

Abstract
BACKGROUND AND PURPOSE
Transient receptor potential vanilloid type 4 (TRPV4) channels are expressed in brain endothelial cells, but their role in regulating cerebrovascular tone under physiological and pathological conditions is still largely unknown.
EXPERIMENTAL APPROACH
Wild-type (WT) mice and mice that overexpress a mutated form of the human amyloid precursor protein (APP mice, model of increased amyloid β), a constitutively active form of TGF-β1 (TGF mice, model of cerebrovascular fibrosis) or both (APP/TGF mice) were used. Dilations to the selective TRPV4 channel opener GSK1016790A (GSK) or to ACh were measured in posterior cerebral artery segments.
KEY RESULTS
Both GSK- and ACh-induced dilations virtually disappeared following endothelium denudation in WT mice. These responses were impaired in vessels from APP, TGF and APP/TGF mice compared with WT. Pre-incubation of WT vessels with the selective TRPV4 channel blocker HC-067047, or with small-conductance (SK channel, apamin) and/or intermediate-conductance (IK channel, charybdotoxin, ChTx) Ca2+-sensitive K+ channel blocker abolished GSK-induced dilations and massively decreased those induced by ACh. These treatments had no or limited effects on ACh-induced dilation in vessels from APP, TGF or APP/TGF mice, and IK and SK channel function was preserved in transgenic mice. Antioxidant superoxide dismutase or catalase normalized GSK- and ACh-mediated dilations only in APP brain arteries.
Conclusion and Implications
We conclude that endothelial TRPV4 channels mediate ACh-induced dilation in cerebral arteries, that they are impaired in models of cerebrovascular pathology and that they are sensitive, albeit in the reversible manner, to amyloid β-induced oxidative stress.
Keywords: cerebral artery, vasodilation, endothelium, oxidative stress, amyloid β peptide, TGF-β1
Introduction
The transient receptor potential vanilloid type 4 (TRPV4) channel, a member of the TRP channel superfamily, is a Ca2+-permeable nonselective cation channel. TRPV4 channels are widely distributed in peripheral and brain tissues (Nilius et al., 2004; Carreno et al., 2009; Hatano et al., 2009; Thorneloe et al., 2012), indicating their involvement in several physiology processes. These channels are activated by stimuli such as osmotic pressure changes, cell swelling, high temperature and mechanical stress (Vriens et al., 2004; O'Neil and Heller, 2005; Clark et al., 2010; Filosa et al., 2013).
TRPV4 channels are expressed in endothelial cells (ECs) from many types of blood vessels including cerebral arteries (Vriens et al., 2005; Kohler et al., 2006; Marrelli et al., 2007; Willette et al., 2008). TRPV4 channels regulate endothelial calcium (Ca2+) influx and endothelium-dependent dilation (Marrelli et al., 2007; Zhang and Gutterman, 2011) in response to flow, shear stress and, as recently demonstrated, ACh (Kohler et al., 2006; Sonkusare et al., 2012). Activation of endothelial TRPV4 channels elicits Ca2+ influx that activates intermediate- (IK; KCa3.1) and small- (SK; KCa2.3) conductance Ca2+-sensitive K+ (KCa) channels (Sonkusare et al., 2012), which results in hyperpolarization of vascular smooth muscle cells and vasodilation (Earley, 2011).
Cerebrovascular dysfunction, characterized by chronic cerebral hypoperfusion and impaired dilatory function, is an important factor in the pathogenesis of Alzheimer's disease (AD; Iadecola, 2004). Studies have demonstrated increased levels of the amyloid β (Aβ) peptide, and of toxic and inflammatory factors such as reactive oxygen species (ROS; Iadecola et al., 1999; Park et al., 2004; Tong et al., 2005) and TGF-β1 in the cerebral circulation of AD mouse models or patients (Wyss-Coray et al., 1997; 2000; Grammas and Ovase, 2001). Correspondingly, neurovascular coupling, cerebral autoregulation and endothelium-dependent dilations are impaired in mice that overexpress mutated forms of the human amyloid precursor protein (APP mice), a constitutively active form of TGF-β1 (TGF mice; Wyss-Coray et al., 2000) or a combined Aβ and TGF-β1 pathology (APP/TGF mice; Wyss-Coray et al., 1997; Ongali et al., 2010). These deficits are remedied by co-expression of APP and the free radical scavenger superoxide dismutase 1 (SOD1), after cortical SOD application (Iadecola et al., 1999), and by in vitro (Tong et al., 2005) or in vivo (Nicolakakis et al., 2008) treatment of APP mouse brain vessels or mice with antioxidants. In contrast, in TGF and APP/TGF models of AD cerebrovascular pathology that includes an inflammatory and pro-fibrotic process, cerebrovascular deficits are unresponsive to antioxidant therapy (Tong et al., 2005; Ongali et al., 2010; Nicolakakis et al., 2011). Yet, limited information is available on the role of TRPV4 channels in brain vessels and whether their function is altered in pathologies of the cerebral circulation that replicate those found in AD.
In the present study, we demonstrate that TRPV4 channels (i) are key mediators of dilatory function in cerebral arteries; (ii) are altered in mouse models of cerebrovascular pathology as seen in APP, TGF and APP/TGF mice; and (iii) are sensitive to ROS.
Materials and methods
Materials
GSK1016790A [(N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}3-hydroxylpropanoyl)-1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide] (GSK), SOD, catalase, phenylephrine (PE), ACh and sodium nitroprusside (SNP) were obtained from Sigma-Aldrich (Oakville, ON, Canada). NS309 (6,7-dicholoro-1H-indole-2,3-dione 3-oxime) and HC-067047 (2-methyl-1-[3-(4-morpholinyl)propyl]-5-phenyl-N-[3-(trifluoro-methyl)phenyl]-1Hpyrrlle-3-carboxamide; HC) were from Tocris Bioscience (Ellisville, MO, USA). Charybdotoxin (ChTx) and apamin were from Abcam (Abcam Ltd, Cambridge, UK).
