

Abstract
BACKGROUND AND PURPOSE
Chronic ethanol abuse and haemorrhagic shock are major causes of global mortality and, separately, induce profound hepato- and immune-toxic effects via activation of NF-κB. Here, we assessed the effects of chronic ethanol intake upon the pathophysiological derangements after haemorrhagic shock with subsequent resuscitation (H/R), with particular attention to the contribution of NF-κB.
EXPERIMENTAL APPROACH
Transgenic NF-κBEGFP mice, expressing the enhanced green fluorescent protein (EGFP) under the transcriptional control of NF-κB cis-elements were fed a Lieber-DeCarli diet containing ethanol (EtOH-diet) or an isocaloric control diet for 4 weeks and were then pairwise subjected to H/R. Liver tissues and peripheral blood were sampled at 2 or 24 h after H/R. Cytokines in blood and tissue and leukocyte activation (as CD11b expression) were measured, along with EGFP as a marker of NF-κB activation.
KEY RESULTS
The EtOH-diet increased mortality at 24 h after H/R and elevated liver injury, associated with an up-regulation of NF-κB-dependent genes and IL-6 release; it also increased production of NF-κB-driven intercellular adhesion molecule 1 (ICAM-1) and EGFP in liver tissue. At 2h after the H/R procedure in ethanol-fed mice we observed the highest proportion of NF-κB activated non-parenchymal cells and an NF-κB-dependent increase in polymorphonuclear leukocyte CD11b expression.
CONCLUSIONS AND IMPLICATIONS
The EtOH-diet exacerbated liver injury after H/R, accompanying an overwhelming hepatic and systemic immune response. Our findings contribute to evidence implicating NF-κB as a key player in the orchestration of the immune response in haemorrhagic shock patients with a history of chronic ethanol abuse.
Keywords: chronic ethanol, haemorrhage/resuscitation, NF-κB, liver injury, inflammation
Introduction
Trauma and chronic ethanol abuse constitute two of the leading causes of morbidity and mortality worldwide (McDonald et al., 2004; Li et al., 2007). Among trauma patients, haemorrhagic shock is a leading cause of death (Cocchi et al., 2007; Angele et al., 2008). Nearly 50% of trauma patients seen in emergency services have also consumed alcohol. This combination is known to increase the risk of subsequent complications (Jurkovich et al., 1993; Maier, 2001). Ethanol abuse is associated with various pathophysiological changes that directly affect the liver, such as steatosis, steatohepatitis, fibrosis and cirrhosis (Mandrekar and Szabo, 2009; Nau et al., 2013). Acute and chronic alcohol intoxication significantly alters the organism's ability to combat infections at a cellular and molecular level (Goral et al., 2008; Lau et al., 2009; Szabo and Mandrekar, 2009).
A profound inflammatory process follows haemorrhagic shock, characterized by expression of cytokines and accumulation of polymorphonuclear leukocytes (PMNL) and recruitment to the site of injury. This, in turn, leads to a systemic inflammatory response syndrome or may even lead to multiple organ dysfunction syndrome (Baue et al., 1998; Akgur et al., 2000; Hierholzer and Billiar, 2001). Haemorrhagic shock also disrupts the microcirculation (Marzi et al., 1996; Partrick et al., 1996; Hietbrink et al., 2006). Production of superoxide anions by NADPH oxidase promotes the generation of highly toxic peroxynitrite after haemorrhagic shock and resuscitation (H/R) (Lehnert et al., 2003). In addition, TNF-α directly causes hepatic dysfunction and stimulates an immune response that triggers an overwhelming inflammatory burst (Wheeler et al., 2001).
Our group and others have demonstrated that the transcription factor NF-κB may play a crucial role in orchestrating an immune response after H/R and chronic ethanol intake (Ono et al., 2004; Lehnert et al., 2008; Relja et al., 2012). NF-κB is involved in cellular responses to stimuli such as free radicals, stress, cytokines, ultraviolet irradiation, oxidized LDL and bacterial or viral antigens. As NF-κB has been additionally shown to play a crucial role in cancer, inflammatory and autoimmune disease and septic shock, it is a key target for drug discovery. In an inactive state, the NF-κB heterotrimer is present in the cytosol and is mostly composed of the subunits p65 (RelA) and p50, whereas p65 contains an active C-terminal transcriptional domain (Baldwin, 1996; Gupta et al., 2010). By binding the regulatory protein IκB (inhibitor of kappa B) to the subunit p65, the translocation of NF-κB to the nucleus is inhibited (Zingarelli, 2005; Gupta et al., 2010). Activating stimuli trigger phosphorylation and subsequent proteasomal degradation of IκB, with subsequent phosphorylation of p65. Once activated, the NF-κB heterodimer translocates to the nucleus and activates various immunomodulatory genes, such as those for the intercellular adhesion molecule 1 (ICAM-1) and the cytokines, IL-6 and TNF-α (Tak and Firestein, 2001). Activation of NF-κB is crucial both after haemorrhagic shock and ethanol consumption. In a previous study from our group, we demonstrated that binge like ethanol exposure prior to H/R blunts acute inflammatory changes associated with NF-κB activation and increases survival (Relja et al., 2012). As NF-κB is involved in the pathophysiology of both H/R and chronic ethanol abuse, we studied the effects of combining both these challenges, paying particular attention to the involvement of NF-κB (Meng et al., 2001; Ono et al., 2004; Lehnert et al., 2008; Relja et al., 2009; 2009,2012).
Methods
Animals
All animal care and experimental protocols were approved by the Veterinary Department of the Regional Council in Darmstadt, Germany. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2001; McGrath et al., 2005). A total of 68 animals were used in the experiments described here. Male NF-κBEGFP (C57BL/6 background, 6–8 weeks old, 20–25 g body weight) were housed in separate individual, filter-top cages in an air flow, light (12 h light/12 h dark cycle) and temperature (21°C) controlled room, with free access to food and water. The cis-NF-κBEGFP mouse strain on a C57BL/6 background was kindly provided by Christian Jobin, Chapel Hill, NC, USA, and bred in our in-house facility. In this mouse strain, expression of the enhanced green fluorescent protein (EGFP) is under the transcriptional control of NF-κB cis-elements; therefore, NF-κB binding results in transcription of EGFP (Magness et al., 2004) and levels of EGFP can be used as a marker of the activation of NF-κB.
Experimental model – duration of feeding diets and H/R protocol
Mice were randomly divided into pairs and assigned to a 4 week pair-feeding regime of standard Lieber-DeCarli diet (Sniff Spezialdiäten, Soest, Germany) supplemented with either maltodextrin (control diet) or ethanol 6.3% (v/v) (EtOH-diet) (DeCarli and Lieber, 1967). The amount of ingested ethanol-supplemented diet of one mouse was determined and an equal volume of maltodextrin-supplemented diet was fed to the other mouse. Accordingly, isocaloric feeding of each individual mouse was warranted.
Haemorrhagic shock protocol
Twenty-eight days after feeding the Lieber-DeCarli diet as described, pair-fed mice were randomly allocated to H/R and sham-operated groups (9–11 pairs and 7 pairs respectively). Mice were anaesthetized (Forene® isofluorane, Abott, Wiesbaden, Germany) and their femoral arteries were exposed and cannulated with polyethylene tubing (Sims Portex, Hythe, UK). Then, haemorrhagic shock was induced by withdrawing blood via one catheter in a heparinized syringe (10 units) over 5 min to a mean arterial pressure (MAP) of 32 mmHg. Systemic BP and heart rate were monitored via the second catheter, connected to a pressure transducer (BPA 400; Micro-Med, Louisville, KY, USA). A MAP of 32 ± 2 mmHg was maintained by reinfusion or withdrawal of small volumes of shed blood for 90 min, and then mice were resuscitated with 60% of the maximal shed blood volume as whole blood plus 50% of the maximal shed blood volume as Ringer's solution through the arterial catheter (Lehnert et al., 2008). Successful resuscitation was defined by the restoration of normal BP. Body temperature was monitored with a rectal probe and maintained at 37°C with a warming lamp. After resuscitation, catheters were removed, vessels were ligated and the wounds were closed. Sham-operated animals underwent the same surgical procedures but without withdrawal of blood.
