

Abstract
INTRODUCTION
Our previous work demonstrated that the transforming-growth factor (TGF) β pathway plays a central role in the liver fibrosis associated with experimental biliary atresia (BA). To confirm these findings in humans, we performed an in silico analysis of publicly available microarray data from liver specimens from children with BA, with the hypothesis that the TGF β pathway would be dysregulated.
METHODS
We analyzed publicly available liver gene expression microarray data from 47 infants with BA. We re-analyzed the microarray image files and clinical data to compare gene expression differences between the fibrogenic and inflammatory cohorts identified in the initial study. Targets from the microarray analysis were confirmed using the animal model of BA.
RESULTS
Analysis of variance (ANOVA) detected 6903 transcripts (2822 distinct genes) differentially regulated between groups (p<0.01; fold change >1.2). We used a targeted approach to identified a subgroup of 24 TGF β-related transcripts. Expressions for procollagen transcripts were increased in the fibrogenic group (1.2 fold to 1.4 fold); expression of matrix metalloproteinase (MMP)-7 was similarly increased 2-fold, while MMP-9 and plasminogen activator inhibitor-1 were decreased 2-fold and 3-fold respectively. Integrins β5 (1.18 fold) and β8 (1.84 fold) also demonstrated increased expression in the fibrogenic group. Increased expression of β5 (3-fold) and β8 (5-fold) as well as Smad-3 (4-fold) and Smad interacting protein (SIP)-1 (3.5 fold) mRNA were confirmed in experimental BA. Phosphorylated Smad 3 protein in the experimental group was also nearly twice that of the control group, further implicating the TGF-β pathway.
CONCLUSION
Gene transcripts for known upstream and downstream TGF-β mediators are differentially expressed in liver specimens from children with BA and a fibrogenic gene signature. The same integrins that were dysregulated in the human specimens were also found to be upregulated in our animal BA model, as were other intermediaries in the TGF-β pathway. Further investigation into whether these mediators may be attractive targets for future therapy in children with BA is warranted.
Keywords: biliary atresia, TGF-beta, liver fibrosis
Introduction
Biliary atresia (BA) is a severe cholestatic disorder of unknown etiology diagnosed in young infants. The pathologic manifestation of this disease is an inflammation and obstruction of the extra-hepatic bile ducts resulting in liver fibrosis, cirrhosis, and eventual liver failure. The exact cause of this abnormality is unknown, however it has been recently shown that transforming growth factor β (TGF β) plays a critical role in other chronic liver diseases (1). TGF-β regulates extracellular matrix formation, degradation, and remodeling, and has proven to be critical in both viral and alcoholic liver dysfunction. Thus, we previously embarked on a series of experiments in attempt to further elucidate the regulation and role of TGF-β in an experimental model of BA. Using the rhesus rotavirus (RRV) model, we demonstrated increased expression of one of the key upstream regulators of TGF-β function, the integrin αv β6.(2) We also found that several downstream end-products of the TGF-β pathway were over expressed including tissue inhibitor of matrix metalloproteinase -1 (TIMP-1) and plasminogen activator inhibitor-1 (PAI-1). However the relevance of these findings to the human condition had not previously been investigated.
To confirm our findings from the animal model in humans with BA would require a large number of liver specimens, more than any single center would possess due to the infrequent incidence and prevalence of the disease. However Moyer et al. published microarray data from liver specimens obtained from 47 infants with BA at the time of portoenterostomy in a prospective study performed by the former Biliary Atresia Research Consortium (BARC) which is now part of the Childhood Liver Diseases Research Network (ChiLDReN) (3). They found that infants with BA could be classified into two fairly distinct categories based on their gene expression and termed them inflammatory and fibrogenic gene signatures. Using their publicly-available microarray data, we performed an in silico analysis of the same liver specimens from children with BA with the hypothesis that the mediators of the TGF β pathway would be dysregualted in patients with fibrotic gene signatures when compared to those with inflammatory gene signatures. We then performed immunohistochemistry (IHC) on liver specimens from patients with BA at our institution to determine whether the mRNA of the mediators identified in the in silico analysis also displayed increased protein expression in the liver. Finally we returned to our animal model of BA to confirm the new findings from our microarray analysis and to evaluate whether the animal model was indeed reflective of the human condition.
