

Abstract
BACKGROUND AND PURPOSE
An increasing body of evidence suggests that the purinergic receptor P2X, ligand-gated ion channel, 7 (P2X7) in the CNS may play a key role in neuropsychiatry, neurodegeneration and chronic pain. In this study, we characterized JNJ-47965567, a centrally permeable, high-affinity, selective P2X7 antagonist.
EXPERIMENTAL APPROACH
We have used a combination of in vitro assays (calcium flux, radioligand binding, electrophysiology, IL-1β release) in both recombinant and native systems. Target engagement of JNJ-47965567 was demonstrated by ex vivo receptor binding autoradiography and in vivo blockade of Bz-ATP induced IL-1β release in the rat brain. Finally, the efficacy of JNJ-47965567 was tested in standard models of depression, mania and neuropathic pain.
KEY RESULTS
JNJ-47965567 is potent high affinity (pKi 7.9 ± 0.07), selective human P2X7 antagonist, with no significant observed speciation. In native systems, the potency of the compound to attenuate IL-1β release was 6.7 ± 0.07 (human blood), 7.5 ± 0.07 (human monocytes) and 7.1 ± 0.1 (rat microglia). JNJ-47965567 exhibited target engagement in rat brain, with a brain EC50 of 78 ± 19 ng·mL−1 (P2X7 receptor autoradiography) and functional block of Bz-ATP induced IL-1β release. JNJ-47965567 (30 mg·kg−1) attenuated amphetamine-induced hyperactivity and exhibited modest, yet significant efficacy in the rat model of neuropathic pain. No efficacy was observed in forced swim test.
Conclusion and Implications
JNJ-47965567 is centrally permeable, high affinity P2X7 antagonist that can be used to probe the role of central P2X7 in rodent models of CNS pathophysiology.
Keywords: purinergic, P2X7, interleukin 1β (IL-1β), autoradiography, microdialysis, depression, mania, neuropathic pain
Introduction
ATP mediates a myriad of critical physiologic functions by activating cell surface purinergic (P2) receptors that are either G-protein coupled (P2Y; metabotropic) or flux non-selective cations via ion channels (P2X; ionotropic) (Abbracchio et al., 2006; Surprenant and North, 2009; Coddou et al., 2011). There are seven subtypes of P2X ion channels, of which ATP has the lowest affinity for purinergic receptor P2X, ligand-gated ion channel, 7 (P2X7); as such the highly regulated ATP never reaches high local concentration to activate P2X7 in normal circumstances rendering P2X7 to be a ‘silent receptor’. On the contrary, local ATP concentration may achieve high micromolar levels (a danger signal) activating P2X7 ion channels and in the process engage the proinflammatory cascade via release of IL-1β (Solle et al., 2001) which in turn may contribute to neuroinflammation and pain (Chessell et al., 2005; North and Jarvis, 2013), mood disorders (Iwata et al., 2013) and neurodegenerative disorders such as Alzheimer's (Diaz-Hernandez et al., 2012), multiple sclerosis (Sharp et al., 2008) and epilepsy (Engel et al., 2012). In the CNS, P2X7 is expressed predominantly within glial cells (Sperlagh et al., 2006) and cross-talks with the neurons via IL-1β signalling. In addition to IL-1β, P2X7 dependent release of glutamate (Ando and Sperlagh, 2013), cathepsins (Clark et al., 2010) and chemokines (Shiratori et al., 2010) have been reported, and all of these mediators can serve as ‘gliotransmitters’ aiding in the neuroimmune cross-talk and modulating synaptic plasticity. The human P2X7 gene (P2RX7) is localized to chromosome 12q24.31 and encodes a 595 amino acid protein encompassing an intracellular N terminal domain, two transmembrane segments, a bulky extracellular domain and a long C terminal tail that is under tight regulation by intracellular messengers (Volonte et al., 2012). This membrane topology was recently verified by the zebra fish P2X4 crystal structure (Hattori and Gouaux, 2012). The extracellular domain hosts the ATP binding site (Young et al., 2007; Browne et al., 2010) and is not highly conserved across P2X subtypes and within P2X7 orthologues. While the former confers P2X7 selectivity, the latter presents a hurdle in developing antagonists with reasonable affinity across rodent and higher mammalian species (Donnelly-Roberts et al., 2009a; Michel et al., 2009; Bhattacharya et al., 2011). In fact, the P2X7 antagonist CE-224,535 that was in clinical development for rheumatoid arthritis (Stock et al., 2012) is a relatively human specific compound with weak potency at the rodent P2X7 channel (Duplantier et al., 2011) and similar speciation has been demonstrated for other P2X7 chemotypes (Baxter et al., 2003; Furber et al., 2007; Guile et al., 2009).
Within the CNS, and in particular within the context of brain neuro(patho)physiology, there has been an explosion of interest in the role of ATP and P2X ion channels in modulating neurophysiology (Burnstock, 2008; Abbracchio et al., 2009; Burnstock et al., 2011a,b; Khakh and North, 2012). As far as mood disorders are concerned, there is a growing interest in the potential role of P2X7 as a therapeutic target for unipolar depression and bipolar disorder (Bennett, 2007; Sperlagh et al., 2012; Iwata et al., 2013). The scientific rationale for P2X7 antagonism as a target for mood disorders is based on the following premises and there is a continuum of emerging science strengthening the hypothesis: (i) several human genetic studies have associated P2RX7 gene with both bipolar and depression (Barden et al., 2006; Lucae et al., 2006; Hejjas et al., 2009; McQuillin et al., 2009; Backlund et al., 2011; Soronen et al., 2011), although there are some reports that have failed to link a strong association as well (Green et al., 2009; Grigoroiu-Serbanescu et al., 2009); (ii) IL-1β, the cytokine that is released by P2X7 activation, is present at higher levels in plasma, CSF and in post-mortem brain tissue from mood disorder patients (Rao et al., 2010; Soderlund et al., 2011; Jones and Thomsen, 2013), suggesting that the tone of P2X7 may be activated under such clinical conditions; (iii) several independent laboratories, have demonstrated a protective phenotype of P2X7 knockout mice in models of depression and mania (Basso et al., 2009; Boucher et al., 2011; Csolle et al., 2013); (iv) in animal models of stress-induced depression, several groups have elegantly demonstrated that IL-1β signalling is critical (Goshen et al., 2008; Koo and Duman, 2008; 2009a,b; Csolle and Sperlagh, 2010) and (v) recent data presented at the Society for Neuroscience annual meeting indicated that P2X7 antagonist A-804598 attenuated chronic stress mediated anhedonia in rats (Iwata et al., 2012).
In this paper, we pharmacologically characterize the novel P2X7 antagonist JNJ-47965567 (Letavic et al., 2013) and describe the molecule against a panel of in vitro and in vivo assays. Since JNJ-47965567 displayed high affinity for the rat P2X7 ion channel, we used in vivo models of target engagement in rats (autoradiography and brain IL-1β microdialysis) to further characterize JNJ-47965567. Lastly, we tested JNJ-47965567 in models of depression, mania and pain in an attempt to test the role of P2X7 in such models of disease. Our data clearly demonstrate that JNJ-47965567 is potent, selective P2X7 antagonist, engages the brain P2X7 channel in vivo and is an excellent tool compound to probe the role of central P2X7 in rats.
Methods
Cell culture
The 1321N1 cells expressing the P2X7 channels (human, mouse, dog, macaque) were cultured in DME–high glucose medium supplemented with 10% FBS and 500 μg·mL−1 G418. The 1321N1 cells expressing the rat P2X7 were grown in same media supplemented with 10% FBS and 100 μg·mL−1 Zeocin. For isolation of primary microglia and astrocytes, rat brains were dissected from 3-day-old neonatal rats. After removal of the meninges, brains were triturated in 10 mL culture medium (DMEM, 10% FBS, pen-strep) and filtered through a 100 μm cell strainer. Cells were pelleted by centrifugation and washed once with culture medium. The mixture of cells was seeded in a 75 cm flask (1 brain/flask) and allowed to grow for 10 days. Medium was replaced at days in vitro (DIV) 10 and grown for 4 more days. Medium was again replaced at DIV 14 and microglial cells were isolated from the mixed cell culture by gentle shaking (100 rpm) on an orbital shaker at 37°C, 5% CO2 for 2 h. Floating microglial cells were spun down, counted and seeded into appropriate assay plates. Remaining adherent monolayer (astrocytes) was trypsinized, spun down, counted and seeded into appropriate assay plates.
Calcium measurement
The 1321N1 cells expressing P2X7 orthologues were dissociated 18–24 h prior to the assay using 0.05% trypsin/EDTA (Invitrogen, Grand Island, NY, USA), and plated at density of 25 000 cells·well−1 into poly-D-lysine coated 96-well black-walled clear bottom plates (Becton-Dickinson, Bedford, MA, USA). On the day of the experiment, cell plates were washed with assay buffer, containing (in mM): 130 NaCl, 2 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 5 glucose; pH 7.40. After the wash, dye loading was achieved by adding a 2× Calcium-4 (Molecular Devices, Sunnyvale, CA, USA) dye solution in the assay buffer. Cells were stained with the Calcium-4 dye in staining buffer for 30 min at room temperature in the dark. Test compounds were prepared at 250× the final test concentration in neat DMSO. Intermediate 96-well compound plates were prepared by transferring 1.2 μL of the compound into 300 μL of assay buffer. A further 3× dilution occurred when transferring 50 μL·well−1 of the compound plate to 100 μL·well−1 in the cell plate. Cells were incubated with test compounds and dye for 30 min. Calcium flux was monitored in FLIPRTetra as the cells were challenged by adding 50 μL·well−1 of BzATP. The final concentration of Bz-ATP was 250 μM (human, rat, dog, macaque) or 600 μM (mouse) and 100 μM (rat primary astrocytes). In experiments where washing of the antagonist was involved (Figure 3), cells were washed repeatedly using a plate washer for three cycles. The wash protocol was kept constant for all experiments that involved a wash cycle.
Figure 3.

Reversibility and protection of the JNJ-47965567 binding site by A-438079. (A and C) Potency shift of JNJ-47965567 and A-438079, with and without washing after 30 min incubation with either the human (A) or the rat (C) P2X7 cells. The effect on the potency of JNJ-47965567 by protecting the binding site by A-438079 (30 μM) is shown for the human (B) and rat (D) P2X7 cells. Data are expressed as percent of control (defined as response in control cells to 250 μM Bz-ATP). Symbols represent mean ± SEM from three independent experiments.
