

Abstract
Stem cell factor (mouse: Kitl, human: KITLG) and insulin-like growth factor-1 (IGF1), acting via KIT and IGF1 receptor (IGF1R), respectively, are critical for the development and integrity of several tissues. Autocrine/paracrine KITLG-KIT and IGF1-IGF1R signaling are also activated in several cancers including gastrointestinal stromal tumors (GIST), the most common sarcoma. In murine gastric muscles, IGF1 promotes Kitl-dependent development of interstitial cells of Cajal (ICC), the non-neoplastic counterpart of GIST, suggesting cooperation between these pathways. Here, we report a novel mechanism linking IGF1-IGF1R and KITLG-KIT signaling in both normal and neoplastic cells. In murine gastric muscles, the microenvironment for ICC and GIST, human hepatic stellate cells (LX-2), a model for cancer niches, and GIST cells, IGF1 stimulated Kitl/KITLG protein and mRNA expression and promoter activity by activating several signaling pathways including AKT-mediated glycogen synthase kinase-3β inhibition (GSK3i). GSK3i alone also stimulated Kitl/KITLG expression without activating mitogenic pathways. Both IGF1 and GSK3i induced chromatin-level changes favoring transcriptional activation at the Kitl promoter including increased histone H3/H4 acetylation and H3 lysine (K) 4 methylation, reduced H3K9 and H3K27 methylation and reduced occupancy by the H3K27 methyltransferase EZH2. By pharmacological or RNA interference-mediated inhibition of chromatin modifiers we demonstrated that these changes have the predicted impact on KITLG expression. KITLG knock-down and immunoneutralization inhibited the proliferation of GIST cells expressing wild-type KIT, signifying oncogenic autocrine/paracrine KITLG-KIT signaling. We conclude that membrane-to-nucleus signaling involving GSK3i establishes a previously unrecognized link between the IGF1-IGF1R and KITLG-KIT pathways, which is active in both physiologic and oncogenic contexts and can be exploited for therapeutic purposes.
Introduction
Stem cell factor (mouse: Kitl; human: KITLG) is the natural ligand of the type 3 receptor tyrosine kinase (RTK) KIT. Kitl/KITLG is widely expressed in stromal tissues and is critical for the differentiation, proliferation, migration, survival and functional activation of germinal, erythroid and mast cells and melanocytes [1], as well as interstitial cells of Cajal (ICC), gastrointestinal mesenchymal cells that generate electrical rhythmicity and mediate nerve-smooth muscle interactions [2]. Kitl/KITLG is a non-covalent homodimer and exists as a membrane-bound or locally secreted peptide [3]. A 164-amino-acid secreted isoform results from proteolytic cleavage of the 248-amino-acid, transmembrane peptide (“soluble” isoform; Kitl248/KITLG248) at a site encoded within exon 6. A 220-amino-acid isoform, which only generates secreted peptide at a slow rate, is produced from an alternatively spliced transcript lacking exon 6 (“membrane-bound” isoform; Kitl220/KITLG220) [3]. Autocrine/paracrine activation of KIT signaling by KITLG plays a role in several tumors and hematologic malignancies [1]. In other cancers including the majority (75-80%) of gastrointestinal stromal tumors (GIST), which originate from cells of the ICC lineage [4,5], KIT signaling is constitutively active due to oncogenic mutations [6]. GIST lacking mutated KIT may harbor activating mutations in PDGF receptor α (PDGFRA) [7] or have neither KIT nor PDGFRA mutations (“wild-type (WT)” GIST) [6]. KIT/PDGFRA inhibitors such as imatinib mesylate are the mainstay of medical treatment for advanced GIST but they are not curative due in part to secondary mutations interfering with drug action [6] or lack of dependence of cancer-initiating cells on KIT/PDGFRA signaling [8]. Since imatinib preferentially targets mutant receptors [6], reduced drug responsiveness [9,10] and aggressive GIST behavior [11] may also reflect activation of WT KIT expressed in the majority of GIST by KITLG originating from the circulation, the tumor cells, or their niche [9,11-13]. However, direct evidence of KITLG-driven GIST cell proliferation is lacking.
Similarly to KIT, PDGFRA and their ligands, insulin-like growth factor (IGF)-1 receptor (IGF1R), a type 2 RTK, and its ligands IGF1 and IGF2 play critical roles in normal growth and development, as well as in cellular stress, aging and cancer by stimulating protein synthesis and the cell cycle [14,15]. IGF1R is expressed and activated in some GIST [16] and is overexpressed in WT GIST [16,17]. Via an autocrine loop, IGF1 stimulates the growth and survival of gastrointestinal smooth muscle cells [18-20], and, thereby, promotes the differentiation of ICC [21] by increasing Kitl availability in their microenvironment [22]. IGF1 also activates gene transcription e.g. via p44/p42 mitogen-activated protein kinase (ERK1/2 MAPK) signaling [15] or by promoting the nuclear translocation and binding to the chromatin of IGF1R [23]. Together, these observations suggest that there may be cooperation between the IGF1-IGF1R and KITLG–KIT signaling pathways both in normal tissues and certain cancers including GIST; these interactions may be mediated by epigenetic control of gene transcription; and increased Kitl/KITLG expression may result in autocrine/paracrine stimulation of proliferation in cells expressing WT KIT. Here, we tested these hypotheses by investigating the effects of IGF1 on endogenous Kitl/KITLG expression and the underlying epigenetic mechanisms and signaling pathways in IGF1R-expressing cells and tissues including gastric smooth muscles [18-20,22], the natural microenvironment for ICC and GIST; in human GIST cells [10,16], and in LX-2 human hepatic stellate cells [24], a model for cancer niches [25]. Our findings indicate that IGF1 stimulates KITLG transcription by inducing coordinated chromatin modifications in part via glycogen synthase kinase (GSK)-3β inhibition. We also provide evidence supporting KITLG-mediated autocrine/paracrine stimulation of cell proliferation in GIST cells expressing WT KIT.
Materials and Methods
Ethics statements
Mice were maintained and the experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol was approved by the Institutional Animal Care and Use Committee of the Mayo Clinic (A64812). De-identified human gastric tissues used for the preparation of primary cell cultures were obtained as surgical waste tissue from patients undergoing bariatric surgery with the approval of the Mayo Clinic Institutional Review Board (07-003371). The Mayo Clinic Institutional Review Board waived the need for written informed consent from the participants.
Tissue preparation
BALB/c mice aged 14-16 days were obtained from breeder pairs purchased from Harlan Laboratories (Madison, WI). LONG R3-recombinant human IGF1 (LR3-rhIGF1; Research Peptides, Orlando, FL) was administered by a single i.p. injection. Mice were anesthetized with isoflurane (AErrane; Baxter Healthcare, Deerfield, IL) inhalation and killed by decapitation. Intact gastric corpus and antrum tunica muscularis tissues were dissected as described [22] and used either as organotypic cultures [22] or primary cell cultures [26]. Primary cell cultures were prepared from human tunica muscularis as described for murine tissues [26].
Tissue culturing and drug treatment
Gastric tunica muscularis organotypic cultures were maintained for up to 24 h. Pharmacological agents were applied as indicated in the Results and Figures. In some experiments tissues were preincubated for 3 h with specific, cell-permeable inhibitors or dimethyl sulfoxide (DMSO) vehicle before exposure to rhIGF1 for 18 h in the continuing presence of the inhibitors. Pharmacological agents included rhIGF1, the ATP-competitive GSK3α/β inhibitors SB415286 and SB216763 [27], the non-ATP-competitive GSK3α/β inhibitor TDZD-8 [28], adenosine dialdehyde (Adox) [29], an inhibitor of S-adenosylhomocysteine hydrolase and indirect inhibitor of S-adenosyl-methionine-dependent methylation reactions including trimethylation of lysine 27 of histone 3 (H3K27me3) by the polycomb repressive complex 2 (PRC2) member enhancer of zeste homolog 2 (EZH2) (Sigma-Aldrich, St. Louis, MO); Tyrphostin AG1024, a specific inhibitor of IGF1 and insulin RTK activity [30]; AKT Inhibitor X (10-(4'-(N-diethylamino)butyl)-2-chlorophenoxazine, HCl), an inhibitor of AKT phosphorylation, in-vitro kinase activity and IGF1-induced nuclear translocation [31]; rapamycin, inhibitor of mechanistic target of rapamycin (MTOR) complex 1 and ribosomal p70 S6 kinase (p70S6K) phosphorylation [20]; and PD98059, inhibitor of MEK1/2 ERK MAPK kinases [32] (EMD Chemicals, Inc., Gibbstown, NJ).