Animals
Heterozygous transgenic mice on a C57BL/6J background and their wild-type (WT) littermates (3–6 months of age, body weight ∼30 g) were bred in-house, with males and females in approximately equal numbers in each group. All experiments were approved by the Animal Ethics Committee of the Montreal Neurological Institute (McGill University, Montréal, QC, Canada) and complied with local and national regulations in accordance to the Canadian Council on Animal Care. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Mice were housed under a 12 h light-dark cycle, in a room with controlled temperature (23°C) and humidity (50%), with food and tap water available ad libitum.
APP mice overexpress the human APP carrying the Swedish (K670N, M671L; APPSwe) and Indiana (V717F; APPInd) familial AD mutations directed by the PDGF β-chain promoter (APP mice, line J20; Mucke et al., 2000). APP mice display Aβ-induced cerebrovascular oxidative stress and early (2–4 months) impairment in endothelial-mediated dilatory function (Tong et al., 2005). TGF mice overexpress a constitutively active form of TGF-β1 under the control of the glial fibrillary acidic protein promoter (TGF mice, line T64), and display the cerebrovascular fibrotic pathology of AD (Wyss-Coray et al., 2000; Tong et al., 2005) with early deficits in dilatory function in brain vessels (Tong et al., 2005). Double-transgenic APP/TGF mice concurrently overexpress both transgenes (Wyss-Coray et al., 2001; Ongali et al., 2010) and are characterized by a combined Aβ- and TGF-β1-induced cerebrovascular pathology with early dilatory deficits (Ongali et al., 2010). Transgene expression was confirmed with touchdown PCR (Wyss-Coray et al., 1997; Tong et al., 2005).
Cerebrovascular reactivity studies
The posterior cerebral artery (PCA; average intraluminal diameter, 80–90 μm) was isolated from WT and transgenic mice euthanized by cervical dislocation and collected in cold and oxygenated (95% O2 and 5% CO2) Krebs solution (4°C, pH 7.4 ± 0.1) containing the following (in mM): 118 NaCl, 4.5 KCl, 2.5 CaCl2, 1 MgSO4, 1 KH2PO4, 25 NaHCO3 and 11 glucose. The reactivity of pressurized PCA segments (∼2 mm in length) mounted in a superfusion chamber system was measured using online videomicroscopy (Living Systems Instrumentation, Burlington, VT, USA), as described before (Tong et al., 2005). Vessels were cannulated on a glass micropipette (∼40 μm diameter) at one end, sealed to another glass micropipette on the other end and filled with oxygenated Krebs' solution (37 ± 1°C, pH 7.4 ± 0.1). A pressure-servo micropump (Living Systems) was used to maintain intraluminal pressure at 60 mmHg. Vessels were superfused (6 mL·min−1) with Krebs solution and allowed to stabilize and acquire basal tone (30–45 min). Online measurements of intraluminal diameter were performed using a closed-circuit video system coupled with a video caliper (Image Instrumentation, Trenton, NJ, USA).
Pharmacological characterization of TRPV4 channels and ACh responses
All compounds were applied extraluminally to the superfusion solution. Dilations induced by GSK (10−11–10−5 M, a selective TRPV4 channel agonist, EC50 value ∼18 nM at mouse TRPV4 channels, Thorneloe et al., 2008), ACh (10−11–10−4 M) or NS309 (10−10–10−5 M, a selective IK/SK channels agonist, EC50 value of ∼10 nM, Strøbaek et al., 2004) were measured on vessels pre-constricted submaximally with PE (2 × 10−7 M). In some vessels, concentration-dependent responses to GSK or ACh were examined before and after treatment with the selective TRPV4 channel antagonist HC (10−9–10−6 M, or 10−6 M, IC50 value of ∼20 nM at mouse TRPV4 channels; Everaerts et al., 2010), the potent and selective SK channel blocker apamin (10−8 M, IC50 value <8 nM; Grunnet et al., 2010), the IK channel blocker ChTx (5 × 10−8 M, IC50 value <28 nM; Grunnet et al., 2010) or a combination of both SK and IK channel blockers. Reversibility of the vascular deficits in response to GSK or ACh was tested before and after pre-incubation (30–60 min) of the arterial segments with the free radical scavenging enzymes, SOD, 120 U mL−1, or catalase (1000 U mL−1). In some arteries, the dilatory responses to GSK (10−11–10−5 M) and ACh (10−11–10−4 M) were tested before and after removing the EC by passing (20–30 s) an air bubble through the lumen. In these vessels, the integrity of the smooth muscle cells was tested with the NO donor SNP (10−11–10−4 M).
Calculations and statistical analysis of cerebrovascular responses
Responses to GSK and ACh were compared on the basis of their dose-dependent and maximal (EAmax) responses (expressed as percentage of change in vessel diameter from basal or pharmacologically induced tone) and/or potency (pD2 values or -log of EC50) expressed as mean ± SEM. When appropriate, the potency of the antagonist was evaluated by the use of pA2 and pD2' values for dual antagonist (competitive and non-competitive; Van den Brink, 1977). Statistical differences were determined by one-way anova followed by a post hoc Dunnett or Newman–Keuls multiple comparison test or, when indicated, by Student t-test for two group comparisons. All statistics and analyses were performed with the software Prism 4 (Graph Pad, San Diego, CA, USA). A P < 0.05 was considered significant.