Radioligand binding
[3H] A-804598 was used as the radioligand (Donnelly-Roberts et al., 2009b). P2 membranes were prepared from recombinant cells; for one 96-well assay, cells were harvested from 10 T225 confluent flasks and frozen at −80°C. On the day of the experiment, cells were thawed for membrane (P2) preparation. A 50 mM Tris-HCl (pH 7.4) was added to the cells and homogenized for ∼30 s at high speed. The homogenate was centrifuge at 1500 rpm for 5 min followed by careful decanting of the supernatant which was centrifuged at 32 000× g for 30 min. A 6 mL of ice-cold assay buffer (50 mM Tris-HCl + 0.1% BSA) was added to the cell pellet (final protein concentration varied between 0.4 and 0.5 mg·well−1). The assay volume of 100 μL was composed of the following: (i) 10 μL compound (10×) + (ii) 40 μL tracer (2.5×) + 50 μL membrane (2×). The reaction was incubated for 1 h at 4°C. The assay was terminated the by filtration (GF/B filters pre-soaked with 0.3% polyethylenimine) and washed with washing buffer (Tris-HCl 50 mM) repeatedly. After drying the plate, Microscint 0 (Perkin Elmer, Waltham, MA, USA) was added to the filters and radioactivity was counted.
IL-1β assay
Human blood, isolated human monocytes or rat microglial cells were primed with LPS followed by addition of the test antagonist or vehicle with the final P2X7 stimulus of Bz-ATP. For the human blood and monocytes, priming concentration of LPS was 30 ng·mL−1. For rat microglial cells, LPS was used at 3 ng·mL−1. Cells were primed with LPS for 1 h. Test compounds were added and incubated for an additional 30 min. The P2X7 agonist, Bz-ATP (1 mM for blood, 0.5 mM for monocytes and 0.3 mM for rat microglia) was finally added and incubated for 1.5 h at 37°C. Post incubation, the plates were centrifuged (low-speed spin) and the supernatant collected for IL-1β ELISA analysis as per as manufacturer's protocol (human IL-1β: Thermo Scientific, Rockford, IL, USA; catalogue # EH2IL1B5; rat IL-1β: R&D Systems, Minneapolis, MN, USA; catalogue # RLB00).
Electrophysiology
The 1321N1 cells stably expressing hP2X7 were maintained in culture as described previously. Twenty-four hours prior to experimentation, cells were washed with divalent-free PBS (Hyclone, Rockford, IL, USA), dissociated with 0.05% trypsin (Gibco, Grand Island, NY, USA) and plated on poly-d-lysine coated cover slips (B-D Biosciences, San Jose, CA, USA) in a 24-well plate at a density of 10 000 cells·well−1. On the day of experimentation, cells were transferred to a recording chamber mounted on a Nikon Eclipse TE2000-U (Melville, NY, USA) and continuously perfused (1 mL·min−1) with a low divalent physiological saline solution (137 mM NaCl, 5.4 mM KCl, 0.5 mM MgCl2, 0 mM CaCl2, 5 mM glucose, 10 mM HEPES, pH = 7.4). Patch pipettes (2–3 MΩ) were visually guided to the surface of individual cells. Patch pipettes contained 135 mM CsF, 10 mM CsCl, 5 mM EGTA, 5 mM NaCl, and 10 mM HEPES, osmolarity adjusted to 290 mOsm·L−1 with sucrose, pH 7.3. Voltage-clamp recordings were made with an Axopatch 200B, and Clampex 9.2 software (Molecular Devices). Data were acquired at 5 kHz and filtered at 2 kHz. Pipette capacitance was cancelled in all recordings. Whole-cell responses to Bz-ATP were measured by rapidly transitioning the cell from being bathed in physiological saline to Bz-ATP (300 μM) for 2 s in successive trials using a large-bore drug applicator (a linear array of glass tubes driven by a Warner Instruments Corp., Hamden, CT, USA; SF-77B Perfusion Fast-Step). Bz-ATP responses were quantified using Clampfit software (Molecular Devices) to calculate the mean current amplitude during the last 100 ms of Bz-ATP or Bz-ATP + JNJ4795567 application. For experiments examining channel block, after establishing a consistent control response by applying Bz-ATP at 60 s intervals, perfusion with 100 nM JNJ4795567 was started 1 min prior to reinitiating the Bz-ATP applications (+100 nM JNJ4795567) at 2 min intervals and block was allowed to proceed to steady state. Upon achieving maximal block, JNJ4795567 was removed from the perfusate and cells were challenged with Bz-ATP at 1 min intervals for 10 min.
Ex vivo receptor binding autoradiography
All animal work performed in this paper was in accordance with the Guide Care for and Use of Laboratory Animals adopted by the US National Institutes of Health. All studies involving animals are in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Animals were allowed to acclimate for 7 days after receipt. They were group housed in accordance with institutional standards, received food and water ad libitum and were maintained on a 12 h light/dark cycle. Male Sprague Daley rats approximately 300–400 g in body weight were used. The animals were killed using carbon dioxide and decapitated at different time points after drug administration. Brains were rapidly frozen on powdered dry ice and stored at −80°C before sectioning. Plasma samples were also collected for bioanalysis. Twenty micron thick tissue sections at the level of the hippocampus were prepared for autoradiography. P2X7 radioligand binding autoradiography was determined at room temperature with 30 nM [3H] A-804598 in 50 mM Tris HCl incubation buffer containing 0.1% BSA as previously described. Non-specific binding was measured with 100 μM A-740003. Sections were incubated for 10 min to minimize dissociation, rinsed four times for 5 min in the 50 mM Tris Buffer with 0.1% BSA, followed by two dips in ice cold water. Sections were allowed to dry before acquisition with β-Imager (BioSpace, Paris, France) for 2 h. Quantitative analysis was performed using the β -imager. Ex vivo receptor labelling was expressed as the percentage of receptor labelling in corresponding brain areas (i.e. hippocampus) of saline-treated animals. The percentage of receptor occupancy was plotted against time or dosage using GraphPad Prism (GraphPad Software, San Diego, CA, USA). Percentage of receptor occupancy was also plotted against drug plasma concentration.
In vivo Bz-ATP induced IL-1β release using microdialysis
Male Sprague Dawley rats (280–380 g) or C57/Bl6 mice (8–12 weeks of age) were implanted with a guide cannula in the hippocampus. Eicom's ATMOS-LM microdialysis sampling system was used for dialysis collection, including the pressure cancelling vented probe (4 mm or 2 mm membrane rat or mouse, respectively) with a 1000kD MW cut-off. Three hours following probe implantation, baseline sample collection began. Dialysate was collected in 60-min samples at 0.5 μL·min−1 and maintained at 4°C. Following 2 h of baseline with 1× aCSF + 0.15% BSA, Bz-ATP (100 mM) was administered via reverse dialysis for 2 h. The perfusate was then returned to the baseline matrix. JNJ-47965567 (30 or 100 mg·kg−1) or vehicle was administered s.c. 30 min prior to Bz-ATP infusion. Samples were analysed for IL-1β using a rat or mouse specific IL-1β ELISA kit from R&D Systems.
Amphetamine hyperactivity, model of mania
Adult, male Sprague Dawley rats (Harlan, Placentia, CA, USA) maintained on a normal 12/12 light/dark cycle (lights on at 0600 h) under standard housing conditions with food and water provided ad libitum (except during testing) were used for this study. Experiments took place during the light-cycle. All methods complied with The Guide for the Care and Use of Laboratory Animals manual and were approved by the local Institutional Animal Care and Use Committee. Animals were single housed with enrichment (sunflower seeds and nylon bone) and were allowed to acclimate to housing conditions for at least 5 days after arrival. On day 1 of the experiment, animals were acclimated to the procedure room for at least 1 h and then each cage was placed onto a MotorMonitor rack (Kinder Scientific, San Diego, CA, USA), which surrounded each animal cage by a grid of photobeams (seven beams along the width of the cage and 15 beams along the length). The photobeams allowed for tracking the distance travelled by each animal. After 1 h, rats were injected s.c. with either vehicle (30% sulfobutylether beta-cyclodextrin) or JNJ-47965567 (30 mg·kg−1) and locomotor activity was recorded for an additional hour. All animals then received an injection of d-amphetamine sulfate (2 mg·kg−1, i.p., weight of salt) and locomotor activity was recorded for two additional hours. On day 2–5, the experiment was repeated in the same manner, with the same treatments provided each day. After 2 days of drug withdrawal, the same procedure was repeated again on day 8 with another amphetamine challenge.
Chung model of neuropathic pain
For creating the neuropathic preparation, the surgical procedure previously described by Kim and Chung (1992) was performed to induce an allodynic state. Briefly, the left L-5 and L-6 spinal nerves were isolated adjacent to the vertebral column and ligated with 6-0 silk suture distal to the dorsal root ganglion while under isoflurane anaesthesia. The rats were allowed a 10–14 days post-operative recovery period and allowed adequate time to develop the neuropathy before being placed in the study. Animals that did not meet a tactile allodynia 50% threshold of <3.0 g were not used. For each drug, six animals were prepared with the Chung model of spinal nerve ligation. Test articles were injected by one individual with all behavioural observations made by a second individual who was unaware of the article received by each animal. Tactile thresholds were assessed using Von Frey hairs and the up/down method as described in the initial Study Protocol at baseline, 30, 60 and 120 min post-compound dosing. Summary statistics were computed and include group means and standard deviations and numbers of animals per group. For time course curves, two-way repeated measures were undertaken and a post hoc analysis performed comparing with time zero. The area under the % hyperalgesic effect curve (not shown) was calculated and referred to as the AUC. Statistical comparison of AUC was carried out with a one-way anova and a post hoc analysis was carried out comparing all value to vehicle. All statistics were performed on spreadsheets using Prism statistical software version 5.0c (GraphPad Software).
Molecular target nomenclature (e.g. receptors, ion channels and so on) used in the manuscript conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011).
Results
In vitro pharmacology: recombinant P2X7 system
JNJ-47965567 is N-((4-(4-Phenyl-piperazin-1-yl)tetrahydro-2H-pyran-4-yl)methyl)-2-(phenyl-thio) nicotinamide (Figure 1). The compound was characterized using a combination of functional and binding assays using the recombinant P2X7 channel, overexpressing in 1321N1 astrocytoma cells. For comparison, three other known P2X7 antagonists have been used in these assays (A-804598, A-438079, AZ-10606120; Figure 1). Bz-ATP induced calcium flux was the primary functional assay. The potency (pIC50 ± SEM) of JNJ-47965567 to attenuate Bz-ATP induced calcium flux was as follows: human (8.3 ± 0.08), macaque (8.6 ± 0.1), dog (8.5 ± 0.2), rat (7.2 ± 0.08) and mouse (7.5 ± 0.1). The data are summarized in Table 1 alongside the three comparator P2X7 antagonists. JNJ-47965567 is a potent P2X7 antagonist across all species tested. JNJ-47965567 did not exhibit any agonist activity (data not shown). Since the calcium flux assay is a system not in equilibrium (Charlton and Vauquelin, 2010), we wanted to test the binding affinity of JNJ-47965567 in a radioligand binding assay under true equilibrium conditions. Our results demonstrate that JNJ-47965567 displaced the binding of [3H]-A-804598 in a concentration dependent manner (Figure 2) both at the human and rat P2X7 cell membranes (other species were not tested routinely in the binding assay as rat was the in vivo species of choice). The rank order potency for displacement of [3H]-A-804598 at the human P2X7 was AZ-10606120 > JNJ-47965567 ∼ A-804598 > A-438079, a profile similar to that observed in the functional assay (Table 1). The human affinity (pKi ± SEM) of JNJ-47965567 was 7.9 ± 0.07. Affinities of AZ-10606120, A-804598 and A-438079 were 8.5 ± 0.08, 8.0 ± 0.04 and 7.1 ± 0.08, respectively. JNJ-47965567 exhibited even higher affinity at the rat P2X7 channel: the affinity was 8.7 ± 0.07, a profile similar to A-804598 (8.8 ± 0.06) but distinctly dissimilar to A-438079 (6.7 ± 0.1) and AZ-10606120 (8.0 ± 0.08). Therefore, JNJ-47965567 was a P2X7 functional antagonist, with high affinity binding at both the human and rat ion channel; moreover, the high affinity rat binding provided JNJ-47965567 as a tool to further evaluate the role of P2X7 in rats.