Cell cultures and antibody/drug treatment
Primary cell cultures prepared from human or murine gastric smooth musculature were maintained as described previously [26]. Stromal cells derived from the fetal hematopoietic microenvironment of Kitl-deficient Sl/Sl 4 mice, Sl/Sl 4 stromal cells genetically modified to express full-length murine Kitl (Sl/Sl 4-Kitl248) (generously donated by Dr. David Williams, Indiana University School of Medicine, Indianapolis, IN) [3], human LX-2 spontaneously immortalized hepatic stellate cells (kindly provided by Dr. Scott Freeman, Mount Sinai School of Medicine, New York, NY) [33], and human GIST-T1 cells derived from metastatic pleural tumor from a gastric GIST containing a heterozygous deletion of 57 bases in exon 11 juxtamembrane domain of KIT (contributed by Dr. Takahiro Taguchi) [34] were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Life Technologies, Carlsbad, CA) containing 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic (Gibco) at 37 °C in the presence of 5% CO2. GIST-T1-5R cells derived from GIST-T1 cells by prolonged in vitro exposure to imatinib and carrying a secondary, imatinib-resistant T670I mutation in exon 14 (contributed by Dr. Anu Gupta and Dr. Brian P. Rubin) were propagated in the presence of 1 µM imatinib mesylate (LC Laboratories, Woburn, MA). Imatinib was removed from the culture media 4 days prior to the experiments. GIST882 human cells from a primary GIST containing a homozygous KIT exon 13 missense mutation leading to K642E substitution in the first part of the split tyrosine kinase domain (contributed by Dr. Jonathan Fletcher) [35] were also cultured in high-glucose DMEM containing 10% FBS and 1% antibiotic-antimycotic but were maintained in a milieu of 4% O2, 5% CO2 and 91% N2. GIST48B KITlow/– human cells derived from GIST48 cells containing KIT exon 11 (homozygous V560D: imatinib-sensitive) and exon 17 phosphotransferase domain (heterozygous D820A: imatinib-resistant) mutations by prolonged heat shock protein 90 inhibition (contributed by Dr. Jonathan Fletcher) [36] were maintained with Iscove’s DMEM (high glucose) containing 15% FBS, 1% l-glutamine and 1% antibiotic-antimycotic (Gibco) at 37 °C in the presence of 5% CO2. LX-2, GIST-T1 and GIST882 cells have previously been demonstrated to express IGF1R [16,24]; results showing IGF1R α and β chain expression in GIST48B cells and IGF1 secretion by LX-2, GIST-T1, GIST882 and GIST48B cells are shown in Figure S1F-G. The role of endogenous KITLG in the proliferation of GIST-T1, GIST-T1-5R, GIST882, GIST48B and LX-2 cells was tested by culturing in the above media in the presence of purified, azide-free goat polyclonal anti-human KITLG antibody (AB-255-NA, R&D Systems, Minneapolis, MN; applied for 4 days at concentrations indicated in the Results), which has been shown to neutralize KITLG-induced proliferation in the TF1 human erythroleukemic cell line [37]. The specificity of KITLG immunoneutralization was verified by preabsorbing the anti-KITLG antibody with rhKITLG (R&D Systems) applied at 10:1 molar ratio overnight at 4 °C. Cell proliferation was assessed by the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) according to the manufacturer’s protocol. To examine the effects of directly modifying the epigenetic status of chromatin on KITLG expression, LX-2 and GIST-T1 cells were treated after 24-h serum starvation with the following specific, cell-permeable drugs or DMSO vehicle for 24-72 h at the concentrations indicated in the text: Adox, BIX-01294, a non-S-adenosyl-methionine analog-based inhibitor of the histone-lysine methyltransferases (HKMT) G9A (EHMT2) and GLP (EHMT1) and the H3K9me1/2 (histone H3 lysine 9 mono/ dimethylation) modification they catalyze [38,39]; chaetocin, an S-adenosyl-methionine-competitive inhibitor of the SUV39 family of H3K9 HKMTs including G9A, GLP and SUV39H1 [39,40]; garcinol, an inhibitor of histone acetyltransferases (HAT) p300 (EP300) and PCAF (KAT2B) (Sigma-Aldrich) [41]; and suberoylanilide hydroxamic acid (SAHA; vorinostat), a class I-II histone deacetylase (HDAC) inhibitor (Santa Cruz Biotechnology, Dallas, TX) [42].
RNA interference (RNAi)
RNAi against KITLG and heterochromatin protein 1 (HP1) homolog α (CBX5) was performed using Dharmacon ON-TARGETplus® SMARTpool® small interfering RNA (siRNA) or corresponding scrambled sequences (25 nM) and DharmaFECT 4 Transfection Reagent (Thermo, Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. HP1β (CBX1) and HP1γ (CBX3) were targeted with short hairpin RNAs (shRNAs) containing 19-mer antisense sequences (HP1β: GAAAGGGAGATGGGTAGCATC; HP1γ: GCAAATCAAAGAAGAAAAG). The sense-loop-antisense-terminator shRNA template inserts were cloned in under the RNA polymerase III H1 promoter in a bicistronic plasmid assembled to express green fluorescent protein from a CMV promoter. Plasmids were transformed into DH5a competent cells (Invitrogen, Carlsbad, CA), expanded and purified using a Plasmid Maxi Kit (QIAGEN, Germantown, MD). Plasmids (30 µg) were electroporated 3 times into LX-2 cells. Transfection efficacy was estimated after 24 h by fluorescence microscopy; cells were harvested after 72 h. Off-target effects were controlled for by transfecting cells with empty vectors.
Reverse transcription-polymerase chain reaction (RT-PCR). Kitl/KITLG transcription was monitored by real-time or traditional RT-PCR (see details including controls in ref [43].) using specific, intron-spanning primers published previously [22] or designed for this study (human KITLG exons 1-2: forward: TGCGCTCGGGCTACCCAATG; reverse: GCAGATCCCTTCAGTTTTGACGAGAG). Transcriptional quantification was obtained by the ΔΔCT method on a Bio-Rad Laboratories (Hercules, CA) CFX96 real-time PCR detector.
KITLG transcriptional activity
A human KITLG promoter-pGL3b luciferase construct was generated in a two-step process. First, a 1452-bp product of the 5' promoter region (-2120 bp to -669 bp) of KITLG was obtained by PCR amplification of human genomic DNA using specific primers. The 5' primer contained a KpnI restriction site at the 5' end for incorporation into the vector. The BglII site contained within the promoter region (-842 bp to -837 bp) was utilized as the 3' restriction site and the 1280-bp KpnI-BglII digestion product was ligated into the KpnI-BglII sites of the pGL3 basic vector (Promega). The 5' KITLG promoter sequence was confirmed and, subsequently, the vector was reopened at the BglII site. A 1280-bp product of the 3' KITLG promoter region (-873 bp to +407 bp) was amplified by PCR, utilizing human genomic DNA and specific primers with the 3' primer containing a BglII site at the 5' end for incorporation into the vector. Again, the BglII site contained within the promoter region was utilized this time as the 5' restriction site and the 1245 bp BglII-BglII digestion product was ligated into the BglII site of the 5' KITLG promoter (-2120 bp to -837 bp)-pGL3b construct. Orientation and the entire promoter sequence was confirmed by sequencing to obtain the full length KITLG promoter (-2120 bp to +407 bp)-pGL3b luciferase construct. Transfection of LX-2 and GIST-T1 cells, as well as primary cell cultures prepared from murine gastric smooth muscles was performed as described above. Cells were harvested, lysed and assayed for luciferase activity 48 h after transfection using the Promega Luciferase Assay System.
Western immunoblotting
Tissue and cell lysates were prepared and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotting as described previously [44] (see antibodies in Table S1). Bound antibodies were visualized using an Odyssey Infrared Imaging System (LI-COR Bioscience, Lincoln, NE) and Bio-Rad Quantity One 4.5.1 software. Protein and phosphoprotein bands of interest were expressed in densitometric units normalized to the loading control (glyceraldehyde-3-phosphate dehydrogenase; Gapdh/GAPDH or β-actin; Actb) and the corresponding total protein, respectively, detected simultaneously in the same sample.
Chromatin immunoprecipitation (ChIP)
DNA–protein complexes from juvenile murine gastric smooth muscle were cross-linked using fresh 1% formaldehyde (Thermo, Fisher) for 10 minutes, followed by glycine quenching. DNA-protein complexes were sheared on cold water using a Bioruptor sonicator (Diagenode, Denville, NJ) at settings optimized for obtaining DNA fragments ranging from ~150 to 600 bp. Histone and chromatin-binding proteins were purified and immunoprecipitated overnight at 4 °C along with bound DNA fragments using reagents and magnetic beads from a EZ-Magna ChIP™ G Chromatin Immunoprecipitation Kit (Millipore, Billerica, MA) and ChIP-grade antibodies against EZH2, H3K27me3, H3K4me2 (dimethylated lysine 4 of histone 3), H3K9me2, H3K9me3, H3K9ac (acetylated lysine 9 of histone 3) and H4ac (acetylated histone 4) (Table S2). The ability of these antibodies to enrich target DNA was verified by PCR and antibodies against RNA polymerase II (positive control), Gapdh (negative control) and “non-immune” mouse IgG (negative control). Bound DNA fragments were isolated by proteinase K digestion for 2 hours at 62 °C, and magnetic beads were dissociated by incubation at 95 °C for 10 minutes. Immunoprecipitated DNA fragments were analyzed by quantitative real-time PCR using primers designed to target the mouse Kitl promoter (-300 bp to -214 bp): forward: GCTGGTGAGCTTGCTGCGGA; reverse: TGAGGCACCGGGAGTCTCGG. PCR results were quantified by the ΔΔCT method using input DNA as reference and the vehicle (DMSO)-treated samples as control.
Statistical analyses
Each data point (n) represents one freshly isolated or cultured stomach or a biological replicate experiment in cultured cells. Data are expressed as mean ± standard error of the mean (SEM) or median and interquartile range. Student’s t test or Mann-Whitney rank sum test were used for comparing two data sets. Three groups or more were compared by one-way analysis of variance (ANOVA) or ANOVA on ranks followed by multiple comparisons. A probability value of P<0.05 was used as a cut-off for statistical significance in all statistical procedures. IC50 values were obtained by nonlinear curve fitting applied to dose-response data using the equation library in SigmaPlot 10.0 (Systat Software, Chicago, IL).
