

Abstract
Stimulation of mast cells through the high affinity IgE receptor (FcεRI) induces degranulation, lipid mediator release, and cytokine secretion leading to allergic reactions. Although various signaling pathways have been characterized to be involved in the FcεRI-mediated responses, little is known about the precious mechanism for the expression of tumor necrosis factor-α (TNF-α) in mast cells. Here, we report that rapamycin, a specific inhibitor of mammalian target of rapamycin (mTOR), reduces the expression of TNF-α in rat basophilic leukemia (RBL-2H3) cells. IgE or specific antigen stimulation of RBL-2H3 cells increases the expression of TNF-α and activates various signaling molecules including S6K1, Akt and p38 MAPK. Rapamycin specifically inhibits antigen-induced TNF-α mRNA level, while other kinase inhibitors have no effect on TNF-α mRNA level. These data indicate that mTOR signaling pathway is the main regulation mechanism for antigen-induced TNF-α expression. TNF-α mRNA stability analysis using reporter construct containing TNF-α adenylate/uridylate-rich elements (AREs) shows that rapamycin destabilizes TNF-α mRNA via regulating the AU-rich element of TNF-α mRNA. The antigen-induced activation of S6K1 is inhibited by specific kinase inhibitors including mTOR, PI3K, PKC and Ca2+chelator inhibitor, while TNF-α mRNA level is reduced only by rapamycin treatment. These data suggest that the effects of rapamycin on the expression of TNF-α mRNA are not mediated by S6K1 but regulated by mTOR. Taken together, our results reveal that mTOR signaling pathway is a novel regulation mechanism for antigen-induced TNF-α expression in RBL-2H3 cells.
Keywords: Mast cells, S6K1, Rapamycin, mTOR, mRNA, Stability
INTRODUCTION
Mast cells and basophils play a central role in the initia-tion of the symptomology associated with asthma and other allergic disorders. Activation of mast cells via aggregation of their high affinity receptor for IgE (FcεRI) leads to activation of different signal transduction pathways as well as production of various cytokine and arachidonic acid or histamine release (Beaven and Metzger, 1993; Galli, 1993; Razin et al., 1995; Boyce, 2003). The release of these potent mediators results in bronch constriction, vasodilation, tissue edema, leukocyte recruitment and other inflammatory reactions in the mucosa (Kopeć et al., 2006). All of these features are common in asthma and allergic diseases. The signal transduction pathways linking these events are important for the development of novel therapies for the treatment of mast cell-driven allergic disorders.
Mast cells bind IgE avidly via specific, high affinity Fc receptors,termed FcεRI. FcεRI are heterotetrameric complexes comprising an IgE-binding, a subunit, a αβ subunit, and two γsubunits (Ravetch and Kinet, 1991). In rat basophilic leukemia cells (RBL-2H3), cross-linking FcεRI has been known to activate the receptor-associated protein tyrosine kinases (PTKs), Lyn and Syk, as well as Bruton’s tyrosine kinase (Eiseman and Bolen, 1992; Hutchcroft et al., 1992; Kawakami et al., 1994) and cause the tyrosine phosphorylation of multiple substrates, including immune receptor tyrosine-based activation motifs in the subunits of the heterotrimeric (αβγ2) FcεRI itself (Li et al., 1992). Activation of these PTKs, in turn, facilitates the translocation and phosphorylation of multiple substrates, including phospholipase Cγ (PLCγ) isozymes, phosphoinositide 3-kinase (PI3K) and the adaptor protein Grb2 (Turner et al., 1995). Protein-tyrosine phosphorylation in turn activates a signaling cascade leading to inositol-1, 4, 5-trisphosphate (Ins (1, 4, 5)P3) (Wilson et al., 1989), Ca2+ mobilization (Millard et al., 1988; Lee and Oliver, 1995), Ras activation (Jabril-Cuenod et al., 1996), and the activation of the ERK and JNK MAP kinases. These biochemical and ionic responses lead to functional responses, including 1. secretion, 2. actin polymerization, 3. membrane ruffling, 4. the assembly of actin plaques implicated in cell spreading and 5. cytokine production.
Although the FcεRI-mediated signaling cascade has been characterized, the regulatory mechanism governing mast cell degranulation and cytokine production is only partially understood. Also there are still unknown signaling cascades including mTOR-S6K1 signaling pathway and their functions in antigen-induced RBL-2H3 cells. The coordinated control of cell growth to produce a genetically predetermined cell size, organ shape or body plan is greatly influenced by mammalian target of rapamycin (mTOR) and its downstream effect or S6 kinase 1 (S6K1) (Um et al., 2006). The anti-fungal macrolide rapamycin blocks mTORcomplex1 function by forming a gain-of-function inhibitory complex with the immunophilin FK506 binding protein 1A (FKBP12) (Brown et al., 1994). Rapamycin is a potent immune suppressive agent which inhibits the pro-liferation and the clonal expansion of interleukin-2-stimulated T cells (Combates et al., 1995). Recent reports show that rapamycin can regulate cycin D3 and Il-3 mRNA levels through their 3'untranslated region (3'UTR) (Banholzer et al., 1997; Pallet et al., 2005). AU-rich elements (AREs) are cis elements that are contained within the 3' UTR of the many short lived mRNAs and constitutes repetitive AU motifs which target the transcript for rapid deadenylation and degradation (Bakheet et al., 2001). AREs promote deadenylation and subsequent degradation of the mRNA body (Shyu et al., 1991; Xu et al., 1997). The targeted deletion of a very similar ARE from the mousegenomic TNF-α locus causes an apparent increase in TNF-α mRNA stability (Kontoyiannis et al., 1999).
