

Abstract
BACKGROUND AND PURPOSE
Paliperidone is an active metabolite of the second-generation atypical antipsychotic, risperidone recently approved for the treatment of schizophrenia and schizoaffective disorder. Because paliperidone differs from risperidone by only a single hydroxyl group, questions have been raised as to whether there are significant differences in the effects elicited between these two drugs.
EXPERIMENTAL APPROACH
We compared the relative efficacies of paliperidone versus risperidone to regulate several cellular signalling pathways coupled to four selected GPCR targets that are important for either therapeutic or adverse effects: human dopamine D2, human serotonin 2A receptor subtype (5-HT2A), human serotonin 2C receptor subtype and human histamine H1 receptors.
KEY RESULTS
Whereas the relative efficacies of paliperidone and risperidone were the same for some responses, significant differences were found for several receptor-signalling systems, with paliperidone having greater or less relative efficacy than risperidone depending upon the receptor–response pair. Interestingly, for 5-HT2A-mediated recruitment of β-arrestin, 5-HT2A-mediated sensitization of ERK, and dopamine D2-mediated sensitization of adenylyl cyclase signalling, both paliperidone and risperidone behaved as agonists.
CONCLUSIONS AND IMPLICATIONS
These results suggest that the single hydroxyl group of paliperidone promotes receptor conformations that can differ from those of risperidone leading to differences in the spectrum of regulation of cellular signal transduction cascades. Such differences in signalling at the cellular level could lead to differences between paliperidone and risperidone in therapeutic efficacy or in the generation of adverse effects.
Keywords: schizophrenia, antipsychotics, GPCR, functional selectivity, biased agonism, dopamine, serotonin, histamine
Introduction
Paliperidone is a second-generation antipsychotic drug (APD) recently approved for the treatment of schizophrenia and schizoaffective disorder. In contrast to first-generation (typical) APDs, second-generation APDs have lower risks of extrapyramidal side effects and tardive dyskinesia and are referred to as ‘atypical’ APDs. In addition, there is some evidence, although controversial, that the atypical APDs may have improved efficacy for the treatment of cognition and some of the negative symptoms of schizophrenia, such as apathy, anhedonia and social withdrawal. However, there is a higher risk of weight gain and metabolic syndrome with atypical APD use. Although antagonism or weak efficacy at dopamine D2 receptors appears essential for antipsychotic activity, the complete molecular mechanism that underlies the therapeutic efficacy of atypical APDs is still unknown. In recent years, several theories have been postulated to account for atypicality, including that atypical APDs have (i) higher affinity for serotonin 2A receptor subtype (5-HT2A) receptors than for dopamine D2 receptors (Meltzer hypothesis), (ii) faster off-rate of binding to D2 receptors and (iii) agonism at presynaptic versus antagonism at post-synaptic D2 receptors (as seen with aripiprazole; for a review, see MacDonald and Bartolome, 2010). Atypical APDs bind to a variety of receptor subtypes [including dopamine D2, 5-HT2A, serotonin 2C receptor subtype (5-HT2C) and histamine H1, among others]. As schizophrenia is a multidimensional disease likely with several aetiologies and pathologies, action at a variety of receptor targets may be an important characteristic for effective therapeutic treatment (Gray and Roth, 2007; Wong et al., 2008).
Paliperidone is the major active metabolite of another, well utilized, atypical APD, risperidone, differing by only a single hydroxyl group. With such high structural similarity, it is reasonable to question whether treatment with paliperidone differs substantively from that of treatment with risperidone (Citrome, 2007; ‘Paliperidone: new drug’, ; Dolder et al., 2008; Dopheide, 2008). Clinical differences between paliperidone versus risperidone may stem from different pharmacokinetic properties and different formulations (extended release vs. oral). Since paliperidone and risperidone display simple, competitive antagonism in radioligand-binding assays with similar affinities for their target receptors (Schotte et al., 1996; Gray and Roth, 2007; Dolder et al., 2008), differences in therapeutic efficacy may result from differences in their ability to regulate receptor-mediated signalling pathways.
It is now well accepted that receptors activate cellular signalling cascades in the absence of a stimulating ligand (agonist) and that this ‘constitutive’ receptor activity can be decreased by inverse agonists. Although risperidone is known to behave as a competitive antagonist, it also has inverse agonist properties at D2, 5-HT2A, 5-HT2C and H1 receptors (Egan et al., 1998; Rauser et al., 2001; Weiner et al., 2001; Akam and Strange, 2004). While the inverse agonist efficacy of risperidone has been well studied, by contrast, there is little known about the efficacy characteristics of paliperidone.
Individual receptor subtypes can regulate the activity of several signal transduction pathways in cells (Perez and Karnik, 2005). A large body of experimental evidence has accumulated recently to suggest that different drugs, acting at a single receptor subtype, can elicit different profiles of signal transduction pathway regulation. This process has been termed ‘functional selectivity’ or ‘biased agonism’ (for reviews see Clarke, 2005; Kenakin, 2007; 2011; Urban et al., 2007). The mechanism underlying functional selectivity is based on the production of ligand-dependent conformational states of the receptor, which have differential ability to interact with and regulate each of the multiple signal transduction molecules [i.e. guanine nucleotide-binding protein (G proteins), β-arrestins, etc.], which couple to the receptor. Thus, drugs have selectivity that extends beyond that afforded by differential affinity for different receptor subtypes. Both agonists and inverse agonists can selectively regulate the activity of distinct signalling pathways coupled to a receptor (Clarke, 2005; Kenakin, 2007; Urban et al., 2007; Kenakin, 2011). It has been suggested that differences in functional selectivity may underlie differences in therapeutic efficacy and/or adverse effect liability of APDs (Heusler et al., 2007).
Importantly, small differences in molecular structure between drugs can lead to pronounced differences in functional selectivity, even for drugs that have similar affinities for a receptor (Moya et al., 2007). Thus, it is possible that even though paliperidone differs from risperidone by a single hydroxyl group and have similar binding affinities for many receptors, they may differ in the profile of cellular signalling pathways they regulate and thus in their therapeutic efficacy or adverse effect profile. In this regard, it is useful to consider that the difference between dopamine and norepinephrine, two drugs with markedly different functional actions, is a single hydroxyl group. Interestingly, risperidone and paliperidone have been found to differ in their effects on the firing rate of serotonergic and noradrenergic neurons in vivo (Dremencov et al., 2007).
The goal of this work was to examine differences in the signalling profiles of paliperidone versus risperidone for each of several cellular responses coupled to a selected group of target receptors that are important in therapeutic action or in the production of adverse effects. We have found and report here that the single hydroxyl group of paliperidone does indeed confer differences in the cellular signalling profiles in comparison to that of risperidone.
Methods
Materials
The following compounds were purchased from Sigma-Aldrich (St. Louis, MO, USA): DOI, SB 206553, ketanserin, quinpirole, butaclamol, mepyramine and histamine. myo-[3H]-inositol and [3H]-arachidonic acid were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA, USA). Risperidone and paliperidone were provided by Janssen Scientific Affairs, LLC (Titusville, NJ, USA). FBS was from Atlanta Biologicals (Atlanta, GA, USA). All other tissue culture reagents were purchased from Invitrogen (Carlsbad, CA, USA). All other drugs and chemicals (reagent grade) were purchased from Sigma-Aldrich.
Cell culture
5-HT2C-mediated responses were measured in CHO-1C19, CHO-1C7 or U2OS cells that express stably the human 5-HT2C receptor. CHO-1C7 cells express the 5-HT2C receptor at high levels and are optimized to detect inverse agonist properties by measuring a reduction in basal effector activities (Berg et al., 1999; Lopez-Giminez et al., 2001; Brea et al., 2003). CHO cell lines were maintained in minimum essential medium, α-formulation, supplemented with 5% FBS and 300 μg·mL−1 hygromycin. U2OS cells (DiscoverRx, Freemont, CA, USA), which stably express the human 5-HT2C receptor tagged with a 42 amino acid fragment of β-galactosidase (ProLink™; DiscoveRx Corporation, Freemont, CA, USA), were used to measure recruitment of β-arrestin to the receptor (see below) were grown according to the manufacturer's protocol.
5-HT2A-mediated responses were measured in CHO-FA4 or HEK-293 cells that stably express the human 5-HT2A receptor (HEK-2A cells). CHO-FA4 cells were maintained as described above for CHO cells. HEK-2A cells were obtained from Bryan Roth (University of North Carolina) and maintained in α-MEM supplemented with 10% FBS. Recruitment of β-arrestin was measured with U2OS cells that stably express the human 5-HT2A receptor tagged with Prolink maintained as described above.
Dopamine D2-mediated responses were measured in HEK-293 and CHO cells stably expressing the human dopamine D2 receptor. CHO-D2 cells were maintained in α-MEM supplemented with 5% FBS and 50 ug·mL−1 G418. HEK-D2 cells were maintained in DMEM supplemented with 10% FBS and 50 ug·mL−1 G418. Recruitment of β-arrestin was measured with CHO cells stably expressing the human dopamine D2 receptor tagged with Prolink.
Histamine H1-mediated responses were measured in HEK cells stably expressing human H1 receptors or in CHO cells transiently expressing human histamine H1 receptors were maintained as described above. For recruitment of β-arrestin, CHO cells expressing the human histamine H1 receptor tagged with Prolink were used and maintained as described above.
All cells were grown in serum-free media for 24 h before each experiment. Parent (non-transfected) cells for each receptor subtype were screened with the ligands to assess possible off-target or non-specific responses. None of the drugs used here altered signalling in the parent cell lines.
Inositol phosphate (IP) accumulation
Measurements of PLC-mediated IP accumulation in response to agonist or constitutive receptor activity were made after 10 min or 30 min of drug exposure, respectively, as described previously (Berg et al., 1998; 1999; 2001b; 2006; 2008).