Results
Kitl/KITLG protein is expressed in IGF target cells and tissues
We investigated Kitl/KITLG protein expression in IGF1R-expressing gastrointestinal smooth muscles [18,20,22], LX-2 hepatic stellate cells [24] and GIST cell lines (see ref [16]. and Figure S1F). In murine gastric muscles, Kitl protein could be readily detected by Western immunoblotting as a ~31-kDa band (Figure S1A) corresponding to uncleaved, cell-associated Kitl220 and secreted Kitl produced from the Kitl248 isoform [3]. Occasionally, we also detected a 43-kDa band likely representing residual, uncleaved Kitl248 and a 21-kDa band, which corresponds to secreted Kitl produced from Kitl220 [3]. However, the low abundance of these minor peptides did not allow quantification. In GIST and LX-2 cells, only the 31-kDa KITLG band was detected (Figure S1B). We validated our method in Sl/Sl 4 fibroblasts lacking full-length Kitl and in Sl/Sl 4 cells engineered to stably express Kitl248 (Figure S1C) or Kitl220 (not shown) [3]. KITLG expression was similar in KIT+ GIST-T1 cells expressing WT KIT, in KIT+ GIST882 and KITlow/– GIST48B cells lacking a WT KIT allele, and in KIT- LX-2 cells (Figure 1D-E). These results demonstrate Kitl/KITLG expression in all our models.
Figure 1. Contribution of KITLG-activated KIT signaling to baseline proliferation of GIST and LX-2 cells.

A, KIT Y721 phosphorylation was activated by exogenous rhKITLG (100 ng/mL for 10 min following 2 h serum deprivation [13,35]) in GIST-T1 cells containing a heterozygous activating KIT mutation (n=4/group) and in GIST-T1-5R cells containing an additional, imatinib-resistant KIT mutation (n=6/group), but not in GIST882 cells lacking a WT KIT allele (n=5/group). B, Culturing with anti-human KITLG neutralizing antibody for 4 days inhibited baseline proliferation of GIST-T1 cells (P<0.001; n=3; regression and 95% confidence band are shown as solid and dashed lines, respectively) and GIST-T1-5R cells (P<0.001; n=6). The effect of KITLG immunoneutralization on GIST-T1 cells was blocked by preabsorbing the antibody with 10-fold molar excess of rhKITLG (see open symbols in the left panel, second row; n=3/group). KITLG immunoneutralization did not inhibit the proliferation of GIST882 or GIST48B cells lacking a WT KIT allele (note that GIST48B cells also express very low to undetectable levels of KIT protein, see Figure S1E) and of LX-2 cells lacking KIT protein (Figure S1E) (n=3/cell line). C, Inhibition of the proliferation of GIST-T1 and GIST-T1-5R cells by siRNA-mediated knock-down of KITLG (n=4/cell line/group; P GIST-T1: day 2: 0.008, days 4, 6 and 8: <0.001; P GIST-T1-5R: day 2: <0.02, day 4: 0.004, days 6 and 8: <0.001).
Endogenous KITLG stimulates the proliferation of GIST cells expressing WT KIT
To investigate the role of autocrine/paracrine KITLG–KIT signaling in GIST proliferation, we examined the effect of KITLG immunoneutralization and RNAi-mediated knock-down in GIST cells expressing or lacking WT KIT. Control experiments verified the induction of KIT phosphorylation on Y721, the docking site for the p85 subunit of PI3K [1], by exogenous KITLG in GIST-T1 cells containing a heterozygous activating KIT mutation (Figure 1A; see reagent concentrations, exposure times, replicate numbers and other statistical details in the figures and their legends). KIT phosphorylation was also increased, albeit to a lesser degree, in GIST-T1-5R cells, derivatives of GIST-T1 cells containing an additional, imatinib-resistant KIT mutation. In contrast, no ligand-dependent KIT phosphorylation was detected in GIST882 cells, which are homozygous for the activating KIT K642E mutation. Culturing with anti-human KITLG neutralizing antibody [37] for 4 days inhibited the proliferation of GIST-T1 cells by ~40% (IC50: ~9 ng/mL) and GIST-T1-5R cells by ~34% (IC50: ~26 ng/mL) (Figure 1B), whereas the same treatment had no effect on KIT+ GIST882 and KITlow/– GIST48B cells lacking WT KIT allele or on KIT- LX-2 cells. The effect of KITLG immunoneutralization on GIST-T1 cells was prevented by preabsorbing the antibody with 10-fold molar excess of rhKITLG (Figure 1B). The proliferation of both GIST-T1 and GIST-T1-5R cells could also be inhibited by RNAi targeting KITLG (Figure 1C). These findings provide direct evidence that activation of KIT signaling by endogenous KITLG contributes to the proliferation of GIST cells expressing WT receptors.
IGF1 stimulates Kitl protein expression
To investigate the direct effects of IGF1 on Kitl expression, we first administered 150 µg/kg LR3-rhIGF1, a potent IGF1 analog with reduced affinity for IGF-binding proteins, to 14-16-day-old BALB/c mice in a single i.p. injection and measured Kitl protein in the gastric tunica muscularis by Western blotting. Kitl expression increased in a time-dependent manner, with maximum effect occurring at 24 h (Figure 2A). In short-term cultures of intact gastric corpus+antrum muscles from 14-16-day-old BALB/c mice, 100 ng/mL rhIGF1 caused maximum stimulation of Kitl protein between 12 and 24 h (Figure 2B). This effect was dose-dependent, plateauing between 100 and 300 ng/mL (Figure 2C). The upregulation of Kitl expression seen in response to 100 ng/mL rhIGF1 applied for 18 h was also verified using Actb as loading control (Figure S2). Tyrphostin AG1024, a specific inhibitor of IGF1R and insulin RTK activity [30], reduced the rhIGF1 effect on Kitl (Figure 2D) indicating that it was likely mediated by IGF1R or insulin receptor/IGF1R heterodimers [15].
Figure 2. Kitl protein expression is stimulated by IGF1 via multiple IGF1R-activated pathways.

A, Time-dependent stimulation of Kitl expression in the gastric tunica muscularis of 14-16-day-old BALB/c mice by the potent IGF1 analog LR3-rhIGF1 administered in a single 150 µg/kg dose i.p.; n=5 mice/group. B, Time-dependent stimulation of Kitl expression by 100 ng/mL rhIGF1 in gastric corpus+antrum tunica muscularis organotypic cultures from 14-16-day-old BALB/c mice; n=4-5/group. C, Concentration-dependent stimulation of Kitl protein expression by 18-h rhIGF1 treatment; n=7 organotypic cultures/group. D, Blockade of the rhIGF1-induced Kitl expression in organotypic cultures by AG1024 (1 µM), a specific inhibitor of IGF1/insulin receptor tyrosine kinase activity; n=4/group. E-F, Effects of 1-h (E) and 3-h (F) rhIGF1treatment on AKT (S473/S474/S472), GSK3β (S9), p70S6K (T389) and ERK1/2 (T202/Y204 and T185/Y187) phosphorylation; n=5 organotypic cultures/group. G, Blockade of rhIGF1-induced Kitl expression in organotypic cultures by specific inhibitors of AKT (AKT Inhibitor X; 150 µM), MTOR–p70S6K (rapamycin; 1.5 nM) and MEK–ERK (PD98059; 50 µM); n=5-16/group. The GSK-3 inhibitor SB415286 (30 µM), which is expected to mimic, rather than inhibit, the effect of the IGF1-induced inhibitory phosphorylation of GSK-3, had no significant effect. Kitl and Gapdh or total (T) and phosphorylated proteins (P) were simultaneously detected in the same samples by two-color immunofluorescence (Figure S1). Representative immunoblots show identical areas of the blots imaged at different wavelengths. Box plots show medians and interquartile ranges; bar graphs indicate means±SEM. Data were normalized to the control groups indicated in the panels (dashed lines). Groups marked by asterisk are different from the control group, and groups not sharing the same superscript letter are different from each other (P<0.05 by post-hoc multiple comparisons). IGF1 stimulated Kitl expression in gastric smooth muscles in vivo and in vitro in a time- and concentration-dependent manner by activating IGFR1 and the AKT–GSK3, MTOR–p70S6K and ERK MAPK pathways.
Multiple signaling pathways mediate IGF1-induced Kitl protein expression
To investigate the mechanisms of IGF1-induced Kitl expression, we first detected the phosphorylation of key IGF1 signaling intermediates [19,20] in murine gastric muscles stimulated by exogenous IGF1 (Figure 2E-F). rhIGF1 elicited time-dependent increase in the phosphorylation of AKT, GSK3β, p70S6K and ERK1/2: After 1 h, we detected increased activating phosphorylation on S473/S474/S472 of AKT isoforms, p70S6K phosphorylation on T389 [19], elevated ERK1 and ERK2 phosphorylation on T202/Y204 and T185/Y187, respectively, and increased inhibitory phosphorylation on S9 of GSK3β [45] (Figure 2E). After 3 h, ERK1/2 phosphorylation returned to baseline but phosphorylated AKT, p70S6K and GSK3β remained elevated (Figure 2F). We then probed the contribution of these intermediates to IGF1-induced Kitl expression by using pathway-specific inhibitors (Figure 2G): rhIGF1-stimulated Kitl expression was reduced by AKT Inhibitor X [31]; by rapamycin, which selectively inhibits the activation of p70S6K by MTOR complex 1 [19]; and by PD98059, a selective MEK inhibitor [32]. SB415286, a selective competitive inhibitor of ATP binding to GSK3α/β and functional mimic of AKT-mediated GSK-3 phosphorylation and inactivation [27], only minimally increased Kitl expression beyond the near-maximal stimulation caused by IGF1. These results indicate cooperation among several major IGF1 intermediate pathways in stimulation of Kitl expression.