In this study, we attempted to investigate possible signaling pathways for TNF-α expression in response to the cross-linking of the high affinity receptor for IgE (FcεRI). Our data show that rapamycin specifically inhibit TNF-α mRNA expression, while other kinase inhibitors have no effect on TNF-α mRNA level. The inhibition of TNF-α mRNA expression by rapamycin results from destabilization of the mRNA through regulating AU-rich element of TNF-α mRNA. However, the effect seems not to be mediated by S6K1, a downstream of mTOR, because antigen-induced TNF-α is reduced only by rapamycin but not by other molecules inhibiting S6K activity.
MATERIALS AND METHODS
Cell culture and reagents
RBL-2H3 cells were grown as monolayer in MEM medium (Gibco BRL) supplemented with 15% heat inactivated fetal bovine serum (HyClone Laboratories, Logan, UT, USA), and 1% penicillin/streptomycin (Gibco BRL). 293T cells were grown as monolayer in DMEM medium (Gibco BRL) supplemented with 10% heat inactivated fetal bovine serum (HyClone Laborato-ries, Logan, UT, USA), and 1% penicillin/streptomycin (Gibco BRL). Calphostin C, wortmannin, LY294002, Rottlerin were obtained from Calbiochem (LA Jolla, CA). Rapamycin, actinomycin D were obtained from Sigma (St. Louis, MO).
Cell stimulation by FcεRI cross-linking
RBL-2H3 cells were incubated overnight in a complete growth medium with 25 ng/ml DNP-specific IgE to achieve 100% occupancy of FcεRI. Cells were incubated with or with-out indicated inhibitor before adding stimulants, 25 ng/ml an-tigen (DNP-BSA).
Immunoblotting
Cell lysates were boiled in Laemmli sample buffer for 3 min, and 30 g of each protein was subjected to SDS-polyacrylamide gel electrophoresis. Proteins were transferred to polyvinylidene difluoride membranes, and the membranes were blocked for 30 min in Tris-buffered saline containing 0.1% Tween 20 and 5% (w/v) dried skim milk powder, and incubated overnight with primary antibodies for with individual monoclonal or polyclonal antibodies. The immunoreactive proteins were detected using horse-radish peroxidase coupled secondary antibodies and through enhanced chemiluminescence,according to the manufacturer’s (Amersham Pharmacia Biotech) instructions.
RT-PCR
Total RNA was extracted using the easy-BLUETM total RNA extraction kit (iNtRON Biotechnology). Integrity of RNA was checked by agarose gel electrophoresis and ethidium bromide staining. One microgram of RNA was used as a template and reversed transcribed with the Superscript first strand synthesis system (Invitrogen) according to the manufacturer’s protocol. PCR was performed at 94℃ for 45 s, 55℃ for 45 s and 72℃ for 60 s for 30 cycles. The following primers were used: rat TNF-α forward 5’-CACCACGCTCTTCTGTCTACTGAA-3’, re verse 5’-CCGGACTCCGTGATGTCTAAGTACT-3’; rat GAPDH forward 5’-GTGGAGTCTACTGGCGTCTTC-3’, reverse 5’-CCAAGGCTGTGGGCAAGGTCA-3’.
Luciferase reporter gene assay
293T cells were seeded into 24-well plates and cultured for 24 h before transfection. TNF-α-luciferase reporter plasmid DNA (0.5 μg) or TNF-α ARE, (nucleotides 1,293-1,383); and ARE deletion (ARE-Del) was transfected using Lipofectamine transfection reagent (Invitrogen) for 24 h. After transfection, the media were freshly replaced and incubated for an additional 24 h following treatment with 20 nM TPA with or without 50 nM rapamycin. The luciferase activity of the cell lysates was measured according to the manufacturer's instructions (Promega). Relative luciferase activity was normalized by total protein content of the extract. TNF-α ARE and ARE deletion plasmids are kindly provided by Dr. William F. C. Rigby (Dart-mouth Hitchcock Medical Center).
RESULTS
Stimulation of RBL-2H3 cells by FcεRI cross-linking activates various signaling pathways and induces TNF-α mRNA level
Downstream phosphorylation events following FcεRI ag-gregation are composed of complex signaling pathways and are responsible for various events such as degranulation, lipid mediator release, and cytokine secretion. Recently we reported the effects of Arecae semen on the phosphorylation of MAP kinase and the expression of TNF-α in RBL-2H3 cells (Lee et al., 2004). To obtain more insight for the effect of FcεRI aggregation, we examined antigen-stimulation in RBL-2H3 cells on the production of TNF-α and various signaling activation.
RBL-2H3 cells were stimulated with various concentration of antigenat various for several times. Consistent with previous report (Gu et al., 2001), activity of AKT, p38 MAP kinase was obviously up-regulated by antigen-stimulation. Interestingly
S6K1 activity also followed similar kinetic patterns by antigen-stimulation. AKT, S6K1, p38 MAP kinase were activated by antigen-stimulation, occurring at a concentration of 10 nM,reaching the maximum at 50 nM of antigen with a gradual reduction after 100 nM (Fig. 1B). The increase in phosphorylation of S6K1, Akt and p38 MAP kinase was apparent at 5 min and sustained for 30 min (Fig. 1A). In parallel to the various signal activation, TNF-α mRNA level reached a maximum in the range of 50 to 100 ng/ml of antigen and the TNF-α mRNA level was apparent as early as 5 min and sustained for 30 min (Fig. 1C and D). Collectively, these findings suggest that FcεRI aggregation up-regulates various signaling pathways including mTOR-S6K1 signaling pathway and induces the expression of TNF-α in RBL-2H3 cells.
