

Abstract
The molecular targets and neural circuits that underlie general anesthesia are not fully elucidated. Here, we directly demonstrate that Kv1-family (Shaker-related) delayed rectifier K+ channels in the central medial thalamic nucleus (CMT) are important targets for volatile anesthetics. The modulation of Kv1 channels by volatiles is network specific as microinfusion of ShK, a potent inhibitor of Kv1.1, Kv1.3, and Kv1.6 channels, into the CMT awakened sevoflurane-anesthetized rodents. In heterologous expression systems, sevoflurane, isoflurane, and desflurane at subsurgical concentrations potentiated delayed rectifier Kv1 channels at low depolarizing potentials. In mouse thalamic brain slices, sevoflurane inhibited firing frequency and delayed the onset of action potentials in CMT neurons, and ShK-186, a Kv1.3-selective inhibitor, prevented these effects. Our findings demonstrate the exquisite sensitivity of delayed rectifier Kv1 channels to modulation by volatile anesthetics and highlight an arousal suppressing role of Kv1 channels in CMT neurons during the process of anesthesia.
Introduction
The mechanisms of general anesthesia involve multiple molecular targets and pathways that are not completely understood. Two-pore-domain K+ channels and Cys-loop receptors are widely accepted as relevant targets (Franks and Lieb, 1991; McKenzie et al., 1995; Jenkins et al., 1996; Patel et al., 1999; Downie et al., 2002; Liu et al., 2004; Andres-Enguix et al., 2007; Solt and Forman, 2007; Brannigan et al., 2010; Garcia et al., 2010; Nury et al., 2011; Moore et al., 2012). However, the complex nature of anesthesia suggests the existence of additional molecular targets in anesthesia- and sleep-related brain regions.
The pathways and neural circuits involved in a sleep-wake cycle regulation provide an important context for understanding the mechanisms of general anesthetic action (Alkire et al., 2009; Moore et al., 2012; Solt, 2012). Thalamocortical circuits are critical for the regulation of arousal and sleep (Steriade et al., 1993; Franks and Zecharia, 2011). Furthermore, the intralaminar and midline thalamic nuclei, including central medial thalamic (CMT) nucleus, are known to be a nonspecific arousal system (Van der Werf et al., 2002). The firing of thalamocortical neurons is regulated by several ion channels, including Kv1-, Kv3-, and Kv4-family channels (Wang et al., 1994; Kasten et al., 2007; Kanyshkova et al., 2011), although the exact role of Kv1 channels in these nuclei has not been defined.
Given the importance of Kv1 channels for fine-tuning and regulation of neuronal firing rate and pattern (Wang et al., 1993, 1994; Gazula et al., 2010), and the ability of sevoflurane to activate Kv1.2 and Kv1.5 channels (Barber et al., 2012), it is compelling to hypothesize that these channels in the CMT might be essential molecular targets for anesthetics. It had been reported earlier that microinfusion into the CMT nucleus of specific antibodies against Kv1.2 channels, mammalian homologues of Shaker and an important component of neuronal excitability in thalamocortical neurons (Lambe and Aghajanian, 2001), restores “consciousness” (measured as righting reflex) in anesthetized rats even in the maintained presence of the anesthetic (Alkire et al., 2009) supporting the role of Kv1.2 channels in general anesthesia. Here, we directly demonstrate a role of Kv1-family channels in anesthesia using both in vivo and in vitro approaches. In rodents, we examined whether infusion of ShK, a potent inhibitor of delayed rectifier Kv1 channels (Castañeda et al., 1995), into the CMT could reverse anesthesia induced by the inhalational anesthetic sevoflurane. We analyzed the effects of sevoflurane, isoflurane, and desflurane on the biophysical properties of Kv1 channels heterologously expressed in mammalian cells. Finally, we used brain slices to characterize the effects of sevoflurane on firing frequency in CMT neurons in the presence or absence of ShK-186, a Kv1.3-selective synthetic analog of ShK (Kalman et al., 1998; Beeton et al., 2005). Together, these studies reveal the extraordinary sensitivity of Kv1 channels to volatiles and demonstrate a crucial role of Kv1 channels in CMT in arousal suppression during inhalational anesthesia.
Materials and Methods
Induction and maintenance of anesthesia in rats.
All experiments were performed in accordance with the Institutional Animal Care and Use Committee at the University of California, Irvine, and were consistent with Federal guidelines. Sprague Dawley male rats were acquired from Charles River Laboratories. The anesthesia was induced as described previously (Alkire et al., 2009). Briefly, the rats with preimplanted guide cannula aimed at the CMT nucleus were placed into an 8 L anesthetic chamber equipped with three ports: (1) for anesthetic inlet connected to 19.1 gas vaporizer, (2) for gas monitoring, and (3) for a microinfusion needle passage. The concentration of sevoflurane in the chamber was monitored continuously using a Datex-Ohmeda Ultima Capnomac.
Potassium channel blockers.
ShK peptide, originally isolated from Caribbean sea anemone Stichodactyla helianthus (Castañeda et al., 1995) and later synthesized (Pennington et al., 1995), is the most potent Kv1.3 inhibitor (IC50, 11 pm) (Kalman et al., 1998; Beeton et al., 2005). For the experiments in vivo, ShK solutions of final concentrations were prepared from frozen aliquots immediately before intrathalamic microinfusion.
Since ShK has also high affinity for Kv1.1 channels (Kalman et al., 1998; Beeton et al., 2005), ShK-186 that has increased specificity for Kv1.3 over Kv1.1 and retains picomolar potency was used in the thalamic slice recordings. ShK-186 is a synthetic analog of the ShK peptide that is modified by (1) attaching a negatively charged l-phosphotyrosine via a hydrophilic linker to ShK-Arg1 (Beeton et al., 2005) and (2) by replacing the N-terminal phosphotyrosine with the nonhydrolyzable phosphate mimetic paraphosphonophenylalanine (Pennington et al., 2009).
Intrathalamic microinfusions.
Once the anesthetized rats past the point of the loss of the righting reflex, the infusion needle (25 gauge; 17 mm; attached to a 10 μl Hamilton syringe via polyethylene tubing that passed through a port in the chamber wall) was placed quickly into the cannula and the rat was placed on its back. The sevoflurane was kept at a steady concentration of 1.3 ± 0.05% atm for at least 15 min before the microinfusion was administered. The ShK toxin (in 0.5 μl of rat serum) was delivered through a minipump-pushed (Harvard Apparatus) syringe over the duration of 1 min. A dose of 1 pm was selected after a few pilot animals demonstrated seizures following the infusions of either 10 or 100 pm solutions.
The righting reflex was considered restored when a rat was able to transition from its back onto its stomach and move around. The involuntary high-frequency shaking movements of the head or limbs without signs of awareness were considered “seizures.” All experiments were monitored and recorded for 10–15 min after the microinfusion.
For confirmation of the microinfusion site, histological analysis of the brain sections was performed as described previously (Alkire et al., 2009). The sections were stained with thionin and the infusion tracts were projected onto a representative coronal brain section from the rat atlas (Paxinos and Watson, 2005) with localization points varying by up to a ±1.0 mm in the anterior–posterior dimension.
Preparation of the aqueous anesthetic solutions.
Aqueous anesthetics solutions were prepared as described previously (Mandal and Pettegrew, 2008). Briefly, 200 μl of neat anesthetics, sevoflurane (Abbott Laboratories), desflurane (Baxter), and isoflurane (Phoenix) were added to 2 ml of extracellular solution, shaken and allowed to equilibrate for ∼10 min. The required volume of saturated anesthetic solution was directly added to the gravity-driven perfusion system of the patch-clamp setup. To confirm the accuracy of the procedure, some sevoflurane samples were prepared by bubbling the aqueous buffer solution with a known concentration of sevoflurane, delivered through an agent-specific 19.1 gas vaporizer. 19F NMR was then used to compare the content of the anesthetic in bubbled samples with that in aqueous sevoflurane samples collected at the end point of the gravity-driven perfusion system used for electrophysiological experiments. All of the concentrations used for generation of the dose–response curves (see Figs. 3, 6) were adjusted accordingly.
Figure 3.