GSK-3 inhibition (GSK3i) stimulates Kitl expression without directly activating pathways involved in cell growth and proliferation
To better understand the role of GSK3i in the stimulation of Kitl expression, we evaluated the effects of SB415286 in the absence of exogenous IGF1 in isolated murine gastric muscles. Under this condition, SB415286 stimulated Kitl expression (Figure 3A). Despite mimicking the effect of IGF1 on Kitl, SB415286 did not stimulate the expression of cyclin D1, a key mediator of IGF1-induced cell cycle progression [15] (Figure 3B), or the phosphorylation of p70S6K and ERK1/2 (Figure 3C) at time points when IGF1 effects on these parameters were prominent. We obtained similar results with SB216763, another ATP-competitive GSK3α/β inhibitor and TDZD-8, a non-ATP-competitive GSK3α/β inhibitor (Figures S3 and S4). Thus, GSK3i alone specifically stimulates Kitl expression offering a pharmacological approach to increase Kitl/KITLG levels in gastrointestinal muscles without reproducing IGF1’s actions promoting cellular stress, aging and cancer [14,15].
Figure 3. GSK3i stimulates Kitl expression without activating cyclin D1 expression and p70S6K and ERK1/2 phosphorylation.

The GSK3α/β inhibitor SB415286 was applied to organotypic cultures of gastric corpus+antrum muscles from of 14-16-day-old BALB/c mice at 30 µM. A, Effect of 18-h application of SB415286 on Kitl expression; n=5/group. B, Effect of the same treatment on cyclin D1 expression; n=3/group. C, Effects of 1-h exposure to SB415286 on p70S6K and ERK1/2 phosphorylation; n=3/group. See Figure 2 for further details.
IGF1 and GSK3i activate Kitl/KITLG transcription
Next, we investigated the role of gene transcription in the IGF1 and GSK3i effects. In primary human smooth muscle cells (Figure 4A), KITLG mRNA was expressed in a serum-dependent fashion (Figure 4B). In intact murine smooth muscles, 12-h exposure to rhIGF1 or SB415286 upregulated Kitl mRNA (Figure 4C). In primary murine smooth muscle cells, SB415286 stimulated endogenous Kitl expression (Figure 4D) and activated transcription from an episomally expressed human KITLG promoter- luciferase construct (Figure 4E). In LX-2 and GIST-T1 cells, both rhIGF1 and SB415286 increased KITLG mRNA (Figure 4F, H), although SB415286 was more effective in LX-2 cells and IGF1 had a greater effect in GIST-T1 cells. KITLG promoter activity was also increased by both IGF1 and SB415286 (Figure 4G, I). In both cell lines, IGF1 displayed more rapid action on KITLG promoter activity than SB415286. Nevertheless, these results indicate that the stimulation of Kitl protein expression by IGF1 and GSK3i primarily occurs at the transcriptional level and led us to investigate the nature of chromatin remodeling events that account for this effect.
Figure 4. IGF1 and GSK3i stimulate Kitl/KITLG transcription.

Results obtained in murine and human smooth muscle cells (A-E), human LX-2 stellate cells (F-G) and human GIST-T1 cells (H-I) are shown. A, Hoffman modulation contrast image of primary human gastric smooth muscle cells. B, KITLG mRNA (total: soluble+membrane-bound) was readily detectable in primary human smooth muscle cells (passage 3) maintained with Smooth Muscle Growth Medium-2 containing insulin, hFGF-B, hEGF and 5% FBS (Lonza) but not in 24-h growth factor- and serum-deficient basal medium. C, Both IGF1 (100 ng/mL) and the GSK3α/β inhibitor SB415286 (30 µM) stimulated Kitl expression in murine gastric tunica muscularis organotypic cultures (n=3/group). D-E, SB415286 stimulated endogenous Kitl expression in murine primary gastric smooth muscle cells (D; n=3/group) and KITLG transcriptional activity in the same cell type transfected with a KITLG promoter (-2120 bp to +407 bp)-pGL3 luciferase construct (E; n=3/group). IGF1 (100 ng/mL; n=3/group) and SB415286 (30 µM; n=6/group) also increased endogenous KITLG mRNA expression in LX-2 (F) and GIST-T1 cells (H) and stimulated KITLG promoter activity in a time-dependent fashion (LX-2: n=6-9/group; G; GIST-T1: n=3-11/group; I). Groups marked by asterisk are different from the control group, and groups not sharing the same superscript letter are different from each other (P<0.05 by post-hoc multiple comparisons).
Coordinated chromatin modifications underlie the activation of Kitl/KITLG transcription by IGF1 and GSK3i
We performed ChIP-PCR targeting the core Kitl promoter in murine gastric smooth muscles exposed for 6 h to rhIGF1 or SB415286 using antibodies against several activating and repressive histone marks and the polycomb group (PcG) HKMT EZH2. Control experiments using “non-immune” mouse IgG and antibodies against the functionally irrelevant Gapdh protein revealed little recovery of Kitl promoter DNA and no variation among treatments (Figure S5). The biological role of the enzymes and other proteins involved in the establishment of the targeted modifications was then validated with the aid of pharmacological inhibitors and RNAi in LX-2 and GIST-T1 cells using KITLG expression as readout. We first studied the role of histone acetylation, which is almost invariably associated with transcriptional activation [46] (Figure 5). In murine gastric smooth muscles, both rhIGF-I and SB415286 increased the occupancy of the Kitl promoter by H4ac (which may include acetylated K5, K8, K12 and K18 [47]) (Figure 5A), whereas increased occupancy by H3K9ac was only detected in response to GSK3i (Figure 5B). Consistent with these findings, inhibition of HDAC classes I-II with SAHA led to a dose-dependent increase in KITLG mRNA in LX-2 cells (Figure 5C). The role of increased H3K9 acetylation in the GSK3i effect was supported by the dose-dependent reduction of SB415286-induced increase in KITLG mRNA by the p300/PCAF HAT inhibitor garcinol (Figure 5D). In GIST-T1 cells, the transcriptional effects of HDAC inhibition could not be determined due to rapid cell death likely reflecting the apoptotic effects of acetylation and consequent loss of function of the KIT chaperone heat shock protein 90 [48].
Figure 5. Role of histone acetylation in the activation of Kitl/KITLG transcription by IGF1 and GSK3i.
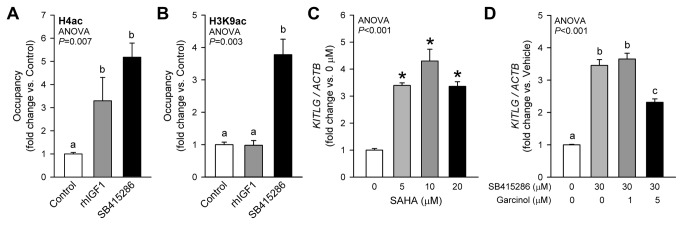
A, Increased occupancy of the Kitl core promoter region by H4ac in response to 6-h rhIGF1 (100 ng/mL) and SB415286 (30 µM) treatment in murine gastric smooth muscles. Only SB415286 increased occupancy by H3K9ac (B). Representatives of two independent experiments, each performed in triplicates, are shown. C, Dose-dependent stimulation of KITLG expression in LX-2 cells by 24-h treatment with the class I-II HDAC inhibitor SAHA (n=3/group). D, Dose-dependent inhibition of the SB415286-induced stimulation of KITLG expression by the p300/PCAF HAT inhibitor garcinol (24 h) in LX-2 cells (n=3/group). Drugs were applied following 24-h serum deprivation. Groups marked by asterisk are different from the control group, and groups not sharing the same superscript letter are different from each other (P<0.05 by post-hoc multiple comparisons).
Next, we examined whether histone methylation, a biochemical mechanism associated with long-term transcriptional memory, also contributes to the regulation of Kitl/KITLG expression. First we studied the activating H3K4me2 mark, which binds enhancer and promoter regions and gene bodies of actively transcribed or transcriptionally poised but repressed, tissue-specific genes [49]. Whereas rhIGF1 increased the level of H3K4me2 on the Kitl promoter ~2.8-fold, SB415286 only had a modest effect (Figure 6A). Since transcriptional activation mediated by H3K4 methylation is often antagonized by PcG complexes [50,51], we also measured the association with the Kitl promoter of the PcG HKMT EZH2 and the repressive H3K27me3 modification it catalyzes [50]. These experiments showed that rhIGF1 and SB415286 reduced the presence of both EZH2 and the H3K27me3 mark on the Kitl promoter (Figure 6B-C). To mechanistically explore the functional impact of the PcG-mediated repression on Kitl/KITLG expression, we inhibited EZH2 activity with the indirect HKMT inhibitor Adox [29]. Adox increased KITLG mRNA in GIST-T1 cells ~2.9-fold (Figure 6F) but had more modest, albeit statistically significant, effects in murine gastric smooth muscles and LX-2 cells (Figure 6D-E). These results indicate that PcG inhibits Kitl/KITLG expression under basal conditions.
Figure 6. Role of trithorax- and polycomb-mediated histone modifications in IGF1- and GSK3i-induced activation of Kitl/KITLG transcription.

