

Abstract
Burkholderia cepacia is an opportunistic human pathogen associated with life-threatening pulmonary infections in immunocompromised individuals. Pathogenesis of B. cepacia infection involves adherence, colonisation, invasion, survival and persistence in the host. In addition, B. cepacia are also known to secrete factors, which are associated with virulence in the pathogenesis of the infection. In this study, the host factor that may be the cause of the infection was elucidated in human epithelial cell line, A549, that was exposed to live B. cepacia (mid-log phase) and its secretory proteins (mid-log and early-stationary phases) using the Illumina Human Ref-8 microarray platform. The non-infection A549 cells were used as a control. Expression of the host genes that are related to apoptosis, inflammation and cell cycle as well as metabolic pathways were differentially regulated during the infection. Apoptosis of the host cells and secretion of pro-inflammatory cytokines were found to be inhibited by both live B. cepacia and its secretory proteins. In contrast, the host cell cycle and metabolic processes, particularly glycolysis/glycogenesis and fatty acid metabolism were transcriptionally up-regulated during the infection. Our microarray analysis provided preliminary insights into mechanisms of B. cepacia pathogenesis. The understanding of host response to an infection would provide novel therapeutic targets both for enhancing the host’s defences and repressing detrimental responses induced by the invading pathogen.
Introduction
Burkholderia cepacia is naturally found in relatively high population in moist soil, in particular the rhizosphere of plants and the freshwater environments [1]. It is a non-pathogenic bacterium found in normal, healthy individuals. However, this organism can become an opportunistic human pathogen associated with life-threatening pulmonary infections that affect immunocompromised individuals, particularly patients with cystic fibrosis (CF) [2] and chronic granulomatous disease [3]. Pathogenesis of B. cepacia has been reported to be multi-factorial, associated with the ability of the bacterium to adhere, colonise, invade, replicate, survive and persist in the host cells, as well as to evade the host immune response [4,5]. These organisms are also able to secrete virulence proteins which aids in bacterial penetration into the epithelial barrier and gain access to the underlying tissue as well as the bloodstream. However, the virulence of B. cepacia infection is dependent on the nature of the bacterial strain in terms of types of virulence factors involved, the number of organisms in the initial exposure, the age of the bacteria and status of the host immune response.
How the lung epithelial cells responds towards B. cepacia infection is still undefined. In recent years, there have been numerous reports of gene expression analyses performed using the microarray platform to study the host’s response to acute bacterial infection in the modulation of different host genes in different target tissues [6,7]. To date, there has been no report on transcriptional changes induced by B. cepacia in the host. In the present study, we determined the factors involved in virulence of B. cepacia i.e., release of the extracellular enzymes and effect of the bacterial multiplicity of infection (MOI) at different stages of growth on invasion and intracellular survival. Investigation was also performed on the transcriptional changes in the host response towards exposure to B. cepacia live bacteria (mid-log phase) and secretory proteins (mid-log and stationary phases) that has previously been identified using 2-DE and MS analyses [5].
The aim of this study was to develop a comprehensive profile of the host transcriptional responses that occur in the lung epithelial cells during an infection with B. cepacia live bacteria or exposure to its secretory proteins. This improves our understanding of the immediate host responses at the early stage of B. cepacia infection.
Materials and Methods
Bacterial strain, growth and culture conditions
B. cepacia CQK, isolated from a non-CF patient at the University Malaya Medical Centre (UMMC), Kuala Lumpur, Malaysia, was cultured on nutrient agar. The bacterial culture was prepared using Luria-Bertani (LB) broth according to protocols described by Mariappan et al. (2011) [5]. Bacterial culture was sampled at every 2 hours interval (0 to 24 hours) and centrifuged at 20,000g for 40 mins at 4°C, after which the supernatant was collected and filtered through a 0.22 µm pore size membrane filter to remove residual bacteria [8].
Exoenzyme profile of B. cepacia secretory proteins
The bacterial free culture supernatant was concentrated 50-fold using ultrafiltration employing 10 kDa centricon ultra-free centrifugal filter units (Millipore, USA). The 10kDa cut-off was selected based on data obtained from our previous studies [5]. Total protein concentrations were determined using Bradford method (1976) [9]; protease was assayed using azacoll according to Chavira et al. (1984) [10]; phospholipase C (PLC) was assayed using p-nitrophenyl phosphorylcholine (NPPC), ( [11]; peroxidase activity was assayed using o-dianisidine [12]; acid and alkaline phosphatase activities were assayed by measuring the release of p-nitrophenol from p-nitrophenol phosphate (p-NPP) at OD410nm [13]; lipase activity was assayed using Tween 80 [14]. The quantification of isocitrate dehydrogenase (ICD) was performed according to Anderson et al. (1991) [15] to determine bacterial cell lysis. In all the experiments, fresh LB was used as control and the assays were performed in triplicates.
Invasion and intracellular survival assays
Approximately 5 x 105 A549 cells were seeded into each well of a 24-well tissue culture plate containing 1 ml of Roswell Park Memorial Institute (RPMI) growth medium containing 10% foetal calf serum and incubated at 37°C overnight. The invasion and intracellular survival assays were performed as described by Mariappan et al. (2011) [5]. In brief, the A549 cells were infected with B. cepacia grown to three different time-points of growth (mid-log, early stationary and stationary) at different MOI -1:10, 1:50 or 1:100 and were incubated for 1, 3, 6, 12, 18 and 24 hours respectively, at 37°C in 5% CO2 to allow bacterial invasion. For the intracellular survival assay, following three hours of invasion, the extracellular bacteria were killed using antibiotics (1 mg/ml ceftazidime and gentamicin, respectively) for 2 hours. The cells were further incubated in the antibiotic-free RPMI medium for 1, 3, 6, 12, 18 and 24 hours. Serial dilutions of the lysate was prepared and plated on nutrient agar to determine the bacterial counts [16]. Non-invasive Escherichia coli was used as a negative control. This experiment was performed in triplicates and the results were averaged.