METHODS
Human Microarray Analysis
In silico analysis of previously published microarray data was performed. Original liver specimens were obtained from 47 infants with Biliary Atresia at the time of portoenterostomy.(3) Total RNA was profiled using Affymetrix Human 133 plus 2.0 microarrays. The publicly available image (CEL) files and meta data were used to compare gene expression differences between the fibrogenic (n=25) and inflammatory (n=18) cohorts predicted by the previous study (Figure 1). There were 4 liver specimens that the prediction analysis models used in the original study did not classify as either inflammatory of fibrotic in terms of their gene signature and these were excluded from our analysis. ANOVA comparing fibrogenic and inflammatory groups was performed using Partek Genomics Suite (Partek Inc., St. Louis, MO). The resulting ANOVA data were filtered at a significance level of p<0.01 and fold change >1.2 or <−1.2. Annotation of transcripts was done using Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA) and a subset of TGF-β-related genes was selected for further analysis.
Figure 1.

Algorithm from Moyer et al manuscript describing the 2 patient cohorts.3
Immunohistochemistry
Liver specimens from patients at our center who had been diagnosed with BA (n=5) were compared to liver tissue from other children who underwent liver biopsy for neonatal hepatitis (n=5). Archived tissue blocks were accessed, and each block of paraffin was cut in 5 sections and immunostained with an antibody to detect protein expression of integrin αv β5 or αv β8 (Sigma-Aldrich, St Louis, MO). The interpretation of the sections was performed by a pediatric pathologist (CR) who was blinded to the original diagnosis of the specimen. The immunohistochemistry (IHC) was graded from 0–4 based on the intensity of protein expression. Standard Ishak scoring for inflammation and fibrosis was also performed.(4) Comparison between the BA and neonatal hepatitis groups was carried out using Fisher’s Exact test for IHC, and Student’s t-test for Ishak scoring. Statistical significance was assigned at p<0.05.
Animal Model
With appropriate IACUC approval, pregnant time dated BALB/c mice obtained from Charles River Labs were housed one per cage with free access to water and the standard laboratory diet. After spontaneous vaginal delivery, the newborn mice were randomly divided into two groups. The first group received intraperitoneally 0.02 ml of 1.5x106 fluorescence forming units (FFU) of rhesus rotavirus (RRV) of serogroup 3 diluted with 0.02 ml of saline within 24 h of birth (n=8). The second group which served as controls received only saline (n=11). The treated mice were returned to their mothers, maintained in normal environment, and housed in a room with a standard 12-hour dark-light cycle. Examination of the development of bilirubinuria, development of acholic stools and icterus in mouse skin (not covered with fur) were performed daily. Subsequent experimental procedures (weighing, subcutaneous saline injection and sacrificing) were performed as previously described (2). Liver specimens were harvested for protein and RNA isolation, and histological and immunohistochemical analysis.
RNA isolation and quantitative RT-PCR
For semi- quantitative analysis, total RNA was isolated from RRV and control samples using TRI Reagent-LS (MRC, Cincinnati, OH) according to the manufacturer’s protocol. Then the total RNA was purified using Rneasy Mini Kit (Qiagen, Valencia, CA). RNA concentrations in all samples were normalized to 500 ng/ul using Nanodrop. A 500 ng aliquot of total RNA was used to generate cDNA with the SuperScript II Reverse Transcriptase (Invitrogen, Grand Island, NY) and Oligo-dT reverse primer. Subsequent semi-quantitative real-time PCR analysis was performed with 2 μl of 1/5 and 1/25 diluted aliquots of RT reaction mixes using iQ SYBR Green Supermix (BioRad, Hercules, CA) with the following profile: 1 cycle of 50°C for 15 s, 95°C for 10 min, 40 cycles of 94°C for 10 s, 58°C for 20 s and 72°C for 20 s. When amplification cycles were completed, a final melting curve was performed according to the dissociation protocol of the ABI 7900HT instrument. The following primers were used:
| Mediator | Left Primer (5′-3′) | Right primer (5′-3′) |
|---|---|---|
| Integrin α v β5 | GGTTTCGGGTCTTTTGTTGA | GCTTCCTCACTTCCTCGTTG |
| Integrin α v β6 | TCTGAGGATGGAGTGCTGTG | GGCACCAATGGCTTTACACT |
| Integrin α v β8 | CATTCTTGATCGGGTTGCTT | CAGGCTTTTCTCGTCGGTAG |
| Smad interacting protein-1 (SIP-1) | TGTTGAGCTTTGTGGACCAG | CCCAAAACGAAGTGCAAAAT |
| Smad-3 | CATCCGTATGAGCTTCGTCA | AGGGTCCATTCAGGTGTAGC |
| GAPDH | GGCATTGCTCTCAATGACAA | CCCTGTTGCTGTAGCCGTAT |
Real-time PCR reactions were carried out at least in triplicate using the ABI 7900 Fast Real-Time PCR system (Applied Biosystems, Carlsbad CA) and SDS 2.3 software. Comparisons of gene expression between the two groups were performed using Student’s t-test. Statistical significance was assigned to any p-value less than 0.05.