Arachidonic acid (AA) release
Measurements of PLA2-mediated AA release in response to agonist or constitutive receptor activity were made after 10 or 30 min of drug exposure, respectively, as described previously (Berg et al., 1998; 1999; 2001b; 2006; 2008).
cAMP accumulation
Measurement of ligand-induced changes in adenylyl cyclase activity (cAMP accumulation) were done as described previously (Berg et al., 1994; 2011; 2003; 2007; Evans et al., 2001).
ERK activity
Changes in ERK activity were measured as described previously (Berg et al., 2011) using the SureFire phospho-ERK (pERK) assay kit (PerkinElmer) according to the manufacturer's directions. The fluorescence signal from pERK was measured in duplicate using a Fluostar microplate reader (BMG Labtech, Offenburg, Germany) with AlphaScreen settings.
β-arrestin recruitment
The PathHunter® β-arrestin GPCR Assay kit (DiscoveRX) was used to measure recruitment of β-arrestin to the receptor according to the manufacturer's directions. Chemiluminescence was read with a Fluostar microplate reader (BMG Labtech).
PLC and ERK sensitization
In the absence of a stimulating ligand, receptor signalling systems can exist in a state of constitutive, partial desensitization as a result of constitutive activation of desensitization signalling pathways (Berg et al., 1999; 2006; Wilbanks et al., 2002). Prolonged (>4 h) treatment with an inverse agonist, which reduces constitutive receptor activity, can promote re-sensitization of the receptor-signalling system that can be visualized by enhanced responsiveness to agonist stimulation. To measure sensitization of receptor-PLC or receptor-ERK signalling pathways, cells were treated for 24 h with ligand or vehicle and washed three times with 5 min incubation intervals at 37°C. Cells were then stimulated with a receptor agonist for 10 min to measure IP accumulation or for 0–30 min to measure ERK activation as described above.
[3H]-raclopride binding
Membranes from CHO cells expressing human dopamine D2 receptors were prepared as described previously (Berg et al., 1999; 2001a; 2008). After ligand treatment (24 h, 100 × Ki), cells grown in 10 cm2 plates, were washed three times with 5 min incubation intervals at 37°C to remove residual drug. Cells were scraped and membranes prepared by trituration through a 26-gauge needle, followed by centrifugation at 39 000× g and sonication at 4°C. Saturation binding assays using [3H]-raclopride in cell membrane preparations (50 μg protein/tube) were done using 15 concentrations of radioligand in duplicate over a 3-log unit range as previously described (Berg et al., 1999; 2001a; 2008). Incubations were carried out for 1 h at 37°C followed by rapid filtration through 0.3% polyethyleneamine coated GF/C glass fiber filters. Non-specific binding was determined in the presence of 10 μM spiperone.
Data analysis
Curve fitting
Concentration-response data were fit with non-linear regression to the model
where E is the measured response at a given concentration (A), Emax is the maximal response, EC50 is the concentration of agonist producing the half-maximal response, and n is the slope index.
For saturation-binding experiments, data were fit with nonlinear regression to the model:
where B is the measured amount of radioligand bound (fmol·mg−1 protein) in the presence of various concentrations of radioligand [D], Bmax is the maximal amount bound, Kd is the concentration of D that produces half-maximal binding, n is the slope factor and m is the slope of the linear regression line for non-specific binding.
Statistical analysis
Data are presented as mean ± SEM from at least three independent experiments. Statistical differences in concentration-response curve parameters (i.e. Emax) or in response to maximal concentrations of ligands followed by Bonferroni's post hoc using Prism software (Graphpad Software, Inc., San Diego, CA, USA). P < 0.05 was considered statistically significant.
Relative efficacy determinations
The ratio of the Emax of a test drug to the Emax of a reference drug was used to characterize the signalling profile of paliperidone and risperidone (Berg and Clarke, 2006; Clarke and Berg, 2010). Measurement of relative efficacy removes the system dependency and permits assessment of differences in the ability of drugs to regulate receptor activity. The relative efficacies of paliperidone and risperidone for selected cellular signalling responses at human 5-HT2A, 5-HT2C, D2 and H1 receptors expressed in CHO or HEK cells were obtained from analysis of concentration-response curves (Emax values) or from measurement of responses to maximal ligands concentrations (100 × Ki), which are expected to produce full receptor occupancy. The reference ligands used were ketanserin, SB 206553, butaclamol and mepyramine for 5-HT2A, 5-HT2C, D2 and H1 receptors respectively.
Results
5-HT2C receptor signalling
The degree of ligand-independent 5-HT2C receptor activity can most easily be measured as the magnitude of the reduction in basal effector activity (i.e. PLC, PLA2) produced by an inverse agonist (Berg et al., 1999). As shown in Figure 1, paliperidone and risperidone were efficacious inverse agonists for reduction in basal IP accumulation in CHO cells expressing stably the human 5-HT2C receptor. The maximal reduction in basal IP accumulation for the reference 5-HT2C inverse agonist, SB 206553, was 72 ± 6%. The maximal reduction in IP accumulation produced by paliperidone (84 ± 1%) was significantly greater than that for risperidone (68 ± 5%). Relative efficacy values, using SB 206553 as the reference ligand, were 1.30 ± 0.03 and 1.02 ± 0.05 for paliperidone and risperidone respectively. The potency values (pEC50) for SB 206553, paliperidone and risperidone were 7.77 ± 0.24 (17 nM), 6.75 ± 0.22 (178 nM) and 6.54 ± 0.05 (286 nM) respectively.
Figure 1.
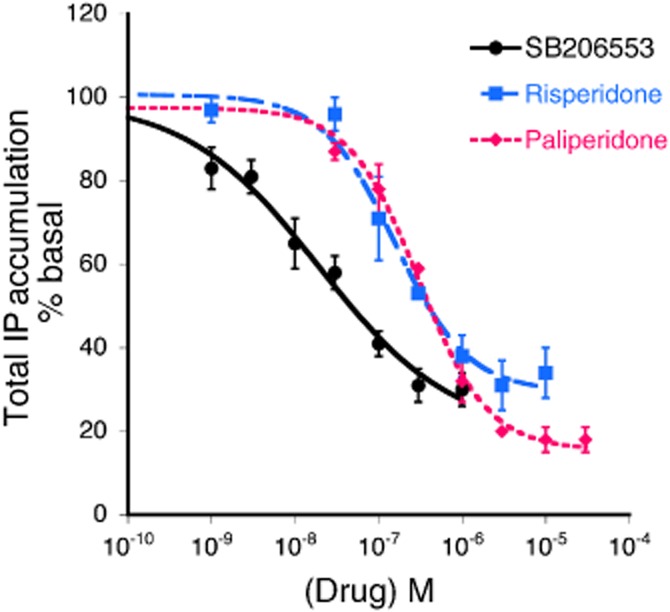
Inverse agonism of risperidone and paliperidone at the 5-HT2C receptor for the PLC-IP pathway. Concentration-response curves to SB 206553 (reference ligand), risperidone and paliperidone for reduction of basal IP accumulation in CHO-1C7 cells expressing the human 5-HT2C receptor. Cells, pre-labelled with [3H]-myo-inositol, were incubated with the indicated concentrations of ligands for 30 min in the presence of LiCl (20 mM). Data are expressed as the percent decrease in basal IP accumulation and represent mean ± SEM of three experiments. Maximal reduction in IP accumulation produced by SB 206553, risperidone and paliperidone were 72 ± 6%, 68 ± 5% and 84 ± 1% respectively. The maximal reduction in basal IP produced by paliperidone was significantly greater than that produced by risperidone (P < 0.05, one-way anova followed by Bonferroni's post hoc test).
Figure 2 shows the effects of maximal concentrations (100 × Ki) of SB 206553 (300 nM), paliperidone (3 μM) and risperidone (1 μM) on IP accumulation, AA release, ERK activity and β-arrestin recruitment. As reported previously (Berg et al., 2005; 2008), the level of constitutive 5-HT2C receptor activity differed for different responses as evidenced by different magnitudes of reduction of basal activity by SB 206553. Constitutive activity of the 5-HT2C receptor towards the PLC pathway was considerably greater than that towards AA release, ERK activity and β-arrestin recruitment. For the PLA2 response, SB 206553 and risperidone were inverse agonists, reducing basal AA release by 28 ± 1% and 14 ± 2% respectively. By contrast, paliperidone behaved as an antagonist and did not reduce basal AA release (maximal reduction = 0.5 ± 8%). For the ERK response, all three ligands were inverse agonists with equal efficacy to reduce basal ERK activity by approximately 25%. Similarly, all three ligands were weak inverse agonists with equal efficacy to reduce basal β-arrestin recruitment to the 5-HT2C receptor.
Figure 2.
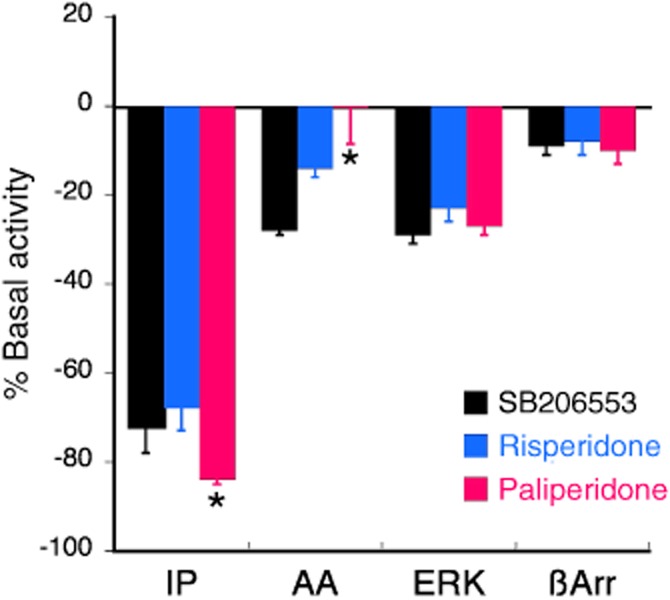
Acute effects of risperidone and paliperidone at the 5-HT2C receptor for various responses compared with the reference ligand, SB 206553. Maximal effects of the ligands on IP accumulation are derived from the Emax values from the concentration-response curves shown in Figure 1. Reduction of basal AA release and ERK activation by maximal concentrations (100 × Ki) of SB 206553 (300 nM), risperidone (1 μM) and paliperidone (3 μM) were measured in CHO-1C7 cells following 30 min (AA release) or 5 min (ERK activity) of incubation with ligand. Recruitment of β-arrestin to the 5-HT2C receptor was measured in U2Os cells following incubation with maximal concentration of ligands for 90 min. DOI-mediated data are expressed as the percent reduction in basal activity and bars represent the mean ± SEM of six experiments. *P < 0.05 for the effect of paliperidone compared with that of risperidone (one-way anova followed by Bonferroni's post hoc test).