A549 epithelial cell viability test
Approximately 1×106 A549 cells were seeded into each well of a 6-well tissue culture plate containing 1 ml of RPMI growth medium and incubated at 37°C in 5% CO2 until approximately 80% confluency. The confluent monolayer of A549 cells were exposed to B. cepacia live bacteria cultured to mid-log phase with ratio of 1:10 to 1:200 and the filter-sterilised mid-log and early-stationary phase secretory proteins at different concentrations ranging from 0-100 µg/ml, for 3 hours at 37°C in 5% CO2. Following 3 hours, 0.1% trypsin was added to detach the cells, after which the cell suspension was centrifuged at 300g for 5 minutes. The pelleted cells were then resuspended in 1 ml of phosphate buffered saline (PBS) and the number of viable cells was enumerated on a haemocytometer using the tryphan blue exclusion method.
Gene expression analyses
Three biological replicates of total cellular RNA from A549 cells were extracted subsequent to 3 hours exposure of B. cepacia culture supernatant (mid-log (ML) and early-stationary (ES) phases) and live bacteria (LBC) using the Qiagen RNeasy Mini Kit with on column DNase treatment according to the manufacturer’s instructions (Qiagen, USA). The integrity of the extracted RNA was assessed using the Agilent 2100 Bioanalyser (Agilent Technologies, USA) and a total of 500 ng RNA was amplified in a single-round of in vitro transcription amplification that allowed incorporation of biotin-labelled nucleotides using the Illumina TotalPrep RNA Amplification Kit according to the manufacturer’s instructions (Ambion, USA). Microarray experiments were performed using the Illumina HumanRef-8 BeadChip, (containing 24,526 distinct genes) according to the instructions provided by the manufacturer (Illumina, USA).
Microarray data analysis
The data was analysed using two different softwares; Illumina’s BeadStudio version 1.0 (Illumina, USA) and GeneSpring GX version 10 (Agilent, California, USA) software. In brief, the BeadStudio was used to generate signal intensity values from the scans, followed by standard normalisation using the GeneSpring (substitution of 0.01 to expression values <0.01, per chip normalisation to 50th percentile, per gene normalisation to median). The normalised data were grouped on the basis of the experimental conditions (treated and non-treated) and filtered using the Scatter Plot. A Venn diagram was generated from the one-way ANOVA results in order to compare the number of genes significantly changed in each set (p value of ≤ 0.05 and an absolute change greater than 2-fold for B. cepacia infected cells relative to the un-infected control cells). The pathways of the differentially expressed genes was analysed using the Kyoto Encyclopaedia of Genes and Genomes (KEGG) mapper database (http://www.genome.jp/kegg/). The web-based software GOTerm Finder (http://go.princeton.edu/cgi-bin/GOTermFinder) and GeneTrail (http://genetrail.bioinf.uni-sb.de/) were used to analyse significant pathways. Selected data were organised by a hierarchical clustering with the web-based software Cluster 3.0. The clustering algorithm is based on an uncentred correlation metric, with average linkage clustering and visualised using Java Treeview V1.1.3.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed in the IQ5 real-time PCR (BioRad Laboratories, USA), to verify and quantify the expression of VAM 8, RGS2, IL-1β, NFKB1A, AKR1B10, PADI2, TNF and LTB. All primers used for the targeted genes including β-actin and GAPDH were obtained from PrimerBank (http://pga.mgh.harvard.edu/primerbank/) (Table S1). All the primers were synthesised at Nano Life Quest Laboratories (Malaysia). Briefly, 25 µl reactions were performed using the iScript™One-Step RT-PCR kit with SYBR green according to the manufacturer’s instruction (BioRad Laboratories, USA), primers at a final concentration of 1 µM and a data acquisition temperature of 56°C. In order to control for variation in RNA concentration, cycle threshold (Ct) values were normalised to β-actin and GAPDH that does not change with infection. The IQ5 real-time PCR software (Biorad, California, USA) was used to generate the quality control of the replicates, data extraction and initial analysis with a manual threshold of 0.6 and an auto baseline applied in order to obtain the threshold cycle (Ct) value for each measurement taken.
Results
Determination of the optimal parameters for host interaction
The secretion profiles of protease, peroxidase, lipase, PLC and acid and alkaline phosphatases were different for each of the enzymes studied at the different time points of bacterial culture (Figure 1). An intracellular enzyme assay, which detects ICD activity as a result of bacterial lysis was performed and insignificant levels of activity (0-0.21 Units/ml) were detected. Maximum enzyme peak activities were observed at 4 hours of growth (mid-log phase) for peroxidase, lipase, acid phosphatase and alkaline phosphatase with 10.96 Units/ml, 12.53 Units/ml, 18.92 Units/ml and 24.81 Units/ml, respectively. In contrast, the activities of protease and PLC increased to their highest levels at 8 hrs of growth (early-stationary phase) and with activities of 6.06 Units/ml and 11.73 Units/ml, respectively.
Figure 1. Exoenzyme profile of isocitrate dehydrogenase, lipase, phospholipase C, peroxidase, protease, acid phosphatase and alkaline phosphatase from B. cepacia secretory proteins at different phases of growth.

The error bars indicate the standard deviation of triplicate values.
The ability of B. cepacia CQK strain to invade the epithelial cells was measured and compared at three different phases of bacterial growth (Table 1). In general, the trend of the invasion was similar for all the phases of growth at MOI 1:10 and 1:50. However, the invasion capacity at MOI 1:50 were comparatively higher than MOI 1:10 at all times of exposure and growth phases. The highest percentage of invasion (2.11%) was observed at MOI 1:50, stationary phase, 24 hours post-infection. At MOI 1:100, the percentage of invasion was moderately higher than MOI 1:50 at 3-12 hours post-infection at all phases of growth. However, the rate of invasion was found to decrease distinctly following longer post-infection time at the respective growth phases.