Killing and collection of tissue and blood samples
Two or 24 h after the end of resuscitation, mice were anaesthetized and killed by exsanguination. From each mouse, portal venous blood was collected; the liver was flushed with normal Ringer solution and then excised and weighed. A section of the liver's median lobe was embedded in Tissue-Tek® O.C.T™ Compound (Sakura Finetek, Helsinki, Finland) for cryo-sectioning. The remaining liver lobes were snap-frozen and stored at −80°C. The left lobe was infused and fixed with 10% neutral buffered formalin and subsequently embedded in paraffin, sectioned (7 μm) and stained with haematoxylin–eosin (HE). Analysis of data at 2 and 24 h after resuscitation from sham-operated animals that were fed either control- or the EtOH-diet revealed no differences between treatments. Therefore, the data from these animals were pooled and only one sham group is presented throughout the manuscript.
Examination of liver injury – steatosis, necrosis and enzyme release
Cryo-sections of liver (5 μm) were fixed with 10% formalin. Hepatic steatosis was assessed using Oil Red O reagent. Photographs were taken using Axio Observer.Z1 microscope (Carl Zeiss MicroImaging, Jena, Germany). The extent of labelling was determined in 10 random fields for each liver by cell explorer software (Bioscitec, Frankfurt, Germany) and was expressed as percentage of the total area of the section. The liver was weighed immediately after excision to calculate liver weight/body weight ratio. Liver necrosis was assessed in HE-stained tissue, Serum was stored at −80°C for measurement of enzyme activities of aspartate aminotransferase (AST) and LDH using a dry chemical system (Kodak Ektachem DT 60 Analyzer and DTSC II, Johnson & Johnson Clinical Diagnostics, Neckargemünd, Germany), as described previously (Prinzinger and Misovic, 2010).
Systemic inflammation
Serum cytokine concentrations
IL-6 and IL-10 in serum samples was measured using flow cytometry with FACSCalibur (BD Biosciences, Heidelberg, Germany) and the Mouse IL-6 and IL-10 Flex Set with a cytometric bead array according to the manufacturer's instructions (BD Biosciences).
EGFP and CD11b cell surface expression in peripheral blood leukocytes
Flow cytometry was performed with RBC-depleted peripheral blood mononuclear cells that were stained with combinations of anti-CD11b, -CD14 and -CD16/32 (BD Biosciences, Heidelberg, Germany) to detect EGFP and CD11b expression on the surface of leukocytes, as described in detail elsewhere (Kalaydjiev et al., 2008). Data were analysed using CellQuest Pro (BD Biosciences). Immediate handling of constantly cooled (4°C) samples and the control-fed sham group decreased the possibility of a false positive result, i.e., green fluorescence not related to H/R or the EtOH-diet.
Local pro-inflammatory changes
Hepatic expression of inflammatory NF-κB target genes and local secretion of IL-6
Snap-frozen liver tissue was homogenized (Precellys24; Bertin Technologies, Montigny-le-Bretonneux, France) and total RNA was extracted using the RNeasy-system (Qiagen, Hilden, Germany), subsequently reverse transcribed with Omniscript® (Qiagen) using the AffinityScript PCR cDNA Synthesis Kit (Strategene, La Jolla, CA, USA) according to the manufacturer's instructions. qRT PCR reactions were performed and analysed as described in Relja et al. (2012) using Stratagene MX3005p QPCR system (Stratagene) with specific primers for target genes (Table 1) and 18S ribosomal RNA as reference gene, all purchased from SABiosciences (SuperArray, Frederick, MD, USA). For assessment of whole-liver IL-6 content, thawed tissue was homogenized (as above) in lysis buffer (Lehnert et al. 2008) at 4°C, followed by centrifugation for 10 min at 4°C at 17 000× g. Supernatants were stored at −80°C for later analysis. Concentrations of hepatic IL-6 were determined using a Quantikine Mouse-IL-6 elisa kit according to the manufacturer's instructions (R&D Systems, Wiesbaden-Nordenstadt, Germany). The elisa 96-well micro titre plates were analysed using a microplate reader Bio-Tek Ceres UV900C (Bio-Tek, Winooski, VT, USA).
Table 1.
Primers used for qRT-PCR in liver tissue
| Gene name | RefSeq accession No. | UniGene No. |
|---|---|---|
| mouse Il6 | NM_031168.1 | Mm.1019 |
| mouse Icam1 | NM_010493.2 | Mm. 435508 |
| mouse Mmp9 | NM_013599.2 | Mm. 4406 |
| mouse Tnf | NM_013693.2 | Mm. 1293 |
Icam1, intercellular adhesion molecule 1.
Expression of adhesion molecules in liver sinusoids
Immunohistochemistry was performed using rat anti-mouse CD54 (ICAM-1) monoclonal antibody (dilution: 1:75, 1 h room temperature; Biozol, Eching, Germany) as described in Relja et al. (2012). Axio Observer.Z1 microscope (Carl Zeiss MicroImaging) was used to capture (×200) and analyse the immunolabelled specimens. The extent of staining in the liver lobule was determined in randomly selected fields (10 per section) by an observer who had no knowledge of the group allocation: 1+ (very low), 2+ (low), 3+ (strong), 4+ (very strong). Data were pooled to determine means.
Visualization and quantification of cis-NF-κBEGFP transcriptional induction and NF-κB/p65 in liver tissue
EGFP in tissue specimens from cis-NF-κB EGFP mice was detected by epifluorescence microscopy. Tissue samples were fixed with 10% buffered zinc-formalin for 24 h and paraffin-embedded. Sections were cut 5 μm and EGFP expression was visualized by using the FITC reflector of Axio Observer.Z1 (Carl Zeiss MicroImaging) with identical exposure times for each data point. Localization and cellular expression pattern of activated NF-κB/GFP was further assessed by immunocytochemistry. Liver sections were fixed and cut as described and then incubated with anti-GFP antibody (1:400, 60 min, RT; Abcam, Cambridge, UK). An anti-rabbit HRP-linked secondary antibody (30 min, RT, Histofine; Nichirei, Tokyo, Japan) and diaminobenzidine (Peroxidase EnVision Kit, DakoCytomation, Hamburg, Germany) was used to detect specific binding, followed by counterstaining with haematoxylin. GFP-positive cells were counted in a total of 25 high power (×500) fields per mouse by observer without knowledge of the group allocation. Non-parenchymal cells (NPCs) were identified by morphology and localization in the liver acinus. Quantification of activated NF-κB was performed by Western blot analysis of the phosphorylated p65 subunit and GFP, using rabbit anti-p-65 antibody (Cell Signaling, Frankfurt, Germany) and the anti-GFP antibody used for immunohistochemistry as described previously (Relja et al., 2012). In brief, total liver proteins were extracted, as described earlier. Lysates (100 μg protein) were separated by electrophoresis on 8% polyacrylamide-SDS gels and transferred to PVDF membranes (Starlab International, Ahrensburg, Germany). Mouse anti-β-actin antibody (Sigma, Taufkirchen, Germany) served as internal control. Proteins were detected, films were digitized using Fusion Fx7 (PEQLAB Biotechnology, Erlangen, Germany), and the integrated density of individual bands was determined using BIO-1D Advanced (PEQLAB Biotechnology). By densitometric measurements using the same software, the amount of protein expression was normalized to β-actin. Phosphorylated p65 from whole cell extracts provides only limited evidence for NF-κB activation, but in the mice used here, the EGFP is under the transcriptional control of NF-κB and thus serves as an indicator of NF-κB activation. Further evidence for NF-κB activation was provided by the quantification of ICAM-1 (Figure 7) and MMP 9 (Figure 10D) that are predominantly under the transcriptional control of NF-κB (Gupta et al., 2010).