Figure 1.
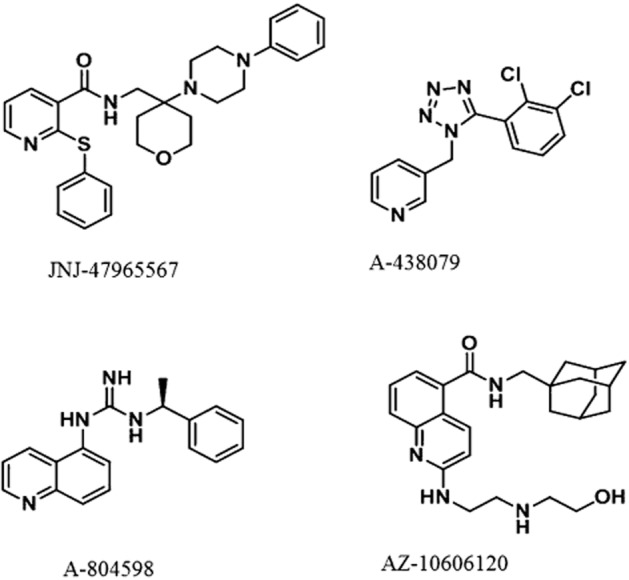
Chemical structure of P2X7 antagonists used in this study.
Table 1.
Potency of JNJ-47965567, in comparison with known P2X7 antagonists, at various orthologues as measured by calcium flux assay (FLIPR)
| Human P2X7 | Macaque P2X7 | Dog P2X7 | Rat P2X7 | Mouse P2X7 | |
|---|---|---|---|---|---|
| Ca2+ flux (TETRAFLIPR) pIC50 ± SEM | |||||
| JNJ-47965567 | 8.3 ± 0.08 | 8.6 ± 0.1 | 8.5 ± 0.2 | 7.2 ± 0.08 | 7.5 ± 0.1 |
| A-804598 | 7.7 ± 0.13 | 7.4 ± 0.1 | 7.5 ± 0.1 | 6.8 ± 0.17 | 7.0 ± 0.06 |
| A-438079 | 6.0 ± 0.02 | 6.0 ± 0.1 | 5.4 ± 0.1 | 5.9 ± 0.2 | 5.5 ± 0.2 |
| AZ-10606120 | 8.9 ± 0.04 | 8.4 ± 0.1 | 7.3 ± 0.04 | 5.7 ± 0.06 | 6.2 ± 0.1 |
Figure 2.
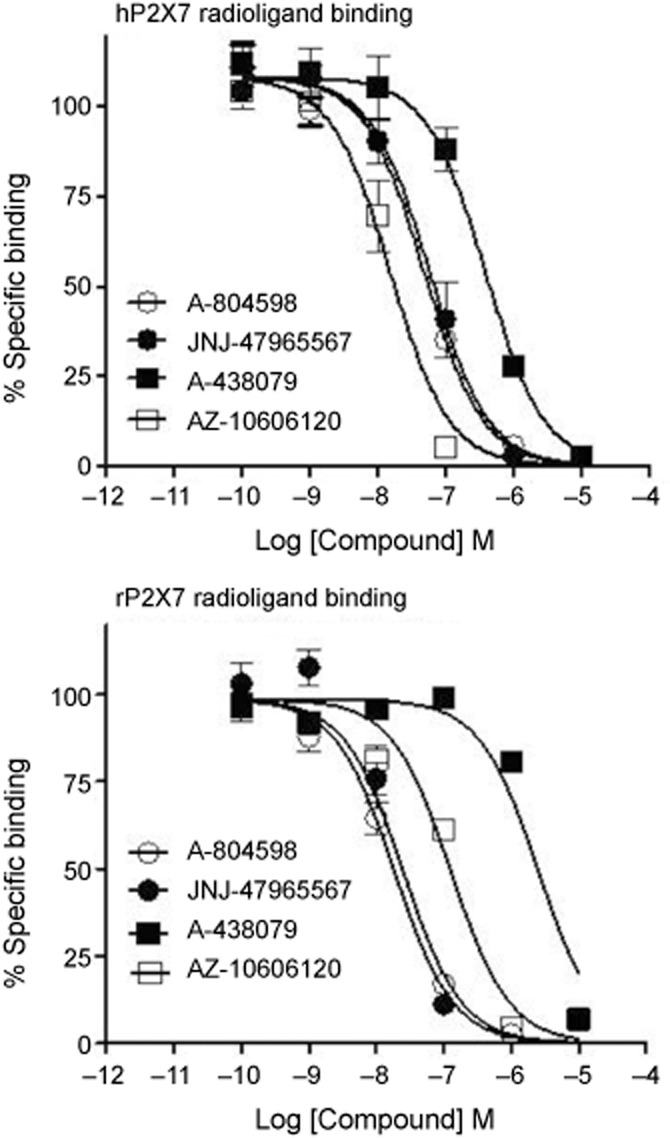
Displacement of [3H] A-804598 by JNJ-47965567, A-804598, A-438079 and AZ-10606120 in membrane preparations of 1321N1 cells expressing either the recombinant human (upper panel) or rat (lower panel) isoform. Data are expressed as percent specific binding. Symbols represent mean ± SEM from five independent experiments. Affinity (pKi) of JNJ-47965567 at the human and rat P2X7 was 7.9 ± 0.07 and 8.7 ± 0.07, respectively.
In vitro pharmacology: mechanism of action studies (recombinant system)
The binding data provided evidence that JNJ-47965567 (and A-438079, AZ-10606120) competed with A-804598 for an identical or mutually non-exclusive binding site(s). In order to elucidate the mechanism further, we decided to use A-438079 to protect the binding of JNJ-47965567. A 30 μM concentration of A-438079 was incubated with P2X7-1321N1 cells (human Figure 3B; and rat Figure 3D) for 5 min (high concentration to achieve a faster on-rate and occupancy of binding site for A-438079), followed by a 25 min incubation with various concentrations of JNJ-47965567. The incubation was terminated by a robotic wash protocol and followed by measurement of calcium flux induced by Bz-ATP. A mutually overlapping binding site of the two compounds would result in significant loss of potency for JNJ-47965567 as A-438079 would protect the accessibility of JNJ-47965567 to the P2X7 binding site during the incubation period. For the human P2X7 expressing cells, the potency of JNJ-47965567 shifted from 8.5 ± 0.05 in the absence of A-438079 to 5.9 ± 0.05 in the presence of the protecting agent A-438079 (Figure 3B). In separate experiments, it was determined that JNJ-47965567 did not wash off in the absence of A-438079 (Figure 3A & C); in fact, compound takes about 6 h to recover from block (Supporting Information Figure S1). These data suggest that JNJ-47965567 and A-438079 share identical to significantly overlapping binding pocket, consistent with the data generated from the radioligand displacement studies. Similar potency shifts, although to a lesser degree, were also seen at the rat P2X7 (Figure 3D).
In vitro pharmacology: native systems expressing P2X7 ion channel
Since P2X7 activation leads to release of the proinflammatory cytokine IL-1β, we sought to characterize JNJ-47965567 in human whole blood and in freshly isolated monocytes. As shown in Figure 4A, JNJ-47965567 attenuated LPS primed BZ-ATP induced IL-1β release in a concentration dependent manner with full block achieved at 1 μM concentrations. The potency of JNJ-47965567 to attenuate IL-1β release was 6.7 ± 0.07 in the human whole blood. Likewise, the pIC50 of JNJ-47965567 was 7.5 ± 0.07 when tested in freshly isolated human monocytes from the blood, the difference in potency probably due to protein binding of JNJ-4796567 in whole blood. Antagonism was surmountable by increasing concentrations of Bz-ATP (Figure 4B), a functional phenomenon that would relate to a similar/overlapping site of action with Bz-ATP. 30 nM JNJ-47965567 shifted the potency of Bz-ATP induced IL-1β release from a pEC50 value of 3.6 ± 0.06 to 3.0 ± 0.04. With all assumptions of Schild analyses intact, an estimate of pA2 for JNJ-47965567 is approximate 8.0, identical to the affinity of JNJ-47965567 obtained by radioligand displacement binding (Figure 2). In addition to IL-1β block, the compound also blocked IL-18 release with a potency of 7.3 ± 0.1. More importantly, JNJ-47965567 did not block IL-6 and TNF-α release, under identical conditions (LPS and BZ-ATP) used for IL-1β and IL-18 release.
Figure 4.
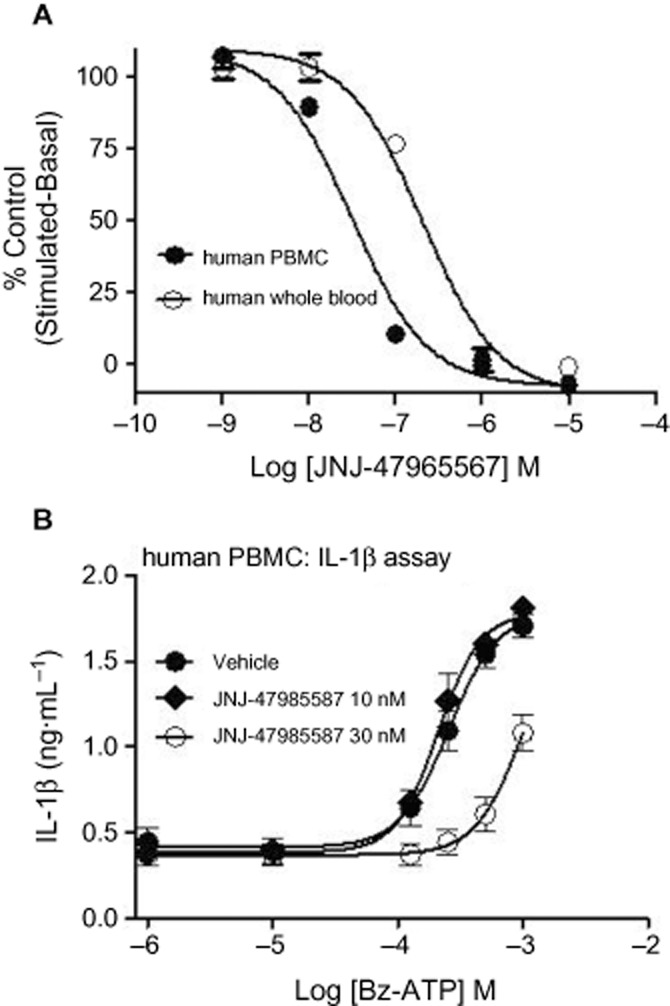
Attenuation of IL-1β release in human blood and human monocytes (PBMC) by JNJ-47965567. LPS primed, Bz-ATP challenged IL-1β release was concentration dependently blocked by JNJ47965567 (A). Potency (pIC50) of block was 6.7 ± 0.07 and 7.5 ± 0.07, in blood and monocytes (PBMC), respectively. Data are expressed as percent of control [defined as net IL-1β release: LPS + Bz-ATP challenged cells (stimulated) minus LPS primed cells (basal)]. (B) Effect of 10 nM and 30 nM JNJ-47965567 on the concentration response of Bz-ATP induced IL-1β release in isolated human peripheral blood mononuclear cells (PBMC). Symbols represent mean ± SEM from three to six independent experiments.