Table 1. Mean percentage of invasion (%) with standard deviation MOI 1:10, 1:50 and 1:100.
| Time (Hours) | Mean percentage of invasion (%) - MOI 1:10 |
Mean percentage of invasion (%) - MOI 1:50 |
Mean percentage of invasion (%) - MOI 1:100 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Mid-log | Early-stationary | Stationary | Mid-log | Early-stationary | Stationary | Mid-log | Early-stationary | Stationary | |
| 3 | 0.0044 ± 0.0016 | 0.0088 ± 0.0032 | 0.0111 ± 0.0042 | 0.0089 ±0.0016 | 0.0155 ± 0.0016 | 0.0177 ± 0.0016 | 0.0144 ± 0.0031 | 0.0211 ± 0.0016 | 0.0244 ± 0.042 |
| 6 | 0.0111 ± 0.0016 | 0.0177 ± 0.0016 | 0.0222 ± 0.0642 | 0.0222 ± 0.0016 | 0.0289 ± 0.0042 | 0.0355 ± 0.0016 | 0.0277 ± 0.0042 | 0.0355 ± 0.0016 | 0.0389 ± 0.0016 |
| 12 | 0.0333±0 | 0.0555 ± 0.0157 | 0.1111 ± 0.0157 | 0.0777 ± 0.0157 | 0.1666 ± 0.0272 | 0.2222 ± 0.0314 | 0.1333 ± 0.0272 | 0.2 ± 0.0272 | 0.2555 ± 0.0157 |
| 18 | 0.2222 ± 0.0157 | 0.2666 ± 0.0272 | 0.3±0 | 0.2666 ± 0.0272 | 0.3555 ± 0.0314 | 0.4111 ± 0.0157 | 0.111 ± 0.1571 | 0.1777 ± 0.0314 | 0.2222 ± 0.0157 |
| 24 | 0.3333±0 | 0.4222 ± 0.1571 | 0.4777 ± 0.1572 | 0.7777 ± 0.1572 | 1.6666±0 | 2.1111 ± 0.1571 | 0.088 ± 0.1571 | 0.1111 ± 0.1571 | 0.1666 ± 0.2722 |
In general, the intracellular survival profiles were similar at all the MOIs (Figure 2). The intracellular survival of B. cepacia at MOI 1:50 showed slow replication of B. cepacia in the epithelial cells from the first hour to 12 hours of incubation. A similar pattern was also observed with the intracellular survival of B. cepacia at MOI of 1:100. The number of bacteria replicated intracellularly peaked following six hours of incubation at all three phases. Nevertheless, it was noticeable that the number of cells living intracellularly decreased at longer time of replication (18 and 24 hours) at all the MOIs, suggesting that intracellular survival in the epithelial cells was saturable.
Figure 2. The intracellular survival assay of B. cepacia (mid-logarithmic, early-stationary and stationary phases of growth) after 3 hours of post-infection with MOI of (panel A) 1:10, (panel B) 1:50 and (panel C) 1:100 at different phases of growth.

The error bars indicate the standard deviation of triplicate values.
Determination of epithelial cells viability upon infection of B. cepacia
The reduction in the percentage of the A549 epithelial cell viability was demonstrated using trypan blue (Figure 3, panels A and B). The cell survival was 100% at a concentration of 0.5 µg/ml culture supernatant harvested at the mid-log and early-stationary phases and decreased gradually as the concentration increased from 1-5 µg/ml. Using 50 µg/ml of the mid-log phase culture supernatant, viability of the host cells were found to be reduced to 55%. Conversely, none of the host cells survived when they were exposed to 50 µg/ml of the early-stationary phase culture supernatant (Figure 3, panel A). In addition, exposure of live B. cepacia to the A549 cells showed 99.2% and 99.05% of cell survival at MOI ratios of 1:10 and 1:50, respectively. However, the percentage of cell survival reduced to 83.02% at an MOI ratio of 1:100. Only 52.07% of the A549 cells were found to further survive at an MOI ratio of 1:200 (Figure 3, panel B).
Figure 3. Percentage of A549 lung epithelial cells exposed to (panel A) secretory proteins (mid-logarithmic and early-stationary phases) ranging from 0 µg/ml-100 µg/ml and (panel B) live B. cepacia (mid- logarithmic phase) for three hours from 1:10 -1:200 using Trypan blue solution.
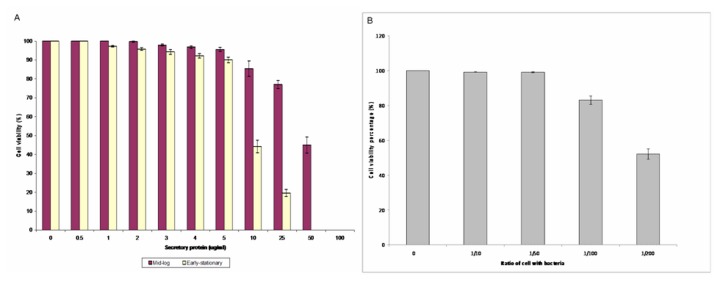
The error bars indicate the standard deviation of triplicate values. The red box indicates the concentration choosen for microarray analysis.