Similarly to agonist stimulation, prolonged ligand-independent receptor activity can lead to a reduction of effector activity (constitutive desensitization; Berg et al., 1999; Marion et al., 2004). Prolonged pretreatment (>4 h) with an inverse agonist can reduce constitutive desensitization of some receptor-signalling systems, which can be visualized as an enhanced response to agonist following washout of the inverse agonist. For the 5-HT2C receptor, prolonged pretreatment (24 h) with SB 206553 enhanced agonist (DOI)-stimulated IP accumulation by approximately 100% and maximal ERK activity by approximately 285% over that of DOI stimulation following vehicle pretreatment (Figure 3). For both responses, the relative efficacy of paliperidone exceeded that of risperidone and for both responses, paliperidone was a more efficacious inverse agonist than was the prototypical 5-HT2C inverse agonist, SB 206553. Prolonged pretreatment with paliperidone or risperidone enhanced DOI-stimulated IP accumulation by 150% [pEC50 = 6.87 ± 0.42 (135 nM)] and 85% [pEC50 = 7.94 ± 0.18 (12 nM)] respectively (Figure 3A). Following vehicle pretreatment, maximal DOI-stimulated ERK activity (at 2 min) was 201 ± 17% over basal. Pretreatment with SB 206553, paliperidone and risperidone enhanced DOI-stimulated ERK activity by 380 ± 36%, 433 ± 12% and 574 ± 94% respectively (Figure 3B).
Figure 3.
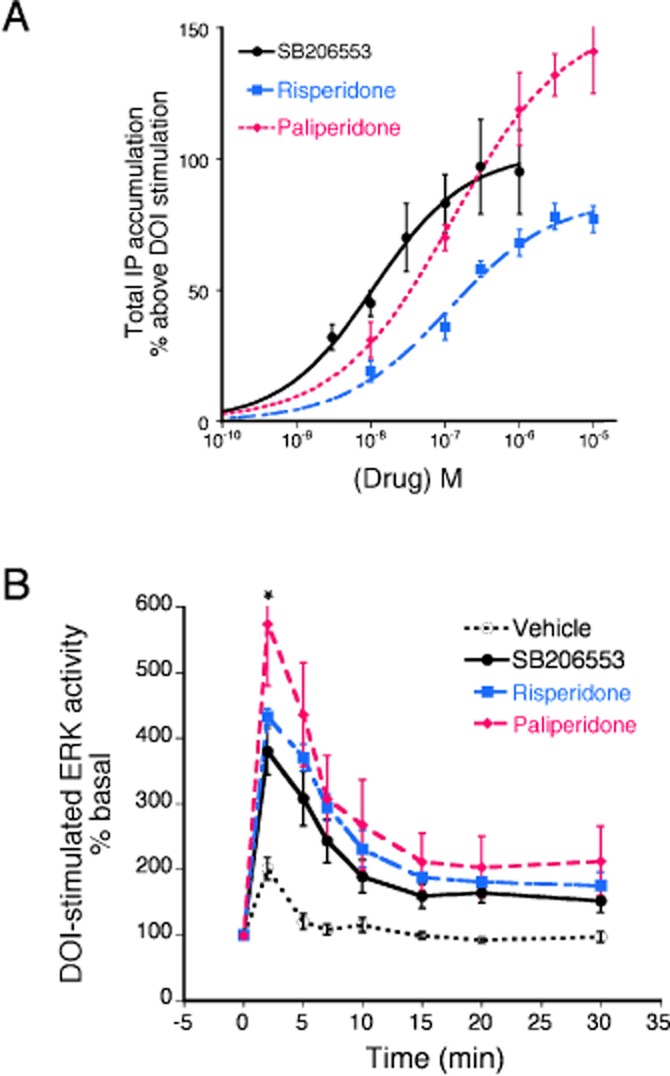
The effect of prolonged (24 h) pretreatment with risperidone and paliperidone compared with the reference ligand, SB 206553, on sensitization (reduction of constitutive activity) of the PLC-IP (A) and the ERK (B) responses. (A) CHO-1C19 cells, stably expressing the human 5-HT2C receptor, were pretreated with the indicated concentrations of SB 206553, risperidone, paliperidone or vehicle for 24 h. Following a wash, cells were treated with a maximal concentration of the 5-HT2A/2C agonist, DOI (1 μM), for 10 min to measure IP accumulation. Data are expressed as the percent above the vehicle-pretreated effect of DOI (94 ± 19% above basal) and represent the mean ± SEM of three experiments. The maximal increases in DOI stimulation by SB 206553, risperidone and paliperidone were 93 ± 5%, 85 ± 7% and 135 ± 11% respectively. The maximal sensitization produced by paliperidone was significantly greater than that produced by risperidone (P < 0.05, one-way anova followed by Bonferroni's post hoc test). (B) CHO-1C19 cells were pretreated with maximal concentrations of SB 206553 (300 nM), risperidone (1 μM) or paliperidone (3 μM) for 24 h. Following a wash, cells were treated with DOI (1 μM) for the indicated periods of time and the levels of pERK were measured. Data are expressed as the percent above basal (0 time) levels and represent the mean ± SEM of five experiments. Pretreatment with all three ligands enhanced DOI-stimulated ERK activity. *P < 0.05 for the effect of paliperidone compared with that of risperidone at the 2 min time point (two-way anova followed by Bonferroni's post hoc test).
5-HT2A receptor signalling
Figure 4 shows the effect of acute treatment of cells expressing the human 5-HT2A receptor with maximal concentrations (100 × Ki) of paliperidone and risperidone, compared to the reference ligand, ketanserin. For the 5-HT2A-PLC and the 5-HT2A-ERK pathways, all three ligands were inverse agonists of comparable efficacy, reducing basal IP accumulation by 38 ± 4%, 43 ± 7% and 39 ± 5% and reducing basal ERK activity by 21 ± 3%, 30 ± 5% and 30 ± 4% for ketanserin, risperidone and paliperidone respectively. Only risperidone significantly reduced AA release (17 ± 4%). Interestingly, all three ligands behaved as weak agonists of comparable efficacy for the recruitment of β-arrestin to the 5-HT2A receptor. Whereas 5-HT (10 μM) produced a robust β-arrestin translocation of 660 ± 50% above basal (mean ± SEM, n = 6), ketanserin, risperidone and paliperidone increased β-arrestin recruitment to the 5-HT2A receptor by 177 ± 29%, 197 ± 23% and 165 ± 25% above basal respectively.
Figure 4.
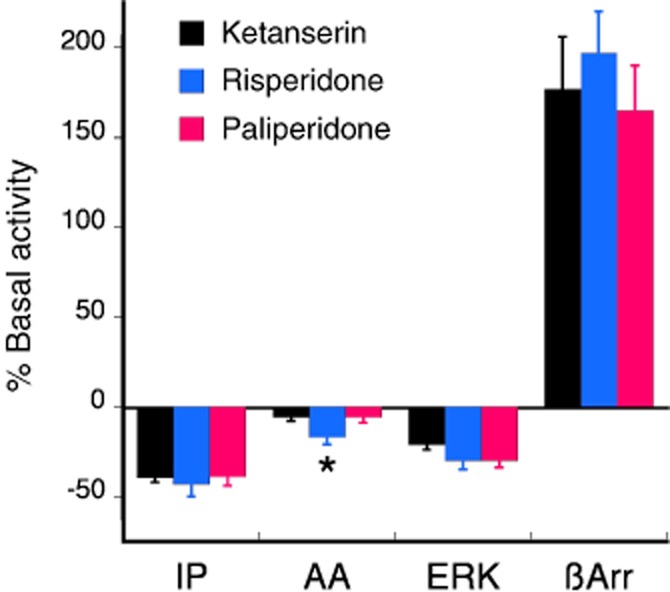
The effect of maximal concentrations (100 × Ki) of risperidone (30 nM) and paliperidone (100 nM) compared with the reference ligand, ketanserin (1 μM), for changes in basal signalling responses coupled to the human 5-HT2A receptor. For the PLC-IP pathway, HEK-2A cells stably expressing the human 5-HT2A receptor (a gift from Bryan Roth, University of North Carolina) were incubated with ligands for 30 min and IP accumulation was measured. All three ligands behaved as inverse agonists, reducing basal IP accumulation. There was no difference in the magnitude of the response between risperidone and paliperidone. For the PLA2-AA pathway, CHO-FA4 cells were incubated with ligands for 30 min. Neither ketanserin, nor paliperidone significantly inhibited basal AA release. CHO-FA4 cells were incubated with ligands for 5 min to measure changes in ERK activity. All three ligands behaved as inverse agonists, reducing basal AA release. There was no difference in the magnitude of the response between risperidone and paliperidone. U2OS cells were treated with ligands for 90 min to measure recruitment of β-arrestin to the 5-HT2A receptor. All three ligands behaved as agonists, increasing the recruitment of β-arrestin to the receptor. There was no difference in the magnitude of the effect between risperidone and paliperidone. Bars represent the mean ± SEM of 3–6 experiments. *P < 0.05 for the effect of paliperidone compared with that of risperidone.