Fig. 1. Stimulation of RBL 2H3 cells by FcεRI crosslinking activates various signaling pathway. (A, C) IgE-sensitized cells were stimulated with DNP-BSA (50 ng/ml) for the indicated period of time. (B, D) IgE-sensitized cells were stimulated with indicated concentrations of DNP-BSA for 15 min. Cell lysates containing total protein were run on 12% SDS-PAGE gel blotted and probed with indicated antibody. The Expression level of TNF-α mRNA was detected by RT-PCR analysis. GAPDH was used to normalize the amount of loaded RNA.

Rapamycin specifically reduces TNF-α mRNA level
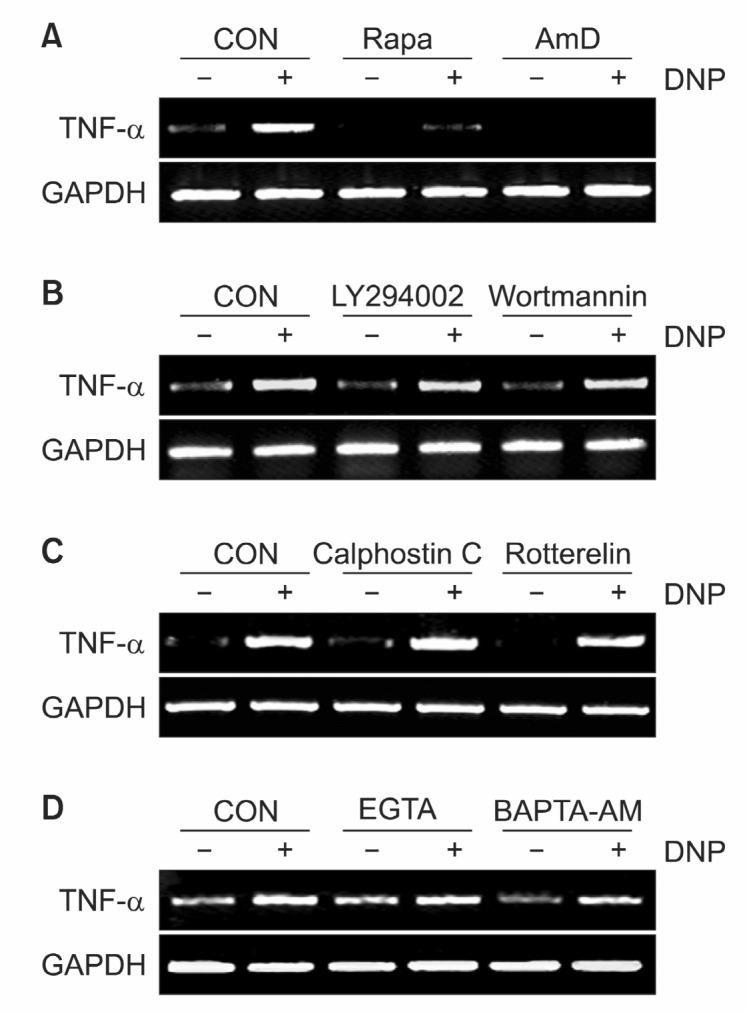
We next tried to confirm the role of mTOR signaling pathways in the transcription of TNF-α, which is one of the most important cytokines in immune reaction by IgE aggregation. Pretreatment of RBL-2H3 cells with rapamycin (10 nM), specific inhibitor for mTOR, or transcriptional inhibitor actinomycin D (10 μg/ml) completely abolished TNF-α transcription down to basal level (Fig. 2A). Interestingly pretreatment with PI3K inhibitor wortmannin (500 nM) or LY294002 (25 nM) had no effect on TNF-α transcription level (Fig. 2B). Likely, other signaling inhibitors, PKC inhibitor and Ca2+chelator had no effect on antigen-stimulated TNF-α mRNA transcription (Fig. 2C and D). These data indicate that the activation of mTOR signal pathway is necessary for antigen-induced TNF-α expression.
Fig. 2. Rapamycin specifically reduces TNF-α mRNA level. (A)RBL 2H3 cells were pretreated with either rapamycin (10 nM) or actinomycin D (10 μg/ml) for 30 min and stimulated with antigen for 15 min. (B) RBL 2H3 cells were pretreated with either wortmannin (500 mM) or LY294002 (25 μM) for 30 min and stimulated with antigen for 15 min. (C) RBL 2H3 cells were pretreated with either calphostin C (0.5 μM) or rottlerin (5 μM) for 30 min and stimulated with antigen for 15 min. (D) RBL 2H3 cells were pretreated with either EGTA (2 mM) or BAPTA-AM (25 μM) for 30 min and stimulated with antigen for 15 min. TNF-α mRNA level was analyzed by RT-PCR. GAPDH was used to normalize the amount of loaded RNA.