The next step in the characterization cascade of JNJ-47965567 was to test the pharmacology in native rat systems; since P2X7 is expressed in glial cells of the CNS, we decided to test the pharmacology of JNJ-47965567 (plus the three reference compounds) in rat primary cultures of astrocytes and microglia. As shown in Figure 5, JNJ-47965567 displayed favourable pharmacology in the rat native system either for block of calcium flux (in astrocytes) or for block of IL-1β release (in microglia). The potency of JNJ-47965567 was 7.1 ± 0.1 in rat microglia cells; likewise, the potency was 7.5 ± 0.4 in rat astrocytes. In both cell types, complete block was achieved at concentrations of 1 μM JNJ-47965567. In summary, JNJ-47965567 is a potent P2X7 antagonist in glial cells, the target population of cells in the CNS with abundant expression of this channel.
Figure 5.
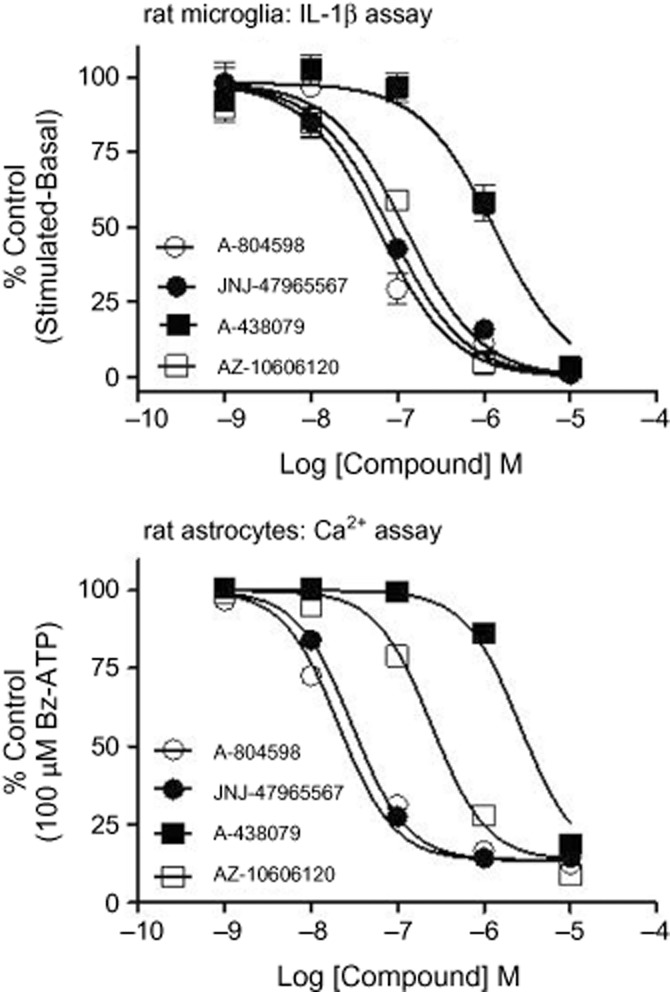
Effect of P2X7 antagonists on net IL-1β release in primary cultures of rat microglia (upper panel) or on calcium flux in rat astrocytes (lower panel). Concentration dependent attenuation of P2X7 mediated IL-1β release or calcium flux by JNJ-47965567. Potency (pIC50) of block for JNJ-47965567 was 7.1 ± 0.1 in rat microglia and 7.5 ± 0.4 in rat astrocytes. Data are expressed as percent of control in rat microglia as defined by net IL-1β release [LPS + Bz-ATP challenged cells (stimulated) minus LPS primed cells (basal)] or in rat astrocytes to a calcium response in control cells to 100 μM Bz-ATP. Symbols represent mean ± SEM from at least three independent experiments.
Electrophysiology of JNJ-47965567 block
About 100 nM JNJ-47965567 rapidly blocked Bz-ATP evoked currents in 1321N1 cells expressing hP2X7 (93.9 ± 1.9% inhibition; Figure 6). Steady-state block was generally achieved in 6 min. Minimal recovery was observed from JNJ-4795567 block in the 10 min recovery period (Figure 6), and it cannot be determined if what little was seen is related to dissociation of antagonist or the aforementioned agonist-induced increase in current amplitude.
Figure 6.
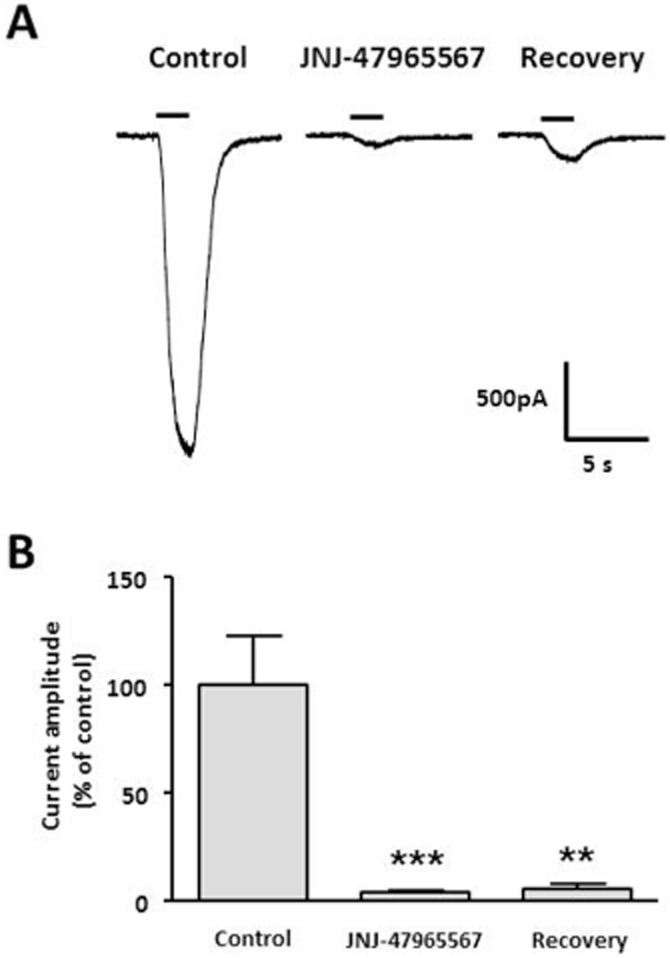
(A) Representative whole-cell patch-clamp recordings of responses to a 2 s application of 300 μM Bz-ATP (solid line) to 1321N1 cells stably expressing recombinant human P2X7. 100 nM JNJ-479655567 rapidly blocked Bz-ATP-evoked currents (middle trace) with minimal recovery being observed after a 10 min wash-out interval (right trace). (B) Normalized mean current amplitude ± SEMs; ***P < 0.001, **P < 0.01 by anova with Dunnet's post hoc test.
Selectivity of JNJ-47965567
The selectivity of JNJ-47965567 was tested in a standard panel of receptors, ion channels and transporters (CEREP, Poitiers, France). Data are summarized in Table 2. The compound was clean at a test concentration of 1 μM. At 10 μM, greater than 50% effect was observed at the human melatonin 1 receptor (65% effect) and the human serotonin transporter (82% effect). Since the effect was more dramatic at the human serotonin transporter, JNJ-47965567 was further evaluated at the rat serotonin transporter radioligand binding assay. The affinity of JNJ-47965567 at the rat transporter was 2 μM (rat P2X7 affinity: 2 nM). The compound was further evaluated for P2X subtype selectivity at the human P2X1, P2X2, P2X3, P2X2/3 and P2X4 channels (Chantest, Cleveland, OH, USA). As exhibited in Table 3, JNJ-47965567 did not block any of these channels up to concentrations of 10 μM. The compound was also characterized against a panel of human CYP450 enzymes; the potency was 7 μM at CYP1A2 with greater than 10 μM IC50 at the other CYPs (2C8, 2C9, 2C19, 2D6, 3A4).
Table 2.