A, Increased occupancy of the Kitl core promoter by the trithorax group-mediated, activating H3K4me2 histone modification in response to 6-h rhIGF1 (100 ng/mL) and SB415286 (30 µM) treatment in murine gastric smooth muscles. B-C, Reduced occupancy of the Kitl core promoter by the PRC2-mediated, repressive H3K27me3 histone modification (B) and by the PRC2 histone methyltransferase EZH2 (C) in response to rhIGF1 and SB415286 in the same tissues. D-F, Stimulation of KITLG expression by the indirect histone methyltransferase inhibitor Adox in murine gastric smooth muscles (D), LX-2 cells (E) and GIST-T1 cells (F) (n=3/group). Adox was applied for 72 h following 24-h serum deprivation. See further details in the legend to Figure 5.
We also studied the involvement H3K9 methylation in the regulation of Kitl/KITLG transcription. The best understood function of H3K9me2/3 is stable gene silencing through heterochromatin formation [52]. This process requires the binding of HP1 proteins and consequent recruitment of the H3K9me1/2 HKMTs G9A and GLP and the H3K9me3 HKMT SUV39H1, which reinforce the silencing of genes located within the constitutive heterochomatin and initiate the repression of genes previously embedded in euchromatin [53]. In murine gastric muscles, we detected both H3K9me2 and H3K9me3 on the Kitl promoter (Figure 7A-B), and both marks were reduced by rhIGF1. GSK3i had less pronounced effects. In LX-2 cells, KITLG mRNA increased in response to RNAi targeting HP1γ (CBX3) but was unaffected by HP1α (CBX5) or HP1β (CBX1) knockdown (Figure 7C-D). These findings are consistent with HP1γ’s role in euchromatic silencing [53]. In response to the G9A/GLP inhibitor BIX-01294 we also detected a dose-dependent increase in KITLG mRNA in LX-2 cells, and a ~100-fold increase in GIST-T1 cells (Figure 7E-F). In LX-2 cells, KITLG expression was also dose-dependently stimulated by chaetocin, a specific inhibitor of SUV39H1 (Figure 7G). The transcriptional effects of chaetocin could not be assessed in GIST-T1 cells due severe cytotoxicity. These results reveal an important role for HP1γ and H3K9me2/3 in IGF1-mediated Kitl/KITLG expression.
Figure 7. Role of reduced H3K9 methylation in the activation of Kitl/KITLG transcription by IGF1 and GSK3i.

A-B, Reduced occupancy of the Kitl core promoter by the repressive H3K9me2 (A) and H3K9me3 (B) histone modifications in response to 6-h rhIGF1 (100 ng/mL) and SB415286 (30 µM) treatment in murine gastric smooth muscles. C-D, Probing the role of HP1 isoforms in transcriptional repression of KITLG in LX-2 cells by siRNA- (CBX5: HP1α; 25 nM, 72 h; C) or shRNA-mediated knock-down (CBX1: HP1β; CBX3: HP1γ; 30 µg plasmid, 72 h; D). Note activation of KITLG expression by shRNA-mediated knock-down of HP1γ (n=3/group). E-F, Stimulation of KITLG expression by the G9A/GLP H3K9me1/2 HKMT inhibitor BIX-01294 in LX-2 cells (E) and GIST-T1 cells (F) (n=3/group). G, Stimulation of KITLG expression by the H3K9 HKMT inhibitor chaetocin in LX-2 cells (n=3/group). Drugs were applied for 24 h following 24-h serum deprivation. See further details in the legend to Figure 5.
In summary, our data support the model that IGF1 promotes autocrine/paracrine Kitl/KITLG–KIT signaling in part via AKT-mediated GSK3i and coordinated chromatin modifications favoring increased Kitl/KITLG transcription, and that these effects can be uncoupled from the direct mitogenic actions of IGF1. To allow Kitl/KITLG expression, IGF target cells must increase the levels of activating H4ac, H3K9ac and H3K4me2 marks and decrease the levels of the repressive marks H3K27me3 and H3K9me2/3. While IGF1 and GSK3i appeared to exert their effects via the same general mechanisms, they also showed differences in their actions. The results presented herein outline a novel RTK cross-talk that regulates ligand-induced KIT activation in both physiologic/developmental and oncogenic contexts.
Discussion
Adequate stimulation of the KIT receptor by its ligand Kitl/KITLG is required for diverse developmental and physiological processes such as hematopoiesis, pigmentation, gametogenesis, spatial learning, cardiomyocyte differentiation and repair, vasculogenesis and angiogenesis, lung function and gastrointestinal motility [1,2]. In contrast, ligand-independent activation of KIT due to oncogenic mutations is associated with several types of cancer such as GIST, seminomas, acute myeloid leukemia, melanomas and systemic mastocytosis [1,6]. However, abnormal ligand-dependent signaling occurring in the absence of mutations and reflecting altered expression of KIT and/or Kitl/KITLG contributes to the pathogenesis of not only non-neoplastic disorders such as gastrointestinal dysmotilities and allergies [1,21,22] but also various cancers including subsets of GIST, acute myeloid leukemia, small-cell lung carcinoma, breast and colorectal cancer, ovarian cancer and neuroblastoma [1,9,11-13]. Therefore, understanding the factors and mechanisms regulating Kitl/KITLG expression will likely provide novel therapeutic tools for both functional disorders and cancers. We have previously shown that long-term IGF1 treatment of intact murine gastric tunica muscularis increases Kitl availability in part by stimulating the growth and survival of smooth muscle cells, the primary source of Kitl in this tissue [22]. Here we demonstrate that IGF1 directly stimulates Kitl/KITLG expression in gastrointestinal smooth muscles, the natural microenvironment for ICC and GIST [8,21,22], in GIST cells and in LX-2 cells derived from hepatic stellate cells [33], the presumed niche for KIT+ hepatic progenitors [54], inflammatory cells [55] and hepatocellular carcinoma [25].
Our study outlines a transcriptionally mediated autocrine/paracrine loop between two distinct RTK signaling systems, the Kitl/KITLG–KIT and IGF–IGF1R systems. Locally produced and circulating IGF1 affects almost all tissues and plays key roles in the regulation of body size, skeletal acquisition, muscle mass, reproduction, metabolism and life/health span [56]. There is also a strong positive correlation between circulating IGF levels and cancer risk and prognosis [14,15]. IGF1R signaling facilitates cell cycle progression mainly at the G1-S transition reflecting increased cyclin D1 transcription and translation stimulated via the ERK MAPK and AKT–MTOR–p70S6K pathways, respectively [15]. As our data show, increased IGF1R activation (in part from autocrine IGF1; Figure S1G) can, in turn, increase Kitl/KITLG expression and thus potentially amplify its oncogenic potential in KIT-expressing cells and tissues. For example, in WT GIST, an IGF2-IGF1R autocrine/paracrine loop sustained by overexpression of both the receptor and its ligand in the same tumor microenvironment [5,16,17,57,58] may activate a secondary autocrine/paracrine loop formed by KITLG and WT KIT leading to increased KIT phosphorylation (refs. 9,10,13 and present results) and cell proliferation. Indeed, our data in the heterozygous KIT mutant GIST-T1 cells indicate that autocrine/paracrine KITLG-mediated KIT signaling may account for ~40% of baseline proliferation even in cells with a secondary, imatinib-resistant mutation and can be inhibited with antibodies or RNAi targeting KITLG. Since imatinib preferentially targets mutant receptors [6,9], inhibiting the IGF–IGF1R and KITLG–KIT coupled autocrine/paracrine loops may be beneficial in GIST, which are in most cases heterozygous for a given KIT mutation and continue expressing WT KIT protein [5]. However, the contribution of this pathway to GIST growth in vivo remains to be established. Interestingly, even in GIST lacking WT KIT, anti-KIT antibodies inhibited tumor growth with an efficacy similar to what we observed in GIST-T1 cells following KITLG immunoneutralization, although the former effect was attributed to increased phagocytosis and stimulation of KIT degradation rather than prevention of KITLG binding [59]. In hepatic stellate cells, KITLG expression stimulated by IGF1R signaling may facilitate the KIT-dependent recruitment of inflammatory cells in injury and fibrosis [55] and progenitor cells in hepatic failure [54], and may thus be associated with both disease progression and tissue regeneration. In gastrointestinal neuromuscular tissues, stimulation of Kitl/KITLG–KIT signaling could prevent or counter ICC loss associated with several motility disorders and conditions including diabetic gastroparesis [22,60] and aging [43,61], where reduced Kitl/KITLG expression is a pathogenetic factor [22,43]. Since ICC differentiation cannot be supported solely by supplying free Kitl/KITLG [2,21], pharmacological interventions will likely require stimulation of local production of both soluble and membrane-associated Kitl/KITLG, which, as our data show, could be accomplished by IGF1 administration or GSK3i. In summary, the crosstalk between the IGF and the Kitl/KITLG pathways revealed by our results is potentially important for understanding and controlling the mechanisms of cell proliferation in both neoplastic and functional gastrointestinal diseases such as GIST, liver disease and gastroparesis. Therefore, it is also important to discuss the information we have obtained on the pathway mediating signal transduction from the IGF1R to the nucleus to regulate Kitl/KITLG expression.