Figure 7.
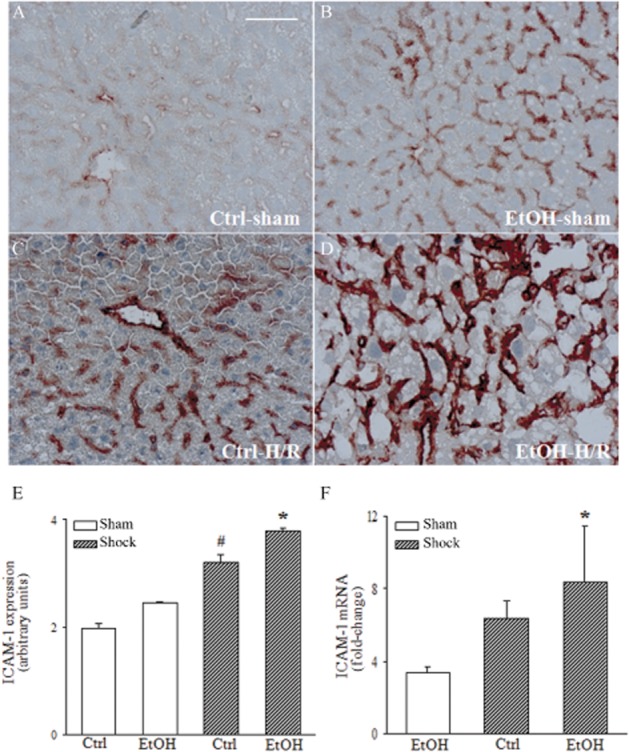
The EtOH-diet (EtOH) intensifies ICAM-1 activity and gene expression at 2 h after H/R. Immunohistochemistry for ICAM-1 was performed on liver cryo-sections (panels A–D). ICAM-1 positive areas were quantified using a score, ranging from 0 (no activity) to 4 (high activity) (panel E). Hepatic mRNA levels were measured as described (F, *P < 0.05 vs. sham). n = 7–9; bar equals 50 μm.
Figure 10.
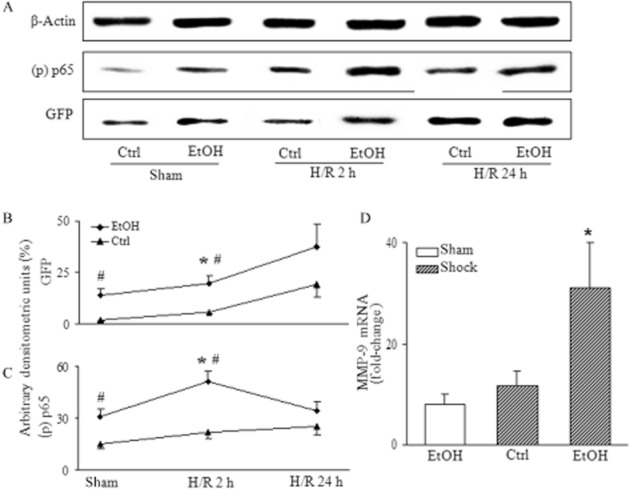
H/R enhanced NF-κB/p65 phosphorylation and subsequent NF-κB-driven GFP expression, in mice fed the EtOH-diet. Total protein was extracted from livers of H/R mice fed the EtOH-diet, 2 h or 24 h after H/R and analysed by Western blot. In panel (A), lanes 1–2 represent mice after sham operation, or at 2 h (lanes 3–4) and at 24 h (lanes 5–6) after H/R. Densitometric measurements of GFP, (p) p65, normalized to β-actin are given (*P < 0.05 vs. 24 h H/R, sham; #P < 0.05 vs. Ctrl, panels B and C). n = 7–11, representative gel from 3 experiments is shown. At 2 h after H/R, MMP-9 gene expression was determined as described in Figure 5 (*P < 0.05 vs. all, panel D); n = 7–9 per group.
Data analysis
Data are presented as mean ± SEM. Differences between groups were determined by anova followed by post hoc analysis (Student–Newman–Keuls), or by Kruskal–Wallis post hoc testing. Fisher's exact test was used for analysis of survival data. A P-value of less than 0.05 was considered significant.
Results
Chronic ethanol feeding results in hepatomegaly and marked steatosis
Feeding mice with the ethanol-containing Lieber-DeCarli diet (EtOH-diet) for 28 days increased the liver/body weight ratio from 4.3 ± 0.1 (mice fed control diet, n = 27) to 5.9 ± 0.2 (ethanol-fed mice, n = 27, P < 0.01) indicating a relative increase in liver weight. This increase of liver weight was due to the marked steatotic changes induced by the EtOH-diet over 4 weeks. These changes were not present after feeding the control diet (32.2 vs. 6.3% of Oil Red O positive area, respectively, P < 0.01; Figure 1).
Figure 1.
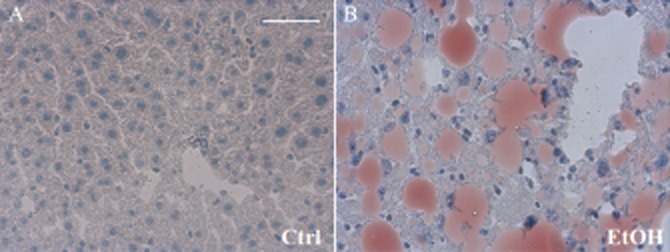
Oil Red O staining reveals hepatic steatosis after the EtOH-diet. Four weeks after feeding mice either an ethanol-containing or a control diet (pair feeding), hepatic cryo-sections were obtained. Marked steatosis was present, as shown by severe accumulation of Oil Red O-stained fat vacuoles in pericentral and periportal zones of liver parenchyma (panel B, Table 2). Bar equals 50 μm.
Haemodynamic characteristics of haemorrhagic shock and survival rate
As ethanol itself may have an effect on vasoregulation (Patterson-Buckendahl et al., 2005), we compared the baseline MAP among the diet groups. Animals on the EtOH-diet showed an elevated MAP before the H/R procedure, compared with pair-fed controls (Table 2); resuscitation protocol restored MAP to identical levels (Table 2). The volume of blood removed to maintain a hypotension of 32 mmHg was lower in ethanol-fed mice, a trend that did not reach statistical significance (P = 0.09; Table 2). Survival rates were determined 24 h after H/R. One out of 15 of the mice fed control diet died within 24 h after H/R, whereas 6 out of 15 of the EtOH-diet mice died after H/R (P < 0.04; Table 2).
Table 2.
Characteristics of BP controlled H/R model
| Ctrl diet | EtOH diet | P-value | |
|---|---|---|---|
| MAP – before shock (mmHg) | 70 ± 1.9 | 75 ± 1.7* | 0.03 |
| MAP – during sham (mmHg) | 75 ± 2.4 | 79 ± 1.9 | 0.16 |
| MAP – during shock (mmHg) | 31 ± 0.4 | 30 ± 0.3 | 0.97 |
| MAP – at 15 min reperfusion (mmHg) | 50 ± 3.9 | 50 ± 3.0 | 0.96 |
| MAP – end of reperfusion (mmHg) | 74 ± 5.2 | 80 ± 4.4 | 0.14 |
| Shed blood volume (mL) | 0.83 ± 0.03 | 0.75 ± 0.03 | 0.09 |
| Mortality at 24 h after H/R (%) | 6.7 (1 out of 15) | 40.0* (6 out of 15) | 0.04 |
P < 0.05 versus Ctrl. Values shown are means ± SEM. MAP (mean arterial BP, in mmHg) was measured during sham operation and H/R protocols. Group sizes: 7–11. Statistical analysis of MAP was performed using Student's t-test. Mortality was analysed with Fisher's exact test.