Results
TRPV4 channels induce dilation in brain arteries via Ca2+-activated K+ channels
To determine whether TRPV4 channels are involved in dilatory function in cerebral arteries, the selective TRPV4 agonist GSK was administered to PCA segments of WT mice. As shown in Figure 1A, GSK potently and dose dependently dilated (EAmax: 59.2 ± 6.0%) pre-constricted PCA segments with a potency (pD2: 8.12 ± 0.16) totally compatible with its affinity at mouse TRPV4 channels (Thorneloe et al., 2008). Furthermore, the GSK-induced cerebral dilation was abolished by pretreatment of the vessels with the selective TRPV4 channel blocker HC (10−6 M; Figure 1A). Ca2+-activated SK and IK channels are considered important targets of Ca2+ influx through TRPV4 channels (Sonkusare et al., 2012). Therefore, we used the SK blocker apamin (10−8 M) to explore the mechanism of TRPV4-mediated vasodilation and found that it almost eliminated (−80.7%, P < 0.001) GSK-induced dilation in PCA segments from WT mice (Figure 1B), confirming a role for SK channel activation in the TRPV4 channel-mediated dilatory response.
Figure 1.
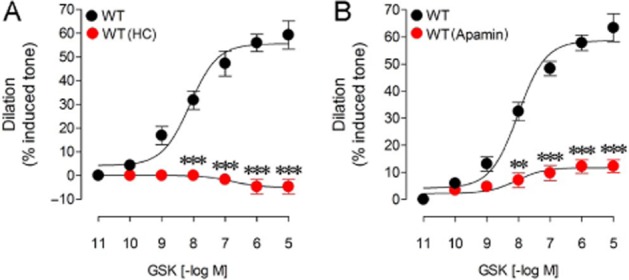
HC and apamin antagonized the GSK-induced cerebrovascular dilations in PCA segments from WT mice. The GSK-induced dilation in WT vessels was eliminated in vessels treated with the TRPV4 channel blocker HC (10−6 M) (A) or was potently reduced by the SK blocker apamin (10−8 M) (B). **P < 0.01, ***P < 0.001, when compared to untreated WT vessels by anova followed by a Newman–Keuls post hoc comparison test. Error bars represent SEM. n = 3–4 per group.
ACh is known to mediate endothelium-dependent dilation of brain arteries in various species (Rosenblum, 1992; Mackert et al., 1997), and TRPV4 channels have recently been shown to contribute to the ACh-induced dilation of mesenteric arteries (Sonkusare et al., 2012). We thus investigated whether TRPV4 channels are involved in the ACh-induced dilation in brain arteries from WT mice. ACh was found to elicit concentration-dependent relaxations (EAmax: 35.8 ± 2%; pD2: 7.86 ± 0.1; Figure 2A) that were inhibited by pretreatment of the vessels with increasing concentrations (10−9–10−6 M) of the selective TRPV4 channel blocker HC, as represented by a rightward shift of the dose-response curve to ACh and a decrease in ACh efficacy (Figure 2A). HC behaved as a mixed competitive/non-competitive antagonist and significantly reduced both the ACh maximal dilation (EAmax) and potency (pD2 value; Figure 2B). HC estimated pA2 value of 7.82 and pD2' value of 6.83 at 10−7 M corresponded well to its reported affinity (pIC50 = 7.7) at TRPV4 receptors (Everaerts et al., 2010). These findings indicate that TRPV4 channels contribute to the ACh-induced cerebrovascular dilation.
Figure 2.
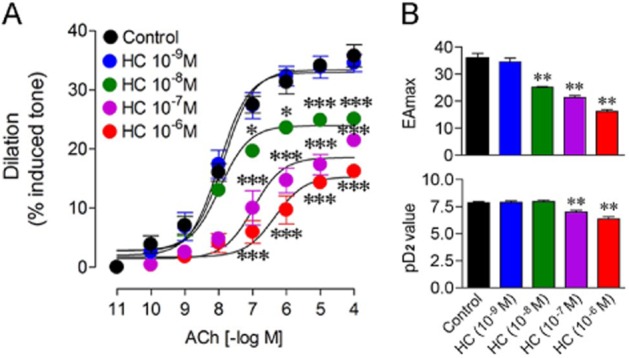
Effects of TRPV4 channel blocker HC on the ACh-induced dilation in WT cerebral arteries. (A) The selective TRPV4 channel blocker HC dose-dependently (10−9–10−6 M) antagonized the ACh-induced dilations compared to untreated WT vessel segments. (B) HC behaved as a mixed competitive non-competitive antagonist and significantly decreased both ACh potency (EAmax, upper panel) and ACh affinity (pD2 value, lower panel) in a dose-dependent manner. *P < 0.05, **P < 0.01, ***P < 0.001, when compared to untreated WT vessels by anova followed by a Dunnett post hoc multiple comparison test. n = 3–6 per group.
Similar to what was observed with GSK, the ACh-mediated cerebral dilation of PCA segments from WT mice was potently inhibited by pretreatment of the arteries with blockers of IK and SK channels (Figure 3A). The SK blocker apamin (10−8 M) and IK blocker ChTx (5 × 10−8 M) significantly reduced the ACh maximal dilation (−57.9%, P < 0.001 and −52.7%, P < 0.001, respectively) with no alteration in potency (Figure 3B). The maximal relaxation induced by ACh was only slightly more reduced (−62.8%, P < 0.001) by a combination of apamin and ChTx (Figure 3A, B), pointing to their possible overlapping role of these Ca2+-sensitive K+ channels in the ACh-induced cerebral vasodilation or the lack of selectivity of the selected antagonists. Future studies with an IK blocker other than ChTx that also blocks large-conductance Ca2+-sensitive K+ channels (BK) and voltage-gated (Kv1.3) channels with high affinity (Grunnet et al., 2010) will help clarify the exact contribution of IK channels in the ACh response.
Figure 3.