Selectivity of JNJ-47965567 against a panel of receptors, transporters and channels
| % control specific binding (@ 1 μM) | ||||
|---|---|---|---|---|
| Assays | First value | Second value | Mean | |
| A1 (h) (antagonist radioligand) | 150.9 | 142 | 146.4 | |
| A2A (h) (agonist radioligand) | 88.6 | 90.5 | 89.6 | |
| A3 (h) (agonist radioligand) | 135.6 | 133.6 | 134.6 | |
| alpha 1 (non-selective) (antagonist radioligand) | 108.6 | 90.1 | 99.4 | |
| alpha 2 (non-selective) (antagonist radioligand) | 98 | 89.1 | 93.5 | |
| beta 1 (h) (agonist radioligand) | 115.4 | 96.8 | 106.1 | |
| AT1 (h) (antagonist radioligand) | 144.3 | 149.6 | 147 | |
| BZD (central) (agonist radioligand) | 129.2 | 113.7 | 121.4 | |
| B2 (h) (agonist radioligand) | 107 | 96.7 | 101.9 | |
| CCK1 (CCKA) (h) (agonist radioligand) | 92.6 | 98 | 95.3 | |
| D1 (h) (antagonist radioligand) | 116.7 | 101.9 | 109.3 | |
| D2S (h) (antagonist radioligand) | 88.8 | 92.1 | 90.5 | |
| ETA (h) (agonist radioligand) | 104.2 | 116.7 | 110.4 | |
| GABA (non-selective) (agonist radioligand) | 128.7 | 109 | 118.8 | |
| GAL2 (h) (agonist radioligand) | 98.7 | 107.9 | 103.3 | |
| CXCR2 (IL- 8β) (h) (agonist radioligand) | 98 | 96.7 | 97.3 | |
| CCR1 (h) (agonist radioligand) | 102.5 | 103.1 | 102.8 | |
| H1 (h) (antagonist radioligand) | 110.6 | 102.4 | 106.5 | |
| H2 (h) (antagonist radioligand) | 116.4 | 129.4 | 122.9 | |
| MC4 (h) (agonist radioligand) | 104.5 | 118.1 | 111.3 | |
| MT1 (ML1A) (h) (agonist radioligand) | 88.7 | 74.3 | 81.5 | >50% at 10 μM |
| M1 (h) (antagonist radioligand) | 112.7 | 111.1 | 111.9 | |
| M2 (h) (antagonist radioligand) | 110.8 | 109.8 | 110.3 | |
| M3 (h) (antagonist radioligand) | 83.4 | 86.6 | 85 | |
| NK2 (h) (agonist radioligand) | 104.3 | 107.7 | 106 | |
| NK3 (h) (antagonist radioligand) | 99.9 | 109.1 | 104.5 | |
| Y1 (h) (agonist radioligand) | 110 | 106.4 | 108.2 | |
| Y2 (h) (agonist radioligand) | 106.2 | 112.2 | 109.2 | |
| NTS1 (NT1) (h) (agonist radioligand) | 113.9 | 112.5 | 113.2 | |
| delta 2 (DOP) (h) (agonist radioligand) | 110.6 | 91.1 | 100.8 | |
| kappa (KOP) (agonist radioligand) | 95.7 | 108.2 | 101.9 | |
| mu (MOP) (h) (agonist radioligand) | 102.5 | 102.3 | 102.4 | |
| NOP (ORL1) (h) (agonist radioligand) | 97.9 | 100.1 | 99 | |
| 5-HT1A (h) (agonist radioligand) | 108.3 | 111.9 | 110.1 | |
| 5-HT1B (antagonist radioligand) | 113.9 | 110.7 | 112.3 | |
| 5-HT2A (h) (antagonist radioligand) | 130.9 | 116.4 | 123.6 | |
| 5-HT2B (h) (agonist radioligand) | 85.5 | 100.8 | 93.1 | |
| 5-HT3 (h) (antagonist radioligand) | 105.2 | 102.9 | 104.1 | |
| 5-HT5a (h) (agonist radioligand) | 111.9 | 100.7 | 106.3 | |
| 5-HT6 (h) (agonist radioligand) | 110.7 | 110.1 | 110.4 | |
| 5-HT7 (h) (agonist radioligand) | 98.5 | 104.5 | 101.5 | |
| SST (non-selective) (agonist radioligand) | 115.4 | 114.3 | 114.9 | |
| VPAC1 (VIP1) (h) (agonist radioligand) | 112.7 | 118.3 | 115.5 | |
| V1a (h) (agonist radioligand) | 104.8 | 114.9 | 109.9 | |
| Ca2+ channel (L, verapamil site) (phenylalkylamine) | 97.7 | 105.6 | 101.6 | |
| KV channel (antagonist radioligand) | 99.1 | 99.5 | 99.3 | |
| SKCa channel (antagonist radioligand) | 104.5 | 103.7 | 104.1 | |
| Na+ channel (site 2) (antagonist radioligand) | 107.5 | 90.1 | 98.8 | |
| Cl− channel (GABA-gated) (antagonist radioligand) | 94.1 | 93.1 | 93.6 | |
| norepinephrine transporter (h) (antagonist radioligand) | 105.3 | 94.5 | 99.9 | |
| dopamine transporter (h) (antagonist radioligand) | 91 | 90.5 | 90.8 | |
| 5-HT transporter (h) (antagonist radioligand) | 56.5 | 53 | 54.7 | >50% at 10 μM |
Data generated in CEREP (http://www.cerep.fr/Cerep/Users/index.asp).
Table 3.
P2X subtype selectivity of JNJ-47965567 (Calcium flux assay; FLIPR)
| hP2X1 | hP2X2 | |||
|---|---|---|---|---|
| Conc (μM) | % Inhibition | SD | % Inhibition | SD |
| 0.003 | 6.59 | 13.86 | 4.79 | 5.9 |
| 0.01 | 7.79 | 10.87 | 8.71 | 2.34 |
| 0.03 | 18.21 | 6.02 | 9.45 | 6.71 |
| 0.1 | 6.49 | 9.59 | 6.2 | 4.52 |
| 0.3 | 17.21 | 10.02 | 9.49 | 3.26 |
| 1 | 20.71 | 7.44 | 14.06 | 10.57 |
| 3 | 20.42 | 4.71 | 18.03 | 5.57 |
| 10 | 2.78 | 7.37 | 16.58 | 9.93 |
| hP2X3 | hP2X2/P2X3 | |||
|---|---|---|---|---|
| Conc (μM) | % Inhibition | SD | % Inhibition | SD |
| 0.003 | 1.31 | 2.07 | 6.91 | 1.53 |
| 0.01 | 1.89 | 2.92 | 7.62 | 1.93 |
| 0.03 | −2.83 | 4.39 | 8.91 | 1.69 |
| 0.1 | 1.48 | 6.86 | 9.85 | 2.25 |
| 0.3 | −2.15 | 5.67 | 8.84 | 1.66 |
| 1 | −0.04 | 1.98 | 10.68 | 1.68 |
| 3 | −1.69 | 2.52 | 10.53 | 1.36 |
| 10 | −3.79 | 3.62 | 11.01 | 0.41 |
| hP2X4 | ||
|---|---|---|
| Conc (μM) | % Inhibition | SD |
| 0.003 | 12.95 | 8.65 |
| 0.01 | 8.68 | 4.23 |
| 0.03 | 10.63 | 3.04 |
| 0.1 | 14.04 | 6.55 |
| 0.3 | 15.37 | 5.5 |
| 1 | 11.76 | 6.81 |
| 3 | 13.54 | 2.09 |
| 10 | 6.79 | 1.45 |
The ability of JNJ-47965567 to act as an antagonist on all P2X channels was evaluated with a calcium influx assay. Each channel was activated with the agonist αβ-meATP (P2X1, P2X3 and P2X2/P2X3) or Bz-ATP (for P2X2 and P2X4). The ability of each test article to inhibit this signal was examined and compared to the positive control antagonist (100 or 300 μM PPADS). The signal, elicited in the presence of the respective positive control agonist, was set to 100% (0% inhibition) and the signal in the presence of the respective positive control antagonist was set to 0% (100% inhibition). Data are presented as normalized inhibition.
Conc, concentration.
Pharmacodynamic properties of JNJ-47965567: target engagement (ex vivo P2X7 brain binding autoradiography and brain IL-1β microdialysis)
Following the in vitro characterization of JNJ-47965567, the next step was to characterize the compound in vivo: to test whether the compound penetrates the CNS space and engages P2X7 in a dose and concentration-dependent manner. An ex vivo P2X7 radioligand binding autoradiography assay was established in house using [3H] A-804598, a technique that has been widely used in general to measure binding of compounds in brain sections and one that has been specifically used for P2X7 (Able et al., 2011). JNJ-479655567 was dosed both via the s.c. and oral route at 10 mg·kg−1 followed by ex vivo receptor binding (see methods) at various time points post dose. As shown in Figure 7A, JNJ-47965567 occupied rat P2X7 receptor following the s.c. dose, but failed to occupy P2X7 receptor following the oral dose. Analysis of exposure data both in plasma and brain tissue revealed that JNJ-47965567 lacked occupancy after oral dosing due to poor oral bioavailability: exposures in the range of 44–98 ng·mL−1 in the plasma and 18–40 ng·mL−1 in the brain for all the time points tested. In contrast, JNJ-47965567 achieved high exposure after s.c. dosing: 720–1200 ng·mL−1 in the plasma and 380–658 ng·mL−1 in the brain at the time points tested. The brain to plasma ratio of JNJ-47965567 was between 0.5 and 0.58, sufficient to result in significant level of P2X7 receptor occupancy in rat brain.
Figure 7.
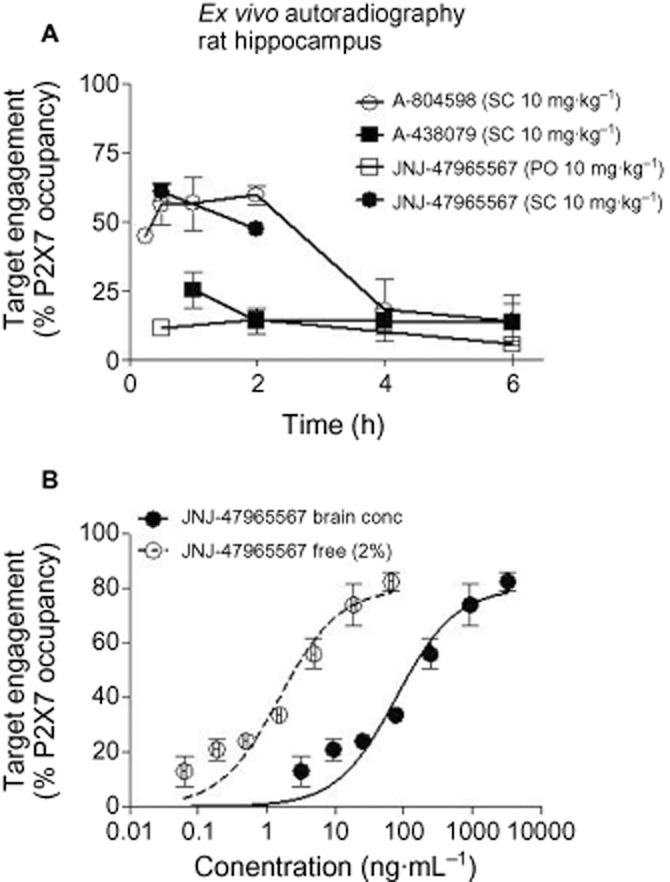
Ex vivo brain P2X7 binding autoradiography by JNJ-47965567, A-438079 and A-804598. (A) Duration of occupancy for P2X7 antagonists post systemic administration in rats. (B) Occupancy versus brain concentration of JNJ-47965567 post-s.c. dosing. The solid line is the true concentration in the brain and the dotted line is extrapolation of free concentration based on 98% protein binding in rat brain tissue. Ex vivo receptor labelling was expressed as the percentage of receptor labelling in corresponding brain areas (i.e. hippocampus) of saline-treated animals. Symbols represent mean ± SEM from at least two to three rats per time point or dose group.
In order to address the specificity of the assay, we decided to test A-804598 and A-438079, two compounds with varying degree of P2X7 pharmacology (Table 1; Figure 1). Both compounds were dosed s.c. at 10 mg·kg−1 and while A-804598 occupied P2X7 receptor (∼65%), A-438079 failed to demonstrate significant level of P2X7 receptor occupancy (Figure 7A), in line with their varying degree of in vitro affinities for P2X7. At 1 h post dose, brain exposures of A-438079 and A-804598 were 1764 ng·mL−1 (5.7 μM) and 183 ng·mL−1 (0.58 μM), respectively. Even though A-438079 achieved a 10-fold higher concentration in the brain than A-804598, it was A-804598 that achieved significant P2X7 receptor occupancy with low levels of receptor occupancy for A-438079. The result obtained was in complete agreement with the in vitro pharmacology of the two compounds: affinity of A-804598 for the rat P2X7 was significantly higher than A-438079 (Figure 1). It was thus clearly demonstrated that the ex vivo binding signal obtained is P2X7 dependent.