Prolonged pretreatment with a maximal concentration (100 × Ki) of the reference inverse agonist, ketanserin (1 μM) increased 5-HT2A agonist (DOI)-stimulated IP accumulation by 28% over that of vehicle pretreatment (Figure 5A). For this response, both risperidone and paliperidone behaved as inverse agonists of comparable efficacy. Pretreatment with risperidone or paliperidone enhanced DOI-stimulated IP accumulation by 38 and 34% respectively. Following vehicle pretreatment, maximal DOI-stimulated ERK activity, measured 2 min following agonist treatment, was 181 ± 13% above basal (Figure 5B). 24 h pretreatment with ketanserin, enhanced DOI-stimulated ERK activity by a maximum of 266 ± 10% over basal, suggesting a 1.8-fold reduction in constitutive desensitization. By contrast, both risperidone and paliperidone appeared to enhance constitutive desensitization (reduced DOI-stimulated ERK activity by approximately 30%), acting as agonists of comparable efficacy. DOI-stimulated ERK activity, measured at the 2 min time point, following risperidone and paliperidone pretreatment was 122 ± 12% and 126 ± 2% over basal.
Figure 5.
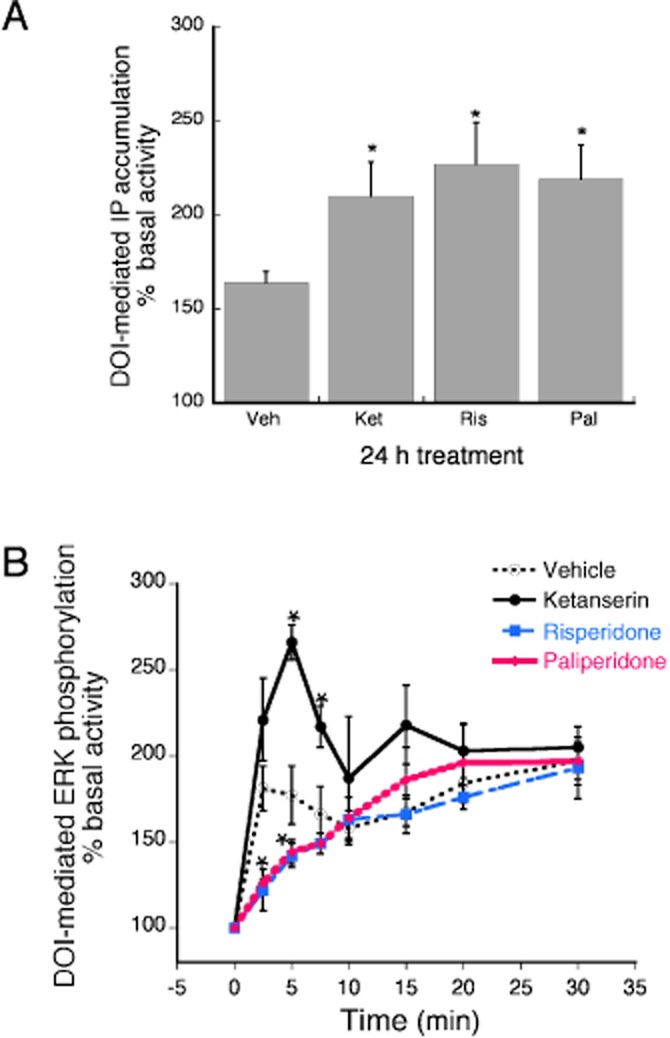
The effect of prolonged (24 h) pretreatment with risperidone and paliperidone compared with the reference ligand, ketanserin, on sensitization (reduction of constitutive activity) of the PLC-IP (A) and the ERK (B) responses. HEK-2A cells, stably expressing the human 5-HT2A receptor, were pretreated with maximal concentrations (100 × Ki) of ketanserin (1 μM), risperidone (30 nM), paliperidone (100 nM) or vehicle for 24 h. Following a wash, cells were treated with a maximal concentration of the 5-HT2A/2C agonist, DOI (1 μM), for 10 min to measure IP accumulation (A) or ERK activity (B). (A) Data are expressed as the percent above the vehicle-pretreated effect of DOI and represent the mean ± SEM of six experiments. All three ligands behaved as inverse agonists, enhancing the stimulation of IP accumulation produced by DOI. There was no difference in the effect of risperidone compared to that of paliperidone. *P < 0.05 for the ligand pretreatment effect compared to that of vehicle pretreatment. (B) Data are expressed as the percent above basal (0 time) levels and represent the mean ± SEM of four experiments. Whereas ketanserin behaved as an inverse agonist, enhancing the effect of DOI, both risperidone and paliperidone acted as agonists, decreasing the effect of DOI. There was no difference in the effect of risperidone compared to that of paliperidone. *P < 0.05 for the ligand pretreatment effect compared to that of vehicle pretreatment.
Dopamine D2 receptor signalling
Agonist activity at dopamine D2 receptors typically results in activation of Gi family G proteins leading to an inhibition of stimulated adenylyl cyclase activity and reduction in cellular cAMP levels (Beaulieu and Gainetdinov, 2011). Consequently, dopamine D2 inverse agonist activity is reflected by an increase in stimulated adenylyl cyclase activity and an increase in cAMP accumulation. As shown in Figure 6A, acute treatment with the reference inverse agonist, butaclamol, increased forskolin-stimulated cAMP accumulation by approximately 300%. Risperidone and paliperidone behaved as inverse agonists, enhancing forskolin-stimulated cAMP accumulation by approximately 400 and 600% respectively. Similarly, both risperidone and paliperidone behaved as inverse agonists for the dopamine D2-ERK pathway (Figure 6B). The inverse agonist activity of risperidone was not different from that of butaclamol, reducing basal ERK activity by approximately 20%. By contrast, the inverse agonist efficacy of paliperidone exceeded that of risperidone, reducing basal ERK activity by approximately 35%. None of the ligands tested altered recruitment of β-arrestin to the dopamine D2 receptor.
Figure 6.
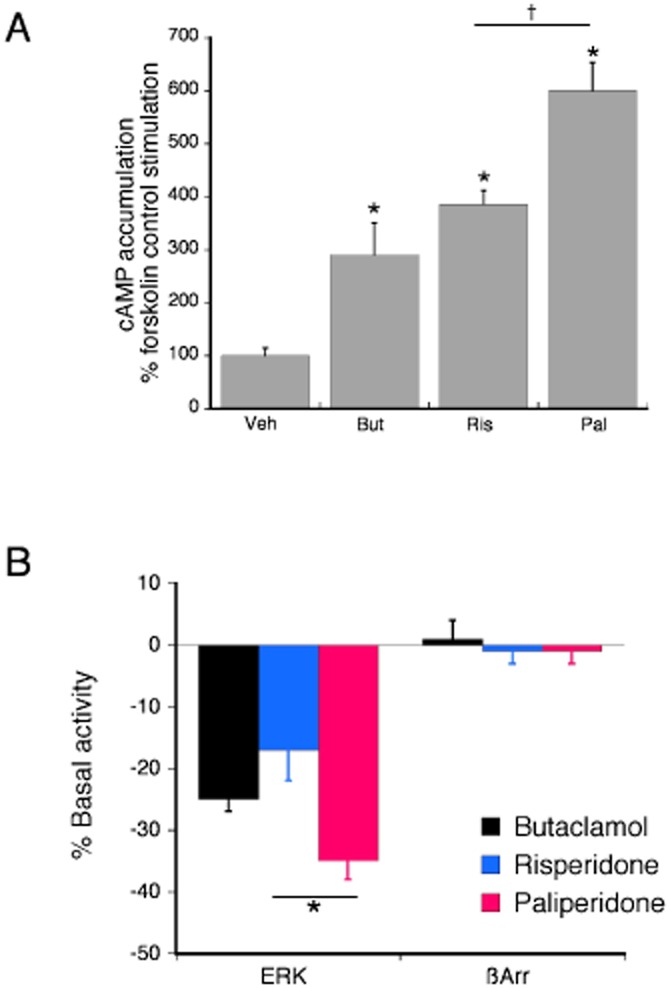
The effect of maximal concentrations (100 × Ki) of risperidone (300 nM) and paliperidone (200 nM) compared with the reference ligand, butaclamol (1 μM), for changes in forskolin-stimulated cAMP accumulation (A), ERK activity and recruitment of β-arrestin (B) in HEK-D2 cells stably expressing the human dopamine D2 receptor. (A) HEK-D2 cells were incubated with ligands or vehicle for 15 min followed by incubation with the adenylyl cyclase activator, forskolin (1 μM) for an additional 15 min. Accumulation of cAMP was measured with RIA. All three ligands behaved as inverse agonists, enhancing forskolin-stimulated cAMP accumulation. The dopamine D2 agonist, quinpirole (1 μM) inhibited forskolin-stimulated cAMP accumulation by 51 ± 3% (not shown). The effect of paliperidone was significantly greater than that of risperidone. Bars represent the mean ± SEM of six experiments. *P < 0.05 compared to vehicle. †P < 0.05 for the effect of paliperidone compared with that of risperidone. (B) For ERK activity, HEK-D2 cells were incubated with ligand for 5 min. All three ligands behaved as inverse agonists, decreasing basal ERK activation. Bars represent the mean ± SEM of five experiments *P < 0.05 for the effect of paliperidone compared with that of risperidone. HEK-D2 cells were incubated for 90 min with ligands to measure recruitment of β-arrestin to the dopamine D2 receptor. None of the ligands altered β-arrestin recruitment. The agonist, quinpirole (1 μM) increased translocation of β-arrestin by approximately 10-fold (984 ± 30% above basal, not shown). Bars represent the mean ± SEM of six experiments.