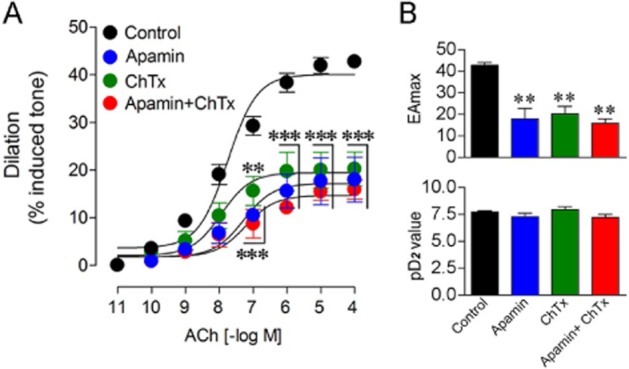
Effects of IK and SK channel blockers on the ACh-induced dilation in WT cerebral arteries. (A) Blockers of SK (apamin) or IK (ChTx) channels alone or in combination inhibited the ACh maximal dilation without any shift in the ACh-dose response curve. (B) Apamin and ChTx decreased the ACh potency (EAmax, upper panel) but not ACh affinity (pD2 value, lower panel). **P < 0.01, ***P < 0.001, when compared to untreated WT vessels by anova followed by a Dunnett post hoc multiple comparison test. Error bars represent SEM. n = 10 for controls and 3 for each drug groups.
TRPV4-mediated GSK and ACh dilation are endothelium dependent
It has been reported that TRPV4 channels do not exist only in ECs, but also in vascular smooth muscle cells on which TRPV4 agonists may act to induce relaxation through activation of BK (KCa1.1) channels (Earley et al., 2005). We thus assessed whether TRPV4-mediated dilation of brain arteries is endothelium dependent or mediated at the level of smooth muscle cells. Both GSK- and ACh-induced dilations of PCA segments in WT mice (43.6 ± 1.4% and 55.8 ± 2.8%, respectively) disappeared after endothelial denudation (<1%, P < 0.001), whereas denuded segments had preserved dilation to the smooth muscle relaxant and NO donor SNP compared to control vessels (EAmax of 34.0 ± 2.6% and 38.0 ± 1.5% respectively). These findings indicated that TRPV4 channel-mediated dilation is endothelium dependent.
Impaired TRPV4, but not SK/IK channel function in brain arteries from APP, TGF and APP/TGF mice
Previous studies have shown that ACh-induced cerebrovascular dilations are impaired in APP, TGF and APP/TGF mice (Iadecola et al., 1999; Niwa et al., 2002; Tong et al., 2005; Nicolakakis et al., 2008; 2011; Ongali et al., 2010), but no attempts were undertaken to investigate the underlying receptor alterations. Therefore, we examined the integrity of TRPV4-SK/IK signalling cascade in PCA segments from these different animal models of cerebrovascular dysfunction at an age when the deficits are established (Tong et al., 2005; Ongali et al., 2010). As expected, APP, TGF and APP/TGF mouse brain vessels had lessened ACh-mediated dilation (Figure 4) with no change in receptor affinity (pD2 values, Table 1). Furthermore, contrary to WT mice, the ACh-induced relaxations of PCA segments in the three transgenic mouse models were not further reduced by pretreatment with the selective TRPV4 channel blocker HC (10−6 M) (Figure 4A–C, Table 1). Similarly, in contrast to WT vessels, pretreatment of the arterial segments with a combined solution of apamin (10−8 M) and ChTx (5 × 10−8 M) had no significant effect on the ACh-induced dilation of cerebral arteries from transgenic mice (Figure 4D–F), despite a small, albeit significant rightward shift in the dose-response curve to ACh in APP mice (Figure 4D, Table 2). Yet, the dilation induced by the IK/SK channel opener NS309 was similar in WT, APP and TGF mice (Figure 5), indicating that TRPV4, but not IK/SK channels are dysfunctional in transgenic mice.
Figure 4.

HC, and combined apamin + ChTx reduced the ACh dilations in WT cerebral arteries, but not in vessels from APP, TGF and APP/TGF mice. Compared with WT vessels, the ACh-induced cerebrovascular dilations were significantly impaired in APP (A), TGF (B) and APP/TGF (C) mice. Treatments with HC (10−6 M) significantly reduced the ACh dilation in WT vessels, but had no or only modest effects in APP (A), TGF (B) or APP/TGF (C) arterial segments. Similar to HC, apamin (10−8 M) and ChTx (5 × 10−8 M) only affected vessels from WT mice, but not those from transgenic APP, TGF or APP/TGF mice (D–F). Only a small albeit significant decrease in the ACh pD2 value was observed in APP mice (D). *P < 0.05, **P < 0.01, ***P < 0.001 and ⋆⋆ P < 0.01, ⋆⋆⋆ P < 0.001, when compared to untreated WT vessels by anova followed by a Newman–Keuls post hoc comparison test. Error bars represent SEM. n = 3–6 per group.
Table 1.
Effects of HC (10−6 M) on cerebrovascular responses to ACh (10−4 M) in PCA segments from WT, APP, TGF and APP/TGF mice
| Treatment | WT (n = 6) | APP (n = 3) | TGF (n = 3) | APP/TGF (n = 3) | |
|---|---|---|---|---|---|
| EAmax | Control | 35.8 ± 1.9 | 25.2 ± 2.9 | 23.4 ± 3.1 | 18.3 ± 2.0 |
| HC | 16.2 ± 0.7a | 19.9 ± 2.0 | 19.3 ± 4.2 | 16.6 ± 2.4 | |
| pD2 | Control | 7.86 ± 0.10 | 7.95 ± 0.21 | 8.06 ± 0.23 | 8.16 ± 0.16 |
| HC | 6.34 ± 0.22a | 7.36 ± 0.21 | 8.02 ± 0.32 | 8.18 ± 0.23 |
Data are mean ± SEM of the number (n) of mice indicated in parentheses and are expressed as the maximal agonist response (EAmax, % dilation from induced tone) or potency [pD2 value or −log (EC50)]. HC had a significant effect only in WT mice.