Having generated confidence in the P2X7 ex vivo target engagement assay, the next step was to define a dose-effect curve for JNJ-47965567. JNJ-47965567 was dosed up from 0.03 to 30 mg·kg−1, followed by ex vivo receptor occupancy at 15 min (time for maximal occupancy established in duration of action studies). Corresponding brain exposures were plotted against receptor occupancy (Figure 7B). The data demonstrate a concentration-dependent P2X7 occupancy as would be expected for a target-ligand interaction with an EC50 of 78 ± 19 ng·mL−1. In a separate measure, it was assessed that the rat brain protein binding of JNJ-47965567 was ∼98%. When the concentrations were adjusted to 2% free concentration, and re-plotted against receptor occupancy (dotted line in Figure 7B), the corresponding EC50 was 1.5 ± 0.4 ng·mL−1 (3 nM). The binding affinity of JNJ-47965567 was close to 2 nM obtained in radioligand displacement assays (Figure 1). The half occupancy obtained in Figure 7B (EC50) is a theoretical definition of binding affinity and it was gratifying to find out that the in vitro affinity of JNJ-47965567 predicted the in vivo EC50, corrected for the free fraction. Not only do we have a reliable P2X7 target engagement assay, we were also confident that the in vitro radioligand binding data could indeed model the concentration needed for half occupancy for other P2X7 compounds.
In an effort to better understand the consequence of P2X7 binding to function, we established an in vivo microdialysis assay to measure brain IL-1β levels in freely moving rats challenged with the P2X7 agonist Bz-ATP (see Methods). As depicted in Figure 8A, Bz-ATP (100 mM) produced a significant increase in measurable IL-1β from hippocampal dialysates (Figure 8). JNJ-47965567 was tested at two dose levels: 30 and 100 mg·kg−1 (s.c. dosing). JNJ-47965567 significantly attenuated IL-1β release at 100 mg·kg−1, with no effect at 30 mg·kg−1 dose group. In a separate study, P2X7 occupancy was measured at these doses, the data for which is shown as an inset in Figure 8A. Although there are no marked differences in peak occupancy between 30 and 100 mg·kg−1 JNJ-47965567, the higher dose tends to sustain P2X7 occupancy for a longer duration and may contribute to a functional block (see Discussion for more details). Moreover, 100 mM Bz-ATP is a super saturating concentration of the agonist and the functional block by a competing antagonist such as JNJ-47965567 can be right shifted (see Discussion for more details), which may account for the apparent discrepancy between full occupancy and lack functional effect at 30 mg·kg−1 dose. In order to rule out any off-target effects of Bz-ATP induced IL-1β release, we tested P2X7 knockout mice and the results shown in Figure 8B demonstrate that Bz-ATP induced IL-1β release is P2X7 dependent, although the magnitude of cytokine release was much lower in mice than rats, probably due to lower affinity of Bz-ATP for the mouse P2X7 channel. Thus, we have demonstrated that the P2X7 antagonist JNJ-47965567 engages the target resulting in functional blockade of IL-1β release.
Figure 8.
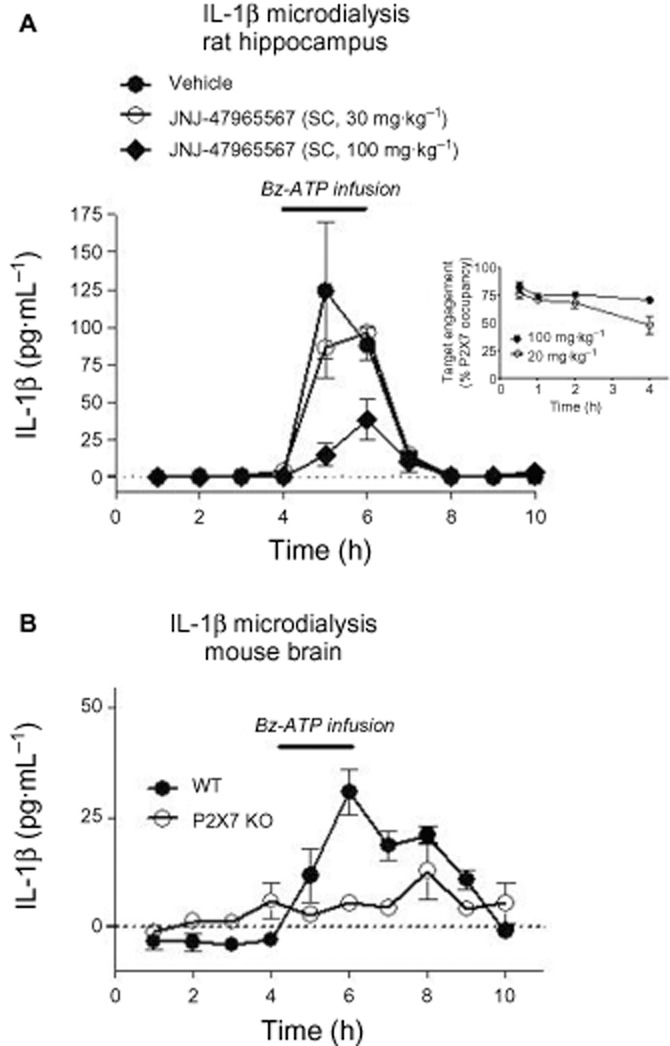
In vivo microdialysis assay to measure brain IL-1β levels in freely moving rats and mice. Bz-ATP infused IL-1β release in rat (A) or mouse (B) brain. Solid line indicates time of Bz-ATP infusion. Data are mean from four (rats) or six (mice) independent experiments. Inset: Duration of P2X7 occupancy, as assessed by ex vivo autoradiography, post systemic dose of JNJ-47965567.
Efficacy of JNJ-47965567 in models of depression, mania and chronic pain
Having established P2X7 occupancy with JNJ-47965567 and blockade of IL-1β release in rat brain, the next series of experiments were designed to address the role of P2X7 in models of depression (forced swim test; Supporting Information Figure S2), mania (amphetamine hyperactivity) and chronic pain (Chung model of neuropathic pain). The compound had no effect on any of the behavioural parameters in the forced swim test (Supporting Information Figure S2).
Amphetamine hyperactivity
Locomotor activity from the acute phase (day 1) and sensitized phase (day 8) are shown in Figure 9A and B, respectively. Amphetamine (2 mg·kg−1) administration (big arrowhead) caused changes in locomotor activity that sensitized between day 1 and day 8 in rats. On day 1, there were no significant differences between the groups (vehicle and JNJ-47965576 treated, small arrowhead) at any time point, indicating that JNJ-47965567 did not have any effect on basal locomotor activity (0–50 min in Figure 9A) nor did the compound modulate amphetamine-induced changes in locomotion. On day 8, JNJ-47965567 (30 mg·kg−1) pretreatment attenuated sensitization; such that on day 8 significant differences between drug-pretreated and vehicle-pretreated groups were revealed. Due to the dramatic effect of JNJ-47965567 at blocking amphetamine-induced hyperactivity, and in order to rule out off-target effects, we sought to test the role of P2X7 by phenotyping knockout animals in the amphetamine model as well. Wild-type mice demonstrated sensitization to amphetamine. Moreover, P2X7 knockout mice demonstrated a protection of sensitization, a behaviour consistent to that observed post-JNJ-47965567 administration (Supporting Information Figure S3). Taken together, we conclude that P2X7 ion channel plays a role in amphetamine-induced sensitization of locomotion.
Figure 9.
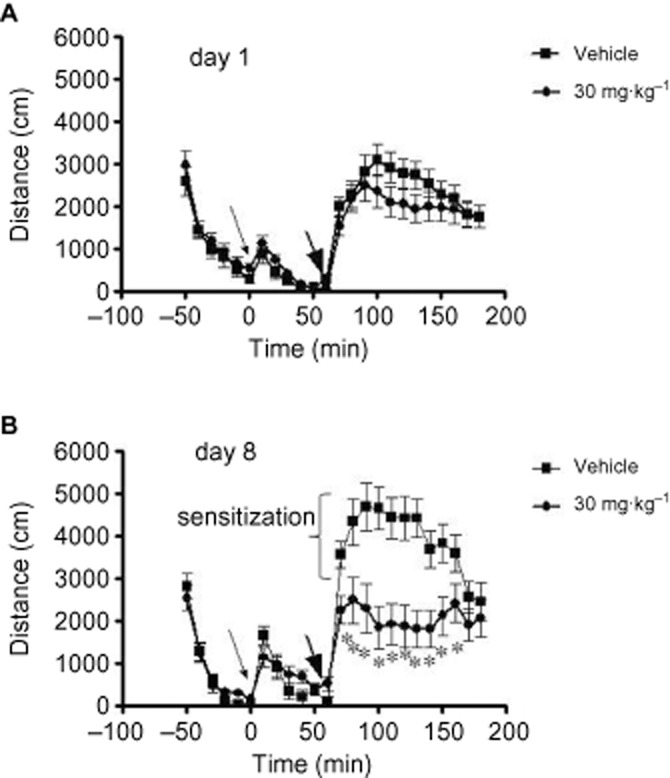
Effect of JNJ-47965567 on amphetamine induced hyperactivity in rats. Distance travelled is shown for day 1 (A) and day 8 (B). Small arrowhead: JNJ/47965567/vehicle (30% SBE) administration; Fat arrowhead: amphetamine administration. On day 8, 30 mg·kg−1 JNJ-47965567 attenuated locomotor activity compared to vehicle at the following 10 min bins: 0–10 min (P 0.02), 10–20 min (P 0.0008), 20–30 min (P 0.000003), 30–40 min (P 0.0000005), 40–50 min (P 0.000001), 50–60 min (P 0.000001), 60–70 min (P 0.000001), 70–80 min (P 0.0007), 80–90 min (P 0.002) and 90–100 min (P 0.04). Asterisks denote statistical significance.
Chung model of neuropathic pain
Animals prepared with Chung lesions demonstrated a prominent tactile allodynia at 10–14 days post-operatively (Figure 10). Animals that exhibited a tactile threshold of 3 g or less were considered significantly allodynic and were selected in the study. Subcutaneous delivery of Gabapentin (200 mg·kg−1) resulted in a significant reversal of the allodynia that lasted in excess of 120 min. While 3 mg·kg−1 dose group had no effect, JNJ-47965567, 30 mg·kg−1 s.c. resulted in a statistically significant numerical increase in tactile thresholds lasting from 30 to 60 min post administration. Interestingly and to our surprise, the 100 mg·kg−1 dose group did not exhibit any efficacy. No effect on contralateral thresholds was noted for any treatment. Animals given Gabapentin showed sign of sedation in two of six animals by 60 min but were easily aroused. These results show that Gabapentin had an anti-allodynic action of an expected magnitude and duration. The test article at appeared to have a modest effect only at 30 mg·kg−1.
Figure 10.
