernández GC. 2009. Hemolytic uremic syndrome: pathogenesis and update of interventions. Expert Rev. Anti Infect. Ther. 7:697–707 [DOI] [PubMed] [Google Scholar]
- 5. Kaper JB, Nataro JP, Mobley HL. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2:123–140 [DOI] [PubMed] [Google Scholar]
- 6. Herold S, Karch H, Schmidt H. 2004. Shiga toxin-converting bacteriophages. Genomes in motion. Int. J. Med. Microbiol. 294:115–121 [DOI] [PubMed] [Google Scholar]
- 7. Wagner PL, Neely MN, Zhang X, Acheson DW, Waldor MK, Friedman DI. 2001. Role for a phage promoter in Shiga toxin 2 expression from a pathogenic Escherichia coli strain. J. Bacteriol. 183:2081–2085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shaikh N, Tarr PI. 2003. Escherichia coli O157:H7 Shiga toxin-encoding bacteriophages: integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 185:3596–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laing CR, Zhang Y, Gilmour MW, Allen V, Johnson R, Thomas JE, Cannon VPJ. 2013. A comparison of Shiga. toxin 2 bacteriophage from classical enterohemorrhagic Escherichia coli serotypes and the German E. coli O104:H4 outbreak strain. Plos One 7:e37362. 10.1371/journal.pone.0037362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kimmitt PT, Harwood CR, Barer MR. 2000. Toxin gene expression by Shiga toxin-producing Escherichia coli: the role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 6:458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmidt H. 2001. Shiga-toxin-converting bacteriophages. Res. Microbiol. 152:687–695 [DOI] [PubMed] [Google Scholar]
- 12. Cornick NA, Helgerson AF, Mai V, Ritchie JM, Acheson DW. 2006. In vivo transduction of an Stx-encoding phage in ruminants. Appl. Environ. Microbiol. 72:5086–5088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Imamovic L, Jofre J, Schmidt H, Serra-Moreno R, Muniesa M. 2009. Phage-mediated Shiga toxin 2 gene transfer in food and water. Appl. Environ. Microbiol. 75:1764–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Strauch E, Lurz R, Beutin L. 2001. Characterization of a Shiga toxin-encoding temperate bacteriophage of Shigella sonnei. Infect. Immun. 69:7588–7595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Sablet T, Chassard C, Bernalier-Donadille A, Vareille M, Gobert AP, Martin C. 2009. Human microbiota-secreted factors inhibit Shiga toxin synthesis by enterohemorrhagic Escherichia coli O157 H7. Infect. Immun. 77:783–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thorpe CM, Smith WE, Hurley BP, Acheson DW. 2001. Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 69:6140–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bentancor LV, Bilen MF, Mejías MP, Fernández-Brando RJ, Panek CA, Ramos MV, Fernández GC, Isturiz M, Ghiringhelli PD, Palermo MS. 2013. Functional capacity of Shiga-toxin promoter sequences in eukaryotic cells. PLoS One 8:e57128. 10.1371/journal.pone.0057128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu F, Song Y, Liu D. 1999. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6:1258–1266 [DOI] [PubMed] [Google Scholar]
- 19. Liu D, Knapp JE. 2001. Hydrodynamics-based gene delivery. Curr. Opin. Mol. Ther. 3:192–197 [PubMed] [Google Scholar]
- 20. Xiao J, Jethanandani P, Ziober BL, Kramer RH. 2003. Regulation of alpha7 integrin expression during muscle differentiation. J. Biol. Chem. 278:49780–49788 [DOI] [PubMed] [Google Scholar]
- 21. Rodriguez-Galán MC, Bream JH, Farr A, Young HA. 2005. Synergistic effect of IL-2, IL-12, and IL-18 on thymocyte apoptosis and Th1/Th2 cytokine expression. J. Immunol. 174:2796–2804 [DOI] [PubMed] [Google Scholar]