Enzyme-linked Immunosorbent Assay (ELISA)
At the time of sacrifice, a portion of the liver was harvested for protein extraction. Total lysates of liver fragments were used for PathScan Phospho-Smad2 sandwich ELISA (Cell Signaling Technology, Danvers, MA). Tissue samples were homogenized in lysis buffer (according to the manufacturer protocol) using homogenizer FastPrep-24 (MP Biomedicals, Santa Ana, CA) at a speed 0.4m/s for 30 sec and then purified by centrifugation at 10,000 RCF for 10 min at 4°C. Supernatants were separated from pellets and applied for Phospho-Smad2 ELISA according to manufacturer protocol. Samples were normalized to the total protein count using DC Protein Assay kit (Bio-Rad, Hercules, CA). The normalized protein levels were compared using Student’s t-test with statistical significance assigned at p<0.05
RESULTS
Microarray analysis
Global expression patterns were significantly different between the groups of infants with fibrogenic and inflammatory gene signatures. We identified 6903 transcripts (representing 2822 distinct genes) with differential gene expression between the two groups whose p value was < 0.01 and fold change >1.2 or <−1.2. We then focused on 24 TGF-β-related transcripts and found that several upstream and downstream TGF-β mediators were differentially expressed between the groups. Expression of the fibrillar collagen-associated genes was increased in the fibrogenic group. Three of 4 probesets for procollagen type 1 α1 were statistically significant between the 2 groups, and the 4th probeset had a p-value of 0.08. The average increase in expression was 34%. All of the probesets for procollagen type 1 α2 (average 35% increase); procollagen type 3 α1 (average 20% increase) and procollagen type 4 α1 (average 28% increase) were also statistically different between the 2 groups (Table 1). Strikingly, matrix metalloproteinase (MMP)-7 expression was two-fold increased, while that of MMP-9 was two-fold decreased in the fibrogenic group. Expression of SERPINE1 was also nearly three-fold decreased in the fibrogenic group (Table 1). Expression of the integrin family members known to mediate TGF-β function was also evaluated. Two of 3 integrin β 5 probesets ( average 18% increase), and 2 of 3 probesets for integrin β 8 (average 84% increase) showed statistically significant increase in expression in the fibrogenic group, (Table 1) while there was no difference in integrin β 6 between the two groups.
Table 1.
Gene expression of pertinent TGF-β related transcripts in Biliary Atresia patients with Fibrogenic or Inflammatory Gene Signatures.
| GENE SYMBOL | MECHANISM OF ACTION | P-VALUE (Fibrogenic v. Inflammatory) | FOLD CHANGE (Fibrogenic v. Inflammatory) |
|---|---|---|---|
| Intβ5 | Activates TGF-β | 0.010 | 1.23 |
| Intβ8 | Activates TGF-β | 0.008 | 1.75 |
| COL1A1 | Increases collagen deposition | 0.002 | 1.57 |
| COL1A2 | Increases collagen deposition | 0.0005 | 1.31 |
| COL3A1 | Increases collagen deposition | 0.0006 | 1.24 |
| COL4A1 | Increases collagen deposition | 0.003 | 1.25 |
| MMP7 | Increases degradation of extracellular matrix | 0.008 | 2.01 |
| MMP9 | Increases degradation of extracellular matrix | 0.0001 | −1.99 |
| SERPINE1 (PAI-1) | Inhibits degradation of extracellular matrix | 0.0002 | −2.91 |
Values shown represent the probeset with the most statistically significant difference between the two groups.