Hepatic injury after H/R – necrosis and cellular damage
Liver sections from mice fed the control diet revealed some areas of coagulative necrosis at 2 h after H/R (Figure 2C). H/R-induced necrosis was augmented in mice on the EtOH-diet, as indicated by nuclear breakdown and cellular enlargement in predominantly midzonal and pericentral areas of liver parenchyma (Figure 2D). In control-fed mice, our H/R procedure raised the serum activity of AST – an indicator of liver damage at 2 h after H/R as compared with sham-operated animals (P < 0.05; Figure 3A). In mice on the EtOH-diet, this raised AST at 2 h after H/R, was increased still further (Figure 3A). Twenty-four hours after H/R, AST declined to near basal levels in both treatment groups. LDH, a marker of general cell damage, was elevated in animals on the EtOH-diet 2 h after H/R compared with mice fed the control diet (P < 0.05; Figure 3B). These results indicated that the EtOH-diet caused cellular injury, soon after H/R.
Figure 2.
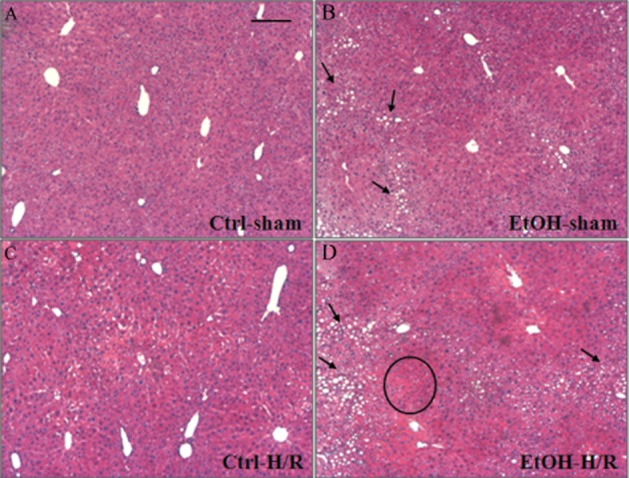
Profound necrosis after H/R in fatty liver from mice on the EtOH-diet. After feeding either the EtOH-diet (EtOH) or the control diet (Ctrl), mice were subjected to H/R or sham operation. Representative photomicrographs of paraffin-embedded liver sections stained with haematoxylin and eosin are given. A typical necrotic area (circle) and cytosolic lipid droplets accumulated as large fat vacuoles in hepatocytes (circle, panels B and D) are marked. No signs of liver damage were observed in sham-operated, control mice (A). Bar equals 100 μm.
Figure 3.
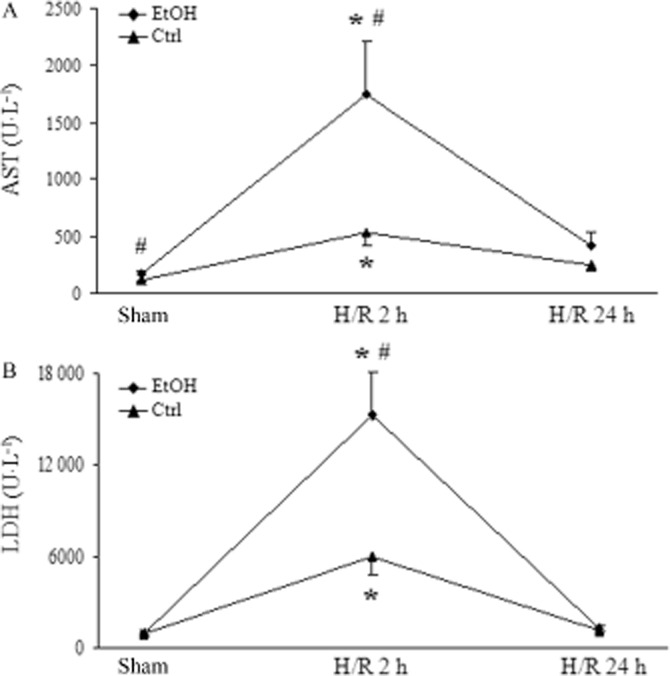
The EtOH-diet increases hepatocellular injury at 2 and 24 h after H/R. Serum activity of AST and LDH was determined in mice on the EtOH-diet, after sham procedure or 2 and 24 h after H/R Ctrl: mice fed control diet, EtOH: the EtOH-diet mice, *P < 0.05 versus 24 h H/R and sham; #P < 0.05 versus Ctrl, n = 7–11.
Systemic inflammatory changes after the EtOH-diet, before H/R
Elevated levels of circulating IL-6 and IL-10
The EtOH-diet and H/R caused a systemic inflammatory and counter-inflammatory response, as determined by circulating levels of IL-6 and IL-10, 2 h after H/R. IL-6 levels rose markedly in the EtOH-diet+H/R group compared with the Ctrl+H/R and EtOH-diet+sham groups (P < 0.05; Figure 4A). Serum IL-6 was also significantly increased in the EtOH-diet group after sham operation (Figure 4A). Systemic levels of the anti-inflammatory cytokine, IL-10, were also elevated 2 h after H/R in EtOH-diet mice, compared with corresponding values from the control+H/R mice and also compared with mice that underwent sham operation (P < 0.05; Figure 4B).
Figure 4.
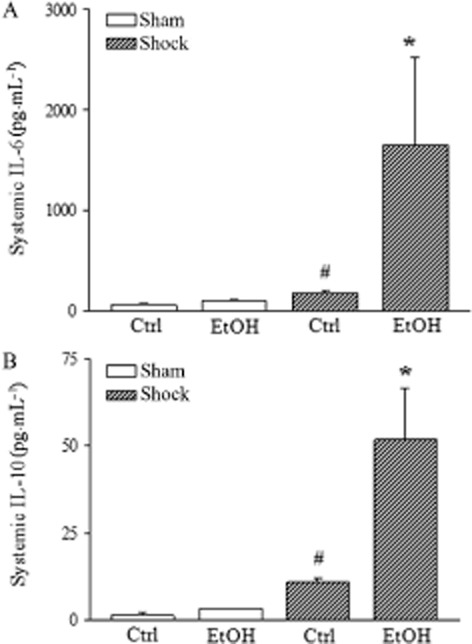
Systemic inflammatory changes are substantially aggravated 2 h after H/R in combination with the EtOH-diet (EtOH). Serum IL-6 and IL-10 levels were measured by CBA after H/R in the EtOH-diet mice and mice fed control diet (Ctrl). Data are presented as means with SEM. (A) serum IL-6 levels; serum IL-10 concentration (panel B); *P < 0.05 versus all, #P < 0.05 versus Ctrl, n = 7–9.
The EtOH-diet primed blood neutrophil granulocytes (PMNL)
Simultaneous analysis of the expression of the integrin Mac-1 (CD11b/CD18) and EGFP in PMNL from peripheral blood samples demonstrated an elevated CD11b expression in EtOH-diet mice after H/R, compared with the Ctrl+H/R group (P < 0.05; Figure 5F, an effect also present after the sham procedure (P < 0.05; Figure 5F). The expression of EGFP was significantly increased after H/R in EtOH-diet mice in the CD11b positive cell population, compared to values in mice fed the control diet (Figure 5E); EGFP was not different after sham operation (Figure 5E). Thus, the EtOH-diet, by itself, caused a considerable pre-activation of circulating neutrophils, an effect that was markedly enhanced by H/R.
Figure 5.