Rapamycin destabilizes TNF-α mRNA at post-transcriptional level
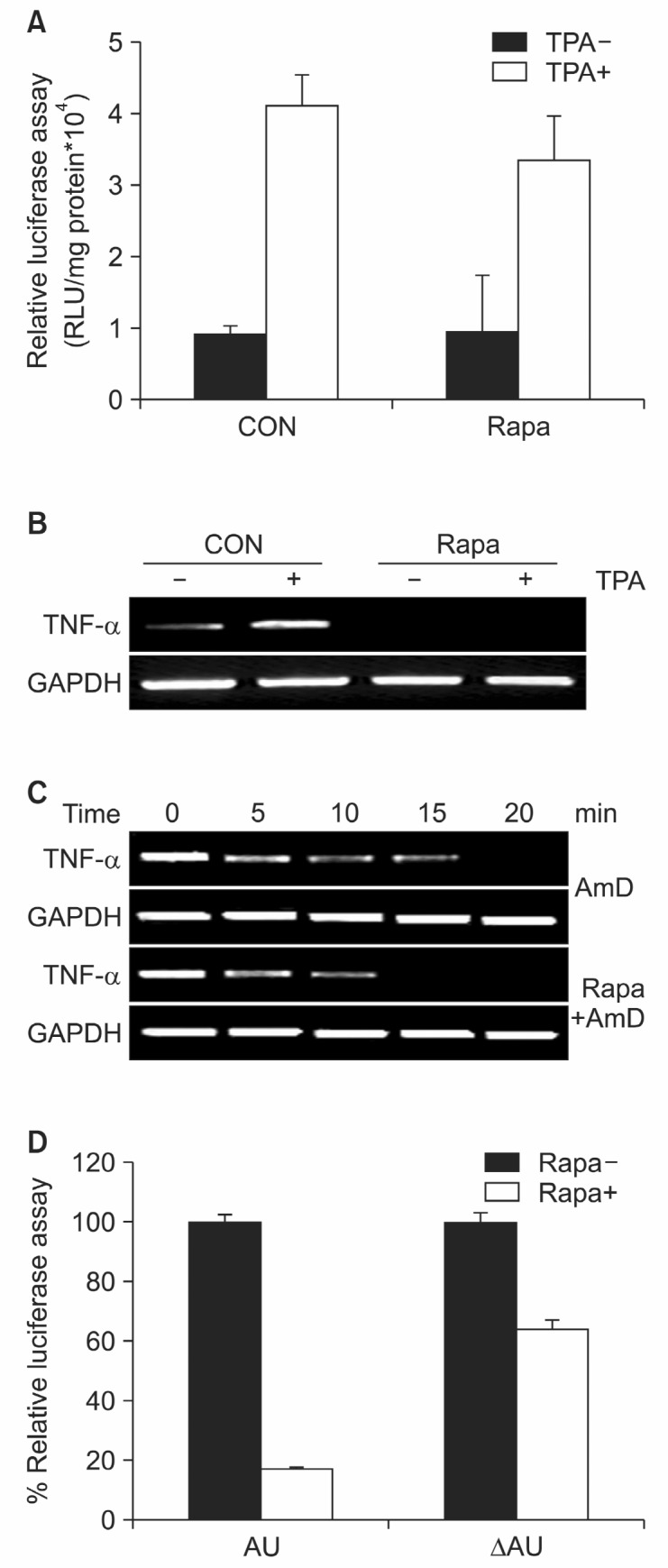
Recent evidence suggests that gene expression may be regulated, at least in part, at the post-transcriptional level by factors inducing the extremely rapid degradation of messenger RNAs. The adenylate/uridylate-rich elements (AREs) present within the 3'-untranslated region (UTR) of mRNAs represent the most characterized and well-conserved group of sequences that are functionally associated with the regulation of mRNA stability and translation (Garneau et al., 2007). Several in vivo and in vitro evidences indicate that the post-transcriptional control of inflammatory transcripts is strongly dependent on ARE-mediated mechanisms (Clark, 2000). Using a TNF-α reporter gene, we investigated the effects of rapamycin on transcriptional activity of TNF-α. Rapamycin reduced TPA-induced TNF-α mRNA level in 293T cells, while it had little effect on TPA-induced TNF-α promoter activity (Fig. 3A and B). To confirm the fact that rapamycin acts at the post-transcriptional level, we compared the decay of TNF-transcripts between rapamycin treated and untreated cells. The decay of transcripts from rapamycin treated cells was more rapid then controls (Fig. 3C). Using reporter construct either has TNF-α AU-rich region or not, we investigated if ARE is the target region of rapamycin induced TNF-α mRNA destabilization. Treatment of rapamycin induced a significant decrease in luciferase activity in 293T cells transfected with ARE contained construct (Fig. 3D). Taken together, rapamycin regulates TNF-α mRNA at the post-transcriptional level and it mediates AU rich element of TNF-α 3’ region.
Fig. 3. Rapamycin destabilizes TNF-α mRNA at post-transcriptional level. (A) RBL 2H3 cells were transfected with pREP luciferase reporter plasmid. After 24 hrs, cells were pretreated with rapamycin (50 nM). 5 mM TPA were added and after 12 hrs incubation cells were harvested. Luciferase activity was normalized to the protein content of each extract. The data were shown as the means (bars, S.E) (n=3). (B) Expression of TNF-α mRNA was detected by RT-PCR. (C) RBL 2H3 cells were stimulated with 10 ng/ml LPS for 4 h, then actinomycin D (10 μg/ml) was added in the absence or presence of rapamycin (50 nM). Cells were harvested at the time intervals as discribed and TNF-α mRNA level were analyzed with RT-PCR method. (D) RBL 2H3 cells were transfected with pGL3 luciferase reporter plasmid. Actinomycin D (10 μg/ml) was added in the absence or presence of rapamycin (50 nM). Luciferase activity was normalized to the protein content of each extract.