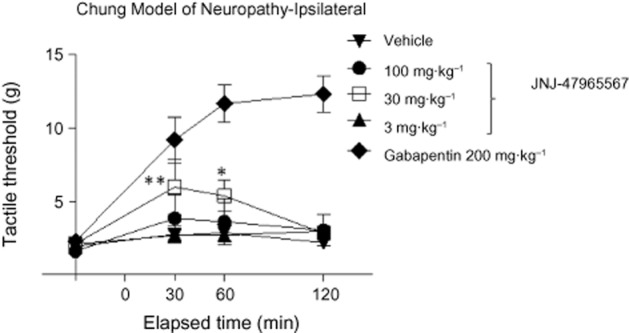
Effect of JNJ-47965567 on reversal of allodynia in a rat model of neuropathic pain. Rats undergoing Chung surgery nerve ligation exhibited prominent allodynia at 10–14 days. Animals (six rats per group) were treated with a single dose of the compounds and allodynic behaviour was assessed at the time points indicated on the X-axis. JNJ-47965567 (30 mg·kg−1) demonstrated modest yet significant efficacy. **P < 0.01; *P < 0.05.
Discussion and conclusions
In this manuscript, we present a novel CNS permeable P2X7 antagonist (JNJ-47965567) that exhibits good affinity/potency for the channel. Pharmacokinetic/pharmacodynamic properties of the compound lend itself suitable as a tool compound to probe the role of P2X7 in rat models that attempt to simulate the pathology of the disease in question. Currently used tool compounds in the literature such as Brilliant Blue G, Evans Blue, TNP-ATP or PPADs are non-selective and often, fail to address the role of P2X7 convincingly due to the poly-pharmacology of these compounds. Recently, several P2X7 antagonists have been available commercially such as A-804598, A-740003, A-839977 and A-438079 that are more potent and selective for P2X7; although, with the exception of A-438079, all of the remaining three compounds exhibit good rat P2X7 potency, our compound, JNJ-47965567 is superior in terms of brain to plasma ratio and hence is a better tool compound to understand the role of P2X7 in CNS models of pathophysiology.
As demonstrated by the results, we have used a battery of assays in both recombinant and native systems to characterize JNJ-47965567. The compound exhibits good pharmacology in vitro with slow wash-off rate that often aids in increased residence time at the target. Our data did not indicate any apparent disconnect between pharmacokinetics and pharmacodynamics (ex vivo target engagement) and as such slow reversibility of the compound did not offer any increased residence time on P2X7, independent of pharmacokinetics. The proinflammatory cytokine IL-1β is one of the major factors that mediate the cross-talk between neuroglia in the CNS (Khairova et al., 2009). P2X7 is expressed predominantly in the glial cells and activation of P2X7, in the presence of high local concentrations of ATP during episodes of stress, injury and inflammation result in activation of the channel. This in turn causes release gliotransmitters (ATP, glutamate/glycine, chemokines, cytokines such as IL-1β, cathepsins) that signal the activation of the ‘neuroinflammatory’ cascade including microglial activation and astrogliosis. Glial activation causes more release of ATP, thereby engaging more number of P2X7 channels, which in turn can spiral into a vicious cycle causing neuroactive cytokine storm and activated glial tone, both of which may contribute to various CNS pathophysiology including mood and neurodegenerative disorders (Khansari and Sperlagh, 2012; Jones and Thomsen, 2013). This is a dynamic process and as such, it is not clear whether P2X7 is critical in the initiation or maintenance of glia derived neuropathophysiology. We hope our compound presents a viable tool to address these important questions.
In this study, we attempted to address the role of P2X7 in three classic models used to gauge the utility of compounds in depression, mania and pain. In the forced swim test, JNJ-47965567 did not exhibit any efficacy in rat forced swim test (Supporting Information) similar to a recent paper that has demonstrated that P2X7 knockout mice do not display a phenotype on day one of testing (Boucher et al., 2011). This is however, in contrast to protective effects of despair behaviour in P2X7 knockout mice in the forced swim test on day 1, observed by other groups (Basso et al., 2009; Csolle et al., 2013). It is however plausible that the compound may have to be dosed chronically (multiple days) to see efficacy or the animals may have to be stressed over time to engage P2X7, or a combination of both. However, JNJ-47965567 did show a significant effect on attenuating the sensitization of amphetamine induced locomotion. While this model has been linked to manic episodes of bipolar disorder, it is not clear to us at this time, the neurochemical mechanism behind the effect of JNJ-47965567. In the amphetamine-challenged rats, the effect of JNJ-47965567 was not due to modulation of norepinephrine, serotonin or dopamine, as these neurotransmitters were relatively unchanged in the two groups (Aluisio et al., unpubl. data). It is possible that amphetamine may produce local burst of ATP as a result of hyperexcitability and our data with the P2X7 antagonist may suggest ATP-induced activation of P2X7 in the rat brain during amphetamine sensitization phase. Nonetheless, this effect of JNJ-47965567 was significant, reproduced in knockout animals and was recently published by an independent laboratory (Csolle et al., 2013). The third and final model was the Chung model of neuropathic pain. To our surprise, JNJ-47965567 did not exhibit robust efficacy; the effect at 30 mg·kg−1 dose was modest (yet significant) and transient effect that did not follow a traditional dose proportional increase in effect size at 100 mg·kg−1. We were surprised at this observation, because P2X7 knockout mice do not develop allodynia in a similar model of neuropathic pain (Chessell et al., 2005) and several publications have indicated a role of P2X7 in models of neuropathic pain. In this study, JNJ-47965567 was dosed acutely (one s.c. dose); in order to maintain sustainable occupancy of P2X7 with this compound, it is important to dose the compound chronically over several days and probably that underpins the discrepancy between our study and the knockout phenotype. We tried to dose JNJ-47965567 at a higher dose to address sustainable occupancy, but the duration of action of the JNJ-47965567 over a 24 h period reveals that the P2X7 occupancy is not significantly different between the 30 and 100 mg·kg−1 doses. The effective dose of JNJ-47965567 was 30 mg·kg−1. Interestingly, at this dose, close to full maximal target engagement (as measured by ex vivo autoradiography) was observed with no effect on brain IL-1β, as assessed by infusion of 100 mM Bz-ATP in the rat brain.
Whether P2X7 intervention will be therapeutically beneficial in neuropsychiatry, neurodegeneration and chronic pain remains an unanswered question. More importantly, the degree and duration of target occupancy required for efficacy is unclear: more P2X7 antagonists need to be profiled in a battery of efficacy models to get a better understanding of this occupancy-efficacy relationship. So far, besides this publication, only a limited number of papers have specially tried to address the role of P2X7 antagonists in mood disorders. There is clearly a growing appreciation of the role of neuroglia cross-talk, and a glial target such as P2X7 would certainly be a novel mechanism to address these unmet medical needs. Our contribution to the field is in disclosing the pharmacology of a novel, high-affinity, selective P2X7 antagonist that will certainly be a valuable tool compound to address probing questions such as those discussed in this manuscript. In the process, we have attempted to establish target engagement with our compound, which too often is not used in the literature prior to running in vivo efficacy models. This paper highlights the use of a spectrum of assays, both in vitro and in vivo, and in the process characterized the P2X7 antagonist JNJ-47965567.
Acknowledgments
The authors acknowledge the contribution of the following individuals for supporting various activities towards the successful completion of the study: Steve W Sutton for assisting in primary glial cultures, Kirsten L. Morton for testing JNJ-47965567 at the serotonin transporter binding assay, Kia Sepassi for guidance on formulation and Brian Scott for analytical support.
Glossary
- P2X7
purinergic receptor P2X, ligand-gated ion channel, 7
Conflict of interest
The authors of this manuscript have no conflicts of interest to declare.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 100 nM of JNJ-47965567 was added at time zero (blue arrow) followed by initiation of the wash cycle at 30 min (black arrow). Wash was repeated every 30 min with reading occurring at 3, 4, 5 and 6 h time point. Bz-ATP was used as stimulus.
Figure S2 Effects of JNJ-47965567 on frequency of immobility, climbing and swimming behaviour (forced swim test) in rats. Bar graphs represent the mean behaviour dosed with desipramine, vehicle and JNJ-47965567. SBE: sulfobutylether beta-cyclodextrin; *P < 0.001.
Figure S3 Phenotype of P2X7 knockout mice in amphetamine induced hyperactivity model. Distance travelled is shown for day 1 (A) and day 4 (B). Small arrowhead: amphetamine administration. Repeated measures anova and Duncan's post hoc test showed that the knockout had a significantly attenuated response to amphetamine (genotype × time interaction; *P < 0.005 at the first four time points after amphetamine, Duncan's post hocs).