P < 0.001 when compared to their respective control groups before HC treatment, by Student t-test.
Table 2.
Effects of a combination of apamin (10−8 M) and ChTx (5 × 10−8 M) on cerebrovascular responses to ACh (10−4 M) in PCA segments from WT, APP, TGF and APP/TGF mice
| Treatment | WT (n = 3) | APP (n = 3) | TGF (n = 3) | APP/TGF (n = 3) | |
|---|---|---|---|---|---|
| EAmax | Control | 45.7 ± 2.5 | 24.3 ± 2.7 | 23.4 ± 2.9 | 18.0 ± 3.1 |
| Apamin +ChTx | 15.9 ± 2.0b | 18.3 ± 3.9 | 20.4 ± 5.3 | 13.4 ± 2.0 | |
| pD2 | Control | 7.67 ± 0.14 | 8.28 ± 0.24 | 8.04 ± 0.25 | 8.02 ± 0.28 |
| Apamin +ChTx | 7.23 ± 0.28 | 7.05 ± 0.35a | 7.93 ± 0.36 | 7.98 ± 0.27 |
Data are mean ± SEM of the number (n) of mice indicated in parentheses and are expressed as the maximal agonist response (EAmax, % dilation from induced tone) or potency [pD2 value or −log (EC50)]. Apamin and ChTx significantly reduced the ACh-mediated response in WT mice, and had a small, albeit significant effect, on the potency of ACh in APP mice.
aP < 0.05 and bP < 0.001, when compared to their respective control groups before treatment, by Student t-test.
Figure 5.
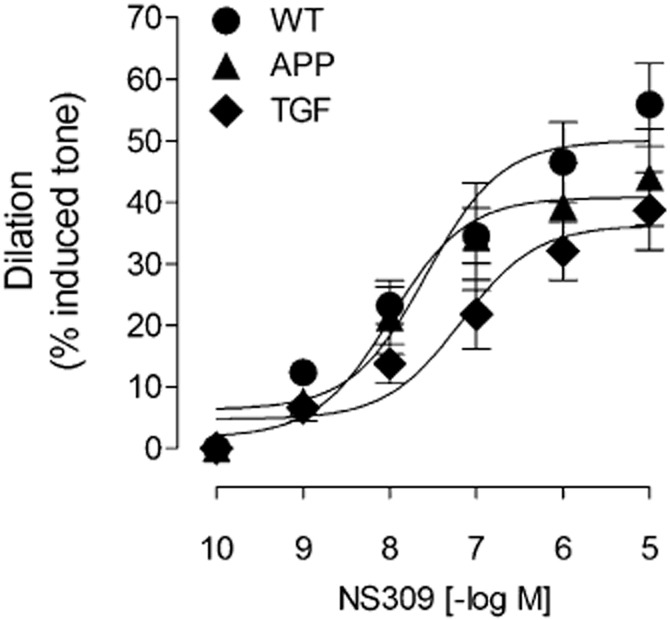
Dilatory responses to the IK/SK channel activator NS309 are preserved in transgenic mice. Concentration response curves of the IK/SK channel activator NS309 in PCA segments from WT, APP and TGF mice were comparable. The affinity of NS309 [pD2 value or −log (EC50)] at cerebrovascular channels was also comparable between all groups, with respective values of 7.58 ± 0.25, 7.99 ± 0.31 and 7.18 ± 0.29. Error bars represent SEM. n = 5 per group.
Antioxidants rescue TRPV4-SK/IK channel function in brain arteries from APP, but not TGF and APP/TGF mice
Impaired endothelial-dependent dilation is an early landmark of APP, TGF and APP/TGF mice cerebrovascular dysfunction. Particularly, this deficit has been imputed to Aβ-induced vascular oxidative stress in APP mice (Iadecola et al., 1999; Tong et al., 2005; Park et al., 2008), whereas in TGF and APP/TGF signalling alterations related to vascular fibrosis have been associated with the lessened dilatory capacity (Tong et al., 2005; Ongali et al., 2010; Nicolakakis et al., 2011). Consistent with these observations, we found that in vitro antioxidant treatment of PCA segments with SOD or catalase in APP mice completely normalized the GSK- and ACh-induced dilations to WT levels (Figure 6A–C). In contrast, superfusion of TGF or APP/TGF vessels with SOD had no beneficial effect, and vessels still exhibited impaired dilations in response to GSK or ACh application (Figure 7). These findings demonstrate that TRPV4 channels are sensitive to Aβ-induced oxidative stress, whereas cerebrovascular fibrosis entails their deregulation through mechanisms not related to oxidative stress.
Figure 6.
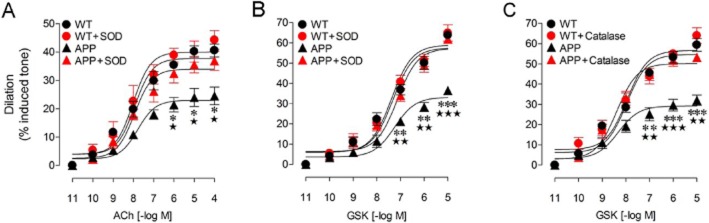
Effects of SOD or catalase on the ACh- and GSK- induced cerebrovascular dilation of the PCA in APP mice. Compared with WT vessels, ACh- (A) and GSK- (B and C) induced cerebrovascular dilations were reduced in APP vessels. Treatment with SOD or catalase completely normalized these responses to levels comparable to those of WT arteries. SOD (A and B) or catalase (C) had no effect on vessels from WT mice. *P < 0.05, **P < 0.01, ***P < 0.001, when compared with untreated WT vessels; ⋆ P < 0.05, ⋆⋆ P < 0.01, ⋆⋆⋆ P < 0.001, compared with SOD- or catalase-treated APP vessels, by anova followed by a Newman–Keuls comparison test. Error bars represent SEM. n = 3 per group.