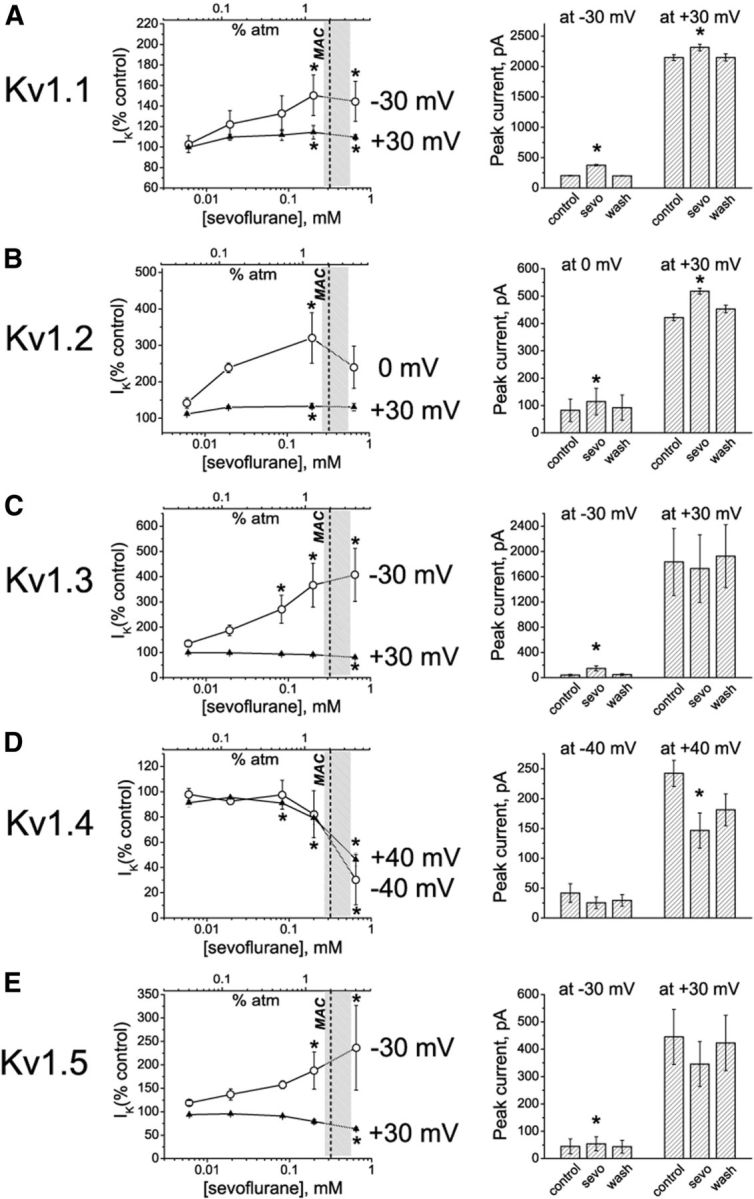
Quantification of the sevoflurane effects on Kv1 currents. A–E, Left, Dose-dependence relationships for sevoflurane effects on potassium currents evoked by depolarizing voltage steps from a −80 mV holding membrane potential to the indicated low and the higher voltages. The light gray area on the graphs shows the surgical range of anesthetic concentrations; the dash line indicates the approximate MAC value for sevoflurane in mice and in human patients of middle age. The row data are normalized to control values (peak IK+) for each cell and are presented as mean ± SEM (n = 3–5 measurements for each concentration; 26 cells total). Statistical analysis was performed on row data before normalization, and the significant differences are indicated by asterisks (pairwise comparisons, Tukey's test). Right, Averaged peak IK+ values in the presence of 0.2 mm sevoflurane compared to peak IK+ values in control conditions and after washing out the anesthetic. The data are presented as mean ± SEM; n = 3 cells for each data point. *p < 0.05 (paired t test).
Figure 6.

Modulation of Kv1.3 channel properties by desflurane and isoflurane. A, D, Representative current traces evoked by depolarizing pulses from a holding potential of −80 mV to the range of potentials from −40 to +30 mV, with 10 mV increments and interpulse intervals of 30 s. Control currents are superimposed with currents recorded in the presence of desflurane (A) or isoflurane (D). B, C, E, F, Dose-dependence relationships for the effects of desflurane (B, C) and isoflurane (E, F) on potassium currents evoked by depolarizing voltage steps from −80 to −30 and +30 mV. The light gray area shows the surgical range of anesthetic concentrations; the dash line indicates the approximate MAC value for the corresponding anesthetic in mice and in human patients of middle age. The row data are normalized to control peak IK+ values and are presented as mean ± SEM (n = 3–5 measurements for each concentration). Statistical analysis was performed on row data before normalization, and the significant differences are indicated by asterisks (pairwise comparisons, Tukey's test). C, F, Averaged peak IK+ values in the presence of desflurane (C) and isoflurane (F) in control conditions and after washing out the anesthetics. The data are presented as mean ± SEM; n = 3 cells. *p < 0.05 (paired t test). The cells were exposed to aqueous anesthetic solutions prepared as described previously (Mandal and Pettegrew, 2008).
19F NMR spectroscopy.
19F NMR spectra were recorded at 298 K using a Bruker DRX400 spectrometer at 376 MHz and a 5 mm QNP (Quattro Nucleus Probe) (1H/13C/31P/19F) NMR probe. XwinNMR (Bruker) was used for data acquisition and processing. A separate CFCl3 sample was used as a reference standard for reporting the 19F chemical shifts. All samples contained 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt, used to manually shim the spectrometer by observation of the proton signal.
Patch-clamp recordings in cell lines.
Patch-clamp experiments in cell lines heterologously expressing Kv1 channels were performed in whole-cell mode using an EPC-9 (HEKA Elektronik) amplifier. All currents were recorded at room temperature (22−24°C). Pipettes pulled from borosilicate glass capillaries (Garner Glass) using a Fleming/Brown micropipette puller (Sutter Instrument) had resistances of 1.8–2.5 MΩ when filled with the internal recording solution that contained the following (in mm): 145 KF, 10 EGTA, 10 HEPES, and 2 MgCl2, pH 7.3. The extracellular solution contained the following (in mm): 2 CaCl2, 1 MgCl2, 4.5 KCl, 155 NaCl, 10 d-glucose, and 5 HEPES. Outward K+ currents were evoked in voltage-clamp mode by depolarizing voltage pulses from −80 mV holding potential. The sampling frequency was 10 kHz. Data files were recorded using PULSE/PULSEFIT (HEKA Elektronik) and analyzed using OriginPro7.5.
For testing the effects of anesthetics on Kv1 currents, the required volumes of freshly prepared aqueous anesthetic stock solutions were directly added to the external solution to achieve the needed concentration (Mandal and Pettegrew, 2008), and the concentrations were gradually increased. The cells were exposed to each concentration of anesthetics for ∼10 min. To ensure that the effects are not due to changes in the electrophysiological parameters, the application of the anesthetics was done 15–20 min after seal formation and only when cell parameters as well as the currents stabilized so that at least three consequent control recordings completely overlapped. For dose-dependence analysis, the peak IK+ values in the presence of the anesthetics were normalized to the control peak IK+. For characterization of the channel activation kinetics, a single exponential function, y = A * (1 − e[−(x − xc)/k]), where A is the Imax, xc is the delay, and k is the rate constant τ, was used to fit the final part of the rising phase of each current starting at the time when current reached 50% of its maximal value (delay time). The inactivation time constants were determined by fitting the rising phase of the current to single-exponential decay function y = A1 * exp(−x/t1) + y0, where t1 is current inactivation time constant, τ. The deactivation time constants were determined from the single-exponential decay function fitted to each tail current. For generation conductance–voltage relationships and steady-state inactivation curves, the data were normalized to maximal control conductance and fitted with Boltzmann function G/Gmax = 1/(1 + exp[(V − Vh)/k]), with Gmax being the maximal conductance, Vh the half maximal in(activation), and k the slope factor.
Cell lines.
Mouse L929, B82, and MEL (Murine erythroleukemia) cell lines stably expressing mKv1.1, rKv1.2, mKv1.3, and hKv1.5 and the cell culture maintenance have been described previously (Grissmer et al., 1994). CHO cells expressing hKv1.4 were obtained from M. Tamkun (University of Colorado, Boulder, CO).
Patch-clamp recordings in brain slices.