Host transcriptional responses to early exposure of B. cepacia live bacteria and the culture supernatant
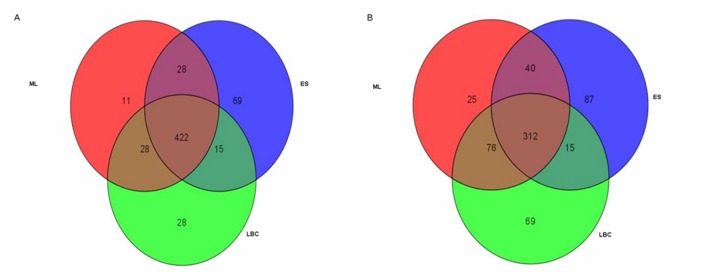
In this study, a total 20,496 of 24,526 genes, (83.57%) were filtered with cut-off values equivalent to Present and Marginal. Using the one-way ANOVA (asymptotic computed p-value <0.01) and Benjamini Hochberg (multiple testing corrections), 9,029 of 20,496 genes (44.02%) were identified as significantly expressed. Additionally, based on the results obtained from the fold change analysis, Venn diagrams were generated to determine common genes of the host that were differentially expressed (Figure 4, panels A and B). From the experiments, 422 genes were identified to be commonly up-regulated under all three experimental conditions (LBC, ML and ES). Conversely, only 312 genes were found to be commonly down-regulated among LBC, ML and ES. Scatter plots were also used to observe the relative distribution of genes that were significantly differentiated between each treated group and the control group.
Figure 4. Venn diagrams showing the number of genes that were up-regulated (panel A) and down-regulated (panel B) with fold changes of ≥2.0-fold.
Heat map analysis was performed and the significantly regulated genes (p-value<0.05) were classified according to the pathways (GeneTrail) and fold-change relative to the control and presented as hierarchical clustering of expression profiles based on functional categories (Figure 5). Several host immune regulatory pathways, cellular processes, metabolic pathways, regulation of cell cycle, apoptosis, inflammatory pathways that include numerous cytokines and chemokines were found to be differentially regulated upon exposure to live B. cepacia and the secretory proteins.
Figure 5. Heat map analysis.

Activation of the host metabolic pathways
Expression of genes involved in the various metabolic pathways was significantly up-regulated under all the three conditions tested. The xenobiotics by cytochrome p450 metabolism and nicotinate and nicotinamide metabolism pathways were found to be enhanced in all the three conditions tested. However, glycolysis/glycogenesis and the arachidonic acid metabolism pathway were increased only in the LBC and ML conditions.
The majority of the genes identified were found to be involved in energy production, lipid metabolic processes, oxidation-reduction, cofactors and electron carrier. Glycolytic/ glycogenesis enzymes were significantly up-regulated including PYGB, ENO 2 and 3, PHKA2, PCK2, ALDH1A3 and HK1, which are the key enzymes involved in the conversion of glucose-6-phosphate to pyruvate. In addition, genes encoding enzymes involved in the metabolism of fatty acids and biosysnthesis of unsaturated fatty acids were also up-regulated. Interestingly, genes associated with the metabolism of xenobiotics by cytochrome P450 (drug metabolism) were up-regulated in all the three conditions. This involves oxidases or detoxification of poisonous compounds i.e., CYP family, MGST2, GSTO2 and AKR1C2.
Suppression of apoptosis and cell death programmes
Cell death related gene sets namely; apoptosis and NOD-like receptors pathway were down-regulated in all the conditions tested. In addition, several NOD-like receptors i.e., CCL 2 and 5, BIRC 2 and 3, IL-6 and IL-1β and NFκB1 were generally down-regulated. However, the subfamily of inflammatory mediator CASP1 was activated together with nucleotide-binding domain and NLRP2 to trigger the pro-inflammatory caspases upon injury, toxins or invasion. The p53 pathway was down-regulated only under the LBC, where SFN, which is the p53-regulated inhibitor of the G2/M progression in cell cycle, was up-regulated. However, the transcriptional of CDKN1A, SERPINE, THBS1, GADD45A and SESN1 were down-regulated in the p53 pathway which resulted in inactivation of apoptosis, genomic stability and inhibition of angiogenesis. The apotosis pathway demonstrated that pro-apoptotic interleukins (IL-1α and IL-1β) associated with IRAK2, which was able to induce the NFκB1, were down-regulated.
Regulation of the host cell cycle and survival
The GeneTrail analysis revealed that most of the genes involved in the cell cycle, VEGF (involved in angiogenesis) and PPAR (involved in lipid oxidation and cell proliferation) signalling pathways were up-regulated. Genes that promote cell growth or arrest, DNA replication or repair and mitosis in the cell cycle pathway i.e., SFN, SKP2, CDKN2D, ESPL1, ORC1L, BUB1 and TTK, were up-regulated (mainly under ES condition). However, the transcript levels of genes involved in stressful growth arrest conditions and treatment with DNA damaging agents including CDKN1A, GADD45A, CCNE2 and E2F5 were down-regulated under the ML and ES conditions.
Alteration of inflammation and cytokine-cytokine receptor interaction
The majority of the inflammatory and signal transduction pathways i.e., MAPK signalling, JAK/STAT, TLR signalling, chemokines and interleukins were generally down-regulated in all the three conditions. Several genes encoding the MAPK signalling pathway proteins of the activator protein-1 (AP-1) complex i.e., JUND, which protect cells from p53-dependent senescence and apoptosis, were up-regulated. Interestingly, MKP (oxidative or heat stress and growth factors gene) was found to be down-regulated by secretory proteins of the ML phase but up-regulated using secretory proteins under the ES and LBC conditions. In addition, it also acts on damaged proteins, causing partial unfolding and possible aggregation which directly inhibits apoptosis.
Under the ES condition, C-FOS and JUND were found to be up-regulated, in addition to the down-regulation of C-JUN. NGF, PDGFB, FGF, TGF and BDNF were also down-regulated under the ES condition. Activation of NFκB appears as one of the key players of the gene regulation coordination. However, there was no significant difference in the expression under the ML and LBC conditions.
On the other hand, under the ES condition, the cytokine transcriptional regulation indicated down-regulation of NFκB. This is in agreement with the down-regulation of TNFα, IL-1α and IL-1β, which are known to be regulated by NFκB. There was up-regulation of interleukin genes (IL-2, IL-3, and IL-6) observed in all the conditions tested. Moreover, genes encoding STAT 4; the Src homology region 2 domain-containing phosphatase-1 (SHP-1; also known as tyrosine-protein phosphatase non-receptor type-6 (PTPN6)), protein inhibitor of activated STAT-1 (PIAS1) and protein 48 (P48; interferon response factor (IRF9)) were also up-regulated under all the experimental conditions.