Figure 7.
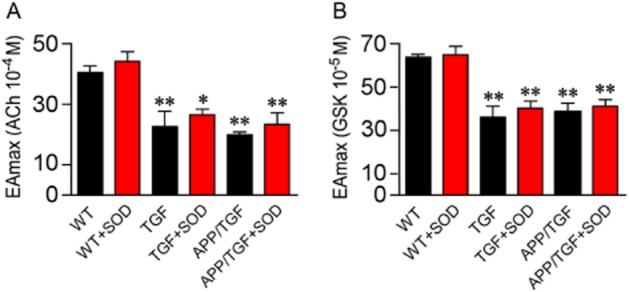
Effects of SOD on the ACh- and GSK- induced dilation in cerebral arteries from TGF and APP/TGF mice. Compared with WT mice, the ACh- (A) and GSK- (B) induced dilations were impaired in TGF and APP/TGF mice. SOD treatment did not rescue these deficits. *P < 0.05, **P < 0.01, when compared to untreated WT vessels, using anova followed by a Dunnett post hoc test. Error bars represent SEM. n = 3 per group.
Discussion
Our findings demonstrate (i) that TRPV4 channels are important mediators of endothelium-dependent dilation in mouse cerebral arteries including that induced by muscarinic ACh receptor (mAChR) activation; (ii) that these channels are altered in transgenic mouse models that recapitulate different aspects of AD cerebrovascular pathology; and (iii) that they are sensitive, albeit in a reversible manner, to Aβ-induced vascular oxidative damage. In contrast, TRPV4 channel dysfunction in pathologies that involve vascular fibrosis as seen in TGF and APP/TGF mice is not remedied by antioxidant therapy.
It has been reported that TRPV4 channels exist in cerebral ECs where they regulate calcium influx and endothelium-dependent hyperpolarizing factor (EDHF)-mediated dilations (Marrelli et al., 2007). Here, we show that TRPV4 channels mediate part of the endothelium-dependent ACh-induced dilation in mouse brain arteries, and that this response results from activation of IK and/or SK channels. In WT cerebral arteries, our results in endothelial-denuded vessels confirmed that TRPV4 channels are endothelial located. Further, direct activation of TRPV4 channels in WT arteries by the TRPV4 channel opener GSK resulted in cerebrovascular dilation that was completely or virtually abolished by HC, a potent and selective TRPV4 channel blocker (Everaerts et al., 2010; Shahidullah et al., 2012) or by the SK blocker apamin. The additional findings that ACh-induced cerebral vasodilation in WT vessels was impaired to a similar extent (−50–60%) by HC, apamin, ChTx or by a combination of apamin and ChTx, further demonstrated interaction between mAChRs and the TRPV4-IK/SK channel signalling cascade. Together, these findings are consistent with endothelial-dependent mAChR acting through TRPV4-mediated stimulation of IK and/or SK channels to promote vasodilation, likely via an EDHF pathway (Marrelli et al., 2007), as recently reported in mesenteric arteries (Sonkusare et al., 2012). In TRPV4–/– mice, the ACh-induced dilation in mesenteric arterial segments was blunted but not eliminated (Zhang et al., 2009), suggesting additional Ca2+ entry pathways in ECs. The findings in WT vessels that the TRPV4 channel antagonist HC completely abrogated the GSK-mediated dilatory response, but only partially reduced (∼55%) that induced by ACh pointed to about half of the mAChR-mediated dilation taking place through other pathways, such as NO signalling (Zhang et al., 2009; Howitt et al., 2012) and, possibly, H2O2 depending on the partial pressure of oxygen (Drouin et al., 2007). Likewise, in mouse mesenteric arteries, the dilation mediated by mAChR had both an NO and an EDHF (76%) components (Sonkusare et al., 2012). Interestingly, in rat carotid artery, Ca2+ entry through TRPV4 channel activation reportedly triggers both NO- and EDHF-mediated vasodilations (Kohler et al., 2006).
An important finding from our study was the exclusive impairment of GSK- and ACh-induced dilations by TRPV4 and SK/IK channel blockers in WT mice. Indeed, these blockers had no or very modest effects on the APP, TGF or APP/TGF mouse brain vessels, indicating an already malfunctioning TRPV4-SK/IK channel signalling cascade in vessels from mice with altered cerebrovascular function or structure. The preserved function of IK/SK channels shown in APP and TGF mice with the selective activator NS309 further pointed to dysfunctional TRPV4 channels. Impaired endothelium-mediated dilation is a key landmark of Aβ-induced cerebrovascular dysfunction in various mouse models of AD, and has been imputed to Aβ-induced oxidative stress (Iadecola et al., 1999; Park et al., 2004; 2008; Tong et al., 2005). Interestingly, antioxidant treatment of isolated arteries with SOD or catalase, which respectively scavenges O2– and hydrogen peroxide, fully normalized both the GSK- and ACh-induced dilations in APP brain arteries, as reported before for ACh dilations in different APP mice (Iadecola et al., 1999; Tong et al., 2005). Consistent with our findings, it has been shown in diabetic rats that O2– can inhibit BK, ATP-sensitive K+ and inwardly rectifying K+ channels in cerebral arteries, and that topical SOD and catalase application can restore these impairments (Erdös et al., 2004).