Figure 7A shows the effect of 24 h pretreatment with vehicle or dopamine D2 ligands on concentration-response curves to the dopamine D2 agonist, quinpirole. The maximal response and pEC50 for quinpirole to inhibit forskolin-stimulated cAMP accumulation was 47 ± 4% and 10.39 ± 0.14 (0.04 nM) following 24 h pretreatment with vehicle. Prolonged treatment with an inverse agonist, by reducing constitutive dopamine D2 receptor activity, would be expected to enhance the effect of quinpirole. By contrast, the reference ligand, butaclamol, and both risperidone and paliperidone behaved as agonists, reducing the potency and/or efficacy of quinpirole to inhibit forskolin-stimulated cAMP accumulation following 24 h pretreatment. Following 24 h pretreatment with risperidone and paliperidone, the potency of quinpirole was reduced by approximately 100-fold to 7.70 ± 0.9 (20 nM) and 7.96 ± 0.04 (11 nM) respectively. The effect of butaclamol to promote desensitization of the dopamine D2-adenylyl cyclase system was equivalent to that of the prototypical agonist, quinpirole. Butaclamol decreased the potency and efficacy of quinpirole [pEC50 = 7.55 ± 0.34 (28 nM), Emax = 17 ± 8%] for inhibition of adenylyl cyclase activity. Similarly, 24 h pretreatment with quinpirole decreased the maximal response of quinpirole to 13 ± 6%.
Figure 7.
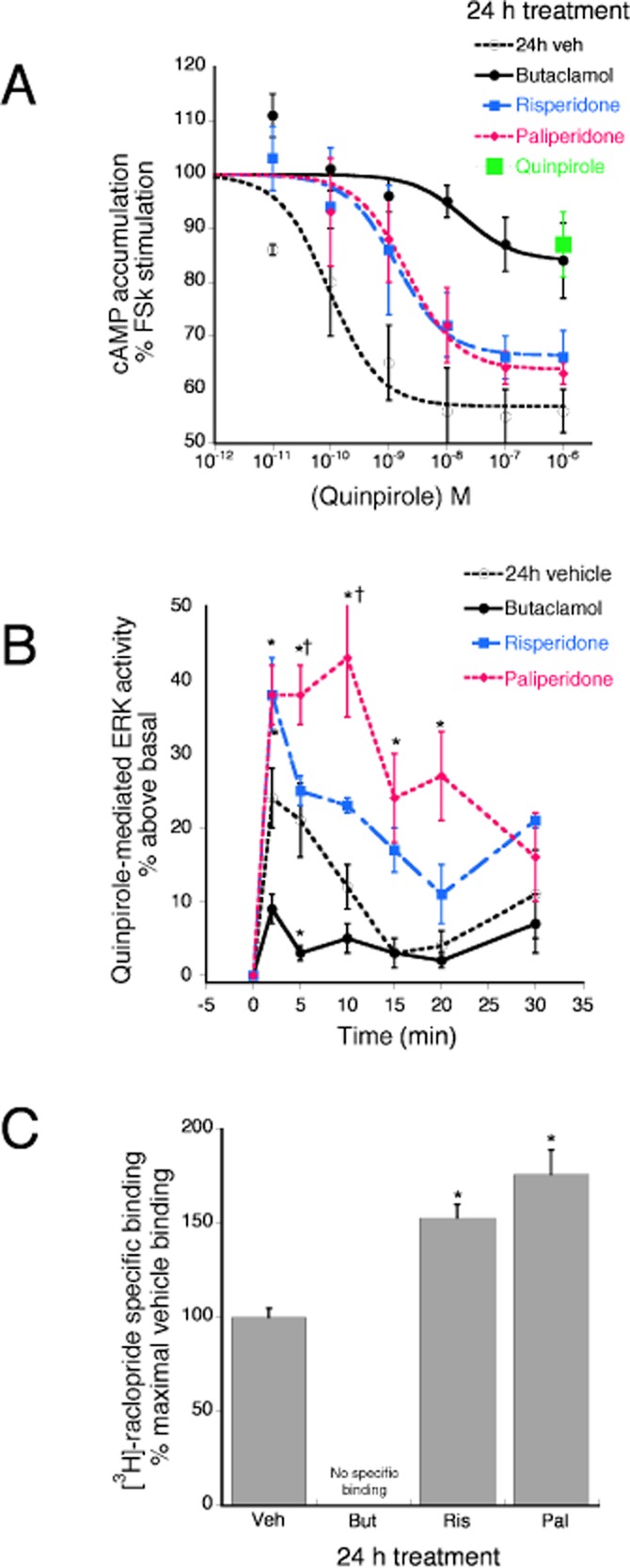
The effect of prolonged pretreatment with risperidone and paliperidone compared with the reference ligand, butaclamol on quinpirole-mediated inhibition of forskolin-stimulated cAMP accumulation, quinpirole-stimulated ERK activity and on dopamine D2 receptor density. (A) CHO-D2 cells were pretreated with maximal concentrations of butaclamol (1 μM), risperidone (300 nM), paliperidone (200 nM) or vehicle. Following a wash, cells were incubated with the indicated concentrations of quinpirole for 15 min followed by forskolin (1 μM) for an additional 15 min. Accumulation of cAMP was measured with RIA. All three ligands acted as agonists, reducing the potency and/or efficacy of quinpirole to inhibit cAMP accumulation. There was no difference in the effect produced between risperidone and paliperidone. Data are expressed as the percent of forskolin-stimulated cAMP accumulation and represent mean ± SEM of four experiments. (B) 24 h pretreatment with paliperidone, but not risperidone, enhanced quinpirole-stimulated ERK activity. CHO-D2 cells were pretreated with maximal concentrations of butaclamol (1 μM), risperidone (300 nM), paliperidone (200 nM) or vehicle for 24 h. Following a wash, cells were incubated with quinpirole (1 μM) for the indicated periods of time and pERK levels were measured. Paliperidone, acting as an inverse agonist, increased and butaclamol, acting as an agonist, decreased quinpirole stimulated ERK activity. *P < 0.05 compared to vehicle pretreatment. Data are expressed as the percent above basal (0 time) levels and represent the mean ± SEM of four experiments. (C) The effect of prolonged treatment with maximal concentrations of butaclamol, risperidone and paliperidone on dopamine D2 receptor density. CHO-D2 cells were treated with the ligands or vehicle as above and [3H]-raclopride binding was measured to determine maximal binding. Pretreatment with risperidone or paliperidone increased maximal [3H]-raclopride binding whereas, pretreatment with butaclamol reduced dopamine D2 receptor density. There was no difference in the magnitude of the effect of risperidone versus paliperidone. Data are expressed as a percent of maximal binding in vehicle pretreated cells and bars represent mean ± SEM of three experiments. *P < 0.05 compared to the vehicle pretreated condition.
Figure 7B shows the effect of 24 h pretreatment with dopamine D2 receptor ligands or vehicle on quinpirole-stimulated ERK activity. Following vehicle pretreatment, quinpirole increased ERK activity a maximum of 24 ± 4% above basal, measured 2 min following agonist stimulation. By contrast, butaclamol decreased quinpirole-stimulated ERK activity by about 50%, to 9 ± 2% above basal. Prolonged pretreatment with risperidone had no effect on maximal ERK stimulation by quinpirole; however, prolonged pretreatment with paliperidone produced a twofold enhancement of the maximal ERK stimulation by quinpirole.
The agonist activity of the prototypical inverse agonist, butaclamol, to act as an agonist to promote desensitization of the dopamine D2-adenylyl cyclase and ERK pathways following 24 h pretreatment was surprising. As discussed above, prolonged treatment with inverse agonists can enhance subsequent agonist responses by reducing constitutive desensitization. In general, two mechanisms have been reported by which prolonged treatment with inverse agonists can reduce constitutive desensitization. One mechanism involves an increase in receptor density due to decreased constitutive internalization (for a review, see Milligan and Bond, 1997). In addition, enhancement of agonist action can occur in the absence of a change in receptor density as a result of changes in receptor–effector coupling efficiency (Berg et al., 1999). To examine the mechanism for butaclamol pretreatment to decrease the response to quinpirole, we measured receptor density following 24 h pretreatment with butaclamol, risperidone and paliperidone. Figure 7C shows that pretreatment with butaclamol reduced [3H]-raclopride binding from 566 ± 44 fmol·mg−1 protein (vehicle pretreatment) to undetectable levels. Interestingly, 24 h pretreatment with risperidone and paliperidone increased [3H]-raclopride binding to 852 ± 117 and 1068 ± 64 fmol·mg−1 protein respectively.
Histamine H1 receptor signalling
Figure 8 shows the acute effects of treatment of cells expressing the human histamine H1 receptor with maximal concentrations (100 × Ki) of the reference ligand mepyramine, risperidone and paliperidone. For the histamine H1-PLC pathway, all three ligands behaved as inverse agonists, reducing basal IP accumulation by 24 ± 2%, 29 ± 6% and 49 ± 7%, for mepyramine, risperidone and paliperidone respectively. The inverse agonist efficacy of paliperidone to reduce basal IP accumulation was significantly greater than that of risperidone. Similarly, all three ligands were inverse agonists on the histamine H1-ERK pathway, again with the inverse agonist efficacy of paliperidone being greater than that of risperidone. Basal ERK activity was reduced by 10 ± 2%, 23 ± 2% and 33 ± 3% by mepyramine, risperidone and paliperidone respectively. For β-arrestin recruitment to the histamine H1 receptor, mepyramine, risperidone and paliperidone were equi-efficacious inverse agonists, reducing basal β-arrestin recruitment by approximately 15%. For the histamine H1-PLA2 pathway, there was no significant change in AA release by any of the three ligands tested. The agonist, histamine, produced a very weak stimulation of AA release (20 ± 4%), suggesting that the histamine H1 receptor couples weakly to the PLA2 signalling pathway in these cells.