TLRs are innate signalling receptors and play an important role in the induction of various immune responses, particularly the inflammatory related genes which were significantly differentiated upon contact of the live B. cepacia or secretory proteins with the host cell. MyD88 (an important regulator of the TLR signalling pathway) and MAP3K8 were significantly up-regulated under all the three conditions tested. However, there is also an apparent decrease in some cytokine end products of the TLR signalling observed under the ES secretory proteins, but no significant difference in expression was observed under the LBC and ML conditions. The expression of the COX-2 isoform was induced after interaction with live B. cepacia and secretory proteins (ML and ES).
Genes of the chemokines C-X-C motif chemokine 5 (CXCL5) as well as IL-11 of the host were up-regulated after interaction with live B. cepacia and secretory proteins (mid-log and early-stationary phases). However, CCL26 and IL-8 were found to be up-regulated only under the LBC condition. On the other hand, CCL2, CCL5 and CCL20 genes along with IL-1β, IL-1α, IL11RA and IL27RA were down-regulated under the ML and ES conditions. In addition, IL10RB was down-regulated under the LBC and ML conditions whereas IL-6 and LIF were down-regulated under the ES condition.
Validation of the microarray data
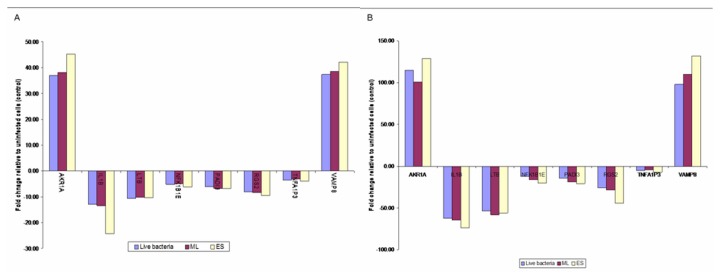
The qRT-PCR assay was used for validation of genes involved in adhesion, cell growth, apoptosis, inflammation, and corresponding to cytokines, receptors, adapters, kinases, phosphatases, and transcription factors that were differentially expressed in the microarray analysis. The data obtained, together with those determined by the microarray experiments are shown in Figure 6 (panels A and B). The gene expressions obtained using the two techniques were comparable. However, differences were observed in the fold-change values whereby the fold-change detected in qRT-PCR assay was higher than the microarray.
Figure 6. The relative relation between (A) microarray and (B) qRT-PCR assay.
Discussion
In the present study, we have modeled an in vitro experiment to compare the host response between the actively dividing and adhering live B. cepacia or their secretory proteins. Previous studies on host-pathogen response of intracellular pathogens have focused primarily on a subset of genes from different pathways or limited gene expression patterns which is inadequate to dissect the host responses to an infection [7]. Our analyses clearly demonstrated that the pathogen had modulated the host metabolism, cell death, cell survival and innate immune system. To the best of our knowledge, the present study is the first to report the transcriptional host response of human airway epithelial cells after an interaction with live B. cepacia or with B. cepacia secretory proteins. This comparison provides a comprehensive genome wide view of the host transcriptional response and a deeper insight into the host response during pathogenesis of B. cepacia.
Various studies have demonstrated the enzyme activities of secreted proteins and their potential role in virulence of B. cepacia [14,17,18]. In this study, the secretion kinetics of six housekeeping exoenzymes of B. cepacia were selected based on their potential roles in the pathogenesis of B. cepacia [19]. The variation in the secretion kinetics of the different enzymes suggests differences in the enzymatic and pathogenic potential of the bacterium at different points of the growth cycle. In an in vivo condition, these enzymes may also be produced in large amounts at the logarithmic phase, in order for the bacteria to obtain nutrients for their multiplication as well as colonisation and subsequent invasion of the host [20].
In addition, invasion and intracellular survival are also important elements in the pathogenesis of B. cepacia whereby many studies have shown that B. cepacia were able to invade and survive within the epithelial cells in vitro and in vivo [4,21-25]. In the present study, the ability of B. cepacia to invade and multiply inside epithelial cells was measured based on the different pathogenic potentials in terms of profiles of exoenzyme activities. Our results suggest that the invasion efficiency of B. cepacia was directly related to the phases of growth and the MOIs. Collectively, these factors may contribute to the virulence potential of pathogenic B. cepacia. In order to survive and replicate efficiently in the host cells, it is necessary for the intracellular pathogens to adapt their metabolism to the available nutrients and physical conditions in the cells (mainly pH, oxygen availability and osmotic pressure), and also coordinate the metabolism with their life cycle.
The host-pathogen response may be reflected by the interaction of host cells with invading bacteria or their products, or to specific host defensive mechanisms. Upon exposure to live B. cepacia and its secretory proteins, genes associated with the host metabolic pathway were demonstrated to be up-regulated. This is compatible with the results shown by Moreilhon and co-workers (2005) [7], which demonstrated that the metabolic pathways were up-regulated in the human airway cells upon contact to live Staphylococcus aureus and its bacterial soluble factors. In our study, among the important metabolic pathways that were up-regulated was the xenobiotic metabolism-cytochrome p450 superfamily related genes, which could be attributed to the presence of toxic secreted proteins. On the contrary, Chin et al. (2010) [6] demonstrated that expression of xenobiotic metabolism was suppressed in the liver of mice with acute melioidosis. The differences observed may be attributed to the longer duration of exposure to Burkholderia pseudomallei and the proteins released during infection in an in vivo condition.