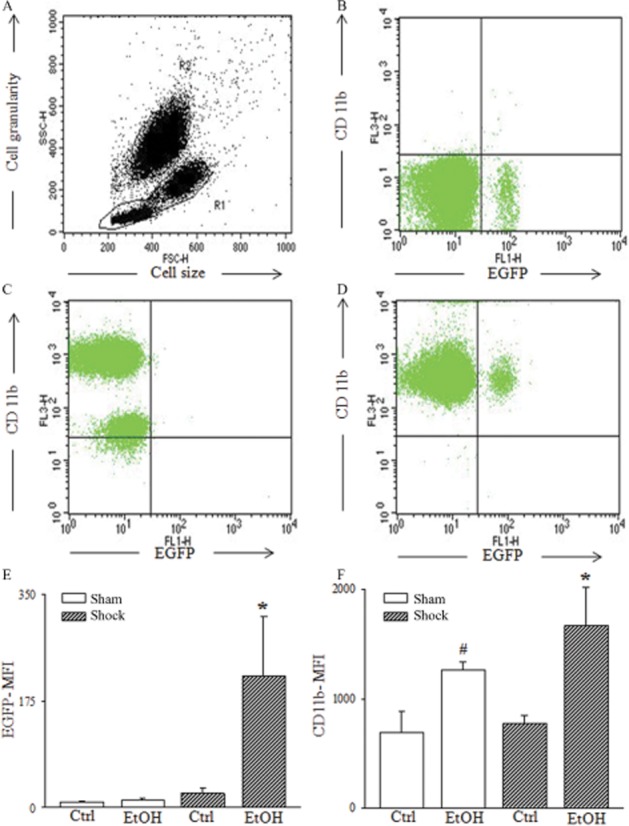
The EtOH-diet increases expression of CD11b on PMNL cell surface and induces cis-NF-κBEGFP transcriptional activity at 2 h following H/R. Representative FACS dot plots are given. In (A), whole leukocyte population was analysed and PMNL were gated (R2) according to the forward/side scatter properties (FSC, SSC) of PMNL. In (B), CD11b (FL-3) versus EGFP (FL-1) of isotype control is presented. In (C), CD11b versus EGFP of Ctrl-H/R is given. A subset of PMNL is both EGFP and CD11b positive, shown in panel (D). Mean fluorescence intensity (MFI) of EGFP (panel E), *P < 0.05 versus all. CD11b-MFI (panel F); *, #P < 0.05 versus Ctrl, n = 7–11.
Hepatic inflammation
Gene expression and release of pro-inflammatory NF-κB responsive cytokines in liver tissue
Semi-quantitative real-time PCR analysis showed a significant elevation of IL-6 in hepatic tissue from mice on the EtOH-diet, after H/R compared with sham-operated mice (Figure 6A). Hepatic gene expression of TNF-α was also increased after EtOH-diet+H/R compared to the control group (P < 0.05; Figure 6B). The EtOH-diet induced a hepatic inflammatory response as indicated by increased hepatic IL-6 release, compared with mice fed control diet (P < 0.05; Figure 6C). Two hours after H/R, EtOH-diet mice had higher hepatic IL-6 levels than those in mice undergoing H/R that were fed control-diet (P < 0.05; Figure 6C).
Figure 6.
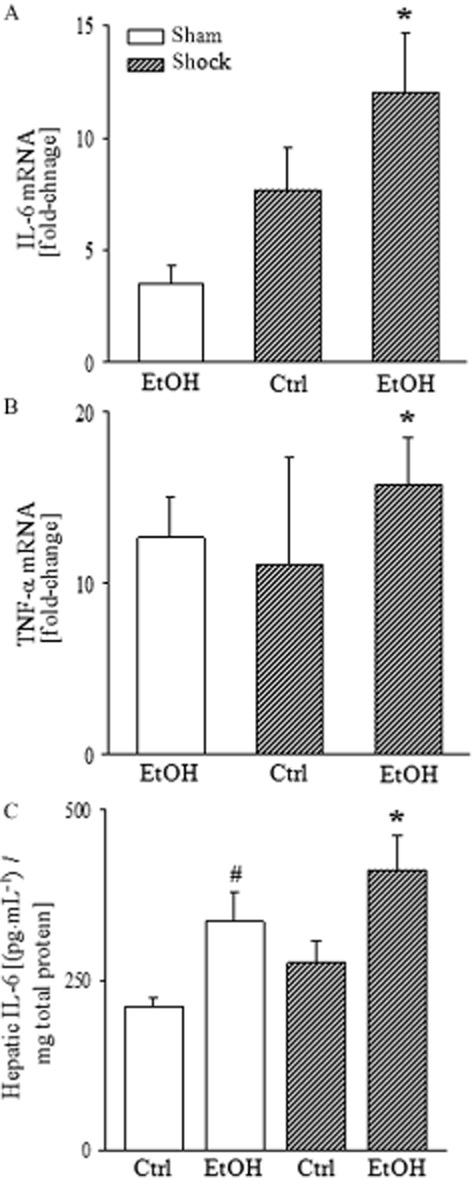
H/R increases the EtOH-diet-induced gene expression and release of pro-inflammatory cytokines. Total RNA was extracted from liver tissue, harvested 2 h after H/R, reverse-transcribed and amplified by RT-PCR using specific IL-6 and TNF-α primer. After normalization to 18S rRNA expression, mRNA levels were determined as fold increase compared with sham-operated group fed the EtOH-diet (EtOH). Gene expressions of IL-6 (panel A, *P < 0.05 vs. sham), and TNF-α (panel B, *P < 0.05 vs. Ctrl) are shown. elisa for mouse IL-6 was performed using isolated total liver proteins at 2 h after H/R (panel C, *P < 0.05 vs. Ctrl; #P < 0.05 vs. Ctrl); n = 7–9.
Hepatic CD54 (ICAM-1) expression increased strongly in EtOH-diet mice after H/R (Figure 7B), compared to sham-operated mice (Figure 7D; P < 0.05) and mice fed control diet after H/R (Figure 7B; P < 0.05; Figure 7E), as demonstrated by immunohistochemistry. Parenchyma surrounding central venules and midzonal areas showed the most intense staining (Figure 7B). Corresponding to these results, levels of mRNA for ICAM-1 were strongly enhanced 2 h after H/R in EtOH-diet mice (P < 0.05; Figure 7F). These results show that the EtOH-diet modulated the local early pro-inflammatory response before H/R, as assessed by the hepatic levels of ICAM-1, IL-6 and TNF-α.
Effect of the EtOH-diet and H/R on NF-κB transcriptional activity in livers from cis-NF-κBEGFP mice
Hepatic topography and cellular activation pattern of EGFP in vivo
To assess the time and site-specific expression of EGFP correlating with NF-κB activity after H/R and the EtOH-diet, untreated liver sections were analysed by epifluorescence microscopy at 2 and 24 h after resuscitation. Low levels of GFP were detected at 2 h after H/R compared with sham-operated mice (Figure 8C vs. 8A), but 24 h after H/R, there was an increase in hepatic fluorescence denoting the presence of NF-κB-dependent EGFP. Feeding with the EtOH-diet further increased the GFP signal (Figure 8F) compared with mice fed control diet (Figure 8E). GFP immunolabelling confirmed these findings and revealed that GFP-stained cells were mainly located in midzonal areas (Figure 9I,J). Hence, GFP expression strongly increased after the EtOH-diet, at 2 and 24 h after H/R, compared to mice fed control diet (P < 0.05; Figure 8K). Morphologically, a considerable amount of GFP-positive cells belonged to the NPC fraction. After the sham procedure, feeding with the EtOH-diet was associated with an enhanced percentage of NPC that expressed GFP, compared with mice fed control diet (P < 0.05; Figure 9). This difference was largely attenuated after H/R, indicating an H/R-dependent activation of NPC in mice that received the control diet (Figure 9). These results indicate that the EtOH-diet increased NF-κB activation in the hepatic NPC fraction.
Figure 8.

Topographic mapping and enhanced induction of the cis-NF-κBEGFP transgene in liver tissue after H/R following pair-feeding with the EtOH-diet (EtOH). NF-κB-dependent EGFP expression in livers from cis-NF-κBEGFP mice, prepared as described in Figure 2, was assessed using fluorescence microscopy. Panels A, C and E depict livers that received control diet (Ctrl) and panels B, D and F depict livers from mice that received the EtOH-diet at 2 or 24 h either after sham or H/R procedures. Immunohistochemistry for GFP mirrored NF-κB transcriptional activity of pair-fed mice at 24 h following sham procedure (panels G and H) or H/R (panels I and J). (K) GFP-positive cells were counted (panel K); *P < 0.05 versus 24 h H/R and sham-operated group; #P < 0.05 vs. group fed control diet; §P < 0.05 versus 2 h H/R and sham operation. Bar equals 100 μm (panels A–F) and 200 μm (panels G–J), n = 7–9.
Figure 9.
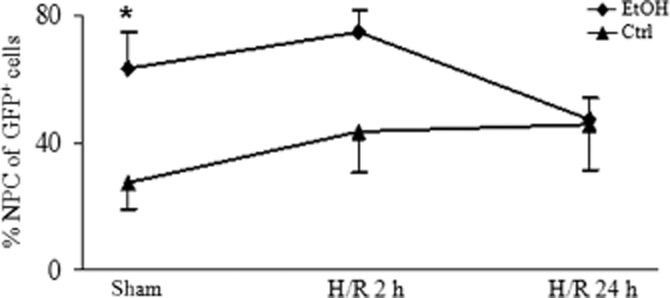
Both feeding the EtOH-diet (EtOH) and H/R independently increases the proportion of NF-κB activated non-parenchymal cells (NPCs). Cellular activation of NF-κB in response to the EtOH-diet and H/R was monitored in cis-NF-κBEGFP mouse livers as described in Figure 8. The percentage of NPC/Kupffer cells in all GFP positive cells is shown. *P < 0.05 versus group fed control diet (Ctrl). Group sizes were 5–9.
Detection and quantification of NF-κB/p65 and NF-κB driven expression of MMP-9
We further characterized the transcriptional activity of NF-κB after H/R by Western blotting. Antibodies specific for GFP and phosphorylated NF-kB/p65 were used in liver homogenates, collected 2 and 24 h after H/R (Figure 10A). After the EtOH-diet, GFP was elevated both after sham and after H/R at 2 and 24 h, compared with mice fed the control diet (P < 0.05; Figure 10B). The early (2 h) and late (24 h) activity of NF-κB/p65 after the EtOH-diet revealed a peak expression of phospho-p65 at 2 h after H/R (P < 0.05; Figure 10C). The EtOH-diet increased the expression of phospho-p65 also after sham procedure and at 2 h and 24 h after H/R, compared with the mice fed control diet (Figure 10C). Analysis of NF-κB controlled MMP-9 gene expression revealed a significant increase in the EtOH-diet group at 2 h after H/R, compared with the control+H/R group and the EtOH-diet group after the sham procedure (Figure 10D). Taken together, these data indicate that NF-κB activation is brought about, at least partially, by phosphorylation of p65 and is markedly enhanced by H/R and by the EtOH-diet. It is predominantly located in NPC at early time points; however, at 24 h after H/R, the activation pattern spreads from NPC to a similar activation in all subsets of hepatic cells and is located mainly midzonally. Thus, H/R strongly modulated the pre-existing effects of feeding with the EtOH-diet on the NF-κB activation pattern.
Discussion and conclusions
There is a large body of evidence that NF-κB plays a crucial role to initiate immune responses after both ethanol consumption and H/R (Ono et al., 2004; Goral et al., 2008; Lau et al., 2009). To evaluate mortality, organ injury, inflammatory response and the time- and cell-/organ-specific activation of NF-κB after both the EtOH-diet and H/R, we applied a NF-κB EGFP reporter gene mouse model, as described previously (Magness et al., 2004; Yang et al., 2005; Kielland and Carlsen, 2010).
We demonstrated that a combination of EtOH-diet and H/R increased mortality and increased liver injury accompanied by elevated levels of systemic and local IL-6 and enhanced hepatic expression of NF-κB-related genes, such as TNF-α, ICAM-1 and MMP-9 (Figures 4A, 6, 7F and 10D). Using the cis-NF-κBEGFP reporter gene mice, we showed a pronounced activation of NF-κB in midzonal and pericentral regions of the liver azinus after H/R (Figures 8F and 9F). NPCs, including Kupffer cells, contribute, at least in part, to this inflammatory response, as 75% of NPC expressed GFP, the indicator of NF-κB activation. However, the EtOH-diet alone induced a profound activation of NPC, even without H/R (Figure 9H). Feeding with the EtOH-diet also contributed to NF-κB associated up-regulation of adhesion molecules in circulating neutrophils compared with mice fed control diet at 2 h after H/R. Therefore, feeding with the EtOH-diet may have a priming effect, accounting for the profound pathophysiological changes observed in this experimental setting (Figure 5). However, despite these ethanol-dependent effects on inflammatory response, the pathophysiological features of the H/R procedure remained unchanged (Table 2).
Feeding with the EtOH-diet is a well-established model of chronic ethanol ingestion (Lieber et al., 1989; Perides et al., 2005; Bradford et al., 2008). This voluntary feeding model reliably causes signs of hepatic steatosis, mimicking hepatic changes in humans after chronic ethanol consumption. We also found steatosis, a characteristic feature of liver injury induced by chronic ingestion of ethanol, with subsequent elevation of the liver to body weight ratio as well as a marked increase in Oil Red O positive droplets when compared with the mice that received the isocaloric ethanol-free diet, as described previously (Table 2 and Figure 1B) (Bradford et al., 2008).
The immunomodulatory effect of the EtOH-diet in our H/R model was characterized by elevated systemic and hepatic IL-6 levels, as well as enhanced gene expression of IL-6 and TNF-α (Figures 4A and 6). Correspondingly, ICAM-1 expression was highest in mice on the EtOH-diet after H/R (Figure 7B,E,F). These changes were associated with a higher mortality and an elevated transaminase and LDH release, signifying hepatic and general cellular damage, after H/R in EtOH-diet mice (Figure 3 and Table 2). IL-6 expression is elevated after H/R in rodents and hepatocellular damage after H/R is largely prevented either by IL-6 antibody treatment or using IL-6 knockout mice (Meng et al., 2001; Ono et al., 2004). Apart from the effects of H/R, diets with ethanol increase systemic as well as hepatic levels of IL-6 in rats (McClain et al., 1993; Iimuro et al., 1997; Yin et al., 1999). TNF-α also contributes substantially to fatty liver after chronic ethanol feeding, an effect that is attenuated after treatment with anti-TNF-α antibodies (Iimuro et al., 1997; Goral et al., 2008; Mandrekar and Szabo, 2009). CD54 (ICAM-1), an NF-κB-related adhesion molecule, is necessary for neutrophil recruitment to the site of tissue injury and is elevated after chronic ethanol ingestion and after H/R (Zingarelli, 2005; Lehnert et al., 2006; 2006,2008; Relja et al., 2012). CD11b/CD18 (Mac-1) mediates adhesion of PMNL to vascular endothelial cells after immune stimulation such as trauma. It is shuttled in specific granules to the neutrophil surface, and after adhesion to the vessel wall and infiltration into tissues, PMNL release different mediators, chemokines or reactive oxygen species that cause accumulation of additional immune cells and further tissue damage (Marzi et al., 1995; Jaeschke, 2006; Ramaiah and Jaeschke, 2007; DiStasi and Ley, 2009; Henrich et al., 2011; Seitz et al., 2011). The EtOH-diet in our model resulted in increased up-regulation of CD11b/CD18 (Mac-1), associated with NF-κB, suggesting a pre-activation or ‘priming’ of PMNL (Figure 5). Chronic ethanol consumption compromises the gut barrier function with concomitant endotoxemia, release of damage-associated molecular patterns and pathogen-associated molecular patterns that may well contribute to the exaggerated inflammatory changes seen in our model (Wheeler, 2003; Goral et al., 2008; Chen and Nunez, 2010; Zhang et al., 2010).
Although NF-κB is known as an important transcription factor after H/R as well as after chronic ethanol feeding, there are only a few studies that further evaluate its role in a model combining both conditions (Uesugi et al., 2001; Ono et al., 2004). This is the first report, to our knowledge, that localizes NF-κB activation predominantly to a midzonal/pericentral region after H/R in mice receiving 4 weeks of the EtOH-diet (Figure 8). In these mice, before H/R, up to 60% of all EGFP-positive cells, signifying NF-κB activation, belong to the NPC fraction (Figure 9) (Thurman, 2000). Adding H/R to the EtOH-diet mice did not induce a further increase, but H/R did raise the proportion of NPCs positive for EGFP after feeding the control diet (Figure 9). These results support the hypothesis that early inflammation after H/R derives from NF-κB activation in NPC, predominantly in more hypoxic regions of the liver lobule. Our topographical characterization of NF-κB activation in midzonal/pericentral regions coincides with the suggestion from previous reports, that both ATP depletion and coagulative necrosis also demonstrate a pericentral pattern (Paxian et al., 2003). Twenty-four hours after H/R, the proportion of EGFP-positive NPCs declined, with an overall increase in the number of EGFP-positive cells, possibly due to the spread of inflammation to hepatocytes, at later time points (Figure 9).
Thus, local and systemic inflammation was exacerbated by the combination of both the EtOH-diet and H/R. By contrast, results from our group using a binge-like, single dose ingestion of ethanol, 12 h before H/R strongly attenuated the early systemic and hepatic inflammation and reduced mortality in a model of H/R (Relja et al., 2012). Taken together, our results clearly identify different and partially opposing effects of chronic and acute alcohol intake on the liver, as an important organ to modulate post-traumatic inflammation, with NF-κB representing an important player to orchestrate the modulation of inflammation, as described in this study. In conclusion, our double-hit model of chronic ethanol ingestion and haemorrhagic shock provides evidence that the NF-κB pathway is strongly involved in the systemic inflammatory changes and the subsequent hepatic injury and immune response. Further studies will be required to elucidate the cell-specific NF-κB driven contribution to these pathophysiological changes, following the combination of chronic ethanol intake with trauma and haemorrhage.
Acknowledgments
The authors are grateful for the outstanding technical assistance of Jasmin E. Bergmann, Birgit Nagel, Elsie Oppermann, Alexander Schaible, Min-Hong Wang and Kerstin Wilhelm. We thank Prof Barker and Dr Relja for critically reading the manuscript. cis-NF-κBEGFP breeding pairs were provided by Christian Jobin based on a material transfer agreement (University of North Carolina, Chapel Hill, USA). The present study was supported by Deutsche Forschungsgemeinschaft (DFG LE 1346/2-1).
Glossary
- AST
aspartate aminotransferase
- EGFP
enhanced green fluorescent protein
- H/R
haemorrhagic shock and resuscitation
- ICAM-1
intercellular adhesion molecule 1
- PMNL
polymorphonuclear leukocytes
Conflict of interest
None of the authors have any conflict of interest regarding the materials used in the present study.
References
- Akgur FM, Brown MF, Zibari GB, McDonald JC, Epstein CJ, Ross CR, et al. Role of superoxide in hemorrhagic shock-induced P-selectin expression. Am J Physiol Heart Circ Physiol. 2000;279:H791–H797. doi: 10.1152/ajpheart.2000.279.2.H791. [DOI] [PubMed] [Google Scholar]
- Angele MK, Schneider CP, Chaudry IH. Bench-to-bedside review: latest results in hemorrhagic shock. Crit Care. 2008;12:218. doi: 10.1186/cc6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Baue AE, Durham R, Faist E. Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure (MOF): are we winning the battle? Shock. 1998;10:79–89. doi: 10.1097/00024382-199808000-00001. [DOI] [PubMed] [Google Scholar]
- Bradford BU, O'Connell TM, Han J, Kosyk O, Shymonyak S, Ross PK, et al. Metabolomic profiling of a modified alcohol liquid diet model for liver injury in the mouse uncovers new markers of disease. Toxicol Appl Pharmacol. 2008;232:236–243. doi: 10.1016/j.taap.2008.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchi MN, Kimlin E, Walsh M, Donnino MW. Identification and resuscitation of the trauma patient in shock. Emerg Med Clin North Am. 2007;25:623–642. doi: 10.1016/j.emc.2007.06.001. vii. [DOI] [PubMed] [Google Scholar]
- DeCarli LM, Lieber CS. Fatty liver in the rat after prolonged intake of ethanol with a nutritionally adequate new liquid diet. J Nutr. 1967;91:331–336. doi: 10.1093/jn/91.3_Suppl.331. [DOI] [PubMed] [Google Scholar]
- DiStasi MR, Ley K. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009;30:547–556. doi: 10.1016/j.it.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goral J, Karavitis J, Kovacs EJ. Exposure-dependent effects of ethanol on the innate immune system. Alcohol. 2008;42:237–247. doi: 10.1016/j.alcohol.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799:775–787. doi: 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrich D, Zimmer S, Seebach C, Frank J, Barker J, Marzi I. Trauma-activated polymorphonucleated leukocytes damage endothelial progenitor cells: probable role of CD11b/CD18-CD54 interaction and release of reactive oxygen species. Shock. 2011;36:216–222. doi: 10.1097/SHK.0b013e3182236eba. [DOI] [PubMed] [Google Scholar]
- Hierholzer C, Billiar TR. Molecular mechanisms in the early phase of hemorrhagic shock. Langenbecks Arch Surg. 2001;386:302–308. doi: 10.1007/s004230100242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietbrink F, Koenderman L, Rijkers G, Leenen L. Trauma: the role of the innate immune system. World J Emerg Surg. 2006;1:15. doi: 10.1186/1749-7922-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iimuro Y, Gallucci RM, Luster MI, Kono H, Thurman RG. Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology. 1997;26:1530–1537. doi: 10.1002/hep.510260621. [DOI] [PubMed] [Google Scholar]
- Jaeschke H. Mechanisms of liver injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1083–G1088. doi: 10.1152/ajpgi.00568.2005. [DOI] [PubMed] [Google Scholar]
- Jurkovich GJ, Rivara FP, Gurney JG, Fligner C, Ries R, Mueller BA, et al. The effect of acute alcohol intoxication and chronic alcohol abuse on outcome from trauma. JAMA. 1993;270:51–56. [PubMed] [Google Scholar]
- Kalaydjiev S, Franz TJ, Busch DH. Mouse phenotyping: immunology. In: de Angelis MH, Chambon P, Brown S, editors. Standards of Mouse Model Phenotyping. Weinheim, Germany: Wiley-VCH Verlag GmbH; 2008. pp. 237–252. [Google Scholar]
- Kielland A, Carlsen H. Molecular imaging of transcriptional regulation during inflammation. J Inflamm (Lond) 2010;7:20. doi: 10.1186/1476-9255-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AH, Szabo G, Thomson AW. Antigen-presenting cells under the influence of alcohol. Trends Immunol. 2009;30:13–22. doi: 10.1016/j.it.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Lehnert M, Arteel GE, Smutney OM, Conzelmann LO, Zhong Z, Thurman RG, et al. Dependence of liver injury after hemorrhage/resuscitation in mice on NADPH oxidase-derived superoxide. Shock. 2003;19:345–351. doi: 10.1097/00024382-200304000-00009. [DOI] [PubMed] [Google Scholar]