Figure 8.
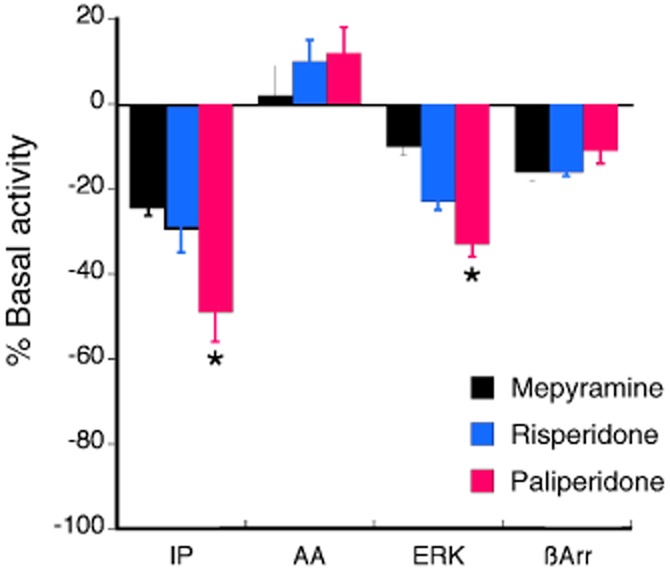
The effect of maximal concentrations (100 × Ki) of risperidone (2 μM) and paliperidone (2 μM) compared with the reference ligand, mepryamine (1 μM), on basal IP accumulation, AA release, ERK activity and β-arrestin recruitment responses coupled to the human histamine H1 receptor. For the PLC-IP pathway, HEK-H1 cells stably expressing the human histamine H1 receptor were incubated with ligands for 30 min and IP accumulation was measured. All three ligands behaved as inverse agonists, reducing basal IP accumulation. The reduction in basal IP accumulation produced by paliperidone was greater than that of risperidone. For the PLA2-AA pathway, CHO-H1 cells were incubated with ligands for 30 min. None of the ligands significantly inhibited basal AA release. HEK-H1 cells were incubated with ligands for 5 min to measure changes in ERK activity. All three ligands behaved as inverse agonists, reducing basal AA release. The effect of paliperidone was greater than that of risperidone. CHO-H1 cells were treated with ligands for 90 min to measure recruitment of β-arrestin to the histamine H1 receptor. All three ligands behaved as agonists, increasing the recruitment of β-arrestin to the receptor. There was no difference in the magnitude of the effect between risperidone and paliperidone. Bars represent the mean ± SEM of 4–6 experiments. *P < 0.05 for the effect of paliperidone compared with that of risperidone.
Figure 9A shows the effect of 24 h pretreatment with maximal concentrations of mepyramine, risperidone and paliperidone on histamine stimulation of the histamine H1-PLC pathway. In cells pretreated with vehicle, histamine stimulated IP accumulation by approximately 120% (twofold) over basal. Pretreatment with mepyramine, risperidone or paliperidone equally enhanced the histamine effect approximately twofold. Twenty-four hours pretreatment also enhanced histamine stimulation of ERK activity. Pretreatment with mepyramine and paliperidone increased histamine stimulation of ERK activity by about 55%, from 22 ± 3% above basal in vehicle pretreated cells to 38 ± 4% and 31 ± 4% respectively. The inverse agonist efficacy of risperidone was greater than that for paliperidone. Pretreatment of cells with risperidone increased histamine stimulation of ERK activity by approximately twofold, to 59 ± 4% above basal.
Figure 9.
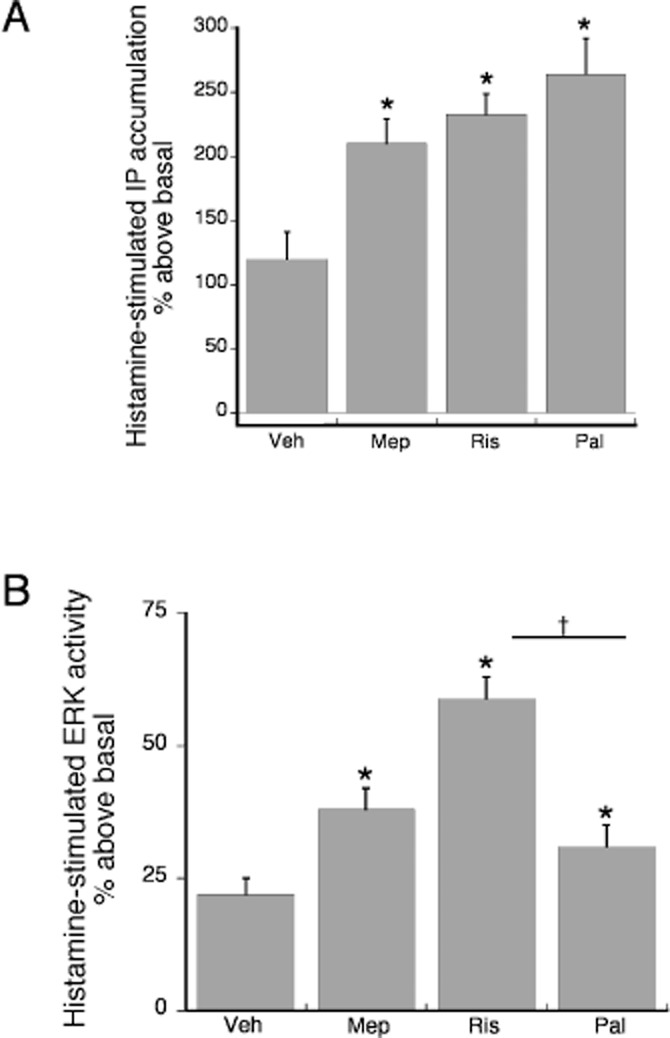
The effect of prolonged pretreatment with risperidone and paliperidone compared with the reference ligand mepyramine on sensitization of the PLC-IP (A) and ERK (B) pathways. Cells were treated with maximal concentrations of mepyramine (1 μM), risperidone (2 μM), paliperidone (2 μM) or vehicle for 24 h. Following a wash, histamine (1 μM)-stimulated IP accumulation or ERK activity was measured. (A) For IP accumulation, all three ligands acted as inverse agonists, enhancing the effect of histamine. There was no difference in the magnitude of the effect of paliperidone versus that of risperidone. *P < 0.05 compared to the vehicle pretreated condition. Bars represent the mean ± SEM of three experiments. (B) For ERK activity, all three ligands acted as inverse agonists, enhancing histamine-stimulated ERK activity. The effect of risperidone was greater than that of paliperidone. *P < 0.05 compared to the vehicle pretreated condition. †P < 0.05 for paliperidone compared to risperidone. Bars represent the mean ± SEM of three experiments.
Discussion
Functional selectivity is a term used to describe the phenomenon whereby different ligands acting at the same receptor subtype can elicit qualitatively different effects on cell function. The underlying mechanism for functional selectivity is believed to be the formation/stabilization of unique spectra of receptor conformations that are dependent upon the structure of the ligand used. Different receptor conformations are expected to have different coupling/activation efficiencies with the various signal transduction molecules (i.e. G proteins, β-arrestins, etc.) coupled to the receptor. Thus, individual drugs have the capacity to generate a unique spectrum of activity of cellular signalling pathways. Given that some cellular pathways may lead to therapeutic effects, whereas others may mediate adverse effects, it is important to understand the functional selectivity profiles of drugs.
In a study exploring the functional selectivity of various derivatives of amphetamine at 5-HT2A and 5-HT2C receptor signalling to PLC and PLA2, we found that relatively small differences in drug structure could result in significant differences in signalling. For example, 2,5-dimethoxyphenylisopropylamine (DMA) differs from 2,5-dimethoxy-4-methylphenylisopropylamine (DOM) by a single methyl group at the 4' position on the phenyl ring. At the 5-HT2C receptor, DOM is a strong agonist for both PLA2 and PLC, whereas DMA has no efficacy at PLA2 (it is an antagonist), but retains relatively strong agonist activity at the PLC pathway.
Paliperidone is a metabolite of risperidone and differs from risperidone by only a single hydroxyl group. Although the receptor-binding characteristics are similar (Gray and Roth, 2007), in this study, we found that the signalling profiles of paliperidone acting at dopamine D2, 5-HT2A, 5-HT2C and histamine H1 receptors differed from those of risperidone. As shown in Figure 10, depending upon the receptor–response pair, the relative efficacy of paliperidone was greater than, less than or not different from that of risperidone. Such response-dependent differences in relative efficacy indicate that as a consequence of a single hydroxyl group, the receptor conformation spectrum promoted by paliperidone differs from that of risperidone. In some cases, the receptor conformations promoted by paliperidone are sufficiently different from those of risperidone that coupling to, and regulation of, cellular signalling pathways differs between the two drugs. Differential regulation of cellular signalling suggests that the physiological and behavioral consequences of paliperidone and risperidone may differ as well.
Figure 10.

Relative efficacy of paliperidone versus risperidone for various cellular signalling responses coupled to human 5-HT2C (A), 5-HT2A (B), dopamine D2 (C) and histamine H1 (D) receptors. For each response, relative efficacy was calculated as the maximal response of paliperidone or risperidone divided by that of the reference ligand. Differences in relative efficacy for responses coupled to a single receptor subtype reflect functional selectivity.
For the majority of responses measured, paliperidone and risperidone displayed either inverse agonism or antagonism. Inverse agonism, the property of a drug that decreases ligand-independent or constitutive receptor activity, is now a well-recognized pharmacological property. Preclinical studies have shown that many atypical APDs display inverse agonism at their target receptors, leading to suggestions that the inverse agonist properties of atypical APDs may be responsible for therapeutic activity (Akam and Strange, 2004; Heusler et al., 2007; Meltzer et al., 2012). However, in general, supportive evidence for therapeutic relevance of inverse agonism is lacking in clinical settings (for review see Parra and Bond, 2007).