The effects of Rapamycin on the expression of TNF-αmRNA are not mediated by S6K1 but regulated by mTOR
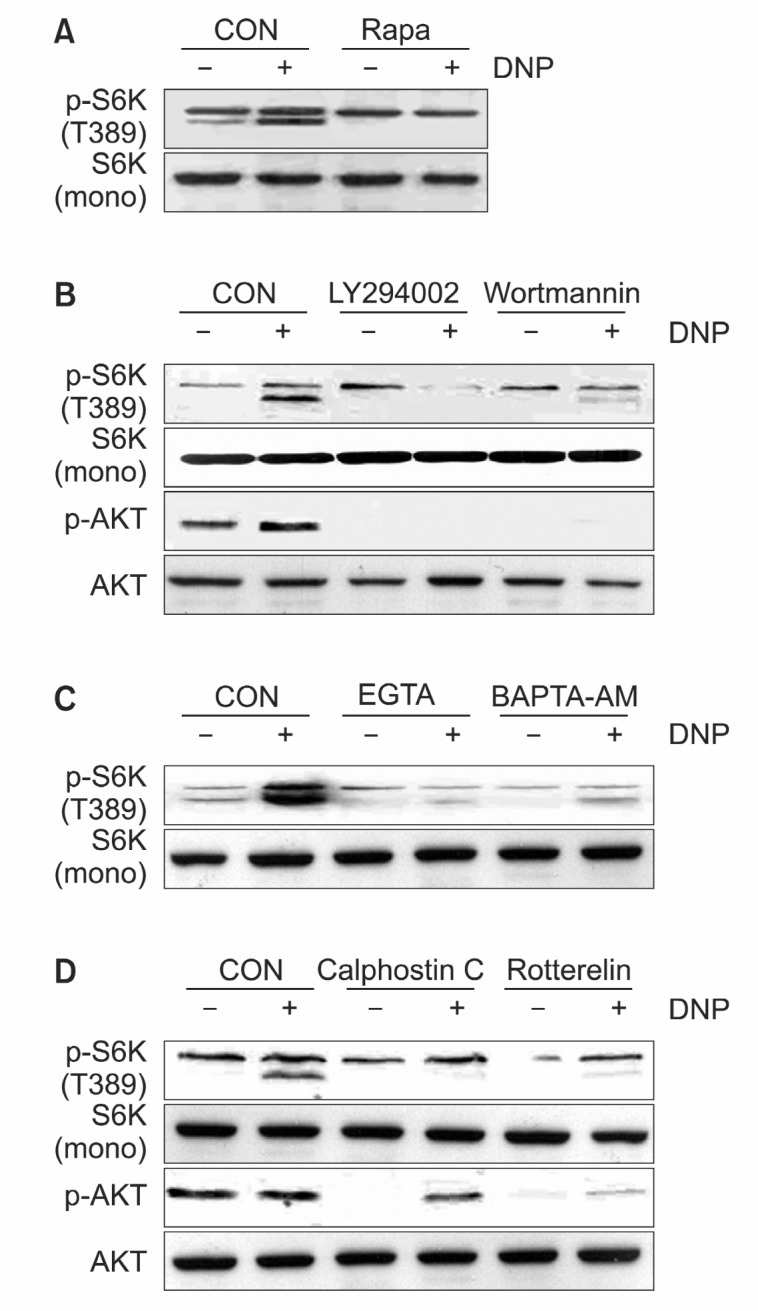
Because S6K1 activation is one of the most important downstream signals of mTOR, we next investigated whether the effects of rapamycin on TNF-α expression are mediated by S6K1. Also we previously reported various signaling molecules including Rac1 and PI3K mediate mitogen activation of S6K1 which plays an important role in cell proliferation and growth (Bae et al., 1999; Jung et al., 2003; Bae et al., 2004). However, little is known about signaling molecules involved in S6K1 activation followed by FcεRI aggregation. So we examined upstream molecules of S6K1 activation using a variety of known inhibitors. Pretreatment of RBL-2H3 cells with rapamycin (10 nM) completely abolished S6K1 phosphorylation (Fig. 4A). Also pretreatment with PI3K inhibitor wortmannin (500 nM) or LY294002 (25 nM) completely abolished S6K1 and Akt phosphorylation (Fig. 4B). These data indicated that mTOR and PI3K were required for the stimulation of S6K1 by IgE sensitization. This was consistent with data that showed the involvement of PI3K and mTOR in growth factor-induced S6K1 signaling pathway (Brown et al., 1994; Weng et al., 1995). Also incubation of RBL-2H3 cells with extracellular Ca2+ chelator EGTA (2 mM) or the cellpermeant Ca2+ chelator BAPTA-AM inhibited antigen-induced S6K1 phosphorylation (Fig. 4C). These results indicate that antigen-stimulated S6K1 signaling pathway is dependent on intracellular Ca2+ in RBL-2H3 cells.
Fig. 4. The effects of rapamycin on TNF-α expression are not mediated by S6K1. (A) RBL 2H3 cells were pretreated with either rapamycin (10 nM) for 30 min or not and stimulated with antigen for 15 min. (B) RBL 2H3 cells were pretreated with either wortmannin (500 mM) or LY294002 (25 μM) for 30 min and stimulated with antigen for 15 min. (C) RBL 2H3 cells were pretreated with either EGTA (2 μmM) or BAPTA-AM (25 μM) for 30 min and stimulated with antigen for 15 min. (D) RBL 2H3 cells were pretreated with either calphostin C (0.5 μM) or rottlerin (5 μM) for 30 min and stimulated with antigen for 15 min. Cell lysates were subjected to 12% SDS-PAGE and analyzed with immunoblotting using indicated antibody.