- Lehnert M, Uehara T, Bradford BU, Lind H, Zhong Z, Brenner DA, et al. Lipopolysaccharide-binding protein modulates hepatic damage and the inflammatory response after hemorrhagic shock and resuscitation. Am J Physiol Gastrointest Liver Physiol. 2006;291:G456–G463. doi: 10.1152/ajpgi.00480.2005. [DOI] [PubMed] [Google Scholar]
- Lehnert M, Relja B, Sun-Young Lee V, Schwestka B, Henrich D, Czerny C, et al. A peptide inhibitor of C-Jun N-terminal kinase modulates hepatic damage and the inflammatory response after hemorrhagic shock and resuscitation. Shock. 2008;30:159–165. doi: 10.1097/SHK.0b013e31815dd623. [DOI] [PubMed] [Google Scholar]
- Li TK, Hewitt BG, Grant BF. Is there a future for quantifying drinking in the diagnosis, treatment, and prevention of alcohol use disorders? Alcohol Alcohol. 2007;42:57–63. doi: 10.1093/alcalc/agl125. [DOI] [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM, Sorrell MF. Experimental methods of ethanol administration. Hepatology. 1989;10:501–510. doi: 10.1002/hep.1840100417. [DOI] [PubMed] [Google Scholar]
- McClain C, Hill D, Schmidt J, Diehl AM. Cytokines and alcoholic liver disease. Semin Liver Dis. 1993;13:170–182. doi: 10.1055/s-2007-1007347. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, 3rd, Wang N, Camargo CA., Jr US emergency department visits for alcohol-related diseases and injuries between 1992 and 2000. Arch Intern Med. 2004;164:531–537. doi: 10.1001/archinte.164.5.531. [DOI] [PubMed] [Google Scholar]
- Magness ST, Jijon H, Van Houten Fisher N, Sharpless NE, Brenner DA, Jobin C. In vivo pattern of lipopolysaccharide and anti-CD3-induced NF-kappa B activation using a novel gene-targeted enhanced GFP reporter gene mouse. J Immunol. 2004;173:1561–1570. doi: 10.4049/jimmunol.173.3.1561. [DOI] [PubMed] [Google Scholar]
- Maier RV. Ethanol abuse and the trauma patient. Surg Infect (Larchmt) 2001;2:133–141. doi: 10.1089/109629601750469456. discussion 141–4. [DOI] [PubMed] [Google Scholar]
- Mandrekar P, Szabo G. Signalling pathways in alcohol-induced liver inflammation. J Hepatol. 2009;50:1258–1266. doi: 10.1016/j.jhep.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi I, Bauer M, Secchi A, Bahrami S, Redi H, Schlag G. Effect of anti-tumor necrosis factor alpha on leukocyte adhesion in the liver after hemorrhagic shock: an intravital microscopic study in the rat. Shock. 1995;3:27–33. [PubMed] [Google Scholar]
- Marzi I, Maier M, Herzog C, Bauer M. Influence of pentoxifylline and albifylline on liver microcirculation and leukocyte adhesion after hemorrhagic shock in the rat. J Trauma. 1996;40:90–96. doi: 10.1097/00005373-199601000-00017. [DOI] [PubMed] [Google Scholar]
- Meng ZH, Dyer K, Billiar TR, Tweardy DJ. Essential role for IL-6 in postresuscitation inflammation in hemorrhagic shock. Am J Physiol Cell Physiol. 2001;280:C343–C351. doi: 10.1152/ajpcell.2001.280.2.C343. [DOI] [PubMed] [Google Scholar]
- Nau C, Wutzler S, Dorr H, Lehnert M, Lefering R, Laurer H, et al. Liver cirrhosis but not alcohol abuse is associated with impaired outcome in trauma patients – a retrospective, multicentre study. Injury. 2013;44:661–666. doi: 10.1016/j.injury.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Ono M, Yu B, Hardison EG, Mastrangelo MA, Tweardy DJ. Increased susceptibility to liver injury after hemorrhagic shock in rats chronically fed ethanol: role of nuclear factor-kappa B, interleukin-6, and granulocyte colony-stimulating factor. Shock. 2004;21:519–525. doi: 10.1097/01.shk.0000126905.75237.07. [DOI] [PubMed] [Google Scholar]
- Partrick DA, Moore FA, Moore EE, Barnett CC, Jr, Silliman CC. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. New Horiz. 1996;4:194–210. [PubMed] [Google Scholar]
- Patterson-Buckendahl P, Kubovcakova L, Krizanova O, Pohorecky LA, Kvetnansky R. Ethanol consumption increases rat stress hormones and adrenomedullary gene expression. Alcohol. 2005;37:157–166. doi: 10.1016/j.alcohol.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Paxian M, Bauer I, Rensing H, Jaeschke H, Mautes AE, Kolb SA, et al. Recovery of hepatocellular ATP and ‘pericentral apoptosis’ after hemorrhage and resuscitation. FASEB J. 2003;17:993–1002. doi: 10.1096/fj.02-0624com. [DOI] [PubMed] [Google Scholar]
- Perides G, Tao X, West N, Sharma A, Steer ML. A mouse model of ethanol dependent pancreatic fibrosis. Gut. 2005;54:1461–1467. doi: 10.1136/gut.2004.062919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinzinger R, Misovic A. Age-correlation of blood values in the rock pigeon (Columba livia) Comp Biochem Physiol A Mol Integr Physiol. 2010;156:351–356. doi: 10.1016/j.cbpa.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Ramaiah SK, Jaeschke H. Hepatic neutrophil infiltration in the pathogenesis of alcohol-induced liver injury. Toxicol Mech Methods. 2007;17:431–440. doi: 10.1080/00952990701407702. [DOI] [PubMed] [Google Scholar]
- Relja B, Schwestka B, Lee VS, Henrich D, Czerny C, Borsello T, et al. Inhibition of c-Jun N-terminal kinase after hemorrhage but before resuscitation mitigates hepatic damage and inflammatory response in male rats. Shock. 2009;32:509–516. doi: 10.1097/SHK.0b013e3181a2530d. [DOI] [PubMed] [Google Scholar]
- Relja B, Hohn C, Bormann F, Seyboth K, Henrich D, Marzi I, et al. Acute alcohol intoxication reduces mortality, inflammatory responses and hepatic injury after haemorrhage and resuscitation in vivo. Br J Pharmacol. 2012;165:1188–1199. doi: 10.1111/j.1476-5381.2011.01595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz DH, Perl M, Liener UC, Tauchmann B, Braumuller ST, Bruckner UB, et al. Inflammatory alterations in a novel combination model of blunt chest trauma and hemorrhagic shock. J Trauma. 2011;70:189–196. doi: 10.1097/TA.0b013e3181d7693c. [DOI] [PubMed] [Google Scholar]
- Szabo G, Mandrekar P. A recent perspective on alcohol, immunity, and host defense. Alcohol Clin Exp Res. 2009;33:220–232. doi: 10.1111/j.1530-0277.2008.00842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RG. Sex-related liver injury due to alcohol involves activation of Kupffer cells by endotoxin. Can J Gastroenterol. 2000;14(Suppl. D):129D–135D. doi: 10.1155/2000/735262. [DOI] [PubMed] [Google Scholar]
- Uesugi T, Froh M, Arteel GE, Bradford BU, Gabele E, Wheeler MD, et al. Delivery of IkappaB superrepressor gene with adenovirus reduces early alcohol-induced liver injury in rats. Hepatology. 2001;34:1149–1157. doi: 10.1053/jhep.2001.29400. [DOI] [PubMed] [Google Scholar]
- Wheeler MD. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res Health. 2003;27:300–306. [PMC free article] [PubMed] [Google Scholar]
- Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31:1544–1549. doi: 10.1016/s0891-5849(01)00748-1. [DOI] [PubMed] [Google Scholar]
- Yang L, Magness ST, Bataller R, Rippe RA, Brenner DA. NF-kappaB activation in Kupffer cells after partial hepatectomy. Am J Physiol Gastrointest Liver Physiol. 2005;289:G530–G538. doi: 10.1152/ajpgi.00526.2004. [DOI] [PubMed] [Google Scholar]
- Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingarelli B. Nuclear factor-kappaB. Crit Care Med. 2005;33:S414–S416. doi: 10.1097/01.ccm.0000186079.88909.94. [DOI] [PubMed] [Google Scholar]