Interestingly, paliperidone and risperidone were both agonists acting at the 5-HT2A receptor for β-arrestin translocation and sensitization of ERK and acting at the dopamine D2 receptor for sensitization of AC signalling. Ligands that act as agonists for some responses coupled to a receptor, but inverse agonists for other responses mediated by the same receptor [e.g. ‘protean’ ligands (Kenakin, 2001; Brink et al., 2004)] have been identified for many GPCRs (for review, see Kenakin, 2007). The existence of protean ligands underscores the importance of specifying the response measured when referring to the pharmacological characteristics of a ligand, as the same ligand, acting at a single receptor subtype, can be both an agonist and an inverse agonist at the same time in the same cells. This also emphasizes that measurement of a single response is not sufficient to pharmacologically characterize a receptor as an agonist, antagonist or inverse agonist.
Interestingly, the dopamine D2 reference inverse agonist ligand, butaclamol, as well as risperidone and paliperidone, behaved as agonists for sensitization of the dopamine D2 receptor-adenylyl cyclase signalling pathway. Prolonged pretreatment resulted in reduced responsiveness to the agonist, quinpirole, to reduce adenylyl cyclase activity. In fact, the reduction in the quinpirole response produced by pretreatment with butaclamol was equivalent to that produced by pretreatment with quinpirole. Similarly, butaclamol behaved as an agonist following prolonged pretreatment when quinpirole stimulation of ERK activity was measured. Changes in receptor-effector responsiveness as a result of treatment with an agonist (desensitization) or with an inverse agonist (sensitization) can occur through changes in receptor density or changes in receptor-effector coupling efficiency as a result of changes in signal transduction components. Pretreatment with butaclamol reduced the expression of dopamine D2 receptors, which is consistent with the reduced inhibition of adenylyl cyclase activity and reduced stimulation of ERK by quinpirole. However, pretreatment with risperidone or paliperidone produced a slight increase in dopamine D2 receptor density. While this increase in dopamine D2 receptor density may underlie the mechanism by which pretreatment with paliperidone enhances quinpirole-stimulated ERK activity, it does not explain why pretreatment with risperidone and paliperidone reduced quinpirole-mediated inhibition of adenylyl cyclase activity. This suggests that additional changes in the dopamine D2 receptor-adenylyl cyclase signalling cascade may occur following risperidone and paliperidone pretreatment that results in an overall reduction in receptor-adenylyl cyclase signalling in the presence of increased receptor density.
Although it is difficult to extrapolate differences in the cellular actions of paliperidone versus risperidone to differences in vivo, Dremencov et al. (2007) reported recently that paliperidone differed from risperidone in the regulation of serotonergic and noradrenergic firing rates in vivo in rats. Unlike risperidone, which was effective at a dose of 0.4 mg·kg−1 (i.v.), acute administration of paliperidone at doses up to 1 mg·kg−1 did not alter the firing rate of 5-HT neurons. Similarly, 5-HT neuronal activity was reduced following 2 day and 14 day regimens (1 mg·kg−1·day−1, s.c.) of risperidone, but not paliperidone, administration. Some differences were also observed in the regulation of noradrenergic neuronal firing activity following selective serotonin reuptake inhibitor (SSRI) administration suggesting that paliperidone, but not risperidone, may be effective in treating SSRI-resistant depression. Even though risperidone is metabolized to paliperidone in vivo, this study suggests that the differences in cellular signalling patterns between risperidone and paliperidone observed in vitro, may have physiological relevance.
For several reasons, it is difficult to attribute differences in clinical effects of paliperidone versus risperidone as due to pharmacodynamic differences. Risperidone is metabolized to paliperidone and therefore paliperidone is present in patients taking risperidone. Moreover, because paliperidone is not metabolized by the liver (CYP2D6), as is risperidone, there is reduced potential for drug interactions and for the consequences of genetic polymorphisms in CYP2D6 (high vs. low metabolizers). Furthermore, the formulations of the drugs used clinically typically are different. Paliperidone is generally administered as an extended release tablet (osmotic pump), which minimizes drug plasma level fluctuations as compared to the oral, immediate release formulation of risperidone. Such different formulations have significant differences in pharmacokinetic properties. The extended release formulation of paliperidone provides relatively stable plasma concentrations as compared to the rapid changes that are characteristic of oral immediate release formulations of risperidone. As it has been suggested that the incidence of extrapyramidal symptoms may be increased by fluctuations in plasma levels of atypical antipsychotics (Ereshefsky and Mascarenas, 2003), the more stable plasma levels of paliperidone may provide for reduced extrapyramidal symptom liability and perhaps improved therapeutic efficacy.
One study has attempted a ‘virtual’ comparison of the effects of risperidone and paliperidone using propensity score matching of data collected from six double-blind, randomized, placebo-controlled, short-term clinical trials that involved both oral, immediate-release risperidone and oral, extended-release paliperidone (Turkoz et al., 2011). When doses of paliperidone (6–12 mg·day−1) calculated to produce similar systemic drug exposure as 2–4 mg·day−1 of risperidone were compared, improvement in the Positive and Negative Syndrome Scale score for paliperidone was greater than that for risperidone. Some differences were also observed in placebo-adjusted adverse events with improved tolerability of paliperidone versus risperidone. However, these results should be interpreted with caution given that comparisons were made between different formulations of the drugs with significant differences in pharmacokinetic profiles and from data collected in studies with different experimental designs.
Two recent studies have made a direct, head-to-head comparison of the effects of long-acting, injectable preparations of risperidone and paliperidone but report conflicting results (Fleischhacker et al., 2011; Pandina et al., 2012). In a 2011 double-blind study designed to compare paliperidone palmitate versus long-acting injectable risperidone (RIS-LAI) in patients with schizophrenia (n = 1220; Pandina et al., 2012), paliperidone was found to be non-inferior to risperidone. Further, the tolerability and safety of paliperidone palmitate was similar to that of RIS-LAI. By contrast, a 2012 phase III, double-blind study, comparing suspensions of paliperidone palmitate and extended-release risperidone microspheres (with oral supplementation), in 749 acutely symptomatic patients (339 patients completed the study), found paliperidone to be less efficacious than risperidone (Fleischhacker et al., 2011). However, measurement of plasma levels of paliperidone and risperidone indicated that the initial dosing regimen of paliperidone in the 2012 study did not lead to a plasma level rapidly enough for therapeutic efficacy as defined as that needed to occupy ≈60–65% of central dopamine D2 receptors (Nyberg et al., 1995; Kapur et al., 2000) as did the risperidone administration procedure.
In summary, the differences in the signalling profiles of paliperidone versus risperidone observed in this study suggest that the overall cellular actions of paliperidone differ from those elicited by risperidone. It is difficult to extrapolate changes in cellular signal transduction pathways to effects on physiological functions and behaviours. Although this study has revealed differences between paliperidone and risperidone in their actions to regulate various signalling systems coupled to individual receptors, attribution of these differences to possible differences in therapeutic efficacy or in generation of adverse effects is currently not possible. Further research is needed to define which of the myriad of cellular signalling pathways coupled to a receptor is/are important in the regulation of a physiological function or behavioral response. However, from a cellular perspective, it seems clear that the effects of paliperidone differ from those of risperidone.
Acknowledgments
The authors wish to thank Christine Dahlhausen for expert help in conducting the experiments. This work was supported in part by a research grant from Janssen Scientific Affairs, LLC.
Glossary
- AA
arachidonic acid
- APD
antipsychotic drug
- G protein
guanine nucleotide-binding protein
- 5-HT2A
serotonin 2A receptor subtype
- 5-HT2C
serotonin 2C receptor subtype
Conflict of interest
None.