Immunohistochemistry
Since there was increased mRNA expression for both the integrin β 5 and integrin β 8, we aimed to evaluate liver specimens from children with BA at our institution for protein expression by IHC. We found that 4 of 5 specimens from patients with neonatal hepatitis had intensity scores of 0 or 1, and the median score was 1.0 with respect to integrin β 5 staining. In the BA patients, 4 of 5 specimens scored 2 or 3 and the median score was 2.0. However, there was no statistical significance between these 2 groups (p=0.2, Fisher’s Exact test). Similarly, there was no statistical difference with respect to integrin β 8 immunoreactivity between the 2 groups (p=0.46) however the specimens with the most intense staining for both integrins were found in BA patients (Figure 2). Integrin β5 staining seems to be stronger in the hepatocytes while β8 staining is strongest in the biliary epithelium. Finally, Ishak scoring revealed a difference between the BA and NH specimens with respect to fibrosis and a trend toward a difference in the inflammation domain. The mean Ishak score for NH specimens was the same for both domains (0.6 ± 0.5). In comparison, the fibrosis score for the BA specimens was 1.8 ± 0.8 (p= 0.03 v NH) and the inflammation score was 1.6 ± 0.9 (p=0.07 v NH).
Figure 2.
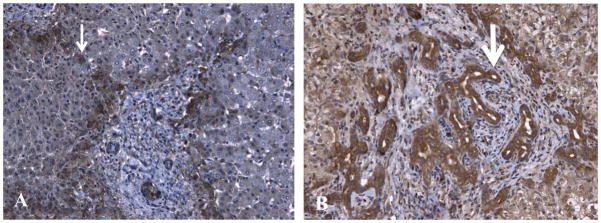
Immunhistochemical staining (brown) in a Biliary Atresia patient for integrins β5 (Panel A) and β8 (Panel B). Integrin β5 staining seems to be stronger in the hepatocytes (thin arrow) while β8 staining is strongest in the biliary epithelium (thick arrow).
Animal Model of Biliary Atresia
Since our human gene chip data suggested an important role for integrins β 5 and 8, and we had never previously evaluated the mRNA expression of these mediators in our animal model, we returned to our experimental BA mice to determine whether these integrins were indeed upregulated in the animals after RRV exposure. We detected significant up-regulation of the both of these members of integrin family in RRV exposed animals when compared with the saline control group 14 days after inocculation. The level of mRNA expression of integrin β5 was 3-fold increased in the experimental BA group when compared to controls (p=0.01). Integrin β8 expression was even more upregulated, the mRNA level was approximately 5-fold increased at day 14 after RRV inoculation (p<0.001). Similarly an approximately 4-fold increase in SMAD3 (p=0.001) and 3.5 fold increase in SIP-1 (p<0.001) mRNA expression was observed compared to control mice. Lasltly, SMAD2 phosphorylation in the RRV group was almost twice the level of that in the control group (0.93 ±0.06 v. 0.50 ±0.13, p<0.001)
DISCUSSION
Our laboratory has previously shown that the TGF-β pathway is important in the pathogenesis of experimental BA, and that this pathway is turned off in animals who recover from the initial viral infection.(2,5) Others have similarly implicated TGF-β in the development of BA in humans.(6) Importantly the regulation of the pathway seems to depend on activation by various integrin family members in our work, in other models of liver disease, and in different organ systems.(7,8,9) Isoforms of TGF-β are first synthesized as precursor molecules containing a propeptide region in addition to a TGF-β homodimer. The newly synthesized TGF-β homodimers interact with a latency associated peptide (LAP) generating a complex called small latent complex.(10) This complex binds with another protein called latent TGF-β-binding protein and forms a larger complex called large latent complex (LLC),(11) which is then secreted into the extracellular matrix. TGF-β activation involves release of the LLC from the matrix followed by further proteolysis of the LAP which enables TGF-β binding with its receptors, all of which may be initiated by various integrins. matrix metalloproteinases (MMPs) and proteases.(8,10,12) Since integrin αvβ6 had been shown to initiate TGF-β pathway activation and subsequent progression of biliary and hepatic fibrosis, (9,13), our work had previously focused on this particular integrin. However the LAP for TGF-β1 and -β 3 contain an RGD motif which is recognized by a majority of αv integrins,(14) suggesting that other family members may also be able to activate the pathway in humans, and thus we wanted to pursue their expression in patients with BA.