In addition, we also observed the acceleration of genes related to glycolysis which may suggest the ability of the host to access high energy during the infection with B. cepacia. However, marked up-regulation of glycolysis observed in the ES condition compared to the ML and LBC could be due to the higher secretion of virulence proteins, which may have contributed to the depletion of the host energy release. Significant up-regulation of the biosynthesis of unsaturated fatty acids was also found only in the ES condition, and this may have served to improve the cell structure due to injury caused by the pathogenic B. cepacia secretory proteins. When taken together, our data suggest that metabolic pathways were generally activated for the pathogen to survive intracellularly. These metabolic alterations are presumed for the maintenance of host homeostasis, which is beneficial to the host as well as the pathogen.
In this study, down-regulation of the host apoptotic pathway was detected under all the conditions investigated, with the maximum effect observed under the ES condition, which correlates with the findings by Moreilhon and co-workers (2005) [7]. We believe that this could be due to an urge for the host to survive under the ES condition, which was more pathogenic compared to the LBC and ML conditions. Inhibition of apoptosis of the A549 cells would ease replication of the bacteria, prolongs their intracellular survival and favours bacterial persistence.
Activation of the p53 signalling pathway can lead to either the cell cycle arrest or apoptosis [26]. In the present study, the significant down-regulation of p53 was only found in the LBC condition. Interaction of the live B. cepacia with the host cells might have caused minimal DNA damage. This was supported by our cell viability test results which showed minimal percentage of cell death. However, an in vitro model for the human meninges has provided evidence that secreted proteins from Neisseria meningitidis also play a role in the induction of the cell cycle and resistance to apoptosis [27].
Cell cycle progression is a highly complex process which requires coordinated activation of several kinases, some of which are activated upon binding of a cyclin subunit [28]. Our analysis consistently demonstrated that the cell cycle was up-regulated. GADD45, through it’s association with CDC2, appears to disrupt interaction between CDC2 and Cyclin B1 and hence induces G2/M arrest. Similar transcriptional profiles were also seen by Zhang et al. (1999) [29], thus suggesting that B. cepacia induces G2/M arrest in response to DNA damage to allow time for DNA repair. In addition, activation of the VEGF signaling pathway promotes angiogenesis, which is involved in the normal processes of wound healing. Therefore, exposure of the host to live B. cepacia and secretory proteins may have caused injuries or cell damage.
Additionally, cyclooxygenase (COX), an inflammation enzyme involved in release of excessive amounts of PGE2 (prostaglandin E2) was also highly up-regulated by the host upon exposure to B. cepacia secretory proteins, suggesting that COX may be an important mediator of B. cepacia pathogenesis. The enhanced expression of COX leads to further proliferation, reduction in apoptosis and angiogenesis [30,31]. Ejima et al. (2003) [32] suggested that COX is an important mediator of the response to bacterial sepsis when they showed that COX deficient mice had increased susceptibly to bacterial peritonitis and toxaemia. Thus, in this study, the induction of COX upon exposure to secreted products may partially explain the resistance of the epithelial cells to apoptosis.
Many researchers have demonstrated that infections by intracellular pathogens alter the expression of genes encoding pro-inflammatory cytokines and chemokines, which have been implicated as principal mediators during infections of the host in both in vitro and in vivo systems [6,7,33]. These cytokines and chemokines also function as central mediators in stimulating various host defences systems such as the cytokine-cytokine receptor interactions pathway, signalling pathways and apoptosis and eventually elicit appropriate adaptive immune system. Moreover, C-JUN, in combination with C-FOS, forms the AP-1 early response transcription factor, which up-regulates transcription of a diverse range of genes involved in proliferation and differentiation to defense against invasion and cell damage. In our study, the pro-inflammatory molecules were found to be down-regulated upon infection, and similar evidences have been observed from in vitro [33] and in vivo [34] studies. However, this is in contrast to the earlier report by Moreilhon and co-workers (2004) [7], which described up-regulation of the interleukins in the human airway cells when in contact with S. aureus supernatant.
Our microarray data demonstrated that B. cepacia interfered with the transcription of genes involved in the production of pro-inflammatory cytokines. Inhibition of these genes by B. cepacia would theoretically result in a reduction of recruitment of the innate immune cells to the site of infection, which in turn could influence the degree of host inflammatory response and bacterial elimination. B. cepacia has been shown to modulate the epithelial bactericidal response in favour of its intracellular survival and persistence in the human host, and this process may be associated with disease relapse. Feezor et al. (2002) [35] demonstrated that the release of TNFα, IL-10 and IL-6 into bactericidal-stimulated whole blood obtained from patients with severe sepsis was significantly decreased compared with the control group. Moreover, Ertel and colleagues (1995) [36] had hypothesised the role of systemic inflammation and an effectively functioning immune system of the host in eliminating invading pathogens. They speculated that during an infection, the pathogens may be made compromised differently depending on the time of exposure. In addition, they have also reported the excessive down-regulation of the synthesis of pro-inflammatory cytokines and their release in the early period after contact with pathogens. This may be an evolutionary process to reduce the incidence of tissue necrosis and the consequential multiple organ failure.
Likewise, Michie et al. (1988) [37] have demonstrated that infusion of high doses of pro-inflammatory cytokines may result in severe host tissue damage, organ failure and death in an in vivo condition. We presume that the response of the host cells to B. cepacia secretory proteins mimics the physiological action of the host towards toxins. This phenomenon may be referred to as ‘toxin tolerance,’ which is due to a reduced capacity of the cells to synthesise and/or secrete pro-inflammatory cytokines. Granowitz et al. (1993) [38] hypothesised that the phenomenon of toxin tolerance may be due to a defect in transcriptional and/or translational regulation of the host. These indicate that the control of the immune response of the host to toxins that is mediated by TNFα and IL-16 may represent a potent protective mechanism against infection with invasive microorganisms. In this light, the reduced capability of the host cells from septic shock to produce and release adequate amounts of pro-inflammatory cytokines after exposure to toxin may indicate the inability of septic patients to adequately respond to repeated or persisting invasion of microorganisms and to maintain an effective defence system.