PKC comprises a large family of multiple isoforms that exhibit
distinct properties, including sensitivities to calcium and the phorbol ester family of tumor promoters. Pretreatment with calphostin C (0.5 μM), a pan-specific inhibitor, and rottlerin (5 μM), a PKCδ-specific inhibitor, significantly attenuated the phosphorylation of S6K1 and Akt. These results suggest that antigen-induced Akt and S6K1 activation requires PKC signaling, and PKC. might be an important factor for Akt and S6K1 activation (Fig. 4D).
Although the antigen-induced activation of S6K1 is inhib-ited by specific kinase inhibitors including mTOR, PI3K, PKC and Ca2+ chelator inhibitor (Fig. 4), TNF-α mRNA level is only reduced by rapamycin treatment (Fig. 2). These data indicate that the effects of rapamycin on antigen-induced TNF-α expression are not mediated by S6K1.
DISCUSSION
FcεRI aggregation elicits the release of three major categories of inflammatory mediators from mast cells; i) presynthesized granule-associated mediators, such as histamine and tryptase, which are released following exocytosis, ii)lipid-derived molecules, such as prostaglandin D2 and leukotriene C4, which are denovo synthesized and then released following cellular activation (Nakamura et al., 1991), iii) cytokines and chemokines which are synthesized and released as a result of enhanced gene expression. TNF-α is expressed by lymphocytes and macrophages and is a critical mediator of joint inflammation in rheumatoid arthritis. Activation of macrophages results in a 10,000-fold increase in TNF-α biosynthesis with only a 3-fold increase in transcription (Kontoyiannis et al., 1999). Thus, the expression of TNF-α is primarily regulated at the level of messenger RNA (mRNA) stability and translation. This post-transcriptional regulation of TNF-α expression is mediated through an adenine-uridine-rich element (ARE) in its 3'-untranslated region (3'-UTR) (Kontoyiannis et al., 1999).
Studies on signal transduction following antigen-mediated aggregation of FcεRI on the mast cell surface have identified the roles of each signaling molecule in the initiation of inflammatory reactions associated with allergic disorders. Several reports have identified requirement of each signaling pathways for various events following FcεRI cross-linking in mast cells. PI3K is known to play a concerted role with PLCγ in the regulation of Ca2+influx in RBL-2H3 mast cells via a PI(3,4,5) P3- sensitive Ca2+entry pathway (Ching et al., 2001). PKCεhas been suggested to be implicated in the suppression of phospholipase (PL) A2 activation in MCs, indicating its involvement in negative regulation of calcium mobilization and/or MAPK activation (Chang et al., 1997). Also PKCε has been shown to positively affect transcription of Fos/Jun transcription factors (Razin et al., 1995). Although the expression of TNF-α is one of the most important cytokines in immune reaction by IgE aggregation, the specific mechanism for antigen-induced TNF-α is yet to be defined.
Recent studies demonstrate that mTOR-S6K1 signaling pathway has a major role in cell growth by integrating growth factor and nutrient cascades. Using various model systems, it was shown that this pathway is essential for cellular homeostasis and that aberrant modulation of this pathway can contribute to obesity, diabetes and cancer (Dann et al., 2007). However, little is known about the role of mTOR-S6K1 signaling pathway in immune responses including antigen-induced RBL 2H3 cells. S6K1 phosphorylates multiple sites of the 40 S ribosomal protein S6 resulting in a specific increase in the translation of a subset of mRNAs containing a polypyrimidine tract in their 5′-untranslated region (5′ TOP mRNAs) (Jefferies et al., 1997). About 20% of the total mRNA in the cell consists of this class of mRNAs which encode many components of the protein synthetic apparatus. Consistent with this finding, inhibition of S6K1 activation by microinjection of neutralizing antibodies into cells, or by treatment of cells with the immu-nosuppressant rapamycin (Kuo et al., 1992), severely suppresses cell cycle progression.
In this study we identified the role of mTOR signaling pathway in response to the cross-linking of FcεRI in RBL 2H3. Treatment of RBL 2H3 cells with IgE and specific antigen (Ag) activates various signaling molecules including AKT, MAPK and S6K1. It is interesting that mTOR-S6K1 was activated by FcεRI cross-linking in mast cells since mTOR was known as an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. In addition, rapamycin specifically inhibited FcεRI-mediated induction of TNF-α expression suggesting mTOR signaling pathway was required for antigen-induced TNF-α expression. Using a actinomycin D or reporter constructs, these effects of rapamycin were post-transcriptional and resulted from destabilization of the mRNA. Transcripts from transgenes lacking AU-rich 3’region were refractory to rapamycin-induced degradation, suggesting that these 3’ sequences were the target of the rapamycin effect. The antigen-induced activation of S6K1 was inhibited by specific kinase inhibitors including mTOR, PI3K, PKC and Ca2+chelator inhibitor, while TNF-α mRNA level was reduced by only rapamycin treatment. These data suggest that the effects of rapamycin on the expression of TNF-α mRNA are not mediated by S6K1 but regulated by mTOR.
Taken together, these results provide a previously unrecognized link between the mTOR signal pathway and antigen-mediated signal pathway in mast cells, and they also show that mTOR signal pathway is a key regulation mechanism for regulating TNF-α expression in immune response.
Acknowledgments
This research was supported by the health fellowship foundation.