Due to the general infrequency of BA, the National Institutes of Health (NIH) funded the formation of a research consortium, BARC, to focus on investigation into the disease. This group was able to gather specimens and perform a microarray analysis on 47 patients with BA and classify them into 2 groups based on the gene expression.(3) Furthermore, they postulated that these gene signatures may predict patient outcome. Access to the specimens is limited to members of BARC, but the microarray data are publically available since it was a NIH-supported study. We decided that an additional analysis of the microarray data was warranted in an attempt to determine the individual expression patterns of the TGF-β pathway members since they have been so strongly implicated in the pathogenesis of BA. Employing our local expertise in genetic medicine, we re-evaluated the raw data with particular emphasis placed on 24 TGF-β related transcripts including the fibrillar collagens. As expected, we found that the different procollagen genes were significantly upregulated to varying degrees in the fibrogenic as defined by Moyer et al group. More interestingly we found that those patients who possessed fibrotic gene signatures had significantly increased expression of integrin β5 (18%) and even higher upregulation of β8 (84%), while the expression of β6 was not different between the 2 groups. When we assessed protein expression by IHC in available BA specimens at our institution, we did not find any statistical difference in β5 or β8 expression, however the number of specimens in our analysis was quite small and we are currently embarking on a collaboration with a BARC member institution in an effort to better assess protein expression in patients and whether protein expression correlates with the degree of fibrosis at the time of diagnosis.
While it was somewhat surprising to find that integrin β6 was not differentially expressed in the BA patients with inflammatory and fibrogenic gene signatures, certainly integrins β5 and β8 have been implicated in activation of the TGF-β pathway in other diseases. The interaction between integrin αvβ5 with Thy-1 has been shown to induce latent TGF-β1 activation and subsequent TGF-β1-dependent lung myofibroblast differentiation (14) It has also been shown that increased integrin αvβ5 expression is linked with myofibroblast differentiation in the skin of patients with scleroderma.(15) Binding to the integrin αvβ8 with subsequent metalloproteolytic cleavage of LAP also represents a major mechanism of TGF-β activation in vivo. Altered expression of the integrin β8 subunit is found in human chronic obstructive pulmonary disease, different cancers, and even brain vascular malformations.(16) Furthermore, increased integrin αvβ8 expression in response to the proinflammatory cytokine interleukin-1β induces TGF-β activation and is associated with pathologic inflammation and fibrosis (17). Perhaps it therefore should not have been unexpected to find that the mRNA for integrin αvβ5 and β8 were upregulated in livers from BA patients, and it suggests that any targeted therapeutic intervention that does not account for the possibility of redundant mechanisms for TGF-β pathway activation may be doomed to failure. Indeed, we also found increased integrin αvβ5 and β8 expression in our animal model where therapeutic administration of anti- αvβ6 antibodies has not been effective in preventing experimental BA despite a myriad of different dosing regimens (data not published).
The other findings in the animal model are important in that they confirm the TGF-β pathway as a likely contributor to the liver fibrosis associated with BA. The TGF-β pathway is essential to fibrogenesis in other liver disease,s and many of these effects are mediated through the Smad protein family.(18) Upon binding with its receptor, TGF-β induces phosphorylation of Smad-2 and Smad-3, which then form a complex with Smad-4.(19) This complex translocates to the nucleus, where it increases expression of extracellular matrix proteins and inhibits production of anti-fibrotic protease inhibitors.(20) Thus our findings that both Smad-3 mRNA expression and phosphorylated Smad-3 protein are increased after RRV exposure implicate activation of the TGF-β pathway. Various authors have suggested that interrupting Smad-3 signaling in rodents can prevent liver fibrosis, which could be an attractive strategy for patients with BA and one we aim to investigate in our animal model.(21,22) We also found that SIP-1 was increased in animals with experimental BA. SIP-1 is a transcription factor that interacts with the TGF-β pathway by binding to activated Smads 1, 2, 3, 5, and 8.(23) SIP-1 binding to activated Smads and Smad complexes causes decreased target gene transcription through binding of zinc fingers to promoter regions.(24) Most research to date has focused on the role of SIP-1 in epithelial-mesenchymal transition,(25) so little is known regarding its importance in liver disease. We found a concomitant increase in both Smad-3 and SIP-1 in our experimental model which may be the animal’s attempt to attenuate the TGF-β pathway once it is activated, but more study would be required in order to validate that hypothesis.
The greatest differences in gene expression that we found in the microarray data were in MMP-7, MMP-9, and PAI-1. MMP-7 was 2 fold increased in the fibrogenic group, while MMP-9 was 2 fold decreased in this group. PAI-1 was nearly 3 fold decreased in the fibrogenic group. It has been well established that MMPs are important in extracellular matrix regulation, facilitate cell migration, and have been implicated in different types of fibrosis (25). Furthermore, MMP2 and MMP9 are known to cleave latent TGF-β.(26) Increased MMP-7 expression has previously been reported in other fibrotic disorders, such as idiopathic pulmonary fibrosis, sarcoidosis, and systemic sclerosis.(27,28,29) Up-regulation of MMP-7 expression has also been identified in human liver fibrosis associated with hepatitis C infection.(30) Evidence of its specific role in the pathogenesis of fibrosis, however, remains limited. The role of MMP-9 in fibrosis is even less clear. MMP-9 expression is increased in chronic hepatitis.(31) Alternately, decreased MMP-9 expression has been demonstrated in moderate liver fibrosis, possibly due to histone deacetylation and decreased promoter activity.(32) The fact that we saw increased MMP-7 but decreased MMP-9 expression in BA patients who were more fibrongenic is thus not unlike that seen in other fibrotic liver diseases. PAI-1 is a serine protease inhibitor that functions to inhibit ECM breakdown. Down-regulation of PAI-1 expression has been established in liver cirrhosis while activity of PAI-1 is increased in acute liver inflammation,(33) which is also consistent with our finding.
In summary, we found that the gene transcripts for known upstream and downstream TGF-β mediators are differentially expressed in liver specimens from children with BA and a fibrogenic gene signature. While TGF-β may be a common intermediary for all fibrotic diseases of the liver, the data presented here demonstrate that BA patients with an inflammatory gene signature may be amenable to anti-TGF-β pathway strategies as the associated mediators are differentially expressed. This suggestion would only be applicable if inflammation precedes and begets fibrosis in patients with BA, which is likely since that is the mechanism with other common forms of chronic liver disease such as non-alchoholic steatohepatitis and viral-mediated liver disease, but has not been definitively shown in human BA. Expression of both integrins β5 and β8, but not β6, were increased in the human specimens and were also found to be upregulated in experimental BA. The differences in MMP-7, MMP-9, and PAI-1 expression were even more profound between groups suggesting that further investigation into all of these mediators as potential targets for future therapy in children with BA is warranted.
Acknowledgments
Support: Funded in part by National Institutes of Health; Grant Number: K08 DK083769
Footnotes
Presented in part at The 97th Clinical Congress of the American College of Surgeons, October 2011.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dooley S, ten Dijke P. TGF-beta in progression of liver disease. Cell Tissue Res. 2011;347:245–256. doi: 10.1007/s00441-011-1246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nadler EP, et al. Integrin alphavbeta6 and mediators of extracellular matrix deposition are up-regulated in experimental biliary atresia. J Surg Res. 2009;154:21–29. doi: 10.1016/j.jss.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 3.Moyer K, et al. Staging of biliary atresia at diagnosis by molecular profiling of the liver. Genome Med. 2010;2:33. doi: 10.1186/gm154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishak KG, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22:696–699. doi: 10.1016/0168-8278(95)80226-6. [DOI] [PubMed] [Google Scholar]
- 5.Nadler EP, et al. Differential expression of hepatic fibrosis mediators in sick and spontaneously recovered mice with experimental biliary atresia. J Surg Res. 2010;159:611–617. doi: 10.1016/j.jss.2009.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Oliveira, et al. Serum and tissue transforming [corrected] growth factor β1 in children with biliary atresia. J Pediatr Surg. 2010 Sep;45(9):1784–90. doi: 10.1016/j.jpedsurg.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 7.Huang XZ, et al. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol. 1996;133:921–928. doi: 10.1083/jcb.133.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munger JS, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 9.Popov Y, et al. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol. 2008;48:453–464. doi: 10.1016/j.jhep.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 10.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 11.Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005;280:7409–7412. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- 12.Ludbrook SB, et al. The integrin alphavbeta3 is a receptor for the latency-associated peptides of transforming growth factors beta1 and beta3. Biochem J. 2003;369:311–318. doi: 10.1042/BJ20020809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang B, et al. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology. 2007;46(5):1404–12. doi: 10.1002/hep.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bader BL, et al. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell. 1998;95:507–519. doi: 10.1016/s0092-8674(00)81618-9. [DOI] [PubMed] [Google Scholar]
- 15.Asano Y, et al. Involvement of alphavbeta5 integrin in the establishment of autocrine TGF-beta signaling in dermal fibroblasts derived from localized scleroderma. J Invest Dermatol. 2006;126:1761–1769. doi: 10.1038/sj.jid.5700331. [DOI] [PubMed] [Google Scholar]
- 16.Mu D, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1 -MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markovics JA, et al. Interleukin-1beta induces increased transcriptional activation of the transforming growth factor-beta-activating integrin subunit beta8 through altering chromatin architecture. J Biol Chem. 2011;286:36864–36874. doi: 10.1074/jbc.M111.276790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol. 2007 Jun 14;13(22):3056–62. doi: 10.3748/wjg.v13.i22.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002 Feb;118(2):211–5. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 20.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–91. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 21.Latella G, et al. Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int. 2009 Aug;29(7):997–1009. doi: 10.1111/j.1478-3231.2009.02011.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee JH, et al. The use of low molecular weight heparin-pluronic nanogels to impede liver fibrosis by inhibition the TGF-β/Smad signaling pathway. Biomaterials. 2011 Feb;32(5):1438–45. doi: 10.1016/j.biomaterials.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 23.Verschueren K, et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 50-CACCT sequences in candidate target genes. J Biol Chem. 1999;274:20489–98. doi: 10.1074/jbc.274.29.20489. [DOI] [PubMed] [Google Scholar]
- 24.Conidi A, et al. Few Smad proteins and many Smad-interacting proteins yield multiple functions and action modes in TGF-β/BMP signaling in vivo. Cytokine Growth Factor Rev. 2011 Oct-Dec;22(5–6):287–300. doi: 10.1016/j.cytogfr.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-de-Alba C, et al. Expression of matrix metalloproteases by fibrocytes: possible role in migration and homing. Am J Respir Crit Care Med. 2010;182:1144–1152. doi: 10.1164/rccm.201001-0028OC. [DOI] [PubMed] [Google Scholar]
- 26.Stetler-Stevenson WG, Aznavoorian S, Liotta LA. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol. 1993;9:541–573. doi: 10.1146/annurev.cb.09.110193.002545. [DOI] [PubMed] [Google Scholar]
- 27.Richards TJ, et al. Allele-specific transactivation of matrix metalloproteinase 7 by FOXA2 and correlation with plasma levels in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302(8):L746–54. doi: 10.1152/ajplung.00319.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piotrowski WJ, et al. The selected genetic polymorphisms of metalloproteinases MMP2, 7, 9 and MMP inhibitor TIMP2 in sarcoidosis. Med Sci Monit. 2011;17(10):CR 598–607. doi: 10.12659/MSM.881987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moinzadeh P, et al. Elevated MMP-7 levels in patients with systemic sclerosis: correlation with pulmonary involvement. Exp Dermatol. 2011;20(9):770–3. doi: 10.1111/j.1600-0625.2011.01321.x. [DOI] [PubMed] [Google Scholar]
- 30.Asselah T, et al. Liver gene expression signature of mild fibrosis in patients with chronic hepatitis C. Gastroenterology. 2005;129(6):2064–75. doi: 10.1053/j.gastro.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Bièche I, et al. Molecular profiling of early stage liver fibrosis in patients with chronic hepatitis C virus infection. Virology. 2005;332(1):130–44. doi: 10.1016/j.virol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 32.Qin L, Han YP. Epigenetic repression of matrix metalloproteinases in myofibroblastic hepatic stellate cells through histone deacetylases 4: implication in tissue fibrosis. Am J Pathol. 2010;177(4):1915–28. doi: 10.2353/ajpath.2010.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beier JI, Arteel GE. Alcoholic liver disease and the potential role of plasminogen activator inhibitor-1 and fibrin metabolism. Exp Biol Med (Maywood) 2012;237(1):1–9. doi: 10.1258/ebm.2011.011255. [DOI] [PMC free article] [PubMed] [Google Scholar]