References
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview. Trends Neurosci. 2009;32:19–29. doi: 10.1016/j.tins.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Able SL, Fish RL, Bye H, Booth L, Logan YR, Nathaniel C, et al. Receptor localization, native tissue binding and ex vivo occupancy for centrally penetrant P2X7 antagonists in the rat. Br J Pharmacol. 2011;162:405–414. doi: 10.1111/j.1476-5381.2010.01025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando RD, Sperlagh B. The role of glutamate release mediated by extrasynaptic P2X7 receptors in animal models of neuropathic pain. Brain Res Bull. 2013;93:80–85. doi: 10.1016/j.brainresbull.2012.09.016. [DOI] [PubMed] [Google Scholar]
- Backlund L, Nikamo P, Hukic DS, Ek IR, Traskman-Bendz L, Landen M, et al. Cognitive manic symptoms associated with the P2RX7 gene in bipolar disorder. Bipolar Disord. 2011;13:500–508. doi: 10.1111/j.1399-5618.2011.00952.x. [DOI] [PubMed] [Google Scholar]
- Barden N, Harvey M, Gagne B, Shink E, Tremblay M, Raymond C, et al. Analysis of single nucleotide polymorphisms in genes in the chromosome 12Q24.31 region points to P2RX7 as a susceptibility gene to bipolar affective disorder. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:374–382. doi: 10.1002/ajmg.b.30303. [DOI] [PubMed] [Google Scholar]
- Basso AM, Bratcher NA, Harris RR, Jarvis MF, Decker MW, Rueter LE. Behavioral profile of P2X7 receptor knockout mice in animal models of depression and anxiety: relevance for neuropsychiatric disorders. Behav Brain Res. 2009;198:83–90. doi: 10.1016/j.bbr.2008.10.018. [DOI] [PubMed] [Google Scholar]
- Baxter A, Bent J, Bowers K, Braddock M, Brough S, Fagura M, et al. Hit-to-Lead studies: the discovery of potent adamantane amide P2X7 receptor antagonists. Bioorg Med Chem Lett. 2003;13:4047–4050. doi: 10.1016/j.bmcl.2003.08.034. [DOI] [PubMed] [Google Scholar]
- Bennett MR. Synaptic P2X7 receptor regenerative-loop hypothesis for depression. Aust N Z J Psychiatry. 2007;41:563–571. doi: 10.1080/00048670701399994. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Neff RA, Wickenden AD. The physiology, pharmacology and future of P2X7 as an analgesic drug target: hype or promise? Curr Pharm Biotechnol. 2011;12:1698–1706. doi: 10.2174/138920111798357429. [DOI] [PubMed] [Google Scholar]
- Boucher AA, Arnold JC, Hunt GE, Spiro A, Spencer J, Brown C, et al. Resilience and reduced c-Fos expression in P2X7 receptor knockout mice exposed to repeated forced swim test. Neuroscience. 2011;189:170–177. doi: 10.1016/j.neuroscience.2011.05.049. [DOI] [PubMed] [Google Scholar]
- Browne LE, Jiang L-H, North RA. New structure enlivens interest in P2X receptors. Trends Pharmacol Sci. 2010;31:229–237. doi: 10.1016/j.tips.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov. 2008;7:575–590. doi: 10.1038/nrd2605. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Fredholm BB, Verkhratsky A. Adenosine and ATP receptors in the brain. Curr Top Med Chem. 2011a;11:973–1011. doi: 10.2174/156802611795347627. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Krugel U, Abbracchio MP, Illes P. Purinergic signalling: from normal behaviour to pathological brain function. Prog Neurobiol. 2011b;95:229–274. doi: 10.1016/j.pneurobio.2011.08.006. [DOI] [PubMed] [Google Scholar]
- Charlton SJ, Vauquelin G. Elusive equilibrium: the challenge of interpreting receptor pharmacology using calcium assays. Br J Pharmacol. 2010;161:1250–1265. doi: 10.1111/j.1476-5381.2010.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–396. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Clark AK, Wodarski R, Guida F, Sasso O, Malcangio M. Cathepsin S release from primary cultured microglia is regulated by the P2X7 receptor. Glia. 2010;58:1710–1726. doi: 10.1002/glia.21042. [DOI] [PubMed] [Google Scholar]
- Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS. Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev. 2011;63:641–683. doi: 10.1124/pr.110.003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csolle C, Sperlagh B. Peripheral origin of IL-1beta production in the rodent hippocampus under in vivo systemic bacterial lipopolysaccharide (LPS) challenge and its regulation by P2X(7) receptors. J Neuroimmunol. 2010;219:38–46. doi: 10.1016/j.jneuroim.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Csolle C, Ando RD, Kittel A, Goloncser F, Baranyi M, Soproni K, et al. The absence of P2X7 receptors (P2rx7) on non-haematopoietic cells leads to selective alteration in mood-related behaviour with dysregulated gene expression and stress reactivity in mice. Int J Neuropsychopharmacol. 2013;16:213–233. doi: 10.1017/S1461145711001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Hernandez JI, Gomez-Villafuertes R, Leon-Otegui M, Hontecillas-Prieto L, Del Puerto A, Trejo JL, et al. In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer's disease through GSK3beta and secretases. Neurobiol Aging. 2012;33:1816–1828. doi: 10.1016/j.neurobiolaging.2011.09.040. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Namovic MT, Han P, Jarvis MF. Mammalian P2X7 receptor pharmacology: comparison of recombinant mouse, rat and human P2X7 receptors. Br J Pharmacol. 2009a;157:1203–1214. doi: 10.1111/j.1476-5381.2009.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Namovic MT, Surber B, Vaidyanathan SX, Perez-Medrano A, Wang Y, et al. [(3)H]A-804598 ([(3)H]2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharmacology. 2009b;56:223–229. doi: 10.1016/j.neuropharm.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Duplantier AJ, Dombroski MA, Subramanyam C, Beaulieu AM, Chang SP, Gabel CA, et al. Optimization of the physicochemical and pharmacokinetic attributes in a 6-azauracil series of P2X7 receptor antagonists leading to the discovery of the clinical candidate CE-224,535. Bioorg Med Chem Lett. 2011;21:3708–3711. doi: 10.1016/j.bmcl.2011.04.077. [DOI] [PubMed] [Google Scholar]
- Engel T, Gomez-Villafuertes R, Tanaka K, Mesuret G, Sanz-Rodriguez A, Garcia-Huerta P, et al. Seizure suppression and neuroprotection by targeting the purinergic P2X7 receptor during status epilepticus in mice. FASEB J. 2012;26:1616–1628. doi: 10.1096/fj.11-196089. [DOI] [PubMed] [Google Scholar]
- Furber M, Alcaraz L, Bent JE, Beyerbach A, Bowers K, Braddock M, et al. Discovery of potent and selective adamantane-based small-molecule P2X(7) receptor antagonists/interleukin-1beta inhibitors. J Med Chem. 2007;50:5882–5885. doi: 10.1021/jm700949w. [DOI] [PubMed] [Google Scholar]
- Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry. 2008;13:717–728. doi: 10.1038/sj.mp.4002055. [DOI] [PubMed] [Google Scholar]
- Green EK, Grozeva D, Raybould R, Elvidge G, Macgregor S, Craig I, et al. P2RX7: a bipolar and unipolar disorder candidate susceptibility gene? Am J Med Genet B Neuropsychiatr Genet. 2009;150B:1063–1069. doi: 10.1002/ajmg.b.30931. [DOI] [PubMed] [Google Scholar]
- Grigoroiu-Serbanescu M, Herms S, Muhleisen TW, Georgi A, Diaconu CC, Strohmaier J, et al. Variation in P2RX7 candidate gene (rs2230912) is not associated with bipolar I disorder and unipolar major depression in four European samples. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:1017–1021. doi: 10.1002/ajmg.b.30952. [DOI] [PubMed] [Google Scholar]
- Guile SD, Alcaraz L, Birkinshaw TN, Bowers KC, Ebden MR, Furber M, et al. Antagonists of the P2X(7) receptor. From lead identification to drug development. J Med Chem. 2009;52:3123–3141. doi: 10.1021/jm801528x. [DOI] [PubMed] [Google Scholar]
- Hattori M, Gouaux E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature. 2012;485:207–212. doi: 10.1038/nature11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejjas K, Szekely A, Domotor E, Halmai Z, Balogh G, Schilling B, et al. Association between depression and the Gln460Arg polymorphism of P2RX7 gene: a dimensional approach. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:295–299. doi: 10.1002/ajmg.b.30799. [DOI] [PubMed] [Google Scholar]
- Iwata M, Li X, Ota KT, Banasr M, Sakaue F, Duman RS. 2012. Signaling pathways underlying the regulation of IL-1β in response to stress: targets for novel antidepressants. In: Society for Neuroscience Annual Meeting. New Orleans, LA.
- Iwata M, Ota KT, Duman RS. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun. 2013;31:105–114. doi: 10.1016/j.bbi.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Thomsen C. The role of the innate immune system in psychiatric disorders. Mol Cell Neurosci. 2013;53:52–62. doi: 10.1016/j.mcn.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Khairova RA, Machado-Vieira R, Du J, Manji HK. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int J Neuropsychopharmacol. 2009;12:561–578. doi: 10.1017/S1461145709009924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, North RA. Neuromodulation by extracellular ATP and P2X receptors in the CNS. Neuron. 2012;76:51–69. doi: 10.1016/j.neuron.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khansari PS, Sperlagh B. Inflammation in neurological and psychiatric diseases. Inflammopharmacology. 2012;20:103–107. doi: 10.1007/s10787-012-0124-x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A. 2008;105:751–756. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Duman RS. Evidence for IL-1 receptor blockade as a therapeutic strategy for the treatment of depression. Curr Opin Investig Drugs. 2009a;10:664–671. [PMC free article] [PubMed] [Google Scholar]
- Koo JW, Duman RS. Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci Lett. 2009b;456:39–43. doi: 10.1016/j.neulet.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letavic MA, Lord B, Bischoff F, Hawryluk NA, Pieters S, Rech JC, et al. Synthesis and pharmacological characterization of two novel, brain penetrating P2X7 antagonists. ACS Med Chem Lett. 2013;4:419–422. doi: 10.1021/ml400040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucae S, Salyakina D, Barden N, Harvey M, Gagne B, Labbe M, et al. P2RX7, a gene coding for a purinergic ligand-gated ion channel, is associated with major depressive disorder. Hum Mol Genet. 2006;15:2438–2445. doi: 10.1093/hmg/ddl166. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillin A, Bass NJ, Choudhury K, Puri V, Kosmin M, Lawrence J, et al. Case-control studies show that a non-conservative amino-acid change from a glutamine to arginine in the P2RX7 purinergic receptor protein is associated with both bipolar- and unipolar-affective disorders. Mol Psychiatry. 2009;14:614–620. doi: 10.1038/mp.2008.6. [DOI] [PubMed] [Google Scholar]
- Michel AD, Ng SW, Roman S, Clay WC, Dean DK, Walter DS. Mechanism of action of species-selective P2X(7) receptor antagonists. Br J Pharmacol. 2009;156:1312–1325. doi: 10.1111/j.1476-5381.2009.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Jarvis MF. P2X receptors as drug targets. Mol Pharmacol. 2013;83:759–769. doi: 10.1124/mol.112.083758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao JS, Harry GJ, Rapoport SI, Kim HW. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry. 2010;15:384–392. doi: 10.1038/mp.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Polak PE, Simonini V, Lin SX, Richardson JC, Bongarzone ER, et al. P2x7 deficiency suppresses development of experimental autoimmune encephalomyelitis. J Neuroinflammation. 2008;5:33. doi: 10.1186/1742-2094-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiratori M, Tozaki-Saitoh H, Yoshitake M, Tsuda M, Inoue K. P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J Neurochem. 2010;114:810–819. doi: 10.1111/j.1471-4159.2010.06809.x. [DOI] [PubMed] [Google Scholar]
- Soderlund J, Olsson SK, Samuelsson M, Walther-Jallow L, Johansson C, Erhardt S, et al. Elevation of cerebrospinal fluid interleukin-1ss in bipolar disorder. J Psychiatry Neurosci. 2011;36:114–118. doi: 10.1503/jpn.100080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, et al. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–132. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- Soronen P, Mantere O, Melartin T, Suominen K, Vuorilehto M, Rytsala H, et al. P2RX7 gene is associated consistently with mood disorders and predicts clinical outcome in three clinical cohorts. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:435–447. doi: 10.1002/ajmg.b.31179. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Vizi ES, Wirkner K, Illes P. P2X7 receptors in the nervous system. Prog Neurobiol. 2006;78:327–346. doi: 10.1016/j.pneurobio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Csolle C, Ando RD, Goloncser F, Kittel A, Baranyi M. The role of purinergic signaling in depressive disorders. Neuropsychopharmacol Hung. 2012;14:231–238. [PubMed] [Google Scholar]
- Stock TC, Bloom BJ, Wei N, Ishaq S, Park W, Wang X, et al. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J Rheumatol. 2012;39:720–727. doi: 10.3899/jrheum.110874. [DOI] [PubMed] [Google Scholar]
- Surprenant A, North RA. Signaling at purinergic P2X receptors. Annu Rev Physiol. 2009;71:333–359. doi: 10.1146/annurev.physiol.70.113006.100630. [DOI] [PubMed] [Google Scholar]
- Volonte C, Apolloni S, Skaper SD, Burnstock G. P2X7 receptors: channels, pores and more. CNS Neurol Disord Drug Targets. 2012;11:705–721. doi: 10.2174/187152712803581137. [DOI] [PubMed] [Google Scholar]
- Young MT, Pelegrin P, Surprenant A. Amino acid residues in the P2X7 receptor that mediate differential sensitivity to ATP and BzATP. Mol Pharmacol. 2007;71:92–100. doi: 10.1124/mol.106.030163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.