C57BL/6J mice [postnatal day 23 (P23)–P29] were anesthetized with halothane and decapitated, and the brains were transferred to an ice-cold, sucrose artificial CSF (ACSF) containing the following (in mm): 85 NaCl, 65 sucrose, 2.5 KCl, 25 glucose, 1.25 NaH2PO4, 4 MgSO4, 0.5 CaCl2, and 24 NaHCO3. Coronal thalamic slices (300 μm) were prepared using a Leica VT1000S vibrating blade microtome (Leica). Slices were incubated at 33°C in oxygenated (95% O2, 5% CO2) standard ACSF containing the following (mm): 126 NaCl, 2.5 KCl, 1.25 NaHPO4, 1.2 MgSO4, 10 glucose, 1.2 CaCl2, and 26 NaHCO3 for at least 1 h before the recordings. For electrophysiological recordings, slices were submerged in a recording chamber and continuously perfused at 2 ml/min with oxygenated ACSF at 33°C during the experiments. Slices were securely held in place using a harp (Warner Instruments). CMT nuclei were identified using mouse atlas (Paxinos and Franklin, 2004). Cells were visualized with an upright microscope (Zeiss Axioskop Plus) with infrared differential interference contrast optics. The CMT neurons with Rs values of <35 MΩ, Cm values of 25 ± 5 pF, and characteristic firing frequencies (>43 Hz firing frequency at 150 pA) were chosen for the experiments. Recording pipettes (2–5 MΩ) were pulled from borosilicate glass (Garner Glass) with a P-97 Flaming-Brown puller (Sutter Instruments) and filled with intracellular solution for current-clamp recordings containing the following (in mm): 126 K-gluconate, 4 KCl, 10 HEPES, 2 Mg-ATP, 0.3 Tris-GTP, 10 phosphocreatine, pH, 7.20–7.25 (270–290 mOsm). The pH was adjusted to 7.2 with KOH. Recordings were performed in whole-cell mode using a MultiClamp 700B amplifier (Molecular Devices), digitized at 4 kHz and sampled at 20 kHz with a Digidata 1322A digitizer (Molecular Devices). Data were acquired and analyzed with pClamp 10.2 software (Molecular Devices). The depolarizing current pulses (10, 20, 30, 40, 50, 100, and 150 pA; 800 ms) from the membrane potential of −60 or −75 mV were applied to evoke tonic or burst firing activity, respectively. Sevoflurane and ShK-186 were perfused for 5 min into the bath to allow for equilibrium to be reached before recording. To determine the reversibility of the compounds, the slices were washed for 10 min with ACSF at the end of the experiments. Input resistance was calculated as V = IR, where V is the change in voltage produced by −20 and −100 pA current injections. ΔV/Δt was calculated as change in voltage (ΔV) divided by change in time (Δt) occurred between the onset of the current step (40 pA) and the time point at which the membrane potential reached the action potential threshold.
Statistical analysis.
The results are presented as mean ± SEM. For electrophysiological experiments, statistical differences were determined by two-way repeated-measures ANOVA or Student's t test for two-group comparisons using OriginPro version 8 (OriginLab). When independent groups were compared, we tested whether or not the means were significantly different from each other using independent t tests. When we compared parameters measured before versus after treatment with anesthetics, we used paired t tests. To confirm that each data set was a normally distributed population of observations, we used the Shapiro–Wilk normality test for each data set before performing a paired t test. For histology and behavior components of the in vivo experiments, statistical analysis was performed using Fisher's exact test for nonparametric data.
Results
ShK, a potent blocker of Kv1 channels, reverses sevoflurane-induced anesthesia in rodents
Since Kv1 channels in the rodent brain are primarily heterotetramers containing Kv1.1, Kv1.3, Kv1.4, and Kv1.6 subunits (Turrigiano et al., 1996; Koch et al., 1997; Coleman et al., 1999; Kupper et al., 2002; Trimmer and Rhodes, 2004; Gazula et al., 2010; Pongs and Schwarz, 2010; Ramirez-Navarro et al., 2011), we examined the effect of ShK, a peptide inhibitor of Kv1.1, Kv1.3, and Kv1.6 channels with picomolar potency (Castañeda et al., 1995), in rats anesthetized with the inhalational anesthetic sevoflurane. Rats were continually exposed to a dose of sevoflurane (1.3 ± 0.05% atm) well past the point required to lose the righting reflex as an index of loss of consciousness. This sevoflurane concentration corresponds to ∼0.6 of the rat's minimum alveolar concentration (MAC), which is defined as the concentration of the inhalational anesthetic in the lungs needed to prevent reaction in 50% of subjects in response to surgical stimuli. Microinfusion of 1 pm ShK directly into the CMT nucleus temporarily restored righting reflex in 66.5% of animals under anesthesia (Fig. 1). ShK-treated rodents righted themselves fully despite being continually exposed to 1.3% sevoflurane (Fig. 1A, 3). Righting occurred in 364 ± 324 s (range, 25–730 s). The arousal response lasted 135 ± 73 s (range, 66–250 s). Infusions that missed the CMT nucleus as revealed by histological analysis (Fig. 1B,C) restored righting reflex only in 4.5% of animals. ShK concentrations of 10 and 100 pm caused seizures in rats, predominantly with direct CMT infusions (data not shown). Control microinfusions of saline into the CMT nucleus were without effect (data not shown), as in previous reports (Alkire et al., 2009).
Figure 1.

ShK toxin restores righting reflex in sevoflurane-anesthetized rats. A, Representative frames from a video showing a typical return to “consciousness” response after thalamic microinfusion of ShK. The infusion started at a time point (1) where the animal is shown unconscious on its back in a chamber filled with sevoflurane (∼1.3% atm). At about 3 min and 31 s after the infusion, the animal started to arouse and then righted itself (2). One minute later (3), the animal is moved across the chamber before the end of arousal response (4). B, Histological localizations of the ShK microinfusions in brain sections. Infusion sites and resulting behavioral responses to ShK are indicated. CMT is highlighted gray (n = 28 rats). C, Thalamic microinfusions of ShK restore righting reflex in rats under sevoflurane anesthesia. The effectiveness of ShK is significantly associated with infusion site (indicated by hash mark). p < 0.005 (Fisher's exact test for nonparametric data).
Collectively, these data indicate that blockade of Kv1 channels in CMT neurons reverses sevoflurane-mediated loss of the righting reflex. The most likely Kv1 channel subunits that contribute to anesthesia-reversal are Kv1.3 [IC50(ShK), 11 pm] and/or Kv1.1 [IC50(ShK), 16 pm], whereas Kv1.6 [IC50(ShK), 165 pm] might be involved in seizure induction resulting from infusion of higher concentrations of ShK.
Sevoflurane modulates heterologously expressed Kv1 channels at subsurgical and surgical concentrations
Because inhibitors of Kv1.1–Kv1.3 (ShK and anti-Kv1.2 antibody; Alkire et al., 2009) reverse sevoflurane-mediated anesthetic effects in vivo, we first explored the effects of inhalational anesthetics on these Kv1-family channels heterologously expressed in mammalian cells. Aqueous solutions of anesthetics of known concentrations were directly applied to the cells during the patch-clamp experiments. Peak K+ current (IK+) through Kv1.1, Kv1.2, and Kv1.3 channels increased in the presence of 0.2 mm sevoflurane at low depolarizing potentials (Fig. 2A–E, left, middle). At higher depolarizing potentials (+30 mV), K+ currents through Kv1.1 and Kv1.2 channels were potentiated by sevoflurane, whereas the Kv1.3 current was inhibited concordant with accelerated C-type inactivation (see Fig. 5B). In addition, we tested whether sevoflurane has similar effect on other Shaker-related channels, rapidly inactivating Kv1.4 channels, and the delayed rectifier Kv1.5 channels. Sevoflurane potentiated current through Kv1.5 channels at low voltages and inhibited it at higher voltages, similar to its effects on Kv1.3 currents, albeit without noticeably altering the inactivation rate (Fig. 2E). In contrast, the Kv1.4 current was inhibited by sevoflurane at depolarizing potentials ranging from −60 to +40 mV (Fig. 2D).
Figure 2.
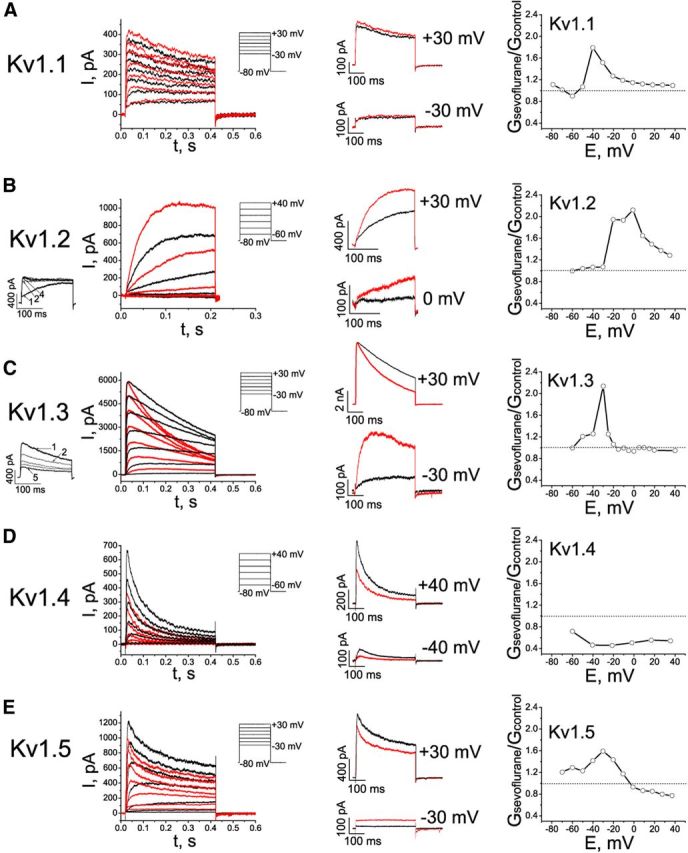
Effects of sevoflurane on potassium currents in cells expressing mammalian Kv1 channels. A–E, Left, Representative outward currents evoked by depolarizing pulses from a holding potential of −80 mV to the range of potentials from −30 to +30 mV, with 10 mV increments and interpulse intervals of 10 s for recording Kv1.1 currents or 30 s for recording Kv1.2, Kv1.3, Kv1.4, and Kv1.5 currents. Control (black) currents are superimposed with currents recorded in the presence of 0.2 mm sevoflurane (red). The cells were exposed to aqueous anesthetic solutions prepared as described previously (Mandal and Pettegrew, 2008). Middle, Comparison of the sevoflurane effects on outward potassium currents at low and higher membrane potentials. The traces shown are the same as in left panels, only scaled as indicated. Right, Voltage dependence of the sevoflurane-mediated conductance changes (n = 3–9 cells).
Figure 5.

Modulation of Kv1.3 channel kinetics by sevoflurane. A–C, The effect of sevoflurane on activation (A), inactivation (B), and deactivation (C) kinetics of Kv1.3 current. D, Effect of 0.2 mm sevoflurane on peak IK+ conductance–voltage relationships and on steady-state inactivation of Kv1.3 channels. The data are fitted with a Boltzmann function, with values for V1/2 given in the Table (right).
To further investigate the effects of sevoflurane on conductance of Kv1-family channels, the ratios of the chord conductance values in the presence of sevoflurane to control conductance values were plotted against the voltage. This analysis revealed the voltage ranges for the potentiating (Gsevoflurane/Gcontrol > 1) and inhibiting (Gsevoflurane/Gcontrol < 1) effects of sevoflurane on each channel type (Fig. 2A–E, right). Based on these results, the concentration dependence of sevoflurane effects was evaluated at voltages that yield most prominent potentiating and inhibiting effects (Fig. 3A–E). In the first set of the experiments (Fig. 3A–E, left) the concentration of sevoflurane was gradually increased to mimic the conditions when the anesthetic was administered in vivo. Sevoflurane potentiated Kv1.1, Kv1.2, Kv1.3, and Kv1.5 channels at concentrations that are significantly lower than surgical anesthetic concentrations (i.e., <0.32 mm or MAC for sevoflurane in rats and in humans; Fig. 3A–C,E), indicating that delayed rectifier Kv1 channels are substantially more sensitive to sevoflurane than other K+ conductances (Patel et al., 1999; Liu et al., 2004). Importantly, the concentrations of sevoflurane at which it inhibited Kv1.4 channels was within the surgical range of concentrations (Fig. 3D), whereas inhibition of Kv1.3 and Kv1.5 was observed only at larger depolarizing potentials and at the concentrations higher than surgical levels (Fig. 3C,E). In the next set of the experiments (Fig. 3A–E, right), to demonstrate the reversibility of the effects, the cells were exposed to a single subsurgical dose (0.2 mm) of sevoflurane followed by washing out the anesthetic. Together, these results indicate that at physiologically relevant voltages (corresponding to small changes in the membrane potential that are sufficient to activate potassium conductance), sevoflurane potentiates Kv1.1, Kv1.2, Kv1.3, and Kv1.5 currents at concentrations ranging from the subsurgical through the surgical level. In contrast, sevoflurane had no effect on lipid bilayer conductance at concentrations that significantly affected Kv1 currents in cells (data not shown), consistent with anesthetics affecting channels, directly or indirectly, but not the membrane conductance.
Sevoflurane interacts with the closed state of Kv1.3 channel and alters Kv1.3 activation, inactivation, and deactivation kinetics
Because our in vivo data suggested an important role for Kv1.3 channel subunits in sevoflurane-mediated loss of the righting reflex, and because sevoflurane potentiated Kv1.3 channels in vitro (Fig. 3C), we characterized sevoflurane effects on Kv1.3 channels in more detail. Gradual increase in the sevoflurane concentration resulted in increased peak K+ current at low depolarizing potentials followed by decreased peak current at higher voltages (Fig. 4A). The current potentiated at low voltages (−30 mV) by sevoflurane in Kv1.3-expressing cells was blocked by ShK and ShK-186 (Fig. 4B), supporting that the channel potentiated by sevoflurane is Kv1.3.
Figure 4.

Modulation of intrinsic Kv1.3 channel properties by sevoflurane. A, Kv1.3 currents in one representative L929 cell activated with 10 mV increments and interpulse intervals of 30 s in control conditions and in the presence of sevoflurane. B, Kv1.3 current evoked by depolarizing voltage pulse to −30 mV is potentiated by sevoflurane and blocked by ShK and ShK-186, specific Kv1.3 channel blockers. C, The potentiation of Kv1.3 current at −30 mV after preexposure to sevoflurane for 10 s, 30 s, and 5 min. D, Sevoflurane modulates Kv1.3 channels in closed configuration (n = 3 experiments). The control current (black) was recorded before exposure to sevoflurane (red). The second current trace (depolarization to either +30 or −30 mV) was evoked 45 s after the addition of sevoflurane without pulsing (channels are closed), and the third current trace was evoked 45 s later. The second trace at −30 mV is superimposed over the third trace (gray over red). The data are presented as mean ± SEM; n = 3 cells for each data point; paired t test. n/s, Not significant.
Application of sevoflurane at the holding potential (−80 mV) significantly enhanced the current evoked by the first depolarizing pulse (to −30 mV) compared to control current recorded before anesthetic addition. The degree of potentiation of Kv1.3 current at −30 mV by sevoflurane was independent of the duration of preexposure (Fig. 4C) and was not affected by the pulse duration, the voltage during the first pulse, or the number of subsequent pulses (Fig. 4D), suggesting that sevoflurane-dependent potentiation is most likely not use dependent (i.e., does not require Kv1.3 opening).
Next, we investigated the effect of sevoflurane on the kinetics of Kv1.3 current. Sevoflurane accelerated Kv1.3 activation compared to control conditions (Fig. 5A), with the most significant acceleration at small depolarizing potentials consistent with the increase of peak current amplitude at these voltages. C-type inactivation of Kv1.3 (Hoshi et al., 1991) was also accelerated in the presence of sevoflurane and coincided with reduction of the peak current amplitude at voltages above 0 mV (Fig. 5B). Sevoflurane slowed deactivation kinetics of Kv1.3 channels (Fig. 5C), suggesting that at the physiologically relevant voltages Kv1.3 channels close slower in the presence of sevoflurane than under control conditions. Both activation and inactivation curves were shifted in the direction of more hyperpolarizing potentials in the presence of sevoflurane compared to control conditions at the voltages ranging from about −40 to −20 mV (Fig. 5D).
Overall, these data demonstrate that sevoflurane interacts with Kv1.3 channels presumably in the closed state, and upon channel activation at the low depolarizing potentials it forces Kv1.3 channels to both open faster and close slower. The consequences of such effects in the brain would be neuronal inhibition during anesthesia by means of increasing the contribution of inhibitory K+ conductance.
Kv1.3 channels are modulated by isoflurane and desflurane
Sevoflurane is a commonly used inhalational anesthetic. If potentiation of Kv1 channels by sevoflurane contributes to the mechanism of inhalational anesthetic action, chemically related inhalational anesthetics should have similar effects on these channels. Indeed, two other commonly used halogenated anesthetics, desflurane and isoflurane, had qualitatively similar effects on Kv1.3 channels (Fig. 6A,D). As in the experiments with sevoflurane, the concentrations of either desflurane or isoflurane were gradually increased to mimic the in vivo anesthesia conditions (Fig. 6B,E). In addition, the subset of cells was exposed to a single dose of either anesthetic to test the reversibility of the effects (Fig. 6C,F). Peak IK+ of Kv1.3 was significantly increased at small depolarizing potentials by subsurgical concentrations of desflurane (Fig. 6B,C) and isoflurane (Fig. 6E,F). At the higher voltages (+30 mV), both anesthetics at concentrations above the surgical concentration-range inhibited Kv1.3 (Fig. 6B,C,E,F). Desflurane and isoflurane also accelerated activation and inactivation of Kv1.3. Potentiation of Kv1.3 saturates and the effect reverses (becoming inhibitory) with increasing concentrations of sevoflurane (>2 MAC; data not shown), desflurane (>1 MAC), and isoflurane (>0.5 MAC; Fig. 6B,E). Thus, all three commonly used inhalational anesthetics (sevoflurane, desflurane, and isoflurane) modulate Kv1.3 channels in a similar fashion.
Sevoflurane inhibits neuronal firing frequency in CMT nucleus
To further elucidate the mechanisms underlying the effect of sevoflurane in the CMT and the role of Kv1 channels in the anesthetic action, we performed current-clamp recordings from rodent brain slices and analyzed firing properties of individual CMT neurons before and after application of sevoflurane. Injections of depolarizing current pulses elicited tonic action potential firing when cells where initially held at −60 mV, or burst activity when holding membrane potential was below −75 mV (Fig. 7A). These results are consistent with previous reported data (Kanyshkova et al., 2011) and demonstrate that the firing properties of CMT neurons are similar to those in the other intralaminar thalamic nuclei. We first tested the effect of sevoflurane on the tonic neuronal activity, which is typically associated with the awake state. Sevoflurane was added to the perfusion system at a concentration of 0.2 mm, which corresponds to 1.3% or ∼0.5 MAC for this anesthetic in mice and is in the subsurgical concentration range. Sevoflurane prominently affected tonic action potential firing properties of CMT neurons, resulting in a significantly increased delay between the onset of the current step and the threshold of the first action potential, as evident from the decreased ΔV/Δt ratio (Fig. 7B,D; Table 1). Sevoflurane also progressively decreased the action potential firing frequency and increased input resistance (Fig. 7B,E; Table 1), whereas the amplitude of the action potential and the half-width were not affected (Table 1). The slight hyperpolarization of the membrane potential was observed in some cells in the presence of sevoflurane, but overall this effect was not statistically significant (Fig. 7C). In addition, sevoflurane reduced the number of action potentials in a burst when applied to phasically firing neurons (Fig. 8). These results are consistent with sevoflurane modulating the action potential properties in CMT neurons by increasing the contribution of the K+ conductance.
Figure 7.

Sevoflurane suppresses intrinsic excitability of central medial thalamic neurons. A, Tonic and phasic activity recorded from CMT neurons in brain slices. CMT neurons were injected with positive current (800 ms) from a membrane potential of −60 or −75 mV. Depolarizing current steps elicited tonic activity in cells with a membrane potential of −60 mV and phasic burst activity in the same cell at −75 mV. B, Representative traces of tonic action potential firing during 800 ms injection of depolarizing currents from a membrane potential of −60 mV in control conditions (left), in the presence of 0.2 mm sevoflurane (center), and after washing out the anesthetic (right). C, Membrane potential before the injection of 150 pA current in control conditions, in the presence of 0.2 mm sevoflurane, and after washing out the anesthetic. D, The effect of 0.2 mm sevoflurane on the mean delay between the onset of the 150 pA current step and the peak of the first action potential. E, The effect of 0.2 mm sevoflurane on firing frequency of the tonic series of action potentials induced by 150 pA current step. Horizontal bars represent the means. n = 9 cells. *p < 0.05 (paired t test).
Table 1.
The effect of 0.2 mm sevoflurane on tonic action potential firing properties
| Resting potential (mV) | AP threshold (mV) | Peak amplitude (mV) | Half-width (ms) | ΔV/Δt | Input resistance (MΩ) | n | |
|---|---|---|---|---|---|---|---|
| Control | −59.4 ± 0.6 | −35.8 ± 0.6 | 23.6 ± 3.1 | 0.55 ± 0.01 | 0.51 ± 0.1 | 211 ± 11 | 9 |
| 0.2 mm sevoflurane | −57.5 ± 1.7 | −35.3 ± 0.9 | 22.8 ± 2.9 | 0.58 ± 0.02 | 0.43 ± 0.1* | 229 ± 10* | 9 |
The series of action potentials was induced by injection of 40 pA current in neurons with membrane potential of −60 mV prior to the application of depolarizing current pulses. The data are presented as mean ± SEM.
*p < 0.05 (vs control, paired t test).
Figure 8.

The effect of sevoflurane on burst activity in CMT neurons in brain slices. A, Representative traces of a phasic firing of action potentials elicited by a depolarizing current pulse (800 ms, 30 pA) from a membrane potential of −75 mV. Sevoflurane reversibly decreased the number of action potentials fired during phasic activity. B, The number of action potentials in bursts during phasic activity in control conditions, in the presence of sevoflurane, and 12 min after washing out the anesthetic with ACSF. Horizontal bar represents the mean. n = 4 cells. *p = 0.01 (paired t test).
ShK-186 prevents sevoflurane's effect on CMT neurons in brain slice recordings
Next, we investigated the contribution of Kv1.3-containing channels in mediating the effects of sevoflurane on neuronal firing properties in the brain slices. We applied ShK-186, a synthetic analog of ShK with 100-fold higher selectivity for Kv1.3 (IC50, 69 pm) over Kv1.1 (IC50, 7000 pm; Beeton et al., 2005; Pennington et al., 2009; Chi et al., 2012), to CMT neurons before application of sevoflurane (Fig. 9A–C) or after application of sevoflurane (D–F).
Figure 9.

ShK-186 toxin prevents the effects of sevoflurane on tonic firing properties in CMT neurons in brain slices. A, Representative traces of tonic action potential firing elicited during 800 ms injection of 150 pA current from a membrane potential of −60 mV in control conditions (left), in the presence of 350 pm ShK-186 (left center), after addition of 0.2 mm sevoflurane (right center), and after washing out the anesthetic (right). B, The effect of ShK-186 (350 pm) on averaged membrane potential and the absence of sevoflurane's effect on the membrane potential before injection of the depolarizing currents. C, The effect of ShK-186 (350 pm) on mean firing frequency of the tonic series of action potentials induced by 150 pA current pulse and the absence of the effect of sevoflurane on firing frequency in the presence of ShK-186. n = 5 cells. *p < 0.05 (paired t test; B, C). D, Representative traces of tonic action potential firing elicited during 800 ms injection of 150 pA current from a membrane potential of −60 mV in control conditions (left), in the presence of 0.2 mm sevoflurane (left center), after the addition of 350 pm ShK-186 (right center), and after washing out the chemicals (right). E, Absence of sevoflurane's effect on averaged membrane potential and the effect of ShK-186 (350 pm) on the membrane potential before injection of the depolarizing currents. F, The effect of 0.2 mm sevoflurane before and after addition of ShK-186 on mean firing frequency of the tonic series of action potentials induced by 150 pA current pulse. n = 4–7 cells. *p < 0.04 (paired t test; E, F). Horizontal bars represent the means. n/s, Not significant.
In the first set of experiments, the slices were exposed to ShK-186 before addition of sevoflurane. Addition of 350 pm ShK-186 to the bath solution caused depolarization of the membrane potential (from −60.1 ± 0.9 mV to −54.7 ± 2 mV) as well as an increase of the tonic action potential firing frequency of CMT neurons (Fig. 9A–C; Table 2). Because 350 pm ShK-186 inhibits Kv1.3 channels without a significant effect on Kv1.1, these results are consistent with a fraction of K+ channels containing one or more Kv1.3 subunits being tonically open at the potentials near the resting and having a role in regulating neuronal excitability in the CMT. Higher concentrations of ShK-186 (7 nm) that block both Kv1.3 and Kv1.1 channels caused strong depolarization, suggesting that Kv1.1 channel subunits also participate in regulating neuronal excitability in the CMT. Under these conditions, however, the membrane potential rose to a level that lowered cell viability, preventing further recordings (data not shown). Addition of sevoflurane produced no further effect on the action potential firing frequency in the presence of 350 pm ShK-186 (Fig. 9A,C), suggesting that the reduction of neuronal firing frequency in the presence of sevoflurane at subsurgical level is, at least partially, due to its effect on Kv1.3-containing channels.
Table 2.
The effect of 350 pm ShK-186 on action potential firing properties of CMT neurons and the lack of sevoflurane's effect in the presence of ShK-186
| Resting potential (mV) | AP threshold (mV) | Peak amplitude (mV) | Half-width (ms) | ΔV/Δt | Input resistance (MΩ) | n | |
|---|---|---|---|---|---|---|---|
| Control | −60.1 ± 0.9 | −39.6 ± 1.2 | 26.9 ± 4.2 | 0.58 ± 0.05 | 0.39 ± 0.1 | 223 ± 24 | 5 |
| 350 pm ShK-186 | −54.7 ± 2.0* | −40 ± 1.2 | 24.4 ± 4 | 0.59 ± 0.06 | 0.49 ± 0.1 | 234 ± 41 | 5 |
| 0.2 mm sevoflurane plus 350 pm ShK-186 | −53.8 ± 2.4 | −36.5 ± 2 | 23.8 ± 3.9 | 0.6 ± 0.04 | 0.57 ± 0.1 | 259 ± 37 | 5 |
The series of tonic action potentials was induced by injection of 40 pA current. The data are presented as mean ± SEM.
*p < 0.05 (vs control, paired t test).
In the second set of the experiments, the slices were first exposed to sevoflurane followed by addition of ShK-186 (Fig. 9D) to mimic the conditions of intrathalamic microinfusion of ShK in anesthetized rats (Fig. 1). Addition of 350 pm ShK-186 to the slices that had already been exposed to sevoflurane partially reversed the sevoflurane-mediated reduction of the action potential firing frequency (Fig. 9D–F), supporting the contribution of ShK-186-sensitive, Kv1.3-containing channels to the sevoflurane-mediated effects in CMT.
Discussion
The mechanisms of general anesthesia involve multiple molecular targets and pathways that are not completely understood. Most of the inhalational anesthetics enhance synaptic and extrasynaptic inhibition by increasing GABAA channel opening through its binding to the proposed cavity between the TM2 and TM3 segments (Krasowski and Harrison, 1999; Jenkins et al., 2001; Hemmings et al., 2005). Furthermore, inhalational anesthetics at clinical concentrations activate different types of two-pore-domain potassium channels, resulting in hyperpolarization and reduced neuronal excitability (Patel et al., 1999; Kindler and Yost, 2005), but inhibit hyperpolarization-activated cyclic nucleotide-gated (HCN) channels (Sirois et al., 2002; Chen et al., 2005), reducing the bursting frequency of certain neurons with pacemaker activity. Among voltage-gated ion channels, Nav and Kv channels are differentially modulated by anesthetics depending on the channel type. Thus, Nav1.2, Nav1.5, Nav1.4, and Nav1.6 channels, but not Nav1.8 channels, are inhibited by inhalational anesthetics, reducing synaptic transmission (Rehberg et al., 1996; Shiraishi and Harris, 2004), whereas certain types of Shaw- and Shaker-related Kv channels are activated by inhalational anesthetics (Correa, 1998; Bhattacharji et al., 2010; Barber et al., 2012). Despite numerous discoveries of anesthetics affecting various receptors and channels, not all of the anesthetic-sensitive molecules are accepted as putative targets. It is essential to establish the neuronal networks that underlie the effects of anesthetics and link the anesthetic–target interaction with behavior endpoints—amnesia, unconsciousness, analgesia, and immobility (Brown et al., 2011).
In the present work, we elucidate the role of Shaker-related K+ channels in anesthesia induced by volatile general anesthetics. Although it has been proposed previously that K+ channels may contribute to regulation of neuronal activity essential for consciousness (Walcourt et al., 2001; Arhem et al., 2003), whether clinical effects of anesthetics are mediated by direct interaction with these channels has long been a subject of debate. Walcourt et al. (2001) demonstrated that Shaker channels are important determinants of halothane anesthesia in a specific brain circuit of Drosophila. We discovered that blocking the mammalian homologues of Shaker channels in the CMT, but not in the nearby nuclei, of lightly anesthetized rats by the ShK toxin restored “consciousness,” as evidenced by an arousal response, strongly supporting the idea that ShK-sensitive Shaker-related K+ channels in the CMT are involved in regulating the arousal state and controlling levels of consciousness. These data are consistent with previously demonstrated altered sensitivity to inhalational anesthetics and/or disrupted sleep cycles in Drosophila melanogaster strains containing mutant Shaker K+ channels (Kaplan and Trout, 1969; Tinklenberg et al., 1991; Walcourt et al., 2001; Cirelli et al., 2005; Weber et al., 2009).
Our results obtained using mouse brain slices further confirm that sevoflurane potentiates potassium currents, and thereby suppresses the neuronal excitability in CMT. The results obtained in vivo using rats and in brain slices using mice are in a good agreement, consistent with well-established equivalence and similar patterns of connectivity of the various intralaminar and midline nuclei of the thalamus between different species including nonprimate mammals, nonhuman primates, and humans (Van der Werf et al., 2002). Using ShK-186, a synthetic ShK analog with high selectivity for Kv1.3, to block the anesthetic effects on neuronal firing properties in CMT in brain slices, we narrowed down the list of K+ channel subunits that could be potential anesthetic targets. In particular, ShK-186 when applied at concentrations as low as 350 pm blocked the effect of sevoflurane on action potential firing frequency, providing pharmacological evidence for the contribution of Kv1.3-containing channels to the effects of sevoflurane. Importantly, the expression of Kv1.1, Kv1.2, and Kv1.4-mRNA had been demonstrated earlier in the rat CMT (Kanyshkova et al., 2011). Therefore, in addition to the previous finding that Kv1.2-containing channels in CMT nucleus are important for maintenance of the unconscious state during inhalational anesthesia (Alkire et al., 2009), our data demonstrate that potassium channels that contain Kv1.3 subunits can also be the targets for inhalational anesthetics in CMT. The other Shaker-related ShK-sensitive potassium channel subunits, such as Kv1.1 and Kv1.6, can play roles in the regulation of neuronal excitability and cannot be excluded as potential targets for anesthetics. Although the other types of potassium channels, distinct from the Shaker-related Kv1 family, that are known to regulate neuronal firing properties in the intralaminar nuclei of the thalamus (e.g., Kv4; Kanyshkova et al., 2011) cannot be excluded as potential anesthetics targets, they are unlikely to contribute to the sevoflurane-mediated reduction of firing frequency, as these channels are shown to be inhibited by inhalational anesthetics (Ishiwa et al., 2008). Finally, high selectivity of ShK-186 peptide over even structurally related Kv1 channels makes it unlikely for two-pore-domain K+ channels to mediate the ShK-sensitive effects of sevoflurane observed in this study.
The Shaker-related Kv1-family of K+ channels comprise eight highly conserved α subunits designated Kv1.1–Kv1.8 (Gutman et al., 2005). Kv1.1, Kv1.2, Kv1.3, Kv1.4, and Kv1.6 are expressed ubiquitously in the mammalian CNS. Of these, Kv1.4 is a rapidly inactivating A-type channel, while the others are delayed rectifiers. Neuronal Kv1 channels coassemble with each other and with auxiliary β subunits forming homomultimers or heteromultimers with diverse biophysical properties (Turrigiano et al., 1996; Koch et al., 1997; Coleman et al., 1999; Kupper et al., 2002; Trimmer and Rhodes, 2004; Gazula et al., 2010; Pongs and Schwarz, 2010; Ramirez-Navarro et al., 2011). In this study, we directly tested the effects of anesthetics on heterologously expressed homomeric Kv1.1, Kv1.2, Kv1.3, Kv1.4, and Kv1.5 channels. In contrast to previous studies (Franks and Lieb, 1991; Arhem et al., 2003), we found that the manner in which inhalational anesthetics modulate Kv1 channels depends markedly on the biophysical properties of the channel in question. Specifically, at the low depolarizing potentials (arguably the most physiologically relevant), these inhalational anesthetics significantly enhanced K+ currents in cells expressing Kv1-family delayed rectifier (Kv1.1, Kv1.2, Kv1.3, and Kv1.5) channels. Furthermore, we found that anesthetics can modulate delayed rectifier Kv1 channels in a biphasic manner, potentiating them at small depolarizing potentials and inhibiting them at higher voltages. Our results are consistent with the previously described isoflurane-mediated increase of K+ conductance of ShakerB mutant channels with delayed rectifier properties (Correa, 1998) and the previously reported activation of Kv1.2 and Kv1.5 channels by sevoflurane (Barber et al., 2012). The potentiation effect was specific to delayed rectifier channels, and fast-inactivating “A-type” Kv1.4 channels were inhibited, in agreement with the results of Franks and Lieb (1991), who demonstrated inhibition by isoflurane of fast A currents in the snail neurons. Critically, potentiation of Kv1 channels by sevoflurane and other volatile anesthetics is achieved at extremely low anesthetic concentrations, significantly below surgical anesthetic levels. At these low subsurgical concentrations, sevoflurane had no effect on the rapidly inactivating Kv1.4 channel, which was inhibited only at surgical concentrations. Anesthetic-mediated inhibition of delayed rectifier Kv1 channels at higher depolarized potentials also occurs at anesthetic concentrations much higher than those required for potentiation. Thus, at subsurgical anesthetic concentrations, potentiation of delayed rectifier Kv1 channels is the predominant effect. Our results obtained using heterologously expressed homomeric channels demonstrate the effects of anesthetics on particular types of Kv1 α subunits. Therefore, the endogenously expressed channels that contain one or more anesthetic-sensitive subunits (e.g., Kv1.1, Kv1.2, or Kv1.3) are likely to be affected by anesthetics as well. The other membrane proteins, including auxiliary β subunits, as well as the stoichiometry and relative expression level of endogenous Kv1 channel subunits cannot be excluded as contributing factors in a complex action of the anesthetics on the native channels in the brain. Although the role of each of these factors remains to be elucidated, the results of the brain slice recordings are consistent with the conclusion that Kv1 channels that contain ShK-sensitive α subunits mediate the effects of sevoflurane on neuronal firing properties in CMT.
Delayed rectifier K+ channels play an important role in membrane repolarization and in setting the threshold for action potentials. The net effect of Kv1 channel modulation by inhalational anesthetics, particularly the facilitation of K+ current at the low depolarizing potentials, acceleration of activation kinetics and slowing of the deactivation, is an increased contribution of the inhibitory potassium conductance. This is consistent with delayed onset and reduced rate of the action potential firing in thalamic slices in the presence of sevoflurane. At the brain level, such changes in the nervous system could contribute to the progressive slowing of the electroencephalogram with increasing doses of anesthesia during induction of the anesthetic state. The potentiation of Kv1 currents in the thalamus by subsurgical anesthetic concentrations might contribute to early loss of consciousness during the behavioral stage of anesthesia, whereas higher doses of anesthetics will inhibit Kv1 channels and affect other molecular targets. This inhibition, in turn, might facilitate the release of inhibitory neurotransmitters during the surgical stage of anesthesia.
In conclusion, our results support the hypothesis that Kv1 channels, including Kv1.3-containing channels, in the central medial thalamus are important for suppressing arousal during anesthesia. The extremely high sensitivity of these channels to volatile anesthetics places them among primary anesthetic targets, such as nicotinic α4β2 ACh receptors (Violet et al., 1997; Yamakura et al., 2000; Yamashita et al., 2005) and GABAA receptors (Wu et al., 1996; Caraiscos et al., 2004; Hall et al., 2004; Weng et al., 2010). Although the molecular mechanisms of Kv1 channel modulation by anesthetics in vivo remain to be determined, the anesthetic sensitivity of Kv1-family channels suggests that this modulation may occur early on during the induction of anesthesia, at around the time when the behavioral loss of consciousness occurs. These findings will contribute to a better understanding of the neural pathways of anesthesia, which in turn should facilitate the design of future anesthetic agents and procedures that will specifically target anesthesia-relevant brain pathways and reduce adverse effects of general anesthesia.
Footnotes
This work was supported by National Institutes of Health Grants 1P01 AG032131 (J.E.H.) and RO1 NS48252 (K.G.C.), a grant from the Larry L. Hillblom Foundation (J.E.H.), NIH Grant NS048336 (A.L.G.), and departmental resources (M.T.A.). We thank Dr. M. D. Cahalan for his critical comments on the earlier version of this manuscript and for access to the patch-clamp setup, Dr. L. Forrest for assistance with cell culture, and Dr. P.R. Dennison and the Nuclear Magnetic Resonance Facility at University of California, Irvine for help and providing access to NMR spectroscopy.
The authors declare no competing financial interests.
References
- Alkire MT, Asher CD, Franciscus AM, Hahn EL. Thalamic microinfusion of antibody to a voltage-gated potassium channel restores consciousness during anesthesia. Anesthesiology. 2009;110:766–773. doi: 10.1097/ALN.0b013e31819c461c. [DOI] [PubMed] [Google Scholar]
- Andres-Enguix I, Caley A, Yustos R, Schumacher MA, Spanu PD, Dickinson R, Maze M, Franks NP. Determinants of the anesthetic sensitivity of two-pore domain acid-sensitive potassium channels. J Biol Chem. 2007;282:20977–20990. doi: 10.1074/jbc.M610692200. [DOI] [PubMed] [Google Scholar]
- Arhem P, Klement G, Nilsson J. Mechanisms of anesthesia: towards integrating network, cellular, and molecular level modeling. Neuropsychopharmacology. 2003;28:S40–S47. doi: 10.1038/sj.npp.1300142. [DOI] [PubMed] [Google Scholar]
- Barber AF, Liang Q, Covarrubias M. Novel activation of voltage-gated K+ channels by sevoflurane. J Biol Chem. 2012;287:40425–40432. doi: 10.1074/jbc.M112.405787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeton C, Pennington MW, Wulff H, Singh S, Nugent D, Crossley G, Khaytin I, Calabresi PA, Chen CY, Gutman GA, Chandy KG. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol Pharmacol. 2005;67:1369–1381. doi: 10.1124/mol.104.008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharji A, Klett N, Go RCV, Covarrubias M. Inhalational anaesthetics and n-alcohols share a site of action in the neuronal Shaw2 Kv channel. British J Pharmacol. 2010;159:1475–1485. doi: 10.1111/j.1476-5381.2010.00642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan G, LeBard DN, Henin J, Eckenhoff RG, Klein ML. Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc Natl Acad Sci U S A. 2010;107:14122–14127. doi: 10.1073/pnas.1008534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EN, Purdon PL, Van Dort CJ. General anesthesia and altered states of arousal: a systems neuroscience analysis. Annu Rev Neurosci. 2011;34:601–628. doi: 10.1146/annurev-neuro-060909-153200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraiscos VB, Newell JG, You-Ten KE, Elliott EM, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Selective enhancement of tonic GABAergic inhibition in murine hippocampal neurons by low concentrations of the volatile anesthetic isoflurane. J Neurosci. 2004;24:8454–8458. doi: 10.1523/JNEUROSCI.2063-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda O, Sotolongo V, Amor AM, Stöcklin R, Anderson AJ, Harvey AL, Engström A, Wernstedt C, Karlsson E. Characterization of a potassium channel toxin from the Caribbean sea anemone Stichodactyla helianthus. Toxicon. 1995;33:603–613. doi: 10.1016/0041-0101(95)00013-C. [DOI] [PubMed] [Google Scholar]
- Chen X, Sirois JE, Lei Q, Talley EM, Lynch C, 3rd, Bayliss DA. HCN subunit-specific and cAMP-modulated effects of anesthetics on neuronal pacemaker currents. J Neurosci. 2005;25:5803–5814. doi: 10.1523/JNEUROSCI.1153-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi V, Pennington MW, Norton RS, Tarcha EJ, Londono LM, Sims-Fahey B, Upadhyay SK, Lakey JT, Iadonato S, Wulff H, Beeton C, Chandy KG. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon. 2012;59:529–546. doi: 10.1016/j.toxicon.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, Tononi G. Reduced sleep in Drosophila Shaker mutants. Nature. 2005;434:1087–1092. doi: 10.1038/nature03486. [DOI] [PubMed] [Google Scholar]
- Coleman SK, Newcombe J, Pryke J, Dolly JO. Subunit composition of Kv1 channels in human CNS. J Neurochem. 1999;73:849–858. doi: 10.1046/j.1471-4159.1999.0730849.x. [DOI] [PubMed] [Google Scholar]
- Correa AM. Gating kinetics of Shaker K+ channels are differentially modified by general anesthetics. Am J Physiol. 1998;275:C1009–C1021. doi: 10.1152/ajpcell.1998.275.4.C1009. [DOI] [PubMed] [Google Scholar]
- Downie DL, Vicente-Agullo F, Campos-Caro A, Bushell TJ, Lieb WR, Franks NP. Determinants of the anesthetic sensitivity of neuronal nicotinic acetylcholine receptors. J Biol Chem. 2002;277:10367–10373. doi: 10.1074/jbc.M107847200. [DOI] [PubMed] [Google Scholar]
- Franks N, Zecharia A. Sleep and general anesthesia. Canadian J Anesthesia. 2011;58:139–148. doi: 10.1007/s12630-010-9420-3. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR. Selective effects of volatile general anesthetics on identified neurons. Ann N Y Acad Sci. 1991;625:54–70. doi: 10.1111/j.1749-6632.1991.tb33829.x. [DOI] [PubMed] [Google Scholar]
- Garcia PS, Kolesky SE, Jenkins A. General anesthetic actions on GABAA receptors. Curr Neuropharmacol. 2010;8:2–9. doi: 10.2174/157015910790909502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazula VR, Strumbos JG, Mei X, Chen H, Rahner C, Kaczmarek LK. Localization of Kv1.3 channels in presynaptic terminals of brainstem auditory neurons. J Comp Neurol. 2010;518:3205–3220. doi: 10.1002/cne.22393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol. 1994;45:1227–1234. [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stühmer W, Wang X. International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- Hall AC, Rowan KC, Stevens RJ, Kelley JC, Harrison NL. The effects of isoflurane on desensitized wild-type and alpha1(S270H) gamma-aminobutyric acid type A receptors. Anesth Analg. 2004;98:1297–1304. doi: 10.1213/01.ane.0000111108.78745.ad. [DOI] [PubMed] [Google Scholar]
- Hemmings HC, Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Ishiwa D, Nagata I, Ohtsuka T, Itoh H, Kamiya Y, Ogawa K, Sakai M, Sekino N, Yamada Y, Goto T, Andoh T. Differential effects of isoflurane on A-type and delayed rectifier K channels in rat substantia nigra. European J Pharmacol. 2008;580:122–129. doi: 10.1016/j.ejphar.2007.10.062. [DOI] [PubMed] [Google Scholar]