In summary, our data is consistent with the interpretation that B. cepacia can manipulate A549 cells for its own advantage, allowing prolonged survival inside the harmless environmental niche of the non-phagocytic cells. The data presented clearly indicate that early exposure of the host cells to either live B. cepacia or their secretory proteins are similar. Contrary to popular belief, we postulate that B. cepacia induced homeostatic responses in the host cell which in turn aid the bacteria to establish persistent infection. Using this knowledge, strategies interfering with essential interactions between B. cepacia and the host cell, like apoptosis inhibition, can be exploited in the future to develop an innovative arsenal of therapeutic compounds.
Supporting Information
(DOC)
Acknowledgments
We are grateful to the Malaysia Genome Institute (MGI) for allowing us to use the microarray equipments and facilities and the Tropical Infectious Disease Research and Education Center (TIDREC), Faculty of Medicine, University of Malaya for allowing us to use the GeneSpring software.
Funding Statement
This study was supported by Ministry of Higher Education (MOHE), Malaysia under the High Impact Research (HIR)-MOHE project (E000013-20001) and Ministry of Science, Innovation and Technology (MOSTI), Malaysia under the Science Fund (55-02-03-1002). The funders had no role in the study design, data collection and analysis or the decision to publish or preparation of the manuscript.
References
- 1. Holt JG, Krieg NR, Sneath PHA, Stately JT, Williams St (1994) Bergey’s Manual of determinative bacteriology, 9th ed. Baltimore: Williams and Wilkins: USA [Google Scholar]
- 2. Jones AM, Dodd ME, Govan JRW, Barcus V, Doherty CJ et al. (2004) Burkholderia cenocepacia and Burkholderia multivorans: influence on survival in cystic fibrosis. Thorax 59: 948–951. doi: 10.1136/thx.2003.017210. PubMed: 15516469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Govan JRW, Hughes JE, Vandamme P (1996) Burkholderia cepacia: medical, taxonomic and ecological issues. J Med Microbiol 45: 395-407. doi: 10.1099/00222615-45-6-395. PubMed: 8958242. [DOI] [PubMed] [Google Scholar]
- 4. Martin DW, Mohr CD (2000) Invasion and intracellular survival of Burkholderia cepacia . Infect Immun 68: 24-29. doi: 10.1128/IAI.68.1.24-29.2000. PubMed: 10603364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mariappan V, Vellasamy KM, Hashim OH, Vadivelu J (2011) Profiling of Burkholderia cepacia secretome at mid-logarithmic and early-stationary phases of growth. PLOS ONE 6(10): e26518. doi: 10.1371/journal.pone.0026518. PubMed: 22046299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chin CY, Monack DM, Nathan S (2010) Genome wide transcriptome profiling of a murine acute melioidosis model reveals new insights into how Burkholderia pseudomallei overcomes host innate immunity. BMC Genomics 11: 672-680. doi: 10.1186/1471-2164-11-672. PubMed: 21110886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moreilhon C, Gras D, Hologne C, Bajolet O, Cottrez F et al. (2005) Live Staphylococcus aureus and bacteria soluble factors induce different transcriptional responses in human airway cells. Physiol Genomics. 20: 244-255. PubMed: 15598879. [DOI] [PubMed] [Google Scholar]
- 8. Mariappan V, Vellasamy KV, Thimma JS, Hashim OH, Vadivelu J (2010) Identification of immunogenic proteins from Burkholderia cepacia secretome using proteomic analysis. Vaccine 13: 1318-1324. PubMed: 19944788. [DOI] [PubMed] [Google Scholar]
- 9. Bradford MM (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-225. doi: 10.1016/0003-2697(76)90527-3. PubMed: 942051. [DOI] [PubMed] [Google Scholar]
- 10. Chavira R Jr., Burnett TJ, Hageman JH (1984) Assaying proteinases with azocoll. Anal Biochem 136: 446-450. doi: 10.1016/0003-2697(84)90242-2. PubMed: 6426343. [DOI] [PubMed] [Google Scholar]
- 11. Geoffroy C, Raveneau J, Beretti JL, Lecroisey A et al. (1991) Purification and characterization of an extracellular 29-kilodalton phospholipase C from Listeria monocytogenes . Infect Immun 59: 2382-2388. PubMed: 1904842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pine L, Hoffman PS, Malcolm GB, Benson RF, Gorman GW (1984) Whole-cell peroxidase test for identification of Legionella pneumophila . J Clin Microbiol 19: 286-290. PubMed: 6365966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Domenech CE, Lisa TA, Salvano MA, Garrido MN (1992) Pseudomonas aeruginosa acid phosphatase. Activation by divalent cations and inhibition by aluminium ion. FEBS Lett 299: 96–98. doi: 10.1016/0014-5793(92)80108-S. PubMed: 1544481. [DOI] [PubMed] [Google Scholar]
- 14. Lonon MK, Woods DE, Straus DC (1988) Production of lipase by clinical isolates of Pseudomonas cepacia . J Clin Microbiol 26: 979-984. PubMed: 3384918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andersen P, Askgaard D, Ljungqvist L, Bennedsen J, Heron I (1991) Proteins released from Mycobacterium tuberculosis during growth. Infect Immun 59: 1905-1910. PubMed: 1903768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miles AA, Misra SS (1938) Estimation of the bactericidal power of the blood. J Hyg (Camb.) 38: 732-749. doi: 10.1017/S002217240001158X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McKenney D, Brown KE, Allison DG (1995) Influence of Pseudomonas aeruginosa exoproducts on virulence factor production in Burkholderia cepacia: evidence of interspecies communication. J Bacteriol 177: 6989-6992. PubMed: 7592496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McKevitt AI, Bajaksouzian S, Klinger JD, Woods DE (1989) Purification and characterization of an extracellular protease from Pseudomonas cepacia . Infect Immun 57: 771–778. PubMed: 2645209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Govan JRW, Vandamme P (1998) Agricultural and medical microbiology: a time for bridging gaps. Microbiol 144: 2373–2375. doi: 10.1099/00221287-144-9-2373. PubMed: 9782485. [DOI] [PubMed] [Google Scholar]