In contrast, in TGF and APP/TGF transgenic mice with structural alterations of the cerebrovascular bed, the reduced TRPV4 channel-mediated dilation was not rescued by antioxidant therapy, as previously reported for the ACh-induced dilation (Tong et al., 2005; Ongali et al., 2010; Papadopoulos et al., 2010; Nicolakakis et al., 2011). These findings suggest that antioxidants through scavenging of non-physiological levels of O2– and H2O2 exert beneficial effects on the TRPV4 channel dysfunction caused by Aβ-induced vascular oxidative stress, but not by pathologies that involve fibrosis of the brain vasculature (Wyss-Coray et al., 2000; Tong et al., 2005; Ongali et al., 2010). Previous studies in TGF mice have imputed the antioxidant-resistant cerebrovascular deficits to altered levels or activities of vasoactive proteins and their downstream signalling cascades (Tong et al., 2005; Tong and Hamel, 2007), which could be rescued in vivo by drugs with anti-inflammatory properties (Nicolakakis et al., 2011). In contrast, APP/TGF vessels resisted to in vitro antioxidant (Ongali et al., 2010) and in vivo anti-inflammatory therapy (Papadopoulos et al., 2013), pointing to their complex Aβ and fibrotic cerebrovascular pathology.
Overall, our results in cerebral blood vessels add to the growing role of TRPV4 channels already described in peripheral blood vessels. Particularly, more than being involved in shear-stress-, epoxyeicosatrienoic acids- and plant-derived flavone-induced dilations (Kohler et al., 2006; Earley, 2011; Ma et al., 2012), endothelial TRPV4 channels appear as important mediators of the ACh-induced dilation in both peripheral (Sonkusare et al., 2012) and brain arteries (this study) through a common IK and/or SK channel cascade. Importantly, we report that cerebrovascular TRPV4 channel function is severely compromised, albeit in a reversible manner, by Aβ-induced vascular oxidative stress, whereas pathologies that involve fibrosis of the cerebral vasculature are insensitive to antioxidant therapy. These findings may be particularly important when trying to rescue cerebrovascular deficits in AD patients who display a combined Aβ and TGF-β1 pathology (Wyss-Coray et al., 2000; Grammas and Ovase, 2001; Tong et al., 2005; Nicolakakis and Hamel, 2011). We suggest that TRPV4 channels may represent a promising target to restore cerebrovascular function in AD.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR, MOP-126001, E. H.). The authors are most thankful to Dr. Xinkang Tong for training and help with the experiments.
Glossary
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- ChTx
charybdotoxin
- EDHF
endothelium-dependent hyperpolarizing factor
- GSK
GSK1016790A
- HC
HC-067047
- IK
intermediate-conductance Ca2+-sensitive K+ (KCa) channels
- KCa
Ca2+-sensitive K+
- mAChR
muscarinic ACh receptor
- PCA
posterior cerebral artery
- PE
phenylephrine
- ROS
reactive oxygen species
- SNP
sodium nitroprusside
- SOD
superoxide dismutase
- SK
small-conductance Ca2+-sensitive K+ (KCa) channels
- TRPV4
transient receptor potential vanilloid type 4
Conflict of interest
The authors declare no conflict of interests.
References
- Carreno FR, Ji LL, Cunningham JT. Altered central TRPV4 expression and lipid raft association related to inappropriate vasopressin secretion in cirrhotic rats. Am J Physiol Regul Integr Comp Physiol. 2009;296:R454–R466. doi: 10.1152/ajpregu.90460.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AL, Votta BJ, Kumar S, Liedtke W, Guilak F. Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: age- and sex-dependent progression of osteoarthritis in Trpv4-deficient mice. Arthritis Rheum. 2010;62:2973–2983. doi: 10.1002/art.27624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin A, Thorin-Trescases N, Hamel E, Falck JR, Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73:73–81. doi: 10.1016/j.cardiores.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Earley S. Endothelium-dependent cerebral artery dilation mediated by transient receptor potential and Ca2+-activated K+ channels. J Cardiovasc Pharmacol. 2011;57:148–153. doi: 10.1097/FJC.0b013e3181f580d9. [DOI] [PubMed] [Google Scholar]
- Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res. 2005;97:1270–1279. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- Erdös B, Simandle SA, Snipes JA, Miller AW, Busija DW. Potassium channel dysfunction in cerebral arteries of insulin-resistant rats is mediated by reactive oxygen species. Stroke. 2004;35:964–969. doi: 10.1161/01.STR.0000119753.05670.F1. [DOI] [PubMed] [Google Scholar]
- Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, et al. Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci U S A. 2010;107:19084–19089. doi: 10.1073/pnas.1005333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa JA, Yio X, Rath G. TRPV4 and the regulation of vascular tone. J Cardiovasc Pharmacol. 2013;61:113–119. doi: 10.1097/FJC.0b013e318279ba42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammas P, Ovase R. Inflammatory factors are elevated in brain microvessels from Alzheimer's disease. Neurobiol Aging. 2001;22:837–842. doi: 10.1016/s0197-4580(01)00276-7. [DOI] [PubMed] [Google Scholar]
- Grunnet M, Strøbaek D, Olesen SO, Christophersen P. KCa1-KCa5 families. In: Kew J, Davies C, editors. Ion Channels, From Structure to Function. New York: Oxford University Press Inc; 2010. pp. 404–418. [Google Scholar]
- Hatano N, Itoh Y, Muraki K. Cardiac fibroblasts have functional TRPV4 activated by 4alpha-phorbol 12,13-didecanoate. Life Sci. 2009;85:808–814. doi: 10.1016/j.lfs.2009.10.013. [DOI] [PubMed] [Google Scholar]
- Howitt L, Grayson TH, Morris MJ, Sandow SL, Murphy TV. Dietary obesity increases NO and inhibits BKCa-mediated, endothelium-dependent dilation in rat cremaster muscle artery: association with caveolins and caveolae. Am J Physiol Heart Circ Physiol. 2012;302:H2464–H2476. doi: 10.1152/ajpheart.00965.2011. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Neurosci Rev. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, et al. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol. 2006;26:1495–1502. doi: 10.1161/01.ATV.0000225698.36212.6a. [DOI] [PubMed] [Google Scholar]