Our initial studies focused on the intracellular kinases that transduce signals downstream of IGF1R. In our models, Kitl/KITLG expression could also be stimulated by inhibiting GSK-3 activity with both ATP-competitive and non-ATP-competitive inhibitors. These results were unexpected because IGF1 effects on Kitl appeared to involve overlapping actions of several major pathways including the ERK MAPK, AKT and MTOR–p70S6K pathways, whereas GSK3i stimulated Kitl expression without increasing ERK1/2 or p70S6K phosphorylation and cyclin D1 expression at time points when IGF1 effects on these parameters were prominent. The potential translational significance of these findings is that GSK3i may allow the stimulation of KITLG expression e.g. in patients with ICC loss without concomitant activation of the pathways that mediate IGF1’s actions promoting cellular stress, aging and cancer [14,15]. While GSK3i may affect ERK1/2 and MTOR–p70S6K at other time points, our results agree with Kuemmerle’s finding that in gastrointestinal smooth muscle cells, the AKT–GSK3i-mediated and p70S6K- and ERK1/2-dependent mechanisms are functionally segregated, with AKT-dependent signaling being involved only in anti-apoptotic but not in the proliferation-stimulating actions of IGF1 [19,20]. Indeed, despite mediating the β-catenin-stabilizing effect of WNT signaling, long-term GSK3i has been associated with reduced, rather than increased, cancer risk possibly due to the concomitant activation of forkhead transcription factors [28], and numerous GSK-3 inhibitors are under investigation for their beneficial effects in diabetes, inflammation, central nervous system injuries, bipolar disorder, Alzheimer’s disease and cancer [45].
Subsequently, we investigated how the IGF1- and GSK3i-induced signaling activates nuclear events to bring about increased Kitl/KITLG expression. By measuring Kitl/KITLG transcript levels and KITLG promoter activity we found that Kitl/KITLG protein expression induced by IGF1 or GSK3i indeed reflected increased gene transcription. However, our data also revealed differences in the time-courses of the IGF1 and GSK3i effects along with preferential stimulation by IGF1 and GSK3i in GIST-T1 and LX-2 cells, respectively. Therefore, we studied the role of several repressive and activating histone modifications, as well as the enzymes and regulatory factors responsible for their establishment by ChIP and pharmacologic and RNAi-mediated interventions. In selecting our ChIP targets we considered that both the mouse and human Kitl/KITLG promoter contain a CpG island but are unlikely to be DNA-methylated, given that we detected significant RNA polymerase II occupancy, baseline expression and transcriptional activity in our models. We found that activation of Kitl expression in murine gastric smooth muscles by both IGF1 and GSK3i elicited coordinated changes in the chromatin involving reduced and increased occupancy of the Kitl promoter by repressive and activating histone marks, respectively. These events involved the activating histone marks H4ac, H3K9ac, H3K4me2, the repressive marks H3K9me2/3 and H3K27me3, as well as related enzymes and regulators such as EZH2, G9A, GLP, SUV39H1, PCAF, HDACs and HP1γ. Our data also indicated some differences between IGF1 and GSK3i-induced changes such as preference for H3K4me2- and H3K9ac-mediated activation by IGF1 and GSK3i, respectively. The complexity of this system raises the question how these changes are orchestrated. There is strong evidence that the process of transcription itself regulates chromatin states [52], and sequence-specific transcription factors, besides transiently modulating transcription, contribute to the recruitment of chromatin modifiers [62]. For example, transcription factors activated in response to IGF1 signaling or GSK3i may bind to the promoter and initiate the recruitment and eviction of the chromatin modifying complexes that establish the activating and repressive marks, respectively [63]. IGF1 may activate transcription via ERK and p70S6K signaling [15], by promoting IGF1R β subunit nuclear translocation and association with lymphoid enhancer-binding factor 1 [23] and via GSK3i. Physiological or pharmacological GSK3i can in turn lead to the loss of inhibitory phosphorylation of several transcription factors in a stimulus-, promoter- and cellular context-dependent manner [45]. Indeed, some of these GSK-3-inhibited transcription factors have putative or verified binding sites in the promoter region of the KITLG gene [1]. Identification of the key transcription factors mediating the observed IGF1 and GSK3i effects on Kitl/KITLG expression is a requisite next step toward understanding the transcriptional mechanisms that couple IGF1-IGF1R and Kitl/KITLG–KIT RTK signaling in physiologic and oncogenic contexts.
Supporting Information
Kitl/KITLG and IGF1R protein expression and IGF1 secretion in murine gastric smooth muscles, human LX-2 stellate cells and GIST cells. A, Detection of Kitl in the lysate of gastric corpus+antrum muscles from a juvenile BALB/c mouse. The membrane was simultaneously probed with antibodies against Kitl and Gapdh (loading control) and appropriate fluorescent secondary antibodies. Note primary Kitl band at ~31 kDa and a weaker band at ~43 kDa. The 21-kDa Kitl band was only borderline detectable. B, In human GIST cell lines, only the 31-kDa KITLG band was detected. B, Validation of the Western immunoblotting method. The 31-kDa Kitl band was also detected in lysates of Sl/Sl 4 hematopoietic stromal cells expressing mKitl248. No Kitl bands were detected in Sl/Sl 4 stromal cells lacking full-length Kitl. D, Expression of KITLG protein in LX-2 human hepatic stellate cells and three human GIST cell lines. E, Expression of KIT protein in the same cell lines. The level reported for LX-2 cells represents background fluorescence as no specific band was detected. Note lack of correlation between KITLG and KIT expression. F, IGF1R α and β chain expression in GIST48B cells. G, IGF1 secretion into culture media by GIST-T1, GIST882, GIST48B and LX-2 cells. 3000 cells/well were plated into 96-well plates and cultured in the presence or absence of FBS (GIST-T1, GIST882, LX-2: 10%; GIST48B: 15%) for 48 h. IGF1 was measured in the harvested cell-free media using the Quantikine Human IGF1 Immunoassay kit (DG100, R&D systems). Note differential responses to FBS.
(TIF)
Kitl protein expression is stimulated by IGF1. Stimulation of Kitl expression, detected using Actb as reference, by 18-h treatment with 100 ng/mL rhIGF1 in gastric corpus+antrum tunica muscularis organotypic cultures from 14-16-day-old BALB/c mice (n=3/group). Kitl and Actb were simultaneously detected in the same samples by two-color immunofluorescence. Representative immunoblots show identical areas of the blots imaged at different wavelengths. The degree of Kitl upregulation was statistically indistinguishable from the increase detected using Gapdh as loading control (1.89±0.21-fold vs. 2.07±0.15-fold, P=0.583).
(TIF)
SB216763 stimulates Kitl expression without activating cyclin D1 expression and p70S6K and ERK1/2 phosphorylation. GSK3α/β inhibitor SB216763 was applied to organotypic cultures of gastric corpus+antrum muscles from of 14-16-day-old BALB/c mice at 3 µM. A, Effect of 18-h application of SB216763 on Kitl expression; n=5/group. B, Effect of the same treatment on cyclin D1 expression; n=3/group. C, Effects of 1-h exposure to SB216763 on p70S6K and ERK1/2 phosphorylation; n=3/group. See Figure 2 for further details.
(TIF)
TDZD-8 stimulates Kitl expression without activating cyclin D1 expression and p70S6K and ERK1/2 phosphorylation. The non-ATP-competitive GSK3α/β inhibitor TDZD-8 was applied to organotypic cultures of gastric corpus+antrum muscles from of 14-16-day-old BALB/c mice at 10 µM. A, Effect of 18-h application of TDZD-8 on Kitl expression; n=6/group. B, Effect of the same treatment on cyclin D1 expression; n=3/group. C, Effects of 1-h exposure to SB216763 on p70S6K and ERK1/2 phosphorylation; n=3/group. See Figure 2 for further details.
(TIF)
Negative controls for the ChIP experiments. A, Low recovery of Kitl promoter DNA by ChIP performed in murine gastric smooth muscles with non-immune mouse IgG (mIgG) or anti-Gapdh antibody relative to input chromatin or ChIP with anti-RNA polymerase II (RNA pol II) antibody. B-C, Unchanged recovery of Kitl promoter sequence in response to 6-h rhIGF1 (100 ng/mL) and SB415286 (30 µM) treatment in murine gastric smooth muscles following ChIP using mIgG (B) or anti-Gapdh antibody (C). Representatives of two independent ChIP experiments, each performed in triplicates, are shown.
(TIF)
Antibodies used in Western immunoblotting studies.
(PDF)
Antibodies used in chromatin immunoprecipitation (ChIP) studies.
(PDF)
Acknowledgments
We thank Dr. Gregory J. Gores (Division of Gastroenterology and Hepatology, Mayo Clinic, Rochester, MN) for granting us access to the LI-COR Odyssey Scanner.