- 20. Lefebre MD, Valvano MA (2001) In vitro resistance of Burkholderia cepacia complex isolates to reactive oxygen species in relation to catalase and superoxide dismutase production. Microbiol 147: 97-109. [DOI] [PubMed] [Google Scholar]
- 21. Caraher E, Duff C, Mullen T, Mc Keon S, Murphy P et al. (2007) Invasion and biofilm formation of Burkholderia dolosa is comparable with Burkholderia cenocepacia and Burkholderia multivorans . J Cyst Fibros 6: 49–56. doi: 10.1016/S1569-1993(07)60184-2. PubMed: 16781896. [DOI] [PubMed] [Google Scholar]
- 22. Duff C, Murphy PG, Callaghan M, McClean S (2006) Differences in invasion and translocation of Burkholderia cepacia complex species in polarised lung epithelial cells in vitro . Microb Pathog 41: 183–192. doi: 10.1016/j.micpath.2006.07.005. PubMed: 16938423. [DOI] [PubMed] [Google Scholar]
- 23. Cieri MV, Mayer-Hamblett N, Griffith A, Burns JL (2002) Correlation between an in vitro invasion assay and a murine model of Burkholderia cepacia lung infection. Infect Immun 70: 1081–1086. doi: 10.1128/IAI.70.3.1081-1086.2002. PubMed: 11854186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chiu CH, Wong S, Hancock REW, Speert DP (2001) Adherence of Burkholderia cepacia to respiratory tract epithelial cells and inhibition with dextrans. Microbiol 147: 2651–2658. PubMed: 11577144. [DOI] [PubMed] [Google Scholar]
- 25. Burns JL, Jonas M, Chi EY, Clark DK, Berger A et al. (1996) Invasion of respiratory epithelial cells by Burkholderia (Pseudomonas) cepacia . Infect Immun 64: 4054–4059. PubMed: 8926068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307-310. doi: 10.1038/35042675. PubMed: 11099028. [DOI] [PubMed] [Google Scholar]
- 27. Wells DB, Tighe PJ, Wooldridge KG, Robinson K, Aldeen DAA (2001) Differential gene expression during meningeal-meningococcal interaction: evidence for self-defense and early release of cytokines and chemokines. Infect Immun 69: 2718–2722. doi: 10.1128/IAI.69.4.2718-2722.2001. PubMed: 11254640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malumbres M, Sotillo R, Santamaría D, Galán J, Cerezo A et al. (2004) Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118: 493–504. doi: 10.1016/j.cell.2004.08.002. PubMed: 15315761. [DOI] [PubMed] [Google Scholar]
- 29. Zhang JM, Zhao X, Wei Q, Paterson BM (1999) Direct inhibition of G1 cdk kinase activity by MyoD promotes myoblast cell cycle withdrawal and terminal differentiation. EMBO J 18: 6983-6993. doi: 10.1093/emboj/18.24.6983. PubMed: 10601020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walduck A, Schmitt A, Lucas B, Aebischer T, Meyer TF (2004) Transcription profiling analysis of the mechanisms of vaccine-induced protection against Helicobacter pylori . FASEB J 18: 1955–1957. PubMed: 15456742. [DOI] [PubMed] [Google Scholar]
- 31. Oshima K, Kakizawa S, Nishigawa H, Jung HY, Wei W et al. (2004) Reductive evolution suggested from the complete genome sequence of a plant-pathogenic Phytoplasma. Nat Genet, 36: 27-29. doi: 10.1038/ng1277. PubMed: 14661021. [DOI] [PubMed] [Google Scholar]
- 32. Ejima K, Layne MD, Carvajal IM, Kritek PA, Baron RM et al. (2003) Cyclooxygenase-2-deficient mice are resistant to endotoxin-induced inflammation and death. FASEB J 17: 1325–1327. PubMed: 12738799. [DOI] [PubMed] [Google Scholar]
- 33. Wongprompitak P, Sirisinha S, Chaiyaroj CS (2008) Differential Gene Expression Profiles of lung epithelial cells exposed to Burkholderia pseudomallei and Burkholderia thailandensis during the initial phase of infection. Asia. Pac. J. Immuno: Allergy 26: 59-70. PubMed: 19548631. [PubMed] [Google Scholar]
- 34. Dinarello CA (1991) The proinflammatory cytokines interleukin- 1 and tumour necrosis factor and treatment of septic shock.syndrome. J Infect Dis 163: 1177-1184. doi: 10.1093/infdis/163.6.1177. PubMed: 2037782. [DOI] [PubMed] [Google Scholar]
- 35. Feezor RJ, Oberholzer C, Baker HV, Novick D, Rubinstein M et al. (2002) Molecular characterization of the acute inflammatory response to infections with Gram-negative versus Gram-positive bacteria. Infect Immun 71: 5803-5813. PubMed: 14500502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ertel W, Kremer JP, Kenney J, Steckholzer U, Jarrar D (1995) Down regulation of proinflammatory cytokine release in whole blood from septic patients. Blood 85: 1341-1347. PubMed: 7858264. [PubMed] [Google Scholar]
- 37. Michie HR, Manogue KR, Spriggs DR, Revhaug A, O’Dwyer S et al. (1988) Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med 318: 1481-1489. doi: 10.1056/NEJM198806093182301. PubMed: 2835680. [DOI] [PubMed] [Google Scholar]
- 38. Granowitz EV, Porat R, Mier JW, Orencole SF, Kaplanski G et al. (1993) Intravenous endotoxin suppresses the cytokine response of peripheral blood mononuclear cells of healthy humans. J Immunol 151: 1637-1645. PubMed: 7687636. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC)