

Abstract
BACKGROUND AND PURPOSE
Endocannabinoids such as anandamide (AEA) are important lipid ligands regulating cell proliferation, differentiation and apoptosis. Their levels are regulated by hydrolase enzymes, the fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MGL). Here, we investigated whether FAAH or AEA are involved in NF (erythroid-derived 2)-like 2 (Nrf2)/antioxidant responsive element (ARE) pathway.
EXPERIMENTAL APPROACH
The aim of this study was to analyse the effects of AEA or FAAH inhibition by the URB597 inhibitor or FAAH/siRNA on the activation of Nrf2-ARE signalling pathway and heme oxygenase-1 (HO-1) induction and transcription.
KEY RESULTS
Endogenous AEA was detected in the immortalized human mammary epithelial MCF-10A cells (0.034 ng per 106 cells) but not in MCF-7 or MDA-MB-231 breast cancer cells. Because breast tumour cells express FAAH abundantly, we examined the effects of FAAH on Nrf2/antioxidant pathway. We found that inhibition of FAAH by the URB597 inhibitor induced antioxidant HO-1 in breast cancer cells and MCF-10A cells. RNAi-mediated knockdown of FAAH or treatment with AEA-activated ARE-containing reporter induced HO-1 mRNA and protein expression, independent of the cannabinoid receptors, CB1, CB2 or TRPV1. Furthermore, URB597, AEA and siRNA-FAAH treatments induced the nuclear translocation of Nrf2, while siRNA-Nrf2 treatment and Keap1 expression blocked AEA, URB597 and si-FAAH from activation of ARE reporter and HO-1 induction. siRNA-HO-1 treatment decreased the viability of breast cancer cells and MCF-10A cells.
CONCLUSIONS AND IMPLICATIONS
These data uncovered a novel mechanism by which inhibition of FAAH or exposure to AEA induced HO-1 transcripts and implicating AEA and FAAH as direct modifiers in signalling mediated activation of Nrf2-HO-1 pathway, independent of cannabinoid receptors.
Keywords: endocannabinoids, oxidative stress, FAAH enzyme, Nrf2, breast cancer, heme oxygenase 1
Introduction
The endocannabinoid system is a complex endogenous signalling system, composed of transmembrane endocannabinoid receptors (the cannabinoid receptors, CB1 and CB2), their endogenous ligands (the endocannabinoids), the proteins involved in endocannabinoid synthesis and inactivation, as well as the intracellular signalling pathways affected by endocannabinoids (Bisogno et al., 2005). Anandamide (AEA) and 2-arachidonoylglycerol (2-AG) are the two best-characterized endocannabinoids identified to date. Accumulating evidence indicated that the endocannabinoid system regulates multiple physiological and pathological conditions, such as food intake, immunomodulation, inflammation, analgesia, addictive behaviour, epilepsy, pain, glaucoma, cancer and neurodegenerative disorders such as Parkinson's disease and multiple sclerosis (Alpini and Demorrow, 2009; Guindon and Hohmann, 2009). Cannabinoids also inhibit cancer cell proliferation, reduce angiogenesis, cell migration and metastasis, and inhibit carcinogenesis and attenuate inflammatory processes (Oesch and Gertsch, 2009). MDA-MB-231 and MCF-7 cells express CB1 and CB2 receptors (Qamri et al., 2009; Nasser et al., 2011). Studies in cancer biology have shown that AEA exerts anti-proliferative effects on various cancer types via receptor-dependent and receptor-independent manners (Alpini and Demorrow, 2009). AEA has been demonstrated to inhibit human breast cancer cell proliferation (De Petrocellis et al., 1998; Melck et al., 1999; Laezza et al., 2006), adhesion and migration (Grimaldi et al., 2006) in CB1 receptor-dependent manner. In comparison, growth-promoting effects of AEA have also been reported in several types of cancer cell lines (Hart et al., 2004). Thus, the specific roles of AEA in tumourogenesis are not clear, and why AEA has opposing effects on tumour cells, and whether these AEA effects are mediated by activation of cannabinoid receptors.
The endocannabinoid ligands exert functions mainly through binding and activation of cannabinoid receptors (McAllister and Glass, 2002). However, other signalling pathways, such as vanilloid receptors (TRPV1) (Di Marzo et al., 2001), PPARs (O'Sullivan, 2007), reactive oxygen species (ROS) signalling and the intracellular Ca2+ signalling (Siegmund et al., 2005; 2006), have also been reported to be activated by AEA. Therefore, it is not known which signalling pathway(s) are affected by the increased activities of AEA in breast cancer cells.
Recent study on comparative gene profiling of gene expression in BV-2 microglial cells treated with either non-psychoactive cannabidiol (CBD) or with psychoactive cannabinoid Δ9-tetrahydrocannabinol (THC) showed differential transcriptional profiles by THC and CBD (Juknat et al., 2012). Specifically, THC and lower levels of CBP induced cellular stress response involving GCN2/e1F2α/p8/ATF-4/CHOP-TR1B3 pathway, which suggested that this effect could underlie its anti-inflammatory activity (Juknat et al., 2012).
Fatty acid amide hydrolase (FAAH) is an integral membrane protein that hydrolyses bioactive amides, such as endocannabinoid AEA, to form free fatty acid and ethanolamine. Genetic or chemical inhibition of FAAH has shown that blocking the degradation pathway results in an increase in AEA levels (Alpini and Demorrow, 2009; Clapper et al., 2009). URB597 is widely regarded as a specific FAAH inhibitor (Kathuria et al., 2003; Piomelli et al., 2006). In pre-clinical laboratory tests, URB597 increased the production of endocannabinoid AEA, resulting in measurable antidepressant and analgesic effects (Gobbi et al., 2005; Clapper et al., 2009). Thus, inhibition of AEA deactivation is considered to be useful in decreasing the susceptibility of cannabis dependence (Clapper et al., 2009). Although TCH and CBD were shown to be involved in cellular stress response (Juknat et al., 2012), it is not clear if FAAH and AEA are also able to induce antioxidant response.
Transcriptional activation of protective genes is mediated by a cis-acting element called the antioxidant response element (ARE). The transcription factor NF-E2-related factor 2 (Nrf2) binds to the ARE and activate this pathway to protect cells from oxidative stress-induced cell death (Kobayashi and Yamamoto, 2005; Kobayashi and Tong, 2006). Nrf2 has been demonstrated to be a key transcriptional factor that activates heme oxygenase-1 (ho-1) gene through the ARE (Martin et al., 2004; Kobayashi and Yamamoto, 2005; Lee and Surh, 2005; Kobayashi and Tong, 2006). In the presence of stimuli, Nrf2 is dissociated from its cytosolic sequestering protein Keap1, leading to subsequent translocation into the nucleus, thereby activating ARE (Kobayashi and Tong, 2006). PKC, PI3K and MAPK (p38, ERK1/2 and JNK) have been shown to regulate Nrf2-ARE activity in the presence of various stimuli (Martin et al., 2004; Kobayashi and Yamamoto, 2005; Lee et al., 2005). Heme oxygenase (HO) is a rate-limiting enzyme of heme catabolism (Maines, 1988). At least three mammalian HO isoforms have been identified. HO-1 is a stress-responsive protein induced by heat, heavy metals, cytokines, endotoxin, oxidants and heme. Compelling evidence revealed that induction of HO-1 protect against a variety of stress conditions such as hydrogen peroxide, cisplatin, UV irradiation and inflammatory cytokine-mediated cell damages (Ewing et al., 2005; Lin et al., 2005; 2007; Kim et al., 2006). In addition, the therapeutic roles of HO-1 in inflammatory and cardiovascular diseases have been proposed (Pae and Chung, 2009; Wang et al., 2009). However, HO-1 has been shown to have anti-apoptotic and pro-angiogenic effects in murine cancer models, such as colon (Fang et al., 2003), haepatoma (Tanaka et al., 2003) and in human pancreatic cancer (Sunamura et al., 2003). HO-1 is usually highly expressed in tumours and can be further increased by many cancer chemopreventive agents (Jozkowicz et al., 2007). In addition, HO-1 expression renders cancer cells growth advantage and cellular resistance against chemotherapy and photodynamic therapy (Fang et al., 2004; Nowis et al., 2006). In contrast, the beneficial roles of HO-1 in inhibiting human breast cancer cell proliferation and invasion have been reported (Hill et al., 2005; Lin et al., 2008). Thus, HO-1 expression may play important roles in normal physiology and pathophysiology of various diseases.
Given that endocannabinoid system regulates multiple physiological and pathological conditions, including inflammation and redox status (Siegmund et al., 2005, 2006; Guindon and Hohmann, 2009), we investigated the relationship between AEA signalling and antioxidants in breast cancer cells. Here, we show that AEA and inhibition of FAAH activate Nrf2 transcription factor, leading to an induction of HO-1 in breast cancer cells, independent of cannabinoid receptors. Depletion of HO-1 resulted in a decrease in cell number in breast cancer cells. Our results suggest that AEA signalling-mediated activation of Nrf2-HO-1 pathway could favour the survival of breast cancer cells.
Methods
Cell culture
MCF-7 and MDA-MB-231 breast cancer cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were maintained in RPMI1640 medium supplemented with 10 % FBS, penicillin/streptomycin and glutamine, in 5% CO2 incubator at 37°C. MCF-10A immortalized human breast epithelial cells were provided by Dr Harikrishna Nakshatri (Indiana University School of Medicine) and cultured in DMEM/F-12 1:1 medium supplemented with 10% FBS, penicillin/streptomycin, insulin, hydrocortisone and EGF, in 5% CO2 incubator at 37°C.
Chemicals, URB597, AEA and N-acetyl cysteine (NAC) were purchased from Sigma (St. Louis, MO, USA). AM251, AM630, THC, CP55940 and R(+)-methanandamide were purchased from Tocris (Ellisville, MO, USA). Capsazepine, oleoylethylamide (OEA) and PF-622 were obtained from Cayman Chemical (Ann Arbor, MI, USA). The chemicals were dissolved in dimethyl sulfoxide (DMSO) or ethanol, and the working solutions for cellular treatment were diluted more than 3000 times.
Anti-HO-1 antibody was purchased from Enzo (Plymouth Meeting, PA, USA). Anti-NQO-1 antibody and anti-Nrf2 polyclonal antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-FAAH antibody was purchased from Cell Signaling (Danvers, MA, USA). Anti-actin antibody and anti-lamin B1 monoclonal antibody were obtained from EMD (Gibbstown, NJ, USA) and Invitrogen (Carlsbad, CA, USA) respectively.
Short interfering (si) RNA transfection
The HiPerFect transfection reagent (QIAGEN, Valencia, CA, USA) was used to deliver siRNA against Nrf2, FAAH, GFP, GL2 and HO-1 into cells. HO-1 FlexiTube GeneSolution siRNA, GL2 and GFP-22 siRNA (Cat. No. 1022064) were purchased from QIAGEN. FAAH siRNA (sc-106807) was obtained from Santa Cruz Biotechnology. siGENOME SMARTpool of human Nrf2 (Cat. No. M-003755-02) was purchased from Thermo Scientific Dharmacon RNAi Technologies (Lafayette, CO, USA).
Luciferase assay
Lipofectamine Plus reagent (Invitrogen) was used to deliver plasmids into mammalian cells. The nqo1-ARE-Luc promoter reporter (Nioi et al., 2003) was provided by Drs Ken Itoh and Masayuki Yamamoto (Center for Tsukuba Advanced Research Alliance and Institute of Basic Medical Sciences, University of Tsukuba, Tsukuba, Japan) and pCMV-β-galactosidase (Clontech, Mountain View, CA, USA) was co-transfected to monitor the transfection efficiency. After transfection with the reporter plasmid and pCMV-β-galactosidase, the cells were treated with URB597 or AEA overnight, followed by luciferase activity assay. siRNAs against Nrf2, FAAH and control GFP were transfected into cells 1 day before the reporter or pCMV-β-galactosidase plasmids were transfected. Keap1 plasmid (Furukawa and Xiong, 2005) was co-expressed with the nqo1-ARE-Luc reporter and pCMV-β-galactosidase in order to determine the effect of Keap1 on the nqo1 promoter activity. The luciferase activity was determined by using luciferase assay kit (Promega, Madison, WI, USA) and normalized by β-galactosidase activity (Promega). The fold increase of luciferase activity was obtained from at least three independent experiments performed in triplicate. Standard deviation was calculated.
Isolation of the nuclear proteins
The cytoplasm and nuclear fractionation kit (Thermo Scientific Pierce, Rockford, IL, USA) were used to separate the nuclear proteins.
SDS-PAGE and Western blotting
In some experiments, total proteins were lysed in 2× Laemmli buffer (125 mM Tris–HCl, 4% SDS, 20% glycerol), followed by boiling for 5 min. Protein assay kit (Bio-Rad, Hercules, CA, USA) was used to determine protein concentration. The nuclear fraction was diluted in 2× Laemmli buffer before being applied to SDS-PAGE. After SDS-PAGE, proteins were transferred to PVDF membrane (Millipore). The membranes were blocked with 5% milk in PBS buffer containing 0.1% Tween 20 for 2 h at room temperature. Then, the membranes and first antibodies diluted with 5% milk in PBS buffer containing 0.1% Tween 20 were incubated for 1–2 h at room temperature or overnight at 4°C. The secondary antibodies conjugated with HRP (GE Healthcare, Piscataway, NJ, USA) (diluted 6000 times) were incubated with membranes for 1 h at room temperature.
Reverse transcriptase (RT)-PCR
Total RNA was isolated by using Trizol reagent (Invitrogen). Furthermore, 20–50 ng of total RNA was used for RT-PCR. Human HO-1 forward primer sequence is 5′-GAGACGGCTTCAAGCTGGTGATG-3′; human HO-1 reverse primer sequence is 5′-GTTGAGCAGGAACGCAGTCTTGG-3′. Human NQO-1 forward primer sequence is 5-AGGCTGGTTTGAGCGAGT-3; NQO-1 reverse primer sequence is 5-ATTGAATTCGGGCGTCTGCTG-3. Human actin forward primer sequence is 5-GCTCGTCGTCGACAACGGCTC-3; actin reverse primer sequence is 5-AAACATGATCTGGGTCATCTTCTC-3. The One-step RT-PCR kit (QIAGEN) was applied to perform semi-quantitative RT-PCR. The One-step RT-PCR program is as follows: 1 cycle of 50°C for 30 min, followed by 1 cycle of 94°C for 15 min, and 27 cycles of 94°C for 0.5 min, 50°C for 0.5 min, 72°C for 2 min, and 1 cycle of 72°C for 10 min then 4°C.
Real-time RT-PCR
In addition, 1 ng of total RNA was used to analyse HO-1 mRNA levels. The reactions were carried out using QuantiFast one-step SYBR Green RT-PCR kit in Applied Biosystems 7300 Real-Time PCR System. HO-1 primers (QT00092645) and β-actin primers (QT00095431) were obtained from QIAGEN.
Crystal violet cell viability assay
Furthermore, (3–4) × 104 cells seeded in 24 well plates were treated with siRNA against GL2, HO-1 or Nrf2. After 3 days, the living cells were stained with crystal violet and the OD570 was determined. Changes in cell numbers were obtained from comparison of OD570 between vehicle-treated cells and si-HO-1- or Nrf2-treated cells.
Endocannabinoid levels in treated breast cancer cells
Endocannabinoid levels in breast cancer cells (MCF-10A cells) were analysed as described (Folch et al., 1957; Williams et al., 2007). The cells were grown in culture for 24 h, collected and washed with PBS. Sample size was 106 cells for analysis. The extraction procedure for the calibration standards, quality control samples in BSA and cell pellets was a modified version of the Folch extraction (Folch et al., 1957; Williams et al., 2007). The cell samples were homogenized in ice-cold acetone : PBS, pH 7.4 (3:1) and internal standard with a handheld homogenizer followed by sonication for 1 min to further disrupt the cells. All samples were centrifuged at 14 000× g for 5 min at 4°C. The resulting supernatants were dried under nitrogen until the acetone was removed. To the remaining supernatant, 100 μL PBS, 1 vol. methanol and 2 vol. chloroform were added for liquid–liquid phase extraction of the lipids. The two phases were separated by centrifugation and the bottom organic layer was evaporated to dryness under nitrogen. Samples were reconstituted in 50 μL of ethanol, vortexed and sonicated briefly, and centrifuged prior to analysis.
Chromatographic separation was then achieved using an Agilent Zorbax SB-CN column (2.1 × 50 mm, 5 mm) on a Finnigan TSQ Quantum Ultra triple quad mass spectrometer (Thermo Electron, San Jose, CA, USA) with an Agilent 1100 HPLC on the front end (Agilent Technologies, Wilmington, DE, USA) as previously described (Williams et al., 2007). The mobile phase consisted of 10 mM ammonium acetate, pH 7.3 (A), and methanol (B) in a flow rate of 0.5 mL min−1; the autosampler was kept at 4°C to prevent analyte degradation. Eluted peaks were ionized via atmospheric pressure chemical ionization in MRM mode. Deuterated internal standards were used for each analyte's standard curves and their levels per 106 cells were determined.
Statistical analysis
Data are reported as mean ± SEM. The Student's t-test and one-way anova test were used to assess the significance of independent experiments. P < 0.05 was used as the criterion to determine statistical significance.
Results
Endocannabinoid levels in breast cancer cells
First, we examined the endogenous levels of endocannabinoids in breast cancer cells. Standard curves for each endocannabinoid were linear with a regression value of ≥0.996. The extraction efficiencies for the quality controls in BSA compared to those in ethanol were greater than 90%. AEA and EEA were only detected in MCF-10A cells (Table 1) at low levels of 0.034 and 0.336 ng per 106 cells. PEA was detected in MCF-10A cells, as well as MDA-MB-231 cells and MCF-7 cells. Additional substrates of FAAH, such as DHEA and EPEA, were not detected in any of the cell lines (Table 1). Although breast cancer cells do not express AEA, these cells express FAAH enzymes abundantly as reported (Di Marzo et al., 2001; Egertova et al., 2004) and FAAH can be induced in these cells in response to oxidative stress (Wei et al., 2009).
Table 1.
Endocannabinoid ethanolamide levels in breast cancer cells and MCF-10A cells
| Cell lines | AEA | PEA | OEA | DHEA | EPEA | EEA |
|---|---|---|---|---|---|---|
| MCF-10A | 0.034 | 0.336 | 0.061 | ND | ND | 0.048* |
| MCF-7 | ND | 0.078 | 0.036 | ND | ND | ND |
| MDA-MB-231 | ND | 0.117 | 0.082 | ND | ND | ND |
Cells (106 cells per group) were collected and analysed for endocannabinoid ethanolamide levels. Values are in ng per 106 cells. ND indicates that the analyte was not detected in the sample. Starred values (*) were quantified lower than standard curve.
Inhibition of FAAH by URB597 induces antioxidants HO-1 in breast cancer cells
In an attempt to investigate the action of URB597 in breast cancer cells, we found that URB597 induces the levels of antioxidants HO-1 and NQO-1 in MCF-7 and MDA-MB-231 cells. As antioxidant enzymes play important roles in a wide variety of physiological and pathological processes, we investigated the mechanism of antioxidant induction through inhibition of FAAH. Induction of HO-1 and NQO-1 mRNA was observed after 6 h of treatment with 5 μM of URB597 in MCF-7 cells and MDA-MB-231 cells, as demonstrated by RT-PCR (Figure 1A). An increase in the levels of HO-1 and NQO-1 proteins was observed in these cells upon URB597 treatment (Figure 1B). Quantitative RT-PCR assay for HO-1 mRNA showed that URB597 increased HO-1 mRNA levels by 1.40 in MCF7 cells (Supporting Information Figure S1A,B). To examine whether HO-1 and NQO-1 were regulated by URB597 at transcriptional levels, we analysed the activation of the ARE sequence-containing nqo1 promoter-driven luciferase reporter (nqo1-ARE-Luc) (Nioi et al., 2003), and HO-1 promoter-driven luciferase reporter in the presence of URB597. URB597 treatment (5 and 10 μM) activated the nqo1-ARE-Luc in MCF-7 and MDA-MB-231 cells in a dose-dependent manner (Figure 1C). URB597 also activated the HO-1 promoter reporter in MCF-7 cells and MDA-MB-231 cells (data not shown). We also demonstrated that the alternative FAAH inhibitor PF-622 also induced HO-1 protein expression in MCF-7 cells after 24 h of treatment (Supporting Information Figure S2). Thus, these data suggest that inhibition of FAAH by chemical inhibitor URB597 induces antioxidants HO-1 and NQO-1 expression at transcriptional levels in breast cancer cells. Because HO-1 has been implicated in the reduction of free radical formation, inflammation and tumour cell resistance to chemotherapy, we then focused on the mechanism by which URB597 regulates the transcription of HO-1 in breast cancer cells.
Figure 1.
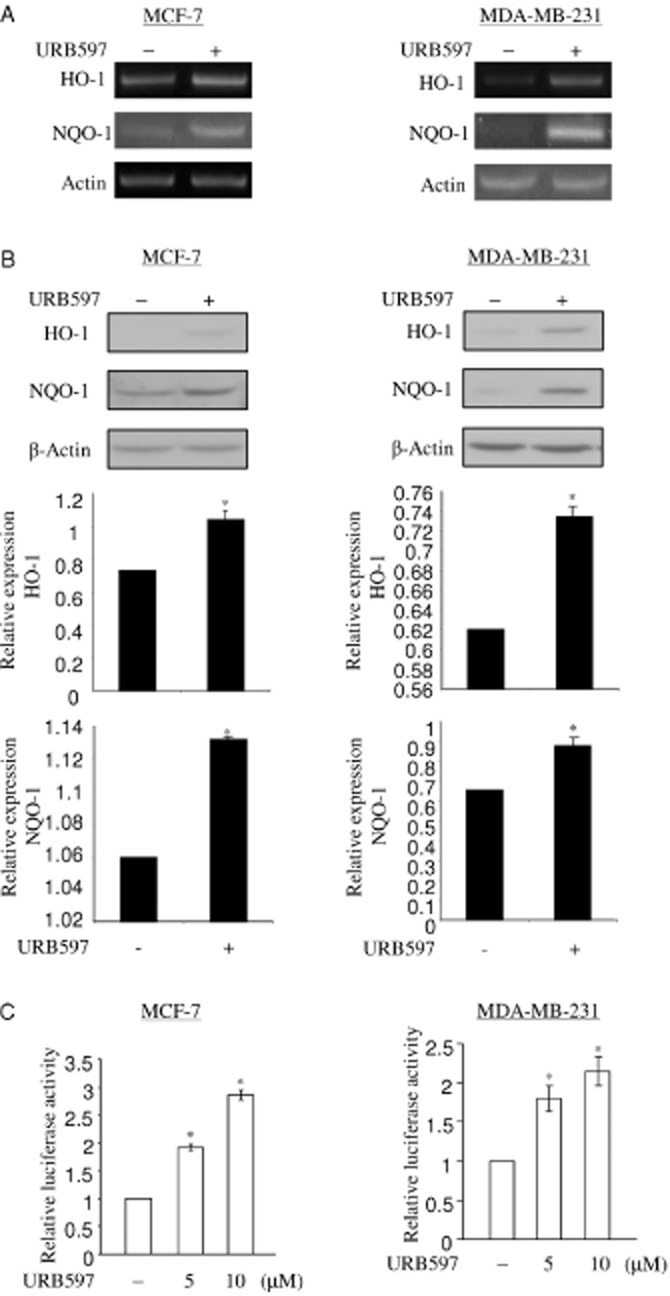
URB597 induces HO-1 and NQO-1 transcripts in breast cancer cell lines. (A) URB597 increases mRNA levels of HO-1 and NQO-1. Furthermore, 50 ng of total RNA of MCF-7 cells or MDA-MB-231 cells treated with vehicle (DMSO) and URB597 (5 μM) for 6 h was subjected to semi-quantitative RT-PCR to determine the levels of HO-1 and NQO-1 mRNA. β-Actin was used as a control for semi-quantitative RT-PCR. (B) URB597 increases protein levels of HO-1 and NQO-1. 25 μg of total protein extract of MCF-7 cells or MDA-MB-231 cells treated with vehicle (DMSO) and URB597 (5 μM) for 16 h was subjected to detect the levels of HO-1 and NQO-1 protein. Equal loading of proteins was demonstrated by anti-actin antibody. The levels of HO-1 and NQO-1 expression were normalized to actin expression and the relative expression was represented graphically. All the figures are representative of three independent experiments. (C) URB597 activates nqo1-ARE-Luc reporter in MCF-7 cells and MDA-MB-231 cells. In 6 well plates, MCF-7 cells or MDA-MB-231 cells were co-transfected with 0.5 μg of nqo1-ARE-Luc reporter and 0.15 μg of pCMV-β-galactosidase plasmids, followed by DMSO (vehicle) and URB597 (5 and 10 μM) treatment for 16 h. URB597-induced luciferase activity was normalized by β-galactosidase activity. The induction of reporter activation was represented as fold increase with respect to the vehicle-treated control. The data represent the mean ± SD of at least three independent experiments performed in triplicate. *P < 0.05 as compared with the control.
AEA activates HO-1 transcription
To analyse whether URB597 acts on HO-1 transcription via AEA, we assessed HO-1 transcripts in the presence of AEA. Addition of 2.5 μM of AEA increased the expression levels of HO-1 mRNA and protein in MCF-7 and MDA-MB-231 cells (Figure 2A). Because the results are similar in both cell lines, we will present data obtained from MCF-7 cells only as detailed below. Quantitative RT-PCR assay for HO-1 mRNA also showed that AEA treatment increases HO-1 mRNA levels by 1.88-fold in MCF7 cells (Supporting Information Figure S1A,B). AEA activated the nqo1-ARE-Luc reporter in MCF-7 cells (Figure 2B) in a dose-dependent manner, suggesting that AEA activates HO-1 transcription. Addition of 1–5 μM of 2-AG, another endocannabinoid ligand, could not activate the basal activity of nqo1-ARE-Luc reporter in MCF-7 cells (Figure 2B). This suggests that AEA specifically regulates HO-1 and NQO-1 transcription. The nqo1-ARE-Luc reporter assay was further performed to investigate the effects of other cannabinoid ligands.
Figure 2.
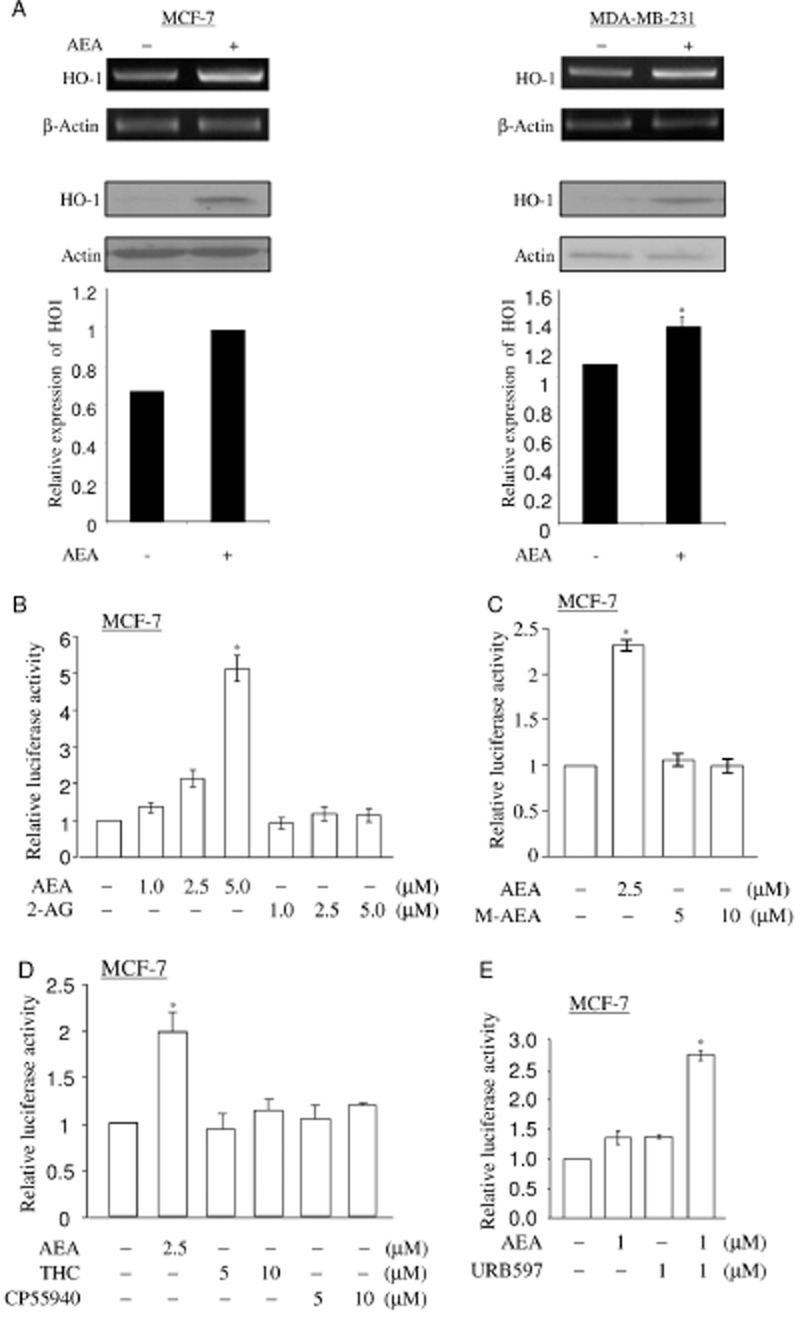
AEA induces HO-1 transcription. (A) AEA induces HO-1 expression. MCF-7 cells and MDA-MB-231 cells were treated with 2.5 μM of AEA for 6 h. The mRNA levels of HO-1 were determined by semi-quantitative RT-PCR as in Figure 1A. Total lysates of MCF-7 cells or MDA-MB-231 cells treated with vehicle (DMSO) and AEA (2.5 μM) for 16 h were applied to detect the levels of HO-1 protein. The levels of HO-1 expression were normalized to actin expression and the relative expression was represented graphically. All figures are representative of three independent experiments. (B) AEA, but not 2-AG, activates nqo1-ARE-Luc reporter in a dose-dependent manner. MCF-7 cells co-transfected with reporter (0.5 μg) and pCMV-β-galactosidase (0.15 μg) were treated with increasing doses (1, 2.5 and 5.0 μM) of AEA or 2-AG for 16 h. Luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. (C) R(+)-Methanandamide fails to activate nqo1-ARE-Luc reporter. MCF-7 cells co-transfected with reporter (0.5 μg) and pCMV-β-galactosidase (0.15 μg) were treated with increasing doses (5 and 10 μM) of R(+)-methanandamide and 2.5 μM of AEA for 16 h. Luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. M-AEA: R(+)-Methanandamide. (D) THC and CP55940 fail to activate nqo1-ARE-Luc reporter. MCF-7 cells co-transfected with reporter (0.5 μg) and pCMV-β-galactosidase (0.15 μg) were treated with increasing doses (5 and 10 μM) of THC and CP55940 and 2.5 μM of AEA for 16 h. Luciferase activity assay and data analysis were performed as shown in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. (E) AEA-mediated nqo1-ARE-Luc reporter activation is augmented by URB597. MCF-7 cells transfected with nqo1-ARE-Luc reporter (0.5 μg) and pCMV-β-galactosidase (0.15 μg) were pretreated with 1 μM of URB597, followed by 1 μM of AEA treatment for 16 h. Luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. *P < 0.05 as compared with the control untreated cells.
R(+)-Methanandamide is a metabolically stable congener of AEA that has higher affinity for the CB receptor and it mimics AEA activity in many cellular events (Abadji et al., 1994). R(+)-Methanandamide could not activate nqo1-ARE-Luc reporter in MCF-7 cells (Figure 2C) at the doses of 5 or 10 μM. The synthetic cannabinoid ligands THC and CP55940 also failed to activate nqo1-ARE-Luc reporter in MCF-7 cells (Figure 2D). However, oleoylethylamide (OEA), another substrate of FAAH, increased the levels of HO-1 protein in MCF 7 cells upon 24 h of treatment (Supporting Information Figure S2). Taken together, these data demonstrate that AEA specifically up-regulates HO-1 transcription.
Next, we investigated if inhibition of FAAH by URB597 potentiates the AEA-dependent activation of nqo1-ARE-Luc reporter. MCF-7 cells transfected with nqo1-ARE-Luc reporter were pretreated with 1 μM of URB597 for 1 h and followed by 1 μM of AEA treatment for 16 h. Pretreatment with 1 μM of URB597 increased the AEA-mediated nqo1-ARE-Luc reporter activity (Figure 2E), suggesting that URB597 enhances the effect of AEA in activating HO-1 transcription.
URB597- and AEA-induced HO-1 expression is independent of CB receptors
It has been demonstrated that AEA exerts varieties of activity in CB1 receptor- and CB2 receptor-dependent, CB receptor-independent manners (Di Marzo et al., 2001; McAllister and Glass, 2002; O'Sullivan, 2007). To analyse if URB597 and AEA regulate HO-1 transcription in a CB receptor-dependent manner, we assessed the URB597- and AEA-dependent nqo1-ARE-Luc reporter activity and HO-1 mRNA expression using chemical inhibitors for CB receptors. Following transfection with nqo1-ARE-Luc reporter plasmid, cells were pretreated with AM251 (CB1 receptor antagonist) or AM630 (CB2 receptor antagonist) at 1 μM for 1 h. The cells were then treated by URB597 (5 and 10 μM) or AEA (2.5 μM) for 16 h. AM251 and AM630 could not block URB597 or AEA from activating nqo1-ARE-Luc reporter in MCF-7 cells (Figure 3A,B). Furthermore, URB597- and AEA-induced HO-1 mRNA expression was not inhibited by AM251 or AM630 in MCF-7 cells (Figure 3C,D). Taken together, these data suggest that URB597 and AEA act on HO-1 transcription in CB1 and CB2 receptor-independent manner.
Figure 3.
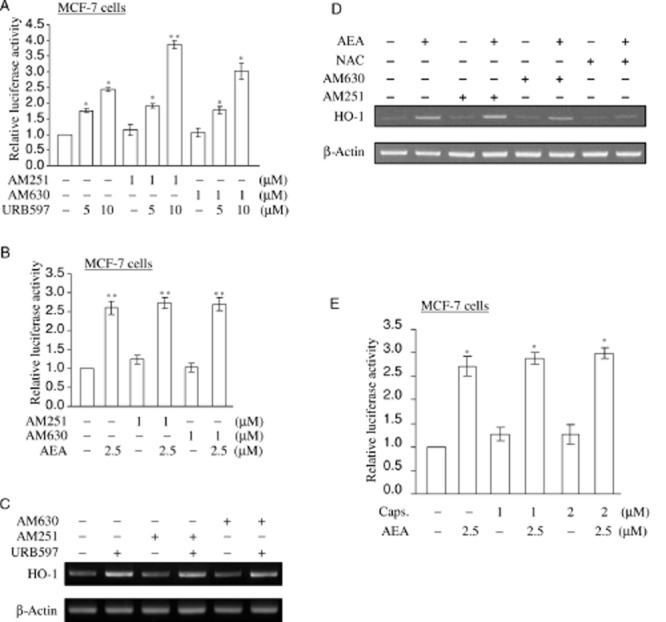
URB597 and AEA induce HO-1 transcripts, independent of CB receptors. (A, B) AM251 or AM630 cannot block URB597 and AEA from activating nqo1-ARE-Luc reporter in MCF-7 cells. MCF-7 cells co-transfected with 0.5 μg of nqo1-ARE-Luc reporter and 0.15 μg of pCMV-β-galactosidase plasmids were pretreated with 1.0 μM of AM251 or AM630 for 1 h, followed by 5 and 10 μM of URB597 or 2.5 μM of AEA treatment for 16 h. Luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. (C, D) AM251 and AM630 fail to block URB597 and AEA from inducing HO-1 mRNA expression. MCF-7 cells were pretreated with vehicle (DMSO), 1 μM of AM251, AM630, or 5 mM of NAC for 1 h, followed by URB597 (5 μM) and AEA (2.5 μM) treatment for 6 h. Semi-quantitative RT-PCR for HO-1 mRNA expression was performed as in Figure 1A. Furthermore, 5 mM of NAC blocks AEA from inducing HO-1 transcripts. All figures are representative of three independent experiments. (E) Capsazepine cannot block AEA from activating nqo1-ARE-Luc reporter. MCF-7 cells co-expressed with 0.5 μg of ARE reporter and 0.15 μg of pCMV-β-galactosidase were pretreated with 1.0 and 2.0 μM of capsazepine for 1 h, followed by 2.5 μM of AEA treatment for 16 h. Luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. Caps.: capsazepine *P < 0.05 as compared with the control untreated cells; **P < 0.001 as compared with the control untreated cells.
Numbers of evidence support the finding that AEA activates the transient receptor potential vanilloid 1 (TRPV1) receptor (Ross, 2003). Therefore, we used capsazepine, a chemical inhibitor for TRPV1 receptor (Walpole et al., 1994), to see if activation of TRPV1 receptor is involved in nqo1-ARE-Luc reporter activation. Capsazepine treatment failed to block URB597 and AEA from activating nqo1-ARE-Luc reporter in MCF-7 cells (Figure 3E), suggesting that TRPV1 receptor is not involved in AEA-mediated activation of HO-1 transcription.
URB597 and AEA induce Nrf2 nuclear translocation
Hepatocytes from Faah-/- knock-out mice displayed increased AEA-induced ROS formation and were susceptible to AEA-mediated death (Siegmund et al., 2006). Given that Nrf2 is a key transcriptional factor that activates ARE promoter in ho-1 gene (Martin et al., 2004) and ROS activate Nrf2 as a transcription factor in the nucleus (Kobayashi and Yamamoto, 2006), we therefore analysed whether URB597 and AEA activate the ROS-Nrf2 signalling pathway and lead to an increase in HO-1 transcription. Cells transfected with nqo1-ARE-Luc reporter plasmid were pretreated with NAC, a specific scavenger of ROS, following with URB597 and AEA treatment respectively. NAC reduced the basal activity of nqo1-ARE-Luc reporter in MCF-7 cells (Figure 4A,B), indicating that the levels of ROS regulate ARE promoter activity. Of note, NAC pretreatment blocked either URB597 or AEA from increasing nqo1-ARE-Luc reporter activity in MCF-7 cells (Figure 4A,B). URB597- and AEA-induced HO-1 mRNA expression was also blocked upon NAC pretreatment in MCF-7 cells (Figure 4C,D). Taken together, these data suggest that URB597 and AEA induce HO-1 by activating the ROS signalling pathway.
Figure 4.
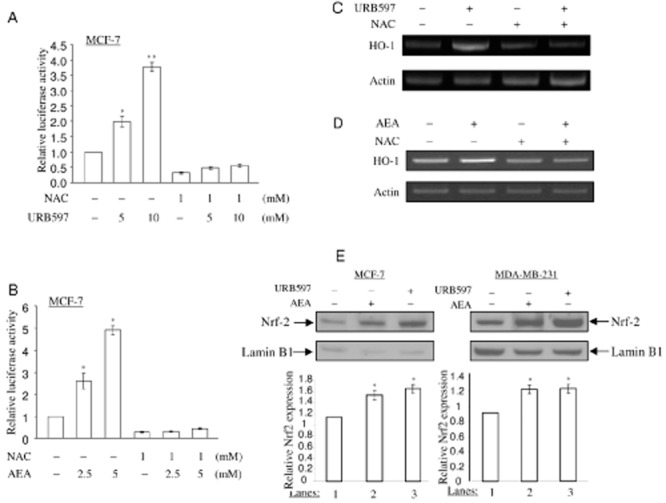
URB597 and AEA induce Nrf2 nuclear translocation. (A, B) NAC blocks URB597 and AEA from activating nqo1-ARE-Luc reporter. MCF-7 cells co-transfected with 0.5 μg of nqo1-ARE-Luc reporter and 0.15 μg of pCMV-β-galactosidase were pretreated with 1.0 mM of NAC for 1 h, followed by URB597 (5.0 and 10.0 μM) and AEA (2.5 and 5.0 μM) treatment for 16 h. Luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. (C, D) NAC inhibits URB597- and AEA-induced HO-1 mRNA expression. MCF-7 cells were pretreated with 5 mM of NAC, followed by 5 μM of URB597 and 2. 5 μM of AEA treatment for 6 h, as indicated. Semi-quantitative RT-PCR for HO-1 mRNA expression was performed as in Figure 1A. (E) URB597 and AEA induce the nuclear translocation of Nrf2 protein in MCF-7 cells and MDA-MB-231 cells. MCF-7 and MDA-MB-231 cells were treated with URB597 (5 μM) and AEA (2.5 μM) for 6 h, followed by the nuclear fractionation. Furthermore, 10 μg of the nuclear proteins were applied to detect the levels of Nrf2 protein. The same membrane was immunoblotted by anti-lamin B1 antibody to monitor equal loading of proteins. The levels of Nrf2 protein were normalized to lamin B1 levels and the relative expression was represented graphically. All the figures are representative of three independent experiments. *P < 0.05 as compared with the control untreated cells; **P < 0.001 as compared with control untreated cells.
In response to ROS signalling, Nrf2 transcription factor is released from Keap1 protein in the cytoplasm and translocates into the nucleus where it activates antioxidant transcription (Kobayashi and Tong, 2006). Therefore, we studied whether URB597 and AEA induce Nrf2 nuclear translocation. In order to determine the levels of Nrf2 protein in the nucleus, the nuclear extracts from URB597- and AEA-treated cells were fractionated and subjected to SDS-PAGE and immunoblotting. URB597 and AEA treatment induced an increase in Nrf2 protein levels in the nucleus of MCF-7 cells and MDA-MB-231 cells (Figure 4E), suggesting that URB597 and AEA induce the Nrf2 nuclear translocation. We further assessed the role of Nrf2 in URB597- and AEA-induced nqo1-ARE-Luc reporter activation and HO-1 transcription using siRNA approach. siRNA-Nrf2 or control siRNA (against GFP) and nqo1-ARE-Luc reporter plasmid were introduced into MCF-7 cells and MDA-MB-231 cells. In order to understand the responsiveness of nqo1-ARE-Luc reporter in the absence of endogenous Nrf2, cells were then treated with URB597 and AEA for 16 h. Reduction of endogenous Nrf2 protein expression levels was observed after siRNA-Nrf2 treatment in MCF-7 cells and MDA-MB-231 cells (Figure 5A). Reduction of Nrf2 protein levels resulted in a significant decrease in the basal levels of nqo1-ARE-Luc reporter activity in MCF-7 cells (Figure 5B), indicating that Nrf2 regulates the basal levels of ARE promoter activity. Furthermore, Nrf2 protein-depleted MCF-7 cells (Figure 5B) failed to respond to URB597 and AEA treatment to induce nqo1-ARE-Luc reporter activation, suggesting that URB597 and AEA activate ARE sequence in an Nrf2-dependent manner. Keap1 acts as an inhibitor to retain Nrf2 protein in the cytoplasm under normal conditions (Kobayashi and Yamamoto, 2005). We then examined the effects of exogenously expressed Keap1 in inhibiting the URB597- and AEA-dependent activation of nqo1-ARE-Luc reporter. Overexpression of Keap1 reduced the basal activity of nqo1-ARE-Luc reporter in MCF-7 cells (Figure 5C), indicating that Keap1 negatively regulated nqo1-ARE-Luc reporter. Upon Keap1 overexpression, URB597 and AEA could no longer activate nqo1-ARE-Luc reporter in MCF-7 cells (Figure 5C). Furthermore, URB597 and AEA failed to induce HO-1 mRNA expression after depletion of Nrf2 in MCF-7 cells (Figure 5D). Taken together, these data support our notion that inhibition of FAAH by URB597 and AEA act on Nrf2 transcription factor to induce antioxidant transcription.
Figure 5.
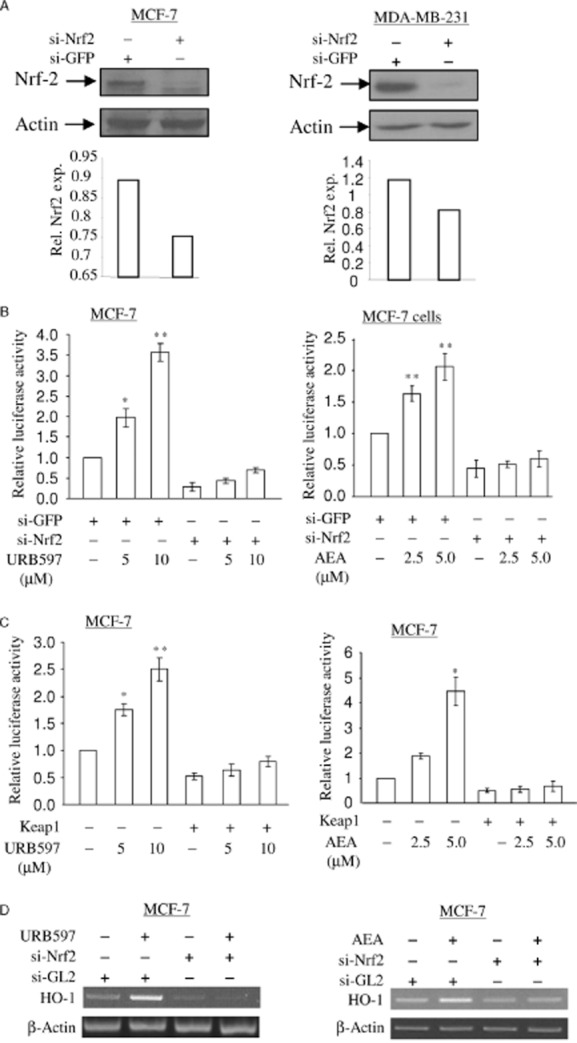
Depletion of Nrf2 abolishes URB597- and AEA-induced nqo1-ARE-Luc reporter activity and HO-1 mRNA expression. (A) Depletion of Nrf2 protein by siRNA approach in MCF-7 cells and MDA-MB-231 cells. MCF-7 cells and MDA-MB-231 cells were treated with 5 nM of si-GFP RNA (control) and si-Nrf2 RNA as indicated. At 48 h, 15 μg of total cell lysates was subjected to immunoblotting in order to detect the levels of Nrf2 protein. The same membrane was blotted with anti-actin antibody to monitor equal loading of proteins. The levels of Nrf2 protein were normalized to actin levels and the relative expression was represented graphically. All the figures are representative of three independent experiments. (B) Depletion of Nrf2 blocks URB597- and AEA-induced nqo1-ARE-Luc reporter activity. MCF-7 cells were treated with 5 nM of si-GFP RNA (control) and si-Nrf2 RNA as indicated. At 16 h, the cells were introduced with 0.5 μg of nqo1-ARE-Luc reporter and 0.15 μg of pCMV-β-galactosidase plasmids, followed by URB597 (5.0 μM) and AEA (2.5 μM) treatment for 16 h. Luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. *P < 0.05 as compared with the cells treated with siGFP; **P < 0.001 as compared with cells treated with si-GFP. (C) Keap1 blocks URB597- and AEA-induced nqo1-ARE-Luc reporter activity. Furthermore, 0.5 μg of nqo1-ARE-Luc reporter and 0.15 μg of pCMV-β-galactosidase plasmid were introduced into MCF-7 cells with or without Keap1, followed by 5.0 μM of URB597 treatment and 2.5 μM of AEA treatment. After 16 h, luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. *P < 0.05 as compared with the untreated cells; **P < 0.001 as compared with untreated cells. D. MCF-7 cells were treated with 5 nM of si-GL2 RNA (control) and si-Nrf2 RNA as indicated. At 48 h, cells were treated with 5 μM of URB597 or 2.5 μM of AEA for 6 h as indicated. Semi-quantitative RT-PCR for HO-1 mRNA was performed as in Figure 1A. All figures are representative of three independent experiments.
Depletion of FAAH increases HO-1 expression
FAAH is responsible for the degradation of endocannabinoid AEA. To determine whether depletion of endogenous FAAH protein exerts an AEA-like effect in inducing HO-1 mRNA expression, we employed siRNA for FAAH. A concentration of 5 nM of siRNA-FAAH treatment reduced the levels of endogenous FAAH protein in MCF-7 cells and MDA-MB-231 cells (Figure 6A), leading to an increase in nqo1-ARE-Luc reporter activity in both cells (Figure 6B). Induction of HO-1 mRNA and protein was observed after siRNA-FAAH treatment in MCF-7 cells and MDA-MB-231 cells, compared with the control siRNA-GL2 treatment (Figure 6C). Quantitative RT-PCR assay also showed that siRNA-FAAH treatment increases HO-1 mRNA levels by approximately twofold in MCF7 cells (Supporting Information Figure S1C,D). This suggests that depletion of FAAH induces HO-1 transcription. Furthermore, siRNA-FAAH treatment increased the levels of Nrf2 protein in the nucleus in MCF-7 cells and MDA-MB-231 cells (Figure 6D), suggesting that depletion of FAAH induces Nrf2 nuclear translocalization. In addition, depletion of Nrf2 and overexpression of Keap1 abolished siRNA-FAAH-induced nqo1-ARE-Luc reporter activity in MCF-7 cells (Figure 6E, left and right respectively). Thus, siRNA-FAAH treatment-induced nqo1-ARE-Luc reporter activation is also Nrf2-dependent, and depletion of FAAH activates Nrf2 and induces HO-1 transcription.
Figure 6.
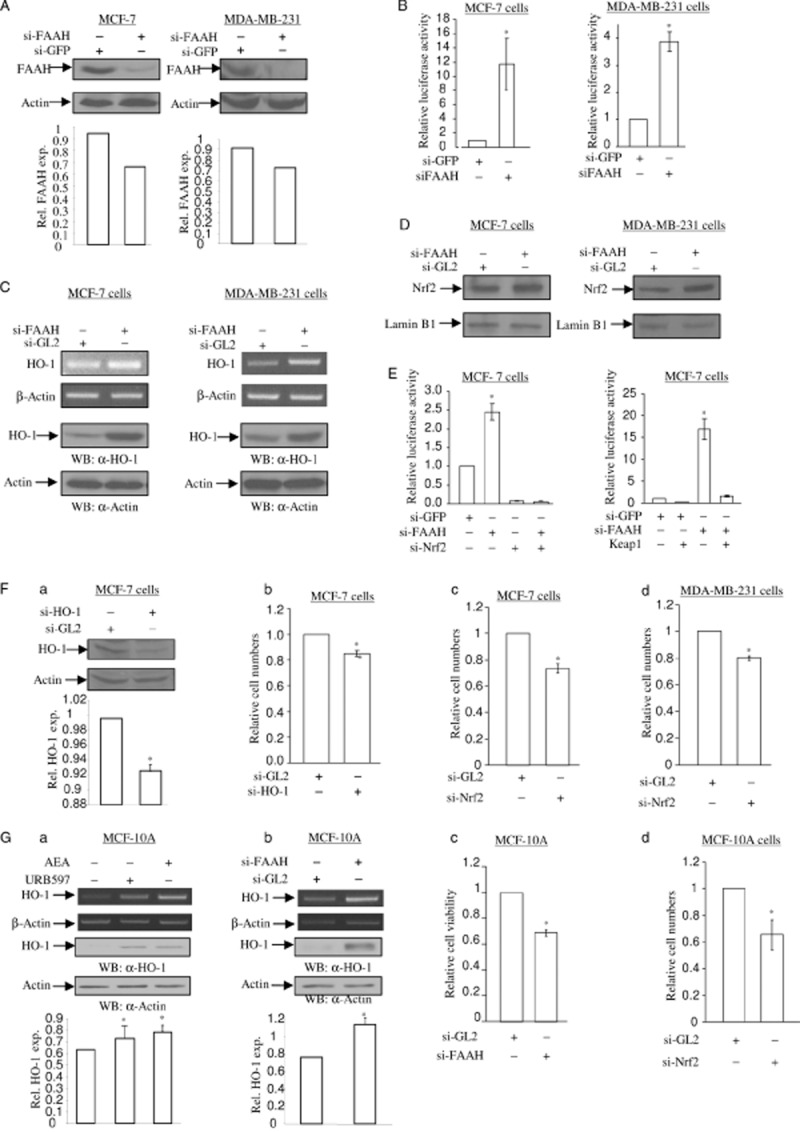
Depletion of FAAH increases HO-1 mRNA and protein expression. (A) Depletion of endogenous FAAH protein after si-FAAH RNA treatment. MCF-7 cells (left) and MDA-MB-231 cells (right) were treated with 5 nM of si-GL2 RNA (control) and si-FAAH RNA. After 72 h, total cell lysates were subjected to SDS-PAGE and immunoblotting to detect the levels of FAAH protein. The same membrane was blotted with anti-actin antibody to monitor equal loading of proteins. The levels of FAAH protein were normalized to actin levels and the relative expression was represented graphically. All the figures are representative of three independent experiments. (B) Depletion of FAAH activates nqo1-ARE-Luc reporter. MCF-7 cells and MDA-MB-231 cells were treated with 5 nM of si-GFP RNA (control) and si-FAAH RNA. After 16 h, cells were introduced with 0.5 μg of nqo1-ARE-Luc reporter and 0.15 μg of pCMV-β-galactosidase plasmids. After 24 h, luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. (C) Depletion of FAAH induces HO-1 mRNA and protein expression. MCF-7 cells (left) and MDA-MB-231 cells (right) were treated with 5 nM of si-GL2 RNA (control) and si-FAAH RNA. After 48 h, semi-quantitative RT-PCR for HO-1 and β-actin mRNA (top two panels) was performed as in Figure 1A. In a parallel experiment, total protein was subjected to SDS-PAGE and immunoblotting in order to detect the levels of HO-1 protein. The same membrane was blotted with anti-actin antibody to monitor equal loading of proteins. (D) Depletion of FAAH induces Nrf2 nuclear translocation. MCF-7 cells (left) and MDA-MB-231 cells (right) were treated with 5 nM of si-GL2 RNA (control) and si-FAAH RNA respectively. After 48 h, the nuclear proteins were separated and 10 μg of protein extract was subjected to SDS-PAGE and immunoblotting in order to detect the levels of Nrf2 protein. The same membrane was blotted with anti-lamin B1 antibody to monitor equal loading of nuclear proteins. All figures are representative of three independent experiments. (E) Nrf2 is involved in si-FAAH treatment-activated nqo1-ARE-Luc reporter activity. On the left side, depletion of Nrf2 abolishes si-FAAH RNA treatment-activated nqo1-ARE-Luc reporter activity. MCF-7 cells were treated with 5 nM of si-GFP RNA, 2.5 nM of si-FAAH RNA and 2.5 nM of si-Nrf2 RNA, in combination as indicated. After 16 h, cells were introduced with 0.5 μg of nqo1-ARE-Luc reporter and 0.15 μg of pCMV-β-galactosidase plasmids. After 24 h, luciferase activity assay and data analysis were performed as Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. On the right side, overexpression of Keap1 blocks si-FAAH RNA treatment-activated nqo1-ARE-Luc reporter activity. MCF-7 cells were co-expressed with 0.5 μg of nqo1-ARE-Luc reporter and 0.15 μg of pCMV-β-galactosidase plasmids, with or without Keap1 plasmid. After 24 h, luciferase activity assay and data analysis were performed as in Figure 1C. The data represent the mean ± SD of at least three independent experiments performed in triplicate. (F) Depletion of HO-1 causes a decrease in cell numbers in MCF-7 and MDA-MB-231 cells. (a) Knockdown of endogenous HO-1 by si-HO-1 RNA treatment. MCF-7 cells and MDA-MB-231 cells were treated with 5 nM of si-GL2 RNA (control) and si-HO-1 RNA. After 72 h, total cell lysates were subjected to determine the levels of HO-1 protein. The same membrane was blotted with anti-actin antibody to monitor equal loading of proteins. The levels of HO-1 protein were normalized to the actin levels and the relative expression was represented graphically. All figures are representative of three independent experiments. (b) Depletion of HO-1 reduces cell numbers. MCF-7 cells treated with 5 nM of si-GL2 RNA (control) and si-HO-1 RNA were stained with crystal violet at 72 h. The cells were extracted in 1% SDS solution and the absorbance was determined at 570 nm. Changes in cell numbers were obtained upon comparison of the OD570 between si-GL2 RNA-treated cells and si-HO-1 RNA-treated cells. The data represent the mean ± SD of at least three independent experiments performed in triplicate. Depletion of Nrf2 decreases cell numbers in MCF-7 (c) and MDA-MB-231 (d) cells. MCF-7 cells and MDA-MB-231 cells treated with 5 nM of si-GL2 RNA (control) and si-Nrf2 RNA were stained with crystal violet at 72 h. The cells number assay was performed as Figure 6F-b. The data represent the mean ± SD of at least three independent experiments performed in triplicate. (G) AEA signalling in MCF-10A immortalized breast epithelial cells. (a, b) URB597, AEA and si-FAAH RNA increase the levels of HO-1 mRNA and protein in MCF-10A cells. In Figure 6G, MCF-10A cells were treated with 5 μM of URB597 and 2.5 μM of AEA for 6 and 16 h for semi-quantitative RT-PCR of HO-1 mRNA and immunoblotting of HO-1 protein respectively. β-Actin primers were used as a control for semi-quantitative RT-PCR and anti-actin antibody was applied to monitor equal loading for immunoblotting. MCF-10A cells treated with 5 nM of si-FAAH RNA and si-GL2 RNA (control) for 3 days were subjected to semi-quantitative RT-PCR for HO-1 mRNA and immunoblotting of HO-1 protein respectively. β-Actin primers were used as a control for semi-quantitative RT-PCR and anti-actin antibody was applied to monitor equal loading for immunoblotting. The levels of HO-1 protein were normalized to actin levels and the relative expression was represented graphically. All the figures are representative of three independent experiments. Depletion of HO-1 and Nrf2 decrease cell numbers in MCF-10A cells. MCF-10A cells were treated with 5 nM of si-FAAH RNA (c), si-Nrf2 RNA (d) and si-GL2 RNA (control) as indicated. At 72 h, the cells were stained with crystal violet as Figure 6F-b. The data represent the mean ± SD of at least three independent experiments performed in triplicate.