- Ma X, He D, Ru X, Chen Y, Cai Y, Bruce IC, et al. Apigenin, a plant-derived flavone, activates transient receptor potential vanilloid 4 cation channel. Br J Pharmacol. 2012;166:349–358. doi: 10.1111/j.1476-5381.2011.01767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackert JL, Parsons AA, Wahl M, Schilling L. Mediation of endothelium-dependent relaxation: different response patterns in rat and rabbit basilar artery. Neurol Res. 1997;19:521–526. doi: 10.1080/01616412.1997.11740851. [DOI] [PubMed] [Google Scholar]
- Marrelli SP, O'Neil RG, Brown RC, Bryan RM., Jr PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol Heart Circ Physiol. 2007;292:H1390–H1397. doi: 10.1152/ajpheart.01006.2006. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, et al. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis N, Hamel E. Neurovascular function in Alzheimer's disease patients and experimental models. J Cereb Blood Flow Metab. 2011;31:1354–1370. doi: 10.1038/jcbfm.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P, et al. Complete rescue of cerebrovascular function in aged Alzheimer's disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci. 2008;28:9287–9296. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis N, Aboulkassim T, Aliaga A, Tong XK, Rosa-Neto P, Hamel E. Intact memory in TGF-beta1 transgenic mice featuring chronic cerebrovascular deficit: recovery with pioglitazone. J Cereb Blood Flow Metab. 2011;31:200–211. doi: 10.1038/jcbfm.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol. 2004;286:C195–C205. doi: 10.1152/ajpcell.00365.2003. [DOI] [PubMed] [Google Scholar]
- Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol. 2002;283:H315–H323. doi: 10.1152/ajpheart.00022.2002. [DOI] [PubMed] [Google Scholar]
- O'Neil RG, Heller S. The mechanosensitive nature of TRPV channels. Pflugers Arch. 2005;451:193–203. doi: 10.1007/s00424-005-1424-4. [DOI] [PubMed] [Google Scholar]
- Ongali B, Nicolakakis N, Lecrux C, Aboulkassim T, Rosa-Neto P, Papadopoulos P, et al. Transgenic mice overexpressing APP and transforming growth factor-beta1 feature cognitive and vascular hallmarks of Alzheimer's disease. Am J Pathol. 2010;177:3071–3080. doi: 10.2353/ajpath.2010.100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos P, Ongali B, Hamel E. Selective in vivo antagonism of endothelin receptors in transforming growth factor-beta1 transgenic mice that mimic the vascular pathology of Alzheimer's disease. Can J Physiol Pharmacol. 2010;88:652–660. doi: 10.1139/Y10-042. [DOI] [PubMed] [Google Scholar]
- Papadopoulos P, Rosa-Neto P, Rochford J, Hamel E. Pioglitazone improves reversal learning and exerts mixed cerebrovascular effects in a mouse model of Alzheimer's disease with combined amyloid-β and cerebrovascular pathology. Plos ONE. 2013;8:e68612. doi: 10.1371/journal.pone.0068612. doi:10.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Anrather J, Forster C, Kazama K, Carlson GA, Iadecola C. Abeta-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J Cereb Blood Flow Metab. 2004;24:334–342. doi: 10.1097/01.WCB.0000105800.49957.1E. [DOI] [PubMed] [Google Scholar]
- Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum WI. Endothelium-derived relaxing factor in brain blood vessels is not nitric oxide. Stroke. 1992;23:1527–1532. doi: 10.1161/01.str.23.10.1527. [DOI] [PubMed] [Google Scholar]
- Shahidullah M, Mandal A, Delamere NA. TRPV4 in porcine lens epithelium regulates hemichannel-mediated ATP release and Na-K-ATPase activity. Am J Physiol Cell Physiol. 2012;302:C1751–C1761. doi: 10.1152/ajpcell.00010.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strøbaek D, Teuber L, Jørgensen TD, Ahring PK, Kjaer K, Hansen RS, et al. Activation of human IK and SK Ca2+-activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime) Biochim Biophys Acta. 2004;1665:1–5. doi: 10.1016/j.bbamem.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP, et al. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1-piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GS K1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: part I. J Pharmacol Exp Ther. 2008;236:432–442. doi: 10.1124/jpet.108.139295. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med. 2012;4:159ra148. doi: 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- Tong X-K, Hamel E. Transforming growth factor-beta 1 impairs endothelin-1-mediated contraction of brain vessels by inducing mitogen-activated protein (MAP) kinase phosphatase-1 and inhibiting p38 MAP kinase. Mol Pharmacol. 2007;72:1476–1483. doi: 10.1124/mol.107.039602. [DOI] [PubMed] [Google Scholar]
- Tong XK, Nicolakakis N, Kocharyan A, Hamel E. Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer's disease. J Neurosci. 2005;25:11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Brink FG. General theory of drug-receptor interactions: drug-receptor interaction models, calculation of drug parameters. In: van Rossum JM, editor. ‘Kinetics of Drug Action’. Berlin Heidelberg: Springer-Verlag; 1977. pp. 169–254. Chapter 4. [Google Scholar]
- Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B. Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci U S A. 2004;101:396–401. doi: 10.1073/pnas.0303329101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, et al. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res. 2005;97:908–915. doi: 10.1161/01.RES.0000187474.47805.30. [DOI] [PubMed] [Google Scholar]
- Willette RN, Bao W, Nerurkar S, Yue TL, Doe CP, Stankus G, et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: part 2. J Pharmacol Exp Ther. 2008;326:443–452. doi: 10.1124/jpet.107.134551. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, et al. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer's disease. Nature. 1997;389:603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E. Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am J Pathol. 2000;156:139–150. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Gutterman DD. Transient receptor potential channel activation and endothelium-dependent dilation in the systemic circulation. J Cardiovasc Pharmacol. 2011;57:133–139. doi: 10.1097/FJC.0b013e3181fd35d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, et al. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension. 2009;53:532–538. doi: 10.1161/HYPERTENSIONAHA.108.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]