References
- 1.Bae G. U. Kim Y. K. Kwon H. K. Park J. W. Lee E. K. Paek S. J. Choi W. S. Jung I. D. Lee H. Y. Cho E. J. Lee H. W. Han J. W. Hydrogen peroxide mediates Rac1 activation of S6K1. Exp. Cell Res. (2004);300:476–484. doi: 10.1016/j.yexcr.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 2.Bae G. U. Seo D. W. Kwon H. K. Lee H. Y. Hong S. Lee Z. W. Ha K. S. Lee H. W. Han J. W. Hydrogen peroxide activates p70(S6k) signaling pathway. J. Biol. Chem. (1999);274:32596–32602. doi: 10.1074/jbc.274.46.32596. [DOI] [PubMed] [Google Scholar]
- 3.Bakheet T. Frevel M. Williams B. R. Greer W. Khabar K. S. ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic. Acids Res. (2001);29:246–254. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banholzer R. Nair A. P. Hirsch H. H. Ming X. F. Moroni C. Rapamycin destabilizes interleukin-3 mRNA in autocrine tumor cells by a mechanism requiring an intact 3' untranslated region. Mol. Cell Biol. (1997);17:3254–3260. doi: 10.1128/mcb.17.6.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beaven M. A. Metzger H. Signal transduction by Fc receptors: the Fc epsilon RI case. Immunol. Today. (1993);14:222–226. doi: 10.1016/0167-5699(93)90167-J. [DOI] [PubMed] [Google Scholar]
- 6.Boyce J. A. The role of mast cells in asthma. Prostaglandins Leukot. Essent. Fatty. Acids. (2003);69:195–205. doi: 10.1016/S0952-3278(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 7.Brown E. J. Albers M. W. Shin T. B. Ichikawa K. Keith C. T. Lane W. S. Schreiber S. L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. (1994);369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 8.Chang E. Y. Szallasi Z. Acs P. Raizada V. Wolfe P. C. Fewtrell C. Blumberg P. M. Rivera J. Functional effects of overexpression of protein kinase C-alpha -beta -delta -epsilon and -eta in the mast cell line RBL-2H3. J. Immunol. (1997);159:2624–2632. [PubMed] [Google Scholar]
- 9.Ching T. T. Hsu A. L. Johnson A. J. Chen C. S. Phosphoinositide 3-kinase facilitates antigen-stimulated Ca2+ influx in RBL-2H3 mast cells via a phosphatidylinositol 3,4,5-trisphosphate-sensitive Ca2+ entry mechanism. J. Biol. Chem. (2001);276:14814–14820. doi: 10.1074/jbc.M009851200. [DOI] [PubMed] [Google Scholar]
- 10.Clark A. Post-transcriptional regulation of pro-inflammatory gene expression. Arthritis. Res. 2000;2:172–174. doi: 10.1186/ar83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Combates N. J. Degiannis D. Raskova J. Raska K. Jr. Direct inhibition of human CD8+ lymphocyte activation by cyclosporine A and Rapamune-Sirolimus. Clin. Immunol. Immunopathol. (1995);77:221–228. doi: 10.1006/clin.1995.1147. [DOI] [PubMed] [Google Scholar]
- 12.Dann S. G. Selvaraj A. Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol. Med. (2007);13:252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Eiseman E. Bolen J. B. Engagement of the high-affinity IgE receptor activates src protein-related tyrosine kinases. Nature. (1992);355:78–80. doi: 10.1038/355078a0. [DOI] [PubMed] [Google Scholar]
- 14.Galli S. J. New concepts about the mast cell. N. Engl. J. Med. (1993);328:257–265. doi: 10.1056/NEJM199301283280408. [DOI] [PubMed] [Google Scholar]
- 15.Garneau N. L. Wilusz J. Wilusz C. J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. (2007);8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 16.Gu H. Saito K. Klaman L. D. Shen J. Fleming T. Wang Y. Pratt J. C. Lin G. Lim B. Kinet J. P. Neel B. G. Essential role for Gab2 in the allergic response. Nature. (2001);412:186–190. doi: 10.1038/35084076. [DOI] [PubMed] [Google Scholar]
- 17.Hutchcroft J. E. Geahlen R. L. Deanin G. G. Oliver J. M. Fc epsilon RI-mediated tyrosine phosphorylation and activation of the 72-kDa protein-tyrosine kinase, PTK72, in RBL-2H3 rat tumor mast cells. Proc. Natl. Acad. Sci. USA. (1992);89:9107–9111. doi: 10.1073/pnas.89.19.9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jabril-Cuenod B. Zhang C. Scharenberg A. M. Paolini R. Numerof R. Beaven M. A. Kinet J. P. Syk-dependent phosphorylation of Shc. A potential link between FcepsilonRI and the Ras/mitogen-activated protein kinase signaling pathway through SOS and Grb2. J. Biol. Chem. (1996);271:16268–16272. doi: 10.1074/jbc.271.27.16268. [DOI] [PubMed] [Google Scholar]
- 19.Jefferies H. B. Fumagalli S. Dennis P. B. Reinhard C. Pearson R. B. Thomas G. Rapamycin suppresses 5'TOP mRNA translation through inhibition of p70s6k. EMBO J. (1997);16:3693–3704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung D. K. Bae G. U. Kim Y. K. Han S. H. Choi W. S. Kang H. Seo D. W. Lee H. Y. Cho E. J. Lee H. W. Han J. W. Hydrogen peroxide mediates arsenite activation of p70s6k and extracellular signal-regulated kinase. Exp. Cell Res. (2003);290:144–154. doi: 10.1016/S0014-4827(03)00320-3. [DOI] [PubMed] [Google Scholar]
- 21.Kawakami Y. Yao L. Miura T. Tsukada S. Witte O. N. Kawakami T. Tyrosine phosphorylation and activation of Bruton tyrosine kinase upon Fc epsilon RI cross-linking. Mol. Cell Biol. (1994);14:5108–5113. doi: 10.1128/mcb.14.8.5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kontoyiannis D. Pasparakis M. Pizarro T. T. Cominelli F. Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. (1999);10:387–398. doi: 10.1016/S1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 23.Kopeć A. Panaszek B. Fal A. M. Intracellular signaling pathways in IgE-dependent mast cell activation. Arch. Immunol.Ther. Exp. (Warsz). (2006);54:393–401. doi: 10.1007/s00005-006-0049-4. [DOI] [PubMed] [Google Scholar]
- 24.Kuo C. J. Chung J. Fiorentino D. F. Flanagan W. M. Blenis J. Crabtree G. R. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. (1992);358:70–73. doi: 10.1038/358070a0. [DOI] [PubMed] [Google Scholar]
- 25.Lee J. H. Chang S. H. Park Y. S. Her E. Lee H. Y. Park J. W. Han J. W. Kim Y. M. Choi W. S. In-vitro and in-vivo anti-allergic actions of Arecae semen. J. Pharm. Pharmacol. (2004);56:927–933. doi: 10.1211/0022357023808. [DOI] [PubMed] [Google Scholar]
- 26.Lee R. J. Oliver J. M. Roles for Ca2+ stores release and two Ca2+ influx pathways in the Fc epsilon R1-activated Ca2+ responses of RBL-2H3 mast cells. Mol. Biol Cell. (1995);6:825–839. doi: 10.1091/mbc.6.7.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W. Deanin G. G. Margolis B. Schlessinger J. Oliver J. M. Fc epsilon R1-mediated tyrosine phosphorylation of multiple proteins, including phospholipase C gamma 1 and the receptor beta gamma 2 complex, in RBL-2H3 rat basophilic leukemia cells. Mol. Cell Biol. (1992);12:3176–3182. doi: 10.1128/mcb.12.7.3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Millard P. J. Gross D. Webb W. W. Fewtrell C. Imaging asynchronous changes in intracellular Ca2+ in individual stimulated tumor mast cells. Proc. Natl. Acad. Sci. USA. (1988);85:1854–1858. doi: 10.1073/pnas.85.6.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura T. Fonteh A. N. Hubbard W. C. Triggiani M. Inagaki N. Ishizaka T. Chilton F. H. Arachidonic acid metabolism during antigen and ionophore activation of the mouse bone marrow derived mast cell. Biochim. Biophys Acta. (1991);1085:191–200. doi: 10.1016/0005-2760(91)90094-x. [DOI] [PubMed] [Google Scholar]
- 30.Pallet N. Thervet E. Le Corre D. Knebelmann B. Nusbaum P. Tomkiewicz C. Meria P. Flinois J. P. Beaune P. Legendre C. Anglicheau D. Rapamycin inhibits human renal epithelial cell proliferation: effect on cyclin D3 mRNA expression and stability. Kidney Int. (2005);67:2422–2433. doi: 10.1111/j.1523-1755.2005.00350.x. [DOI] [PubMed] [Google Scholar]
- 31.Ravetch J. V. Kinet J. P. Fc receptors. Annu. Rev. Immunol. (1991);9:457–492. doi: 10.1146/annurev.iy.09.040191.002325. [DOI] [PubMed] [Google Scholar]
- 32.Razin E. Pecht I. Rivera J. Signal transduction in the activation of mast cells and basophils. Immunol. Today. (1995);16:370–373. doi: 10.1016/0167-5699(95)80003-4. [DOI] [PubMed] [Google Scholar]
- 33.Shyu A. B. Belasco J. G. Greenberg M. E. Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mRNA decay. Genes Dev. (1991);5:221–231. doi: 10.1101/gad.5.2.221. [DOI] [PubMed] [Google Scholar]
- 34.Turner H. Reif K. Rivera J. Cantrell D. A. Regulation of the adapter molecule Grb2 by the Fc epsilon R1 in the mast cell line RBL2H3. J. Biol. Chem. (1995);270:9500–9506. doi: 10.1074/jbc.270.16.9500. [DOI] [PubMed] [Google Scholar]
- 35.Um S. H. D'Alessio D. Thomas G. Nutrient overload, insulin resistance and ribosomal protein S6 kinase 1, S6K1. Cell Metab. (2006);3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Weng Q. P. Andrabi K. Klippel A. Kozlowski M. T. Williams L. T. Avruch J. Phosphatidylinositol 3-kinase signals activation of p70 S6 kinase in situ through site-specific p70 phosphorylation. Proc. Natl. Acad. Sci. USA. (1995);92:5744–5748. doi: 10.1073/pnas.92.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson B. S. Deanin G. G. Standefer J. C. Vanderjagt D. Oliver J. M. Depletion of guanine nucleotides with mycophenolic acid suppresses IgE receptor-mediated degranulation in rat basophilic leukemia cells. J. Immunol. (1989);143:259–265. [PubMed] [Google Scholar]
- 38.Xu N. Chen C. Y. Shyu A. B. Modulation of the fate of cytoplasmic mRNA by AU-rich elements: key sequence features controlling mRNA deadenylation and decay. Mol. Cell Biol. (1997);17:4611–4621. doi: 10.1128/mcb.17.8.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]