Depletion of HO-1 decreases cell numbers
To understand the significance of AEA signalling-inducible HO-1 in breast cancer cells, we depleted HO-1 by employing siRNA approach in MCF-7 cells and MDA-MB-231 cells. A specific siRNA-HO-1 was applied to knock down endogenous HO-1 protein (Figure 7A). Cells treated with siRNA-HO-1 were cultured for 72 h and stained with crystal violet. Differences in cell counts were obtained in comparison to siRNA-GL2 (control)-treated cells. There was a decrease in cell numbers after HO-1 depletion in MCF-7 cells (approximately 15 %) (Figure 7B), suggesting that HO-1 plays a pro-proliferative role in breast cancer cell lines. Depletion of Nrf2 also decreased cell numbers by approximately 25 and 20% in MCF-7 and MDA-MB-231 cells, respectively (Figure 7C,D), indicating that loss of cellular stress protective role of Nrf2 causes cell growth arrest. Taken together, depletion of HO-1 results in cell number decreases due to an accumulation of cellular stress.
Figure 7.
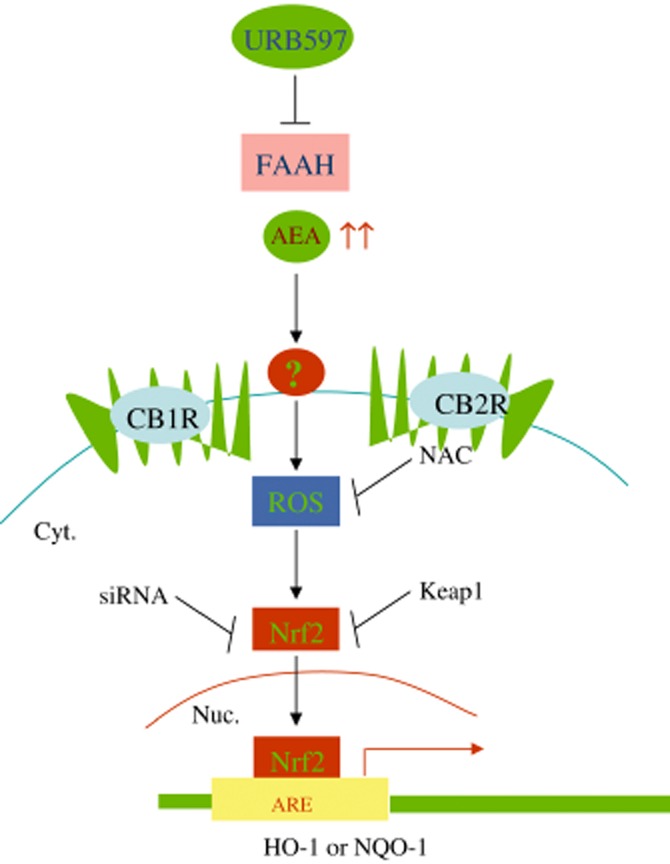
Proposed mechanisms for AEA-HO-1 signalling. AEA accumulates ROS, which induces Nrf2 nuclear translocation. Nrf2 transcriptionally activates ARE promoter of HO-1 or NQO-1. Under physiological conditions, HO-1 plays an important role in anti-inflammation, tissue injury and pain relief.
MCF-10A cells are immortalized normal breast epithelial cells. To understand whether the AEA signalling induces HO-1 in normal breast epithelial cells, we analysed the levels of HO-1 mRNA and protein expression in the presence of URB597, AEA and siRNA-FAAH in MCF-10A cells. URB597, AEA and siRNA-FAAH treatment induced HO-1 mRNA and protein expression, demonstrating that AEA signalling induces HO-1 expression in normal breast epithelial cells. Furthermore, siRNA-mediated depletion of HO-1 protein and Nrf2 protein in MCF-10A cells reduced cell numbers by approximately 30% and 35%, respectively, suggesting that HO-1 expression is associated with cell proliferation in normal breast epithelial cells.
Discussion
In this study, we demonstrated that inhibition of FAAH or AEA treatment induced HO-1 transcripts and unveiled the molecular mechanism in activating Nrf2 transcription factor and inducing HO-1 mRNA expression in breast cancer cell lines. AEA, URB597 and siRNA-mediated depletion of FAAH induced HO-1 transcripts in MCF-7 and MDA-MB-231 breast cancer cell lines. Inhibition of FAAH and AEA induced Nrf2 nuclear translocation and activated nqo1-ARE-Luc reporter. As AEA, URB597 and siRNA-mediated depletion of FAAH induced both constructs of NQO1 (Figure 1C) and HO-1 promoters (data not shown) in a similar manner, we therefore provided results with NQO-1 promoter activation only. The induction of HO-1 by FAAH inhibition and AEA treatment was independent of cannabinoid receptors, CB1, CB2 or TRPV1 receptors. Depletion of endogenous HO-1 protein resulted in a decrease in cell numbers in MCF-7 cells and MDA-MB-231 cells, suggesting that HO-1 is required for cell proliferation. The inhibition of FAAH and AEA treatment also induced HO-1 transcripts in MCF-10A normal breast epithelial cells and depletion of HO-1 protein decreased cell numbers in MCF-10A cells. Our findings revealed a novel function that inhibition of FAAH and increased AEA levels induce antioxidant expression (see Figure 7 for schematic presentation), independent of cannabinoid receptors. Furthermore, these results strongly suggest that accumulation of AEA or other fatty amides of FAAH substrates can also activate Nrf2 pathway independent of CB1, CB2 or TRPV1 receptors.
URB597 is a well-known inhibitor of FAAH, an enzyme responsible for AEA degradation. Inhibition of FAAH enzyme by URB597 or knockdown of FAAH has been demonstrated to increase the levels of endogenous AEA ligand (Alpini and Demorrow, 2009; Clapper et al., 2009). Our results showed that URB597 and siRNA-FAAH, like AEA, activated nqo1-ARE-Luc reporter and induced HO-1 transcripts and protein expression. Inhibition of FAAH by another FAAH inhibitor PF622 also showed an induction of HO-1 protein (Supporting Information Figure S2), further suggesting that inhibition of FAAH is responsible for HO-1 induction. FAAH is also responsible for other fatty acid amides. We found that treatment of OEA, another substrate of FAAH, also resulted in an increase of HO-1 protein in MCF 7 cells (Supporting Information Figure S2), suggesting that other substrates of FAAH might be involved in inducing HO-1 transcripts.
R(+)-Methanandamide, a metabolically stable congener of AEA, has been shown to mimic AEA activity in many cellular events. However, we found that R(+)-methanandamide does not possess an activity in activating nqo1-ARE-Luc reporter. AEA has been demonstrated to induce ROS formation, possibly mediated by its binding to membrane and lipid peroxidation (Siegmund et al., 2005). Therefore, it is possible that the stereochemical properties of AEA from its structure, but not R(+)-methanandamide, causes ROS formation, which contributes to an induction of HO-1 transcripts.
AEA exerts various biological activities through cannabinoid receptors, CB1, CB2 and TRVP1 (Di Marzo et al., 2001; McAllister and Glass, 2002), although other CB receptor-independent biological activities have also been reported (Siegmund et al., 2005; 2006; O'Sullivan, 2007). In our study, we found that HO-1 transcriptional activation by inhibition of FAAH or AEA ligand is independent of CB receptors. The chemical inhibitors AM251 (CB1 antagonist), AM630 (CB2 antagonist) and capsazepine (TRPV1 antagonist) could not inhibit AEA, URB597 and siRNA-FAAH from activating nqo1-ARE-Luc reporter and inducing HO-1 transcripts. Furthermore, when the inhibitors for CB1 and CB2 were added directly to MDA-MB-231 and MCF-7 cells in the presence or absence of AEA, we observed induction of HO-1 mRNA that was independent of CB1 and CB2 receptors (data not shown). As mentioned earlier, AEA can cause ROS formation in liver cells and lead to cell death (Siegmund et al., 2005). It has been demonstrated that AEA binds to cell membrane of primary hepatic stellate cells. Therefore, it has been proposed that AEA, like its metabolite arachidonic acid, increases ROS formation through membrane perturbation followed by lipid peroxidation (Siegmund et al., 2005). ROS signalling is known to transcriptionally up-regulate the ARE sequence in antioxidants, such as HO-1 and NQO-1 (Martin et al., 2004; Kobayashi and Yamamoto, 2005; Lee et al., 2005; Kobayashi and Tong, 2006). We observed that NAC, a scavenger of ROS, blocked URB597 and AEA from activating nqo1-ARE-Luc reporter and inducing HO-1 transcripts, suggesting that ROS signalling is involved in AEA-induced HO-1 transcriptional activation. Nrf2 transcription factor plays a key role in cellular defence against stress insults (Lee et al., 2005). ROS can cause the Nrf2 nuclear translocation, where it activates the ARE sequence in antioxidants (Kobayashi and Tong, 2006). AEA, URB597 and siRNA-FAAH treatment induced the nuclear translocation of Nrf2. However, depletion of Nrf2 protein by both siRNA approaches and Nrf2 protein cytosolic retention by overexpressing Keap1 protein inhibited the AEA, URB597 and siRNA-FAAH-induced nqo1-ARE-Luc reporter activation or HO-1 transcriptional activation. Thus, these observations support the notion that activation of ROS-Nrf2 signalling pathway is involved in inhibition of FAAH and AEA-induced transcriptional activation of HO-1. In human primary coronary artery endothelial cells, AEA also induced ROS in CB1 receptor-dependent and receptor-independent manners (Rajesh et al., 2010). Therefore, it is possible that AEA and URB597 induce HO-1 transcriptional activation in cell-type-dependent manner. Besides, 15d-PGJ2, a metabolite of AEA by COX2, has been reported to up-regulate endoplasmic reticulum-stress associated genes, such as CHOP in breast cancer cells (Stuhlmeier, 2000). Thus, it is of great interests to understand whether AEA metabolites induce cellular stresses to activate Nrf2-HO-1 pathway.