References
- Akam E, Strange PG. Inverse agonist properties of atypcial antipsychotic drugs. Biochem Pharmacol. 2004;67:2039–2045. doi: 10.1016/j.bcp.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Beaulieu J, Gainetdinov R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Berg K, Rowan M, Sanchez T, Silva M, Patwardhan A, Milam S, et al. Regulation of kappa-opioid receptor signaling in peripheral sensory neruons in vitro and in vivo. J Pharmacol Exp Ther. 2011;338:92–99. doi: 10.1124/jpet.110.177493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Clarke WP. Development of functionally selective agonists as novel therapeutic agents. Drug Discov Today Ther Strat. 2006;3:421–428. [Google Scholar]
- Berg KA, Clarke WP, Sailstad C, Saltzman A, Maayani S. Signal transduction differences between 5-hydroxytryptamine type 2A and type 2C receptor systems. Mol Pharmacol. 1994;46:477–484. [PubMed] [Google Scholar]
- Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- Berg KA, Stout BD, Cropper JD, Maayani S, Clarke WP. Novel actions of inverse agonists on 5-HT2C receptor systems. Mol Pharmacol. 1999;55:863–872. [PubMed] [Google Scholar]
- Berg KA, Cropper JD, Niswender CM, Sanders-Bush E, Emeson RB, Clarke WP. RNA-editing of the 5-HT(2C) receptor alters agonist-receptor-effector coupling specificity. Br J Pharmacol. 2001a;134:386–392. doi: 10.1038/sj.bjp.0704255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Stout BD, Maayani S, Clarke WP. Differences in rapid desensitization of 5-hydroxytryptamine2A and 5-hydroxytryptamine2C receptor-mediated phospholipase C activation. J Pharmacol Exp Ther. 2001b;299:593–602. [PubMed] [Google Scholar]
- Berg KA, Evans KL, Cropper JD, Clarke WP. Temporal regulation of agonist efficacy at 5-hydroxytryptamine (5-HT)1A and 5-HT 1B receptors. J Pharmacol Exp Ther. 2003;304:200–205. doi: 10.1124/jpet.102.042564. [DOI] [PubMed] [Google Scholar]
- Berg KA, Harvey JA, Spampinato U, Clarke WP. Physiological relevance of constitutive activity of 5-HT2A and 5-HT2C receptors. Trends Pharmacol Sci. 2005;26:625–630. doi: 10.1016/j.tips.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Berg KA, Navailles S, Sanchez TA, Silva YM, Wood MD, Spampinato U, et al. Differential effects of 5-methyl-1-[[2-[(2-methyl-3-pyridyl)oxyl]-5-pyridyl]carbamoyl]-6-trifluoro methylindone (SB 243213) on 5-hydroxytryptamine(2C) receptor-mediated responses. J Pharmacol Exp Ther. 2006;319:260–268. doi: 10.1124/jpet.106.104448. [DOI] [PubMed] [Google Scholar]
- Berg KA, Patwardhan AM, Sanchez TA, Silva YM, Hargreaves KM, Clarke WP. Rapid modulation of mu-opioid receptor signaling in primary sensory neurons. J Pharmacol Exp Ther. 2007;321:839–847. doi: 10.1124/jpet.106.116681. [DOI] [PubMed] [Google Scholar]
- Berg KA, Dunlop J, Sanchez T, Silva M, Clarke WP. A conservative, single amino acid substitution in the second cytoplasmic domain of the h5-HT2C receptor alters both ligand-dependent and ligand-independent receptor signaling. J Pharmacol Exp Ther. 2008;324:1084–1092. doi: 10.1124/jpet.107.131524. [DOI] [PubMed] [Google Scholar]
- Brea J, Masaguer C, Villazon M, Cadavid M, Ravina E, Fontaine F, et al. Conformationally constrained butyrophenones as new pharmacological tools to study 5-HT2A and 5-HT2C receptor behaviors. Eur J Med Chem. 2003;38:433–440. doi: 10.1016/s0223-5234(03)00054-0. [DOI] [PubMed] [Google Scholar]
- Brink C, Harvey B, Bodenstein J, Venter D, Oliver D. Recent advances in drug action and therapeutics: relevance of novel concepts in G-protein-coupled receptor and signal transduction pharmacology. Br J Clin Pharmacol. 2004;57:373–387. doi: 10.1111/j.1365-2125.2003.02046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citrome L. Paliperidone: quo vadis? Int J Clin Pract. 2007;61:653–662. doi: 10.1111/j.1742-1241.2007.01321.x. [DOI] [PubMed] [Google Scholar]
- Clarke W, Berg K. Use of functional assays to detect and quantify functional selectivity. Drug Discov Today Technol. 2010;7:e31–e36. doi: 10.1016/j.ddtec.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Clarke WP. What's for lunch at the conformational cafeteria? Mol Pharmacol. 2005;67:1819–1821. doi: 10.1124/mol.105.013060. [DOI] [PubMed] [Google Scholar]
- Dolder C, Nelson M, Deyo Z. Paliperidone for schizophrenia. Am J Health Syst Pharm. 2008;65:403–413. doi: 10.2146/ajhp070261. [DOI] [PubMed] [Google Scholar]
- Dopheide JA. Paliperidone: an improvement over risperidone? Am J Health Syst Pharm. 2008;65:401. doi: 10.2146/ajhp070505. [DOI] [PubMed] [Google Scholar]
- Dremencov E, El Mansari M, Blier P. Distinct electrophysiological effects of paliperidone and risperidone on the firing activity of rat serotonin and norepinephrine neurons. Psychopharmacology (Berl) 2007;194:63–72. doi: 10.1007/s00213-007-0818-8. [DOI] [PubMed] [Google Scholar]
- Egan CT, Herrick-Davis K, Teitler M. Creation of a constitutively activated state of the 5-hydroxytryptamine2A receptor by site-directed mutagenesis: inverse agonist activity of antipsychotic drugs. J Pharmacol Exp Ther. 1998;286:85–90. [PubMed] [Google Scholar]
- Ereshefsky L, Mascarenas C. Comparison of the effects of different routes of antipsychotic administration on pharmacokinetics and pharmacodynamics. J Clin Psychiatry. 2003;64(Suppl. 16):18–23. [PubMed] [Google Scholar]
- Evans KL, Cropper JD, Berg KA, Clarke WP. Mechanisms of regulation of agonist efficacy at the 5-HT(1A) receptor by phospholipid-derived signaling components. J Pharmacol Exp Ther. 2001;297:1025–1035. [PubMed] [Google Scholar]
- Fleischhacker W, Gopal S, Lane R, Gassmann-Mayer C, Lim R, Hough D, et al. A randomized trial of paliperidone palmitate and risperidone long-acting injectable in schizophrenia. Int J Neuropsychopharmacol. 2011;15:107–118. doi: 10.1017/S1461145711001076. [DOI] [PubMed] [Google Scholar]
- Gray JA, Roth BL. Molecular targets for treating cognitive dysfunction in schizophrenia. Schizophr Bull. 2007;33:1100–1119. doi: 10.1093/schbul/sbm074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusler P, Newman-Tancredi A, Castro-Fernandez A, Cussac D. Differential agonist and inverse agonist profile of antipsychotics at D2L receptors coupled to GIRK potassium channels. Neuropharmacology. 2007;52:1106–1113. doi: 10.1016/j.neuropharm.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Kapur S, Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D2 occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157:514–520. doi: 10.1176/appi.ajp.157.4.514. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. Faseb Journal. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Lopez-Giminez J, Villazon M, Brea J, Loza M, Palacios J, Mengod G, et al. Multiple conformations of native and recombinant human 5-hydroxytrptamine (2A) receptors are labeled by aognists and discriminated by antagonists. Mol Pharmacol. 2001;60:690–699. [PubMed] [Google Scholar]
- MacDonald G, Bartolome J. A decade of progress in the discovery and development of ‘atypical’ antipsychotics. Prog Med Chem. 2010;49:37–80. doi: 10.1016/S0079-6468(10)49002-5. [DOI] [PubMed] [Google Scholar]
- Marion S, Weiner DM, Caron MG. RNA editing induces variation in desensitization and trafficking of 5-hydroxytryptamine 2C receptor isoforms. J Biol Chem. 2004;279:2945–2954. doi: 10.1074/jbc.M308742200. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Massey BW, Horiguchi M. Serotonin receptors as targets for drugs useful to treat psychosis and cognitive impairment in schizophrenia. Curr Pharm Biotechnol. 2012;13:1572–1586. doi: 10.2174/138920112800784880. [DOI] [PubMed] [Google Scholar]
- Milligan G, Bond RA. Inverse agonism and the regulation of receptor number. Trends Pharmacol Sci. 1997;18:468–474. doi: 10.1016/s0165-6147(97)01139-5. [DOI] [PubMed] [Google Scholar]
- Moya PR, Berg KA, Gutierrez-Hernandez MA, Saez-Briones P, Reyes-Parada M, Cassels BK, et al. Functional selectivity of hallucinogenic phenethylamine and phenylisopropylamine derivatives at human 5-hydroxytryptamine (5-HT)2A and 5-HT2C receptors. J Pharmacol Exp Ther. 2007;321:1054–1061. doi: 10.1124/jpet.106.117507. [DOI] [PubMed] [Google Scholar]
- Nyberg S, Farde L, Halldin C, Dahl M, Bertilsson L. D2 dopamine receptor occupancy during low-dose treatment with haloperidol decanoate. Am J Psychiatry. 1995;152:173–178. doi: 10.1176/ajp.152.2.173. [DOI] [PubMed] [Google Scholar]
- Paliperidone: new drug. Just a metabolite of risperidone, a neuroleptic soon off-patent. Prescrire Int. 2007;16:236–237. [PubMed] [Google Scholar]
- Pandina G, Lane R, Gopal S, Gassmann-Mayer C, Hough D, Remmerie B, et al. A double-blind study of paliperidone palmitate and risperidone long-acting injectable in adults with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2012;35:218–226. doi: 10.1016/j.pnpbp.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Parra S, Bond RA. Inverse agonism: from a curiosity to accepted dogma, but is it clinically relevant? Curr Opin Pharmacol. 2007;7:146–150. doi: 10.1016/j.coph.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Perez DM, Karnik SS. Multiple signaling states of G-protein-coupled receptors. Pharmacol Rev. 2005;57:147–161. doi: 10.1124/pr.57.2.2. [DOI] [PubMed] [Google Scholar]
- Rauser L, Savage JE, Meltzer HY, Roth BL. Phsyiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2001;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- Schotte A, Janssen P, Gommeren W, Luyten W, Van Gompet P, Lesage A, et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berl) 1996;124:57–73. doi: 10.1007/BF02245606. [DOI] [PubMed] [Google Scholar]
- Turkoz I, Bossie C, Lindenmayer J, Scooler N, Canuso C. Paliperidone ER and oral risperidone in patients with schizophrenia: a compoarative database analysis. BMC Psychiatry. 2011;11:21. doi: 10.1186/1471-244X-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Weiner DM, Burstein ES, Nash N, Croston GE, Currier EA, Vanover KE, et al. 5-hydroxytryptamine2A inverse agonists as antipsychotics. J Pharmacol Exp Ther. 2001;299:268–276. [PubMed] [Google Scholar]
- Wilbanks AM, Laporte SA, Bohn LM, Barak LS, Caron MG. Apparent loss-of-function mutant GPCRs revealed as constitutively desensitized receptors. Biochemistry. 2002;41:11981–11989. doi: 10.1021/bi020275m. [DOI] [PubMed] [Google Scholar]
- Wong E, Nikam S, Shahid M. Multi- and single-target agents for major psychiatric diseases: therapeutic opportunities and challenges. Curr Opin Investig Drugs. 2008;9:28–36. [PubMed] [Google Scholar]