Funding Statement
This work was supported, in part, by National Institutes of Health (http://www.nih.gov/) grants R01 DK058185, P01 DK068055 (Project 2), P30 DK084567, P30 CA015083, P50 CA127003, as well as by the GIST Cancer Research Fund (http://www.gistinfo.org/) and the Mayo Clinic Center for Individualized Medicine http://mayoresearch.mayo.edu/center-for-individualized-medicine/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lennartsson J, Rönnstrand L (2012) Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev 92: 1619-1649. doi: 10.1152/physrev.00046.2011. PubMed: 23073628. [DOI] [PubMed] [Google Scholar]
- 2. Sanders KM, Ward SM (2007) Kit mutants and gastrointestinal physiology. J Physiol 578: 33-42. PubMed: 17095561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Majumdar MK, Feng L, Medlock E, Toksoz D, Williams DA (1994) Identification and mutation of primary and secondary proteolytic cleavage sites in murine stem cell factor cDNA yields biologically active, cell-associated protein. J Biol Chem 269: 1237-1242. PubMed: 7507105. [PubMed] [Google Scholar]
- 4. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T et al. (1998) Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279: 577-580. doi: 10.1126/science.279.5350.577. PubMed: 9438854. [DOI] [PubMed] [Google Scholar]
- 5. Corless CL, Barnett CM, Heinrich MC (2011) Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer 11: 865-878. PubMed: 22089421. [DOI] [PubMed] [Google Scholar]
- 6. Antonescu CR (2011) The GIST paradigm: lessons for other kinase-driven cancers. J Pathol 223: 251-261. PubMed: 21125679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ et al. (2003) PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299: 708-710. doi: 10.1126/science.1079666. PubMed: 12522257. [DOI] [PubMed] [Google Scholar]
- 8. Bardsley MR, Horváth VJ, Asuzu DT, Lorincz A, Redelman D et al. (2010) Kitlow stem cells cause resistance to Kit/platelet-derived growth factor alpha inhibitors in murine gastrointestinal stromal tumors. Gastroenterology 139: 942-952. doi: 10.1053/j.gastro.2010.05.083. PubMed: 20621681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Negri T, Bozzi F, Conca E, Brich S, Gronchi A et al. (2009) Oncogenic and ligand-dependent activation of KIT/PDGFRA in surgical samples of imatinib-treated gastrointestinal stromal tumours (GISTs). J Pathol 217: 103-112. doi: 10.1002/path.2450. PubMed: 18973210. [DOI] [PubMed] [Google Scholar]
- 10. Théou-Anton N, Tabone S, Brouty-Boyé D, Saffroy R, Ronnstrand L et al. (2006) Co expression of SCF and KIT in gastrointestinal stromal tumours (GISTs) suggests an autocrine/paracrine mechanism. Br J Cancer 94: 1180-1185. doi: 10.1038/sj.bjc.6603063. PubMed: 16570044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hirano K, Shishido-Hara Y, Kitazawa A, Kojima K, Sumiishi A et al. (2008) Expression of stem cell factor (SCF), a KIT ligand, in gastrointestinal stromal tumors (GISTs): a potential marker for tumor proliferation. Pathol Res Pract 204: 799-807. doi: 10.1016/j.prp.2008.05.002. PubMed: 18602222. [DOI] [PubMed] [Google Scholar]
- 12. Bono P, Krause A, von Mehren M, Heinrich MC, Blanke CD et al. (2004) Serum KIT and KIT ligand levels in patients with gastrointestinal stromal tumors treated with imatinib. Blood 103: 2929-2935. doi: 10.1182/blood-2003-10-3443. PubMed: 15070666. [DOI] [PubMed] [Google Scholar]
- 13. Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D et al. (2004) Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 23: 3999-4006. doi: 10.1038/sj.onc.1207525. PubMed: 15007386. [DOI] [PubMed] [Google Scholar]
- 14. Guevara-Aguirre J, Balasubramanian P, Guevara-Aguirre M, Wei M, Madia F et al. (2011) Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med 3: 70ra13 PubMed: 21325617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samani AA, Yakar S, LeRoith D, Brodt P (2007) The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev 28: 20-47. PubMed: 16931767. [DOI] [PubMed] [Google Scholar]
- 16. Tarn C, Rink L, Merkel E, Flieder D, Pathak H et al. (2008) Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci U S A 105: 8387-8392. doi: 10.1073/pnas.0803383105. PubMed: 18550829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janeway KA, Zhu MJ, Barretina J, Perez-Atayde A, Demetri GD et al. (2010) Strong expression of IGF1R in pediatric gastrointestinal stromal tumors without IGF1R genomic amplification. Int J Cancer 127: 2718-2722. doi: 10.1002/ijc.25247. PubMed: 20162573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Williams KL, Fuller CR, Fagin J, Lund PK (2002) Mesenchymal IGF-I overexpression: paracrine effects in the intestine, distinct from endocrine actions. Am J Physiol Gastrointest Liver Physiol 283: G875-G885. PubMed: 12223347. [DOI] [PubMed] [Google Scholar]
- 19. Kuemmerle JF (2003) IGF-I elicits growth of human intestinal smooth muscle cells by activation of PI3K, PDK-1, and p70S6 kinase. Am J Physiol Gastrointest Liver Physiol 284: G411-G422. PubMed: 12444011. [DOI] [PubMed] [Google Scholar]
- 20. Kuemmerle JF (2005) Endogenous IGF-I protects human intestinal smooth muscle cells from apoptosis by regulation of GSK-3 beta activity. Am J Physiol Gastrointest Liver Physiol 288: G101-G110. doi: 10.1152/ajpgi.00032.2004. PubMed: 15297258. [DOI] [PubMed] [Google Scholar]
- 21. Lorincz A, Redelman D, Horváth VJ, Bardsley MR, Chen H et al. (2008) Progenitors of interstitial cells of Cajal in the postnatal murine stomach. Gastroenterology 134: 1083-1093. doi: 10.1053/j.gastro.2008.01.036. PubMed: 18395089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horváth VJ, Vittal H, Lörincz A, Chen H, Almeida-Porada G et al. (2006) Reduced stem cell factor links smooth myopathy and loss of interstitial cells of Cajal in murine diabetic gastroparesis. Gastroenterology 130: 759-770. doi: 10.1053/j.gastro.2005.12.027. PubMed: 16530517. [DOI] [PubMed] [Google Scholar]
- 23. Warsito D, Sjöström S, Andersson S, Larsson O, Sehat B (2012) Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. EMBO Rep 13: 244-250. doi: 10.1038/embor.2011.251. PubMed: 22261717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sokolović A, Sokolović M, Boers W, Elferink RP, Bosma PJ (2010) Insulin-like growth factor binding protein 5 enhances survival of LX2 human hepatic stellate cells. Fibrogenesis Tissue Repair 3: 3. doi: 10.1186/1755-1536-3-3. PubMed: 20163708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coulouarn C, Corlu A, Glaise D, Guénon I, Thorgeirsson SS et al. (2012) Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res 72: 2533-2542. doi: 10.1158/1538-7445.AM2012-2533. PubMed: 22419664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ordög T, Redelman D, Horowitz NN, Sanders KM (2004) Immunomagnetic enrichment of interstitial cells of Cajal. Am J Physiol Gastrointest Liver Physiol 286: G351-G360. doi: 10.1152/ajpgi.00281.2003. PubMed: 14563669. [DOI] [PubMed] [Google Scholar]
- 27. Cross DA, Culbert AA, Chalmers KA, Facci L, Skaper SD et al. (2001) Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J Neurochem 77: 94-102. doi: 10.1046/j.1471-4159.2001.t01-1-00251.x. PubMed: 11279265. [DOI] [PubMed] [Google Scholar]
- 28. Zhou Y, Uddin S, Zimmerman T, Kang JA, Ulaszek J et al. (2008) Growth control of multiple myeloma cells through inhibition of glycogen synthase kinase-3. Leuk Lymphoma 49: 1945-1953. doi: 10.1080/10428190802304966. PubMed: 18728964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M et al. (2009) DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther 8: 1579-1588. doi: 10.1158/1535-7163.MCT-09-0013. PubMed: 19509260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Flynn RS, Mahavadi S, Murthy KS, Kellum JM, Kuemmerle JF (2009) Insulin-like growth factor-binding protein-5 stimulates growth of human intestinal muscle cells by activation of Gαi3. Am J Physiol Gastrointest Liver Physiol 297: G1232-G1238. doi: 10.1152/ajpgi.00323.2009. PubMed: 19808657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thimmaiah KN, Easton JB, Germain GS, Morton CL, Kamath S et al. (2005) Identification of N10-substituted phenoxazines as potent and specific inhibitors of Akt signaling. J Biol Chem 280: 31924-31935. doi: 10.1074/jbc.M507057200. PubMed: 16009706. [DOI] [PubMed] [Google Scholar]
- 32. Kuemmerle JF, Bushman TL (1998) IGF-I stimulates intestinal muscle cell growth by activating distinct PI 3-kinase and MAP kinase pathways. Am J Physiol 275: G151-G158. PubMed: 9655695. [DOI] [PubMed] [Google Scholar]
- 33. Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM et al. (2005) Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 54: 142-151. doi: 10.1136/gut.2004.042127. PubMed: 15591520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T et al. (2002) Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab Invest 82: 663-665. doi: 10.1038/labinvest.3780461. PubMed: 12004007. [DOI] [PubMed] [Google Scholar]
- 35. Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T et al. (2000) KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 156: 791-795. doi: 10.1016/S0002-9440(10)64946-2. PubMed: 10702394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bauer S, Yu LK, Demetri GD, Fletcher JA (2006) Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res 66: 9153-9161. doi: 10.1158/0008-5472.CAN-06-0165. PubMed: 16982758. [DOI] [PubMed] [Google Scholar]
- 37. Kitamura T, Tange T, Terasawa T, Chiba S, Kuwaki T et al. (1989) Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. J Cell Physiol 140: 323-334. doi: 10.1002/jcp.1041400219. PubMed: 2663885. [DOI] [PubMed] [Google Scholar]
- 38. Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q et al. (2007) Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 25: 473-481. doi: 10.1016/j.molcel.2007.01.017. PubMed: 17289593. [DOI] [PubMed] [Google Scholar]