HO-1 is a 32 kDa inducible heat shock protein, which is found at low levels in most mammalian tissues but is highly induced by a variety of stress stimuli, including heat shock (Stuhlmeier, 2000), UV irradiation (Doi et al., 1999), hydrogen peroxide (Lautier et al., 1992), heavy metals (Eyssen-Hernandez et al., 1996), hypoxia (Motterlini et al., 2000) and cytokines (Rizzardini et al., 1998; Terry et al., 1998). Recent findings indicate that HO-1 and its products possess anti-inflammatory and anti-apoptotic functions (Berberat et al., 2003; Liu et al., 2003; Pae et al., 2004). Moreover, new studies suggest that HO-1 influences cell growth and proliferation (Durante, 2003). In addition to inhibiting inflammation, HO-1 induction could also prevent or ameliorate the course of the disease (Otterbein et al., 2003). However, elevated HO-1 expression and activity was found in various tumours such as human renal cell carcinoma (Goodman et al., 1997) and prostate tumours (Maines and Abrahamsson, 1996). In human gliomas and melanomas, HO-1 is linked to angiogenesis (Nishie et al., 1999; Torisu-Itakura et al., 2000; Sunamura et al., 2003). In an experimental mouse model, HO-1 accelerates pancreatic cancer growth by promoting tumour angiogenesis (Sunamura et al., 2003). These findings suggest that HO-1 may also have pro-angiogenic and growth-regulative properties in the development and progression of cancers. Moreover, its anti-inflammatory and anti-apoptotic activities imply that HO-1 may enhance radioresistance and chemoresistance in cancer cells. In this study, we found that depletion of HO-1 by siRNA approaches decreases the cell numbers of MCF-7, MDA-MB-231 breast cancer cells and MCF-10A immortalized normal breast epithelial cells. Nrf2 plays an important role in cell stress defence (Kaspar et al., 2009). We observed that depletion of Nrf2 causes significant cell number decrease in these cells. HO-1, like Nrf2, may play a cytoprotective role in these cells. The present study examined the effects of Nrf2 on breast cancer cells in vitro, demonstrating that accumulation of AEA or other fatty acid amide of FAAH substrates can activate Nrf2 pathway independent of receptors in these cells. However, activation of Nrf2 pathway can be also induced by AEA or other FAAH substrates present in the microenvironment niche surrounding the tumour cells in vivo, such as endothelial cells, tumour macrophages and tumour fibroblasts. Therefore, the contribution of AEA and FAAH substrates on Nrf2 pathway and its application to resistance chemotherapy should be examined in vivo. Regardless, the previous studies, combined with our observations, imply that activation of AEA-HO-1 signalling may be beneficial for normal cells in order to protect against insults but in favour of radioresistance and chemoresistance in cancer treatment.
AEA inhibits human breast cancer cell proliferation (De Petrocellis et al., 1998; Melck et al., 1999; Laezza et al., 2006), adhesion and migration (Grimaldi et al., 2006), in CB1 receptor-dependent manner. Our findings suggest that induction of HO-1 by AEA favours cell growth, which counteract with CB1 receptor-dependent growth inhibitory activity of AEA. We also observed a significant reduction of cell numbers after siRNA-mediated knockdown of FAAH for 72 h (unpubl. data). ROS can initiate cell growth promotion, cell cycle arrest and apoptosis, depending on the extent of its accumulation (Chatterjee et al., 2009). Therefore, besides CB1 receptor-dependent growth inhibition by AEA, accumulation of ROS after inhibition of FAAH may in part contribute to cell growth inhibition. ROS also play an important role in the development of cancer (Benhar et al., 2002; Sander et al., 2004). Accumulating evidence has demonstrated that Nrf2 exerts dual roles in toxicology and pharmacology (Pi et al., 2010). Therefore, better understanding of the cellular events regulated by inhibition of FAAH and increase of AEA should provide insights into the role of the endocannabinoid system in prevention and treatment of diseases.
In summary, we observed that inhibition of FAAH and AEA induce HO-1 and NQO-1 antioxidants. This study revealed a novel mechanism by which inhibition of FAAH or AEA treatment activates ROS-Nrf2 signalling pathway and induces HO-1 transcripts (Figure 7). Furthermore, understanding of the role of HO-1 induced by inhibition of FAAH and AEA treatment in vivo is required to increase the therapeutic potential of the endocannabinoid system and improve treatment in breast cancer.
Acknowledgments
The authors want to thank Lili Wang for editing the manuscript, Dr Harikrishna Nakshatri for providing MCF-10A cells, and Drs Ken Itoh, Masayuki Yamamoto for providing nqo1-ARE-Luc reporter plasmid, Dr. Shalom Avraham for his input and comments during the study and for critical reading of the manuscript. This research was supported in part by the National Institutes of Health Training (H. L.) T32DA007312 and CA135226, and DOD Idea Awards BC094909 and BC102246 (H. K. A.).
Glossary
- AEA
‘anandamide’, N-arachidonoylethanolamine
- AA
arachidonic acid
- AG
2-arachidonoylglycerol
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- DHEA
docosahexaenoylethanolamine
- EEA
eicosanoylethanolamine
- EPEA
eicosapentaenoylethanolamine
- FAAH
fatty acid amide hydrolase
- MGL
monoacylglycerol lipase
- NAPE
N-acyl-phosphatidylethanolamine
- OEA
oleoylethanolamine
- PEA
palmitoylethanolamine
Conflict of interest
No conflict of interest on behalf of all the authors.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Demonstration of HO-1 mRNA induction by semi-quantitative and real-time RT-PCR. (A) MCF7 cells were treated with 5 μM of AEA and URB597 for 6 h respectively. Total RNAs were used to perform Semi-quantitative RT-PCR and real-time RT-PCR for HO-1 mRNA. β-Actin was used as controls for both assays. (B) 1 ng of total RNAs was used to perform real-time RT-PCR for HO-1 mRNA. The assay was analysed by ddCt method. Real-time RT-PCR for each RNA sample was performed in triplicate. The fold increase in the levels of HO-1 mRNA was obtained in comparison with control. (C) MCF7 cells were treated with si-GL2 (control) and si-FAAH for 48 h. Total RNAs were used to detect the levels of HO-1 mRNA by Semi-quantitative RT-PCR and real-time RT-PCR. β-actin was used as controls for both assays. (D) The real-time RT-PCR for HO-1 mRNA was performed as Fig. S1B. *P < 0.05 as compared to control.
Figure S2 OEA and PF622 induce HO-1 protein in MCF7 cells. MCF7 cells were treated with 5 μM of OEA and PF622 for 24 h respectively. Total cell lysates were applied to detect the levels of HO-1 protein by Western blotting with anti-HO-1 antibody. The same membrane was reprobed with anti-actin antibody to monitor equal loading. The levels of HO-1 protein were normalized to actin levels and the relative expression was represented graphically. *P < 0.05 as compared to control.
References
- Abadji V, Lin S, Taha G, Griffin G, Stevenson LA, Pertwee RG, et al. (R)-methanandamide: a chiral novel anandamide possessing higher potency and metabolic stability. J Med Chem. 1994;37:1889–1893. doi: 10.1021/jm00038a020. [DOI] [PubMed] [Google Scholar]
- Alpini G, Demorrow S. Changes in the endocannabinoid system may give insight into new and effective treatments for cancer. Vitam Horm. 2009;81:469–485. doi: 10.1016/S0083-6729(09)81018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 2002;3:420–425. doi: 10.1093/embo-reports/kvf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberat PO, Katori M, Kaczmarek E, Anselmo D, Lassman C, Ke B, et al. Heavy chain ferritin acts as an antiapoptotic gene that protects livers from ischemia reperfusion injury. FASEB J. 2003;17:1724–1726. doi: 10.1096/fj.03-0229fje. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Ligresti A, Di Marzo V. The endocannabinoid signalling system: biochemical aspects. Pharmacol Biochem Behav. 2005;81:224–238. doi: 10.1016/j.pbb.2005.01.027. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Kundu S, Sengupta S, Bhattacharyya A. Divergence to apoptosis from ROS induced cell cycle arrest: effect of cadmium. Mutat Res. 2009;663:22–31. doi: 10.1016/j.mrfmmm.2008.12.011. [DOI] [PubMed] [Google Scholar]
- Clapper JR, Mangieri RA, Piomelli D. The endocannabinoid system as a target for the treatment of cannabis dependence. Neuropharmacology. 2009;56(Suppl. 1):235–243. doi: 10.1016/j.neuropharm.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Melck D, Palmisano A, Bisogno T, Laezza C, Bifulco M, et al. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci U S A. 1998;95:8375–8380. doi: 10.1073/pnas.95.14.8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Orlando P, Bisogno T, Zagoory O, Bifulco M, et al. Palmitoylethanolamide inhibits the expression of fatty acid amide hydrolase and enhances the anti-proliferative effect of anandamide in human breast cancer cells. Biochem J. 2001;358(Pt 1):249–255. doi: 10.1042/0264-6021:3580249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi K, Akaike T, Fujii S, Tanaka S, Ikebe N, Beppu T, et al. Induction of haem oxygenase-1 nitric oxide and ischaemia in experimental solid tumours and implications for tumour growth. Br J Cancer. 1999;80:1945–1954. doi: 10.1038/sj.bjc.6690624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante W. Heme oxygenase-1 in growth control and its clinical application to vascular disease. J Cell Physiol. 2003;195:373–382. doi: 10.1002/jcp.10274. [DOI] [PubMed] [Google Scholar]
- Egertova M, Michael GJ, Cravatt BF, Elphick MR. Fatty acid amide hydrolase in brain ventricular epithelium: mutually exclusive patterns of expression in mouse and rat. J Chem Neuroanat. 2004;28:171–181. doi: 10.1016/j.jchemneu.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Ewing P, Wilke A, Eissner G, Holler E, Andreesen R, Gerbitz A. Expression of heme oxygenase-1 protects endothelial cells from irradiation-induced apoptosis. Endothelium. 2005;12:113–119. doi: 10.1080/10623320500189814. [DOI] [PubMed] [Google Scholar]
- Eyssen-Hernandez R, Ladoux A, Frelin C. Differential regulation of cardiac heme oxygenase-1 and vascular endothelial growth factor mRNA expressions by hemin, heavy metals, heat shock and anoxia. FEBS Lett. 1996;382:229–233. doi: 10.1016/0014-5793(96)00127-5. [DOI] [PubMed] [Google Scholar]
- Fang J, Sawa T, Akaike T, Akuta T, Sahoo SK, Khaled G, et al. In vivo antitumor activity of pegylated zinc protoporphyrin: targeted inhibition of heme oxygenase in solid tumor. Cancer Res. 2003;63:3567–3574. [PubMed] [Google Scholar]
- Fang J, Sawa T, Akaike T, Greish K, Maeda H. Enhancement of chemotherapeutic response of tumor cells by a heme oxygenase inhibitor, pegylated zinc protoporphyrin. Int J Cancer. 2004;109:1–8. doi: 10.1002/ijc.11644. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley G. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, et al. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AI, Choudhury M, da Silva JL, Schwartzman ML, Abraham NG. Overexpression of the heme oxygenase gene in renal cell carcinoma. Proc Soc Exp Biol Med. 1997;214:54–61. doi: 10.3181/00379727-214-44069. [DOI] [PubMed] [Google Scholar]