- 22. Chen TH, Yeh CT, Ho YP, Hsu CM, Huang CC, Shiau SS, Liang CK, Chang ML. 2009. Hydrodynamics-based transfection of pancreatic duodenal homeobox 1 DNA improves hyperglycemia and is associated with limited complications in diabetic mice. Endocr. J. 56:783–790 [DOI] [PubMed] [Google Scholar]
- 23. Bentancor LV, Bilen M, Brando RJ, Ramos MV, Ferreira LC, Ghiringhelli PD, Palermo MS. 2009. A DNA vaccine encoding the enterohemorragic Escherichia coli Shiga-like toxin 2 A2 and B subunits confers protective immunity to Shiga toxin challenge in the murine model. Clin. Vaccine Immunol. 16:712–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H. 1985. The association between idiopathic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 151:775–782 [DOI] [PubMed] [Google Scholar]
- 25. Fernandez GC, Lopez MF, Gomez SA, Ramos MV, Bentancor LV, Fernandez-Brando RJ, Landoni VI, Dran GI, Meiss R, Isturiz MA, Palermo MS. 2006. Relevance of neutrophils in the murine model of haemolytic uraemic syndrome: mechanisms involved in Shiga toxin type 2-induced neutrophilia. Clin. Exp. Immunol. 146:76–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brando RJ, Miliwebsky E, Bentancor L, Deza N, Baschkier A, Ramos MV, Fernández GC, Meiss R, Rivas M, Palermo MS. 2008. Renal damage and death in weaned mice after oral infection with Shiga toxin 2-producing Escherichia coli strains. Clin. Exp. Immunol. 153:297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tironi-Farinati C, Loidl CF, Boccoli J, Parma Y, Fernandez-Miyakawa ME, Goldstein J. 2010. Intracerebroventricular Shiga toxin 2 increases the expression of its receptor globotriaosylceramide and causes dendritic abnormalities. J. Neuroimmunol. 222:48–61 [DOI] [PubMed] [Google Scholar]
- 28. Eng LF, Ghirnikar RS. 1994. GFAP and astrogliosis. Brain Pathol. 4:229–237 [DOI] [PubMed] [Google Scholar]
- 29. Landoni VI, Schierloh P, de Campos Nebel M, Fernández GC, Calatayud C, Lapponi MJ, Isturiz MA. 2012. Shiga toxin 1 induces on lipopolysaccharide-treated astrocytes the release of tumor necrosis factor-alpha that alters brain-like endothelium integrity. PLoS Pathog. 8:e1002632. 10.1371/journal.ppat.1002632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boccoli J, Loidl CF, Lopez-Costa JJ, Creydt VP, Ibarra C, Goldstein J. 2008. Intracerebroventricular administration of Shiga toxin type 2 altered the expression levels of neuronal nitric oxide synthase and glial fibrillary acidic protein in rat brains. Brain Res. 1230:320–333 [DOI] [PubMed] [Google Scholar]
- 31. Paetau A, Elovaara I, Paasivuo R, Virtanen I, Palo J, Haltia M. 1985. Glial filaments are a major brain fraction in infantile neuronal ceroid-lipofuscinosis. Acta Neuropathol. 65:190–194 [DOI] [PubMed] [Google Scholar]
- 32. Fujii J, Kita T, Yoshida S, Takeda T, Kobayashi H, Tanaka N, Ohsato K, Mizuguchi Y. 1994. Direct evidence of neuron impairment by oral infection with verotoxin-producing Escherichia coli O157:H-7 in mitomycin-treated mice. Infect. Immun. 62:3447–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goldstein J, Loidl CF, Creydt VP, Boccoli J, Ibarra C. 2007. Intracerebroventricular administration of Shiga toxin type 2 induces striatal neuronal death and glial alterations: an ultrastructural study. Brain Res. 1161:106–115 [DOI] [PubMed] [Google Scholar]
- 34. Robertson CL, Puskar A, Hoffman GE, Murphy AZ, Saraswati M, Fiskum G. 2006. Physiologic progesterone reduces mitochondrial dysfunction and hippocampal cell loss after traumatic brain injury in female rats. Exp. Neurol. 197:235–243 [DOI] [PubMed] [Google Scholar]
- 35. Mejias MP, Ghersi G, Craig PO, Panek CA, Bentancor LV, Baschkier A, Goldbaum FA, Zylberman V, Palermo MS. 2013. Immunization with a chimera between the B subunit of Shiga toxin type 2 and Brucella lumazine synthase confers total protection against Shiga toxins. J. Immunol. 191:2403–2411 [DOI] [PubMed] [Google Scholar]