- 39. Shinkai Y, Tachibana M (2011) H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev 25: 781-788. doi: 10.1101/gad.2027411. PubMed: 21498567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A (2005) Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat Chem Biol 1: 143-145. doi: 10.1038/nchembio721. PubMed: 16408017. [DOI] [PubMed] [Google Scholar]
- 41. Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A et al. (2004) Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem 279: 33716-33726. doi: 10.1074/jbc.M402839200. PubMed: 15155757. [DOI] [PubMed] [Google Scholar]
- 42. Marks PA, Breslow R (2007) Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 25: 84-90. doi: 10.1038/nbt1272. PubMed: 17211407. [DOI] [PubMed] [Google Scholar]
- 43. Izbeki F, Asuzu DT, Lorincz A, Bardsley MR, Popko LN et al. (2010) Loss of Kitlow progenitors, reduced stem cell factor and high oxidative stress underlie gastric dysfunction in progeric mice. J Physiol 588: 3101-3117. doi: 10.1113/jphysiol.2010.191023. PubMed: 20581042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Asuzu DT, Hayashi Y, Izbeki F, Popko LN, Young DL et al. (2011) Generalized neuromuscular hypoplasia, reduced smooth muscle myosin and altered gut motility in the klotho model of premature aging. Neurogastroenterol Motil 23: e309-e323. doi: 10.1111/j.1365-2982.2011.01730.x. PubMed: 21605285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Beurel E, Michalek SM, Jope RS (2009) Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol 31: 24-31. PubMed: 19836308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kouzarides T (2007) Chromatin modifications and their function. Cell 128: 693-705. doi: 10.1016/j.cell.2007.02.005. PubMed: 17320507. [DOI] [PubMed] [Google Scholar]
- 47. Kouzarides T (2007) SnapShot: Histone-modifying enzymes. Cell 131: 822. doi: 10.1016/j.cell.2007.11.005. PubMed: 18022374. [DOI] [PubMed] [Google Scholar]
- 48. Mühlenberg T, Zhang Y, Wagner AJ, Grabellus F, Bradner J et al. (2009) Inhibitors of deacetylases suppress oncogenic KIT signaling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors. Cancer Res 69: 6941-6950. doi: 10.1158/0008-5472.CAN-08-4004. PubMed: 19706776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pekowska A, Benoukraf T, Ferrier P, Spicuglia S (2010) A unique H3K4me2 profile marks tissue-specific gene regulation. Genome Res 20: 1493-1502. doi: 10.1101/gr.109389.110. PubMed: 20841431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sauvageau M, Sauvageau G (2010) Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 7: 299-313. doi: 10.1016/j.stem.2010.08.002. PubMed: 20804967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schuettengruber B, Martinez AM, Iovino N, Cavalli G (2011) Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol 12: 799-814. doi: 10.1038/nrm3230. PubMed: 22108599. [DOI] [PubMed] [Google Scholar]
- 52. Zhou VW, Goren A, Bernstein BE (2011) Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 12: 7-18. doi: 10.1038/nri3147. PubMed: 21116306. [DOI] [PubMed] [Google Scholar]
- 53. Lomberk G, Bensi D, Fernandez-Zapico ME, Urrutia R (2006) Evidence for the existence of an HP1-mediated subcode within the histone code. Nat Cell Biol 8: 407-415. doi: 10.1038/ncb1383. PubMed: 16531993. [DOI] [PubMed] [Google Scholar]
- 54. Baumann U, Crosby HA, Ramani P, Kelly DA, Strain AJ (1999) Expression of the stem cell factor receptor c-kit in normal and diseased pediatric liver: identification of a human hepatic progenitor cell? Hepatology 30: 112-117. doi: 10.1002/hep.510300140. PubMed: 10385646. [DOI] [PubMed] [Google Scholar]
- 55. Gaça MD, Pickering JA, Arthur MJ, Benyon RC (1999) Human and rat hepatic stellate cells produce stem cell factor: a possible mechanism for mast cell recruitment in liver fibrosis. J Hepatol 30: 850-858. doi: 10.1016/S0168-8278(99)80139-1. PubMed: 10365812. [DOI] [PubMed] [Google Scholar]
- 56. Yakar S, Adamo ML (2012) Insulin-like growth factor 1 physiology: lessons from mouse models. Endocrinol Metab Clin North Am 41: 231-247. doi: 10.1016/j.ecl.2012.04.008. PubMed: 22682628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Janeway KA, Kim SY, Lodish M, Nosé V, Rustin P et al. (2011) Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A 108: 314-318. doi: 10.1073/pnas.1105216108. PubMed: 21173220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pantaleo MA, Astolfi A, Indio V, Moore R, Thiessen N et al. (2011) SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst 103: 983-987. doi: 10.1093/jnci/djr130. PubMed: 21505157. [DOI] [PubMed] [Google Scholar]
- 59. Edris B, Willingham SB, Weiskopf K, Volkmer AK, Volkmer JP et al. (2013) Anti-KIT monoclonal antibody inhibits imatinib-resistant gastrointestinal stromal tumor growth. Proc Natl Acad Sci U S A 110: 3501-3506. doi: 10.1073/pnas.1222893110. PubMed: 23382202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Grover M, Farrugia G, Lurken MS, Bernard CE, Faussone-Pellegrini MS et al. (2011) Cellular changes in diabetic and idiopathic gastroparesis. Gastroenterology 140: 1575-1585. doi: 10.1053/j.gastro.2011.01.046. PubMed: 21300066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gomez-Pinilla PJ, Gibbons SJ, Sarr MG, Kendrick ML, Shen KR et al. (2011) Changes in interstitial cells of Cajal with age in the human stomach and colon. Neurogastroenterol Motil 23: 36-44. doi: 10.1111/j.1365-2982.2010.01590.x. PubMed: 20723073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xiong Y, Khanna S, Grzenda AL, Sarmento OF, Svingen PA et al. (2012) Polycomb antagonizes p300/CREB-binding protein-associated factor to silence FOXP3 in a Krüppel-like factor-dependent manner. J Biol Chem 287: 34372-34385. doi: 10.1074/jbc.M111.325332. PubMed: 22896699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128: 707-719. doi: 10.1016/j.cell.2007.01.015. PubMed: 17320508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kitl/KITLG and IGF1R protein expression and IGF1 secretion in murine gastric smooth muscles, human LX-2 stellate cells and GIST cells. A, Detection of Kitl in the lysate of gastric corpus+antrum muscles from a juvenile BALB/c mouse. The membrane was simultaneously probed with antibodies against Kitl and Gapdh (loading control) and appropriate fluorescent secondary antibodies. Note primary Kitl band at ~31 kDa and a weaker band at ~43 kDa. The 21-kDa Kitl band was only borderline detectable. B, In human GIST cell lines, only the 31-kDa KITLG band was detected. B, Validation of the Western immunoblotting method. The 31-kDa Kitl band was also detected in lysates of Sl/Sl 4 hematopoietic stromal cells expressing mKitl248. No Kitl bands were detected in Sl/Sl 4 stromal cells lacking full-length Kitl. D, Expression of KITLG protein in LX-2 human hepatic stellate cells and three human GIST cell lines. E, Expression of KIT protein in the same cell lines. The level reported for LX-2 cells represents background fluorescence as no specific band was detected. Note lack of correlation between KITLG and KIT expression. F, IGF1R α and β chain expression in GIST48B cells. G, IGF1 secretion into culture media by GIST-T1, GIST882, GIST48B and LX-2 cells. 3000 cells/well were plated into 96-well plates and cultured in the presence or absence of FBS (GIST-T1, GIST882, LX-2: 10%; GIST48B: 15%) for 48 h. IGF1 was measured in the harvested cell-free media using the Quantikine Human IGF1 Immunoassay kit (DG100, R&D systems). Note differential responses to FBS.
(TIF)
Kitl protein expression is stimulated by IGF1. Stimulation of Kitl expression, detected using Actb as reference, by 18-h treatment with 100 ng/mL rhIGF1 in gastric corpus+antrum tunica muscularis organotypic cultures from 14-16-day-old BALB/c mice (n=3/group). Kitl and Actb were simultaneously detected in the same samples by two-color immunofluorescence. Representative immunoblots show identical areas of the blots imaged at different wavelengths. The degree of Kitl upregulation was statistically indistinguishable from the increase detected using Gapdh as loading control (1.89±0.21-fold vs. 2.07±0.15-fold, P=0.583).
(TIF)
SB216763 stimulates Kitl expression without activating cyclin D1 expression and p70S6K and ERK1/2 phosphorylation. GSK3α/β inhibitor SB216763 was applied to organotypic cultures of gastric corpus+antrum muscles from of 14-16-day-old BALB/c mice at 3 µM. A, Effect of 18-h application of SB216763 on Kitl expression; n=5/group. B, Effect of the same treatment on cyclin D1 expression; n=3/group. C, Effects of 1-h exposure to SB216763 on p70S6K and ERK1/2 phosphorylation; n=3/group. See Figure 2 for further details.
(TIF)
TDZD-8 stimulates Kitl expression without activating cyclin D1 expression and p70S6K and ERK1/2 phosphorylation. The non-ATP-competitive GSK3α/β inhibitor TDZD-8 was applied to organotypic cultures of gastric corpus+antrum muscles from of 14-16-day-old BALB/c mice at 10 µM. A, Effect of 18-h application of TDZD-8 on Kitl expression; n=6/group. B, Effect of the same treatment on cyclin D1 expression; n=3/group. C, Effects of 1-h exposure to SB216763 on p70S6K and ERK1/2 phosphorylation; n=3/group. See Figure 2 for further details.
(TIF)
Negative controls for the ChIP experiments. A, Low recovery of Kitl promoter DNA by ChIP performed in murine gastric smooth muscles with non-immune mouse IgG (mIgG) or anti-Gapdh antibody relative to input chromatin or ChIP with anti-RNA polymerase II (RNA pol II) antibody. B-C, Unchanged recovery of Kitl promoter sequence in response to 6-h rhIGF1 (100 ng/mL) and SB415286 (30 µM) treatment in murine gastric smooth muscles following ChIP using mIgG (B) or anti-Gapdh antibody (C). Representatives of two independent ChIP experiments, each performed in triplicates, are shown.
(TIF)
Antibodies used in Western immunoblotting studies.
(PDF)
Antibodies used in chromatin immunoprecipitation (ChIP) studies.
(PDF)