- Grimaldi C, Pisanti S, Laezza C, Malfitano AM, Santoro A, Vitale M, et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp Cell Res. 2006;312:363–373. doi: 10.1016/j.yexcr.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. The endocannabinoid system and pain. CNS Neurol Disord Drug Targets. 2009;8:403–421. doi: 10.2174/187152709789824660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart S, Fischer OM, Ullrich A. Cannabinoids induce cancer cell proliferation via tumor necrosis factor alpha-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res. 2004;64:1943–1950. doi: 10.1158/0008-5472.can-03-3720. [DOI] [PubMed] [Google Scholar]
- Hill M, Pereira V, Chauveau C, Zagani R, Remy S, Tesson L, et al. Heme oxygenase-1 inhibits rat and human breast cancer cell proliferation: mutual cross inhibition with indoleamine 2,3-dioxygenase. FASEB J. 2005;19:1957–1968. doi: 10.1096/fj.05-3875com. [DOI] [PubMed] [Google Scholar]
- Jozkowicz A, Was H, Dulak J. Heme oxygenase-1 in tumors: is it a false friend? Antioxid Redox Signal. 2007;9:2099–2117. doi: 10.1089/ars.2007.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juknat A, Pietr M, Kozela E, Rimmerman N, Levy R, Coppola G, et al. Differential transcriptional profiels mediated by exposure to the cannabinoids cannabidiol and Δ9-tetrahydrocannabinol in BV-2 microglial cells. Br J Pharmacol. 2012;165:2512–2528. doi: 10.1111/j.1476-5381.2011.01461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Kim HJ, So HS, Lee JH, Park C, Park SY, Kim YH, et al. Heme oxygenase-1 attenuates the cisplatin-induced apoptosis of auditory cells via down-regulation of reactive oxygen species generation. Free Radic Biol Med. 2006;40:1810–1819. doi: 10.1016/j.freeradbiomed.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Tong KI. [Oxidative/electrophilic stress sensor Keap1 regulates the rapid turnover of transcripition factor Nrf2] Tanpakushitsu Kakusan Koso. 2006;51(10 Suppl):1304–1308. [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Laezza C, Pisanti S, Crescenzi E, Bifulco M. Anandamide inhibits Cdk2 and activates Chk1 leading to cell cycle arrest in human breast cancer cells. FEBS Lett. 2006;580:6076–6082. doi: 10.1016/j.febslet.2006.09.074. [DOI] [PubMed] [Google Scholar]
- Lautier D, Luscher P, Tyrrell RM. Endogenous glutathione levels modulate both constitutive and UVA radiation/hydrogen peroxide inducible expression of the human heme oxygenase gene. Carcinogenesis. 1992;13:227–232. doi: 10.1093/carcin/13.2.227. [DOI] [PubMed] [Google Scholar]
- Lee JS, Surh YJ. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005;224:171–184. doi: 10.1016/j.canlet.2004.09.042. [DOI] [PubMed] [Google Scholar]
- Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, et al. Nrf2, a multi-organ protector? FASEB J. 2005;19:1061–1066. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- Lin CW, Shen SC, Hou WC, Yang LY, Chen YC. Heme oxygenase-1 inhibits breast cancer invasion via suppressing the expression of matrix metalloproteinase-9. Mol Cancer Ther. 2008;7:1195–1206. doi: 10.1158/1535-7163.MCT-07-2199. [DOI] [PubMed] [Google Scholar]
- Lin HY, Shen SC, Chen YC. Anti-inflammatory effect of heme oxygenase 1: glycosylation and nitric oxide inhibition in macrophages. J Cell Physiol. 2005;202:579–590. doi: 10.1002/jcp.20160. [DOI] [PubMed] [Google Scholar]
- Lin HY, Shen SC, Lin CW, Yang LY, Chen YC. Baicalein inhibition of hydrogen peroxide-induced apoptosis via ROS-dependent heme oxygenase 1 gene expression. Biochim Biophys Acta. 2007;1773:1073–1086. doi: 10.1016/j.bbamcr.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Liu H, Nowak R, Chao W, Bloch KD. Nerve growth factor induces anti-apoptotic heme oxygenase-1 in rat pheochromocytoma PC12 cells. J Neurochem. 2003;86:1553–1563. doi: 10.1046/j.1471-4159.2003.01978.x. [DOI] [PubMed] [Google Scholar]
- McAllister SD, Glass M. CB(1) and CB(2) receptor-mediated signalling: a focus on endocannabinoids. Prostaglandins Leukot Essent Fatty Acids. 2002;66:161–171. doi: 10.1054/plef.2001.0344. [DOI] [PubMed] [Google Scholar]
- Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557–2568. [PubMed] [Google Scholar]
- Maines MD, Abrahamsson PA. Expression of heme oxygenase-1 (HSP32) in human prostate: normal, hyperplastic, and tumor tissue distribution. Urology. 1996;47:727–733. doi: 10.1016/s0090-4295(96)00010-6. [DOI] [PubMed] [Google Scholar]
- Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, et al. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279:8919–8929. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- Melck D, Rueda D, Galve-Roperh I, De Petrocellis L, Guzman M, Di Marzo V. Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999;463:235–240. doi: 10.1016/s0014-5793(99)01639-7. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Foresti R, Bassi R, Calabrese V, Clark JE, Green CJ. Endothelial heme oxygenase-1 induction by hypoxia. Modulation by inducible nitric-oxide synthase and S-nitrosothiols. J Biol Chem. 2000;275:13613–13620. doi: 10.1074/jbc.275.18.13613. [DOI] [PubMed] [Google Scholar]
- Nasser MW, Qamri Z, Deol YS, Smith D, Shilo K, Zou X, et al. Crosstalk between chemokine receptor CXCR4 and cannabinoid receptor CB2 in modulating breast cancer growth and invasion. Plos One. 2011;6:e23901. doi: 10.1371/journal.pone.0023901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD. Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence. Biochem J. 2003;374(Pt 2):337–348. doi: 10.1042/BJ20030754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishie A, Ono M, Shono T, Fukushi J, Otsubo M, Onoue H, et al. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin Cancer Res. 1999;5:1107–1113. [PubMed] [Google Scholar]
- Nowis D, Legat M, Grzela T, Niderla J, Wilczek E, Wilczynski GM, et al. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene. 2006;25:3365–3374. doi: 10.1038/sj.onc.1209378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesch S, Gertsch J. Cannabinoid receptor ligands as potential anticancer agents – high hopes for new therapies? J Pharm Pharmacol. 2009;61:839–853. doi: 10.1211/jpp/61.07.0002. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- Pae HO, Chung HT. Heme oxygenase-1: its therapeutic roles in inflammatory diseases. Immune Netw. 2009;9:12–19. doi: 10.4110/in.2009.9.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pae HO, Choi BM, Oh GS, Lee MS, Ryu DG, Rhew HY, et al. Roles of heme oxygenase-1 in the antiproliferative and antiapoptotic effects of nitric oxide on Jurkat T cells. Mol Pharmacol. 2004;66:122–128. doi: 10.1124/mol.66.1.122. [DOI] [PubMed] [Google Scholar]
- Pi J, Leung L, Xue P, Wang W, Hou Y, Liu D, et al. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J Biol Chem. 2010;285:9292–9300. doi: 10.1074/jbc.M109.093955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, et al. Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597) CNS Drug Rev. 2006;12:21–38. doi: 10.1111/j.1527-3458.2006.00021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamri Z, Preet A, Nasser MW, Bass CE, Leone G, Barsky SH, et al. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol Cancer Ther. 2009;8:3117–3129. doi: 10.1158/1535-7163.MCT-09-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Hasko G, Liaudet L, Mackie K, Pacher P. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br J Pharmacol. 2010;160:688–700. doi: 10.1111/j.1476-5381.2010.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzardini M, Zappone M, Villa P, Gnocchi P, Sironi M, Diomede L, et al. Kupffer cell depletion partially prevents hepatic heme oxygenase 1 messenger RNA accumulation in systemic inflammation in mice: role of interleukin 1beta. Hepatology. 1998;27:703–710. doi: 10.1002/hep.510270311. [DOI] [PubMed] [Google Scholar]
- Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003;140:790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander CS, Chang H, Hamm F, Elsner P, Thiele JJ. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int J Dermatol. 2004;43:326–335. doi: 10.1111/j.1365-4632.2004.02222.x. [DOI] [PubMed] [Google Scholar]
- Siegmund SV, Uchinami H, Osawa Y, Brenner DA, Schwabe RF. Anandamide induces necrosis in primary hepatic stellate cells. Hepatology. 2005;41:1085–1095. doi: 10.1002/hep.20667. [DOI] [PubMed] [Google Scholar]
- Siegmund SV, Seki E, Osawa Y, Uchinami H, Cravatt BF, Schwabe RF. Fatty acid amide hydrolase determines anandamide-induced cell death in the liver. J Biol Chem. 2006;281:10431–10438. doi: 10.1074/jbc.M509706200. [DOI] [PubMed] [Google Scholar]
- Stuhlmeier KM. Activation and regulation of Hsp32 and Hsp70. Eur J Biochem. 2000;267:1161–1167. doi: 10.1046/j.1432-1327.2000.01112.x. [DOI] [PubMed] [Google Scholar]
- Sunamura M, Duda DG, Ghattas MH, Lozonschi L, Motoi F, Yamauchi J, et al. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis. 2003;6:15–24. doi: 10.1023/a:1025803600840. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Akaike T, Fang J, Beppu T, Ogawa M, Tamura F, et al. Antiapoptotic effect of haem oxygenase-1 induced by nitric oxide in experimental solid tumour. Br J Cancer. 2003;88:902–909. doi: 10.1038/sj.bjc.6600830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry CM, Clikeman JA, Hoidal JR, Callahan KS. Effect of tumor necrosis factor-alpha and interleukin-1 alpha on heme oxygenase-1 expression in human endothelial cells. Am J Physiol. 1998;274(3 Pt 2):H883–H891. doi: 10.1152/ajpheart.1998.274.3.H883. [DOI] [PubMed] [Google Scholar]
- Torisu-Itakura H, Furue M, Kuwano M, Ono M. Co-expression of thymidine phosphorylase and heme oxygenase-1 in macrophages in human malignant vertical growth melanomas. Jpn J Cancer Res. 2000;91:906–910. doi: 10.1111/j.1349-7006.2000.tb01033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walpole CS, Bevan S, Bovermann G, Boelsterli JJ, Breckenridge R, Davies JW, et al. The discovery of capsazepine, the first competitive antagonist of the sensory neuron excitants capsaicin and resiniferatoxin. J Med Chem. 1994;37:1942–1954. doi: 10.1021/jm00039a006. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhang J, Liu H, Huang H, Wang C, Shen Y, et al. Melatonin, a potent regulator of hemeoxygenase-1, reduces cardiopulmonary bypass-induced renal damage in rats. J Pineal Res. 2009;46:248–254. doi: 10.1111/j.1600-079X.2008.00624.x. [DOI] [PubMed] [Google Scholar]
- Wei Y, Wang X, Wang L. Presence and regulation of cannabinoid receptors in human retinal pigment epithelial cells. Mol Vis. 2009;15:1243–1251. [PMC free article] [PubMed] [Google Scholar]
- Williams J, Wood J, Pandarinathan L, Karanian DA, Bahr BA, Vouros P, et al. Quantitative method for the profiling of the endocannabinoid metabolome by LC-atmospheric pressure chemical ionization-MS. Anal Chem. 2007;79:5582–5593. doi: 10.1021/ac0624086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.