- 36. Zhang X, McDaniel AD, Wolf LE, Keusch GT, Waldor MK, Acheson DW. 2000. Quinolone antibiotics induce Shiga toxin encoding bacteriophages, toxin production, and death in mice. J. Infect. Dis. 181:664–670 [DOI] [PubMed] [Google Scholar]
- 37. Imamovic L, Muniesa M. 2012. Characterizing RecA-independent induction of shigatoxin 2-encoding phages by EDTA treatment. PLoS One 7:e32393. 10.1371/journal.pone.0032393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guo ZS, Wang LH, Eisensmith RC, Woo SL. 1996. Evaluation of promoter strength for hepatic gene expression in vivo following adenovirus-mediated gene transfer. Gene Ther. 3:802–810 [PubMed] [Google Scholar]
- 39. Karmali MA. 2004. Infection by Shiga toxin-producing Escherichia coli: an overview. Mol. Biotechnol. 26:117–122 [DOI] [PubMed] [Google Scholar]
- 40. Cimolai N, Carter JE. 1998. Bacterial genotype and neurological complications of Escherichia coli O157:H7-associated haemolytic uraemic syndrome. Acta Paediatr. 87:593–594 [DOI] [PubMed] [Google Scholar]
- 41. Eriksson KJ, Boyd SG, Tasker RC. 2001. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch. Dis. Child. 84:434–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sheth KJ, Swick HM, Haworth N. 1986. Neurologic involvement in hemolytic uremic syndrome. Ann. Neurol. 19:90–99 [DOI] [PubMed] [Google Scholar]
- 43. Hahn JS, Havens PL, Higgins JJ, O’Rourke PP, Estroff JA, Strand R. 1989. Neurological complications of hemolytic uremic syndrome. J. Child Neurol. 4:108–113 [DOI] [PubMed] [Google Scholar]
- 44. Karch H, Denamur E, Dobrindt U, Finlay BB, Hengge R, Johannes L, Ron EZ, Tonjum T, Sansonetti PJ, Vincete M. 2012. The enemy within us: lessons from the 2011 European Escherichia coli O104:H4 outbreak. EMBO Mol. Med. 4:841–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petruzziello-Pellegrini TN, Marsden PA. 2012. Shiga toxin-associated hemolytic uremic syndrome: advances in pathogenesis and therapeutics. Curr. Opin. Nephrol. Hypertens. 21:433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Milford D, Taylor CM, Rafaat F, Halloran E, Dawes J. 1989. Neutrophil elastases and haemolytic uraemic syndrome. Lancet ii:1153. [DOI] [PubMed] [Google Scholar]
- 47. Fitzpatrick MM, Shah V, Filler G, Dillon MJ, Barratt TM. 1992. Neutrophil activation in the haemolytic uraemic syndrome: free and complexed elastase in plasma. Pediatr. Nephrol. 6:50–53 [DOI] [PubMed] [Google Scholar]
- 48. Unal-Cevik I, Kilinç M, Gürsoy-Ozdemir Y, Gurer G, Dalkara T. 2004. Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res. 1015:169–174 [DOI] [PubMed] [Google Scholar]
- 49. Capozzo AV, Pistone Creydt V, Dran G, Fernández G, Gómez S, Bentancor LV, Rubel C, Ibarra C, Isturiz M, Palermo MS. 2003. Development of DNA vaccines against hemolytic-uremic syndrome in a murine model. Infect. Immun. 71:3971–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mukherjee J, Chios K, Fishwild D, Hudson D, O’Donnell S, Rich SM, Donohue-Rolfe A, Tzipori S. 2002. Human Stx2-specific monoclonal antibodies prevent systemic complications of Escherichia coli O157:H7 infection. Infect. Immun. 70:612–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parma YR, Chacana PA, Lucchesi PM, Rogé A, Granobles Velandia CV, Kruger A, Parma AE, Fernandez-Miyakawa ME. 2012. Detection of Shiga toxin-producing Escherichia coli by sandwich enzyme-linked immunosorbent assay using chicken egg yolk IgY antibodies. Front Cell. Infect Microbiol. 2:84. 10.3389/fcimb.2012.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]