- Jenkins A, Franks N, Lieb W. Actions of general anaesthetics on 5-HT3 receptors in N1E-115 neuroblastoma cells. British J Pharmacol. 1996;117:1507–1515. doi: 10.1111/j.1476-5381.1996.tb15314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, Andreasen A, Viner A, Trudell JR, Harrison NL. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21:RC136. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalman K, Pennington MW, Lanigan MD, Nguyen A, Rauer H, Mahnir V, Paschetto K, Kem WR, Grissmer S, Gutman GA, Christian EP, Cahalan MD, Norton RS, Chandy KG. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J Biol Chem. 1998;273:32697–32707. doi: 10.1074/jbc.273.49.32697. [DOI] [PubMed] [Google Scholar]
- Kanyshkova T, Broicher T, Meuth SG, Pape HC, Budde T. A-type K+ currents in intralaminar thalamocortical relay neurons. Pflugers Arch. 2011;461:545–556. doi: 10.1007/s00424-011-0953-2. [DOI] [PubMed] [Google Scholar]
- Kaplan WD, Trout WE., 3rd The behavior of four neurological mutants of Drosophila. Genetics. 1969;61:399–409. doi: 10.1093/genetics/61.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten MR, Rudy B, Anderson MP. Differential regulation of action potential firing in adult murine thalamocortical neurons by Kv3.2, Kv1, and SK potassium and N-type calcium channels. J Physiol. 2007;584:565–582. doi: 10.1113/jphysiol.2007.141135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler CH, Yost CS. Two-pore domain potassium channels: new sites of local anesthetic action and toxicity. Regional Anesth Pain Med. 2005;30:260–274. doi: 10.1016/j.rapm.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Koch RO, Wanner SG, Koschak A, Hanner M, Schwarzer C, Kaczorowski GJ, Slaughter RS, Garcia ML, Knaus HG. Complex subunit assembly of neuronal voltage-gated K+ channels. J Biol Chem. 1997;272:27577–27581. doi: 10.1074/jbc.272.44.27577. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Harrison NL. General anaesthetic actions on ligand-gated ion channels. Cell Mol Life Sci. 1999;55:1278–1303. doi: 10.1007/s000180050371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupper J, Prinz AA, Fromherz P. Recombinant Kv1.3 potassium channels stabilize tonic firing of cultured rat hippocampal neurons. Pflugers Arch. 2002;443:541–547. doi: 10.1007/s00424-001-0734-4. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Aghajanian GK. The role of Kv1.2-containing potassium channels in serotonin-induced glutamate release from thalamocortical terminals in rat frontal cortex. J Neurosci. 2001;21:9955–9963. doi: 10.1523/JNEUROSCI.21-24-09955.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Au JD, Zou HL, Cotten JF, Yost CS. Potent activation of the human tandem pore domain K channel TRESK with clinical concentrations of volatile anesthetics. Anesth Analg. 2004;99:1715–1722. doi: 10.1213/01.ANE.0000136849.07384.44. table of contents. [DOI] [PubMed] [Google Scholar]
- Mandal P, Pettegrew J. Clinically relevant concentration determination of inhaled anesthetics (halothane, isoflurane, sevoflurane, and desflurane) by 19F NMR. Cell Biochem Biophys. 2008;52:31–35. doi: 10.1007/s12013-008-9022-7. [DOI] [PubMed] [Google Scholar]
- McKenzie D, Franks N, Lieb W. Actions of general anaesthetics on a neuronal nicotinic acetylcholine receptor in isolated identified neurones of Lymnaea stagnalis. British J Pharmacol. 1995;115:275–282. doi: 10.1111/j.1476-5381.1995.tb15874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JT, Chen J, Han B, Meng QC, Veasey SC, Beck SG, Kelz MB. Direct activation of sleep-promoting VLPO neurons by volatile anesthetics contributes to anesthetic hypnosis. Curr Biol. 2012;22:2008–2016. doi: 10.1016/j.cub.2012.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M, Corringer PJ. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M. Inhalational anesthetics activate two-pore-domain background K + channels. Nat Neurosci. 1999;2:422–426. doi: 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Amsterdam: Elsevier; 2004. [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Amsterdam: Elsevier; 2005. [DOI] [PubMed] [Google Scholar]
- Pennington MW, Byrnes ME, Zaydenberg I, Khaytin I, de Chastonay J, Krafte DS, Hill R, Mahnir VM, Volberg WA, Gorczyca W, Kem WR. Chemical synthesis and characterization of ShK toxin: a potent potassium channel inhibitor from a sea anemone. Int J Pept Protein Res. 1995;46:354–358. doi: 10.1111/j.1399-3011.1995.tb01068.x. [DOI] [PubMed] [Google Scholar]
- Pennington MW, Beeton C, Galea CA, Smith BJ, Chi V, Monaghan KP, Garcia A, Rangaraju S, Giuffrida A, Plank D, Crossley G, Nugent D, Khaytin I, Lefievre Y, Peshenko I, Dixon C, Chauhan S, Orzel A, Inoue T, Hu X, et al. Engineering a stable and selective peptide blocker of the Kv1.3 channel in T lymphocytes. Mol Pharmacol. 2009;75:762–773. doi: 10.1124/mol.108.052704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010;90:755–796. doi: 10.1152/physrev.00020.2009. [DOI] [PubMed] [Google Scholar]
- Ramirez-Navarro A, Glazebrook PA, Kane-Sutton M, Padro C, Kline DD, Kunze DL. Kv1.3 channels regulate synaptic transmission in the nucleus of solitary tract. J Neurophysiol. 2011;105:2772–2780. doi: 10.1152/jn.00494.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehberg B, Xiao YH, Duch DS. Central nervous system sodium channels are significantly suppressed at clinical concentrations of volatile anesthetics. Anesthesiology. 1996;84:1223–1233. doi: 10.1097/00000542-199605000-00025. discussion 27A. [DOI] [PubMed] [Google Scholar]
- Shiraishi M, Harris RA. Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels. J Pharmacol Exp Ther. 2004;309:987–994. doi: 10.1124/jpet.103.064063. [DOI] [PubMed] [Google Scholar]
- Sirois JE, Lynch C, 3rd, Bayliss DA. Convergent and reciprocal modulation of a leak K+ current and I(h) by an inhalational anaesthetic and neurotransmitters in rat brainstem motoneurones. J Physiol. 2002;541:717–729. doi: 10.1113/jphysiol.2002.018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt K. General anesthesia: activating a sleep switch? Curr Biol. 2012;22:R918–R919. doi: 10.1016/j.cub.2012.09.033. [DOI] [PubMed] [Google Scholar]
- Solt K, Forman SA. Correlating the clinical actions and molecular mechanisms of general anesthetics. Curr Opin Anaesthesiol. 2007;20:300–306. doi: 10.1097/ACO.0b013e32816678a5. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Tinklenberg JA, Segal IS, Guo TZ, Maze M. Analysis of anesthetic action on the potassium channels of the Shaker mutant of Drosophila. Ann N Y Acad Sci. 1991;625:532. doi: 10.1111/j.1749-6632.1991.tb33884.x. [DOI] [PubMed] [Google Scholar]
- Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol. 2004;66:477–519. doi: 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Marder E, Abbott LF. Cellular short-term memory from a slow potassium conductance. J Neurophysiol. 1996;75:963–966. doi: 10.1152/jn.1996.75.2.963. [DOI] [PubMed] [Google Scholar]
- Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Rev. 2002;39:107–140. doi: 10.1016/S0165-0173(02)00181-9. [DOI] [PubMed] [Google Scholar]
- Violet JM, Downie DL, Nakisa RC, Lieb WR, Franks NP. Differential sensitivities of mammalian neuronal and muscle nicotinic acetylcholine receptors to general anesthetics. Anesthesiology. 1997;86:866–874. doi: 10.1097/00000542-199704000-00017. [DOI] [PubMed] [Google Scholar]
- Walcourt A, Scott RL, Nash HA. Blockage of one class of potassium channel alters the effectiveness of halothane in a brain circuit of Drosophila. Anesth Analg. 2001;92:535–541. doi: 10.1213/00000539-200102000-00047. [DOI] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Martin TM, Schwartzkroin PA, Tempel BL. Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature. 1993;365:75–79. doi: 10.1038/365075a0. [DOI] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci. 1994;14:4588–4599. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber B, Schaper C, Bushey D, Rohlfs M, Steinfath M, Tononi G, Cirelli C, Scholz J, Bein B. Increased volatile anesthetic requirement in short- sleeping Drosophila mutants. Anesthesiology. 2009;110:313–316. doi: 10.1097/ALN.0b013e3181942df2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y, Yang L, Corringer PJ, Sonner JM. Anesthetic sensitivity of the Gloeobacter violaceus proton-gated ion channel. Anesth Analg. 2010;110:59–63. doi: 10.1213/ANE.0b013e3181c4bc69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Harata N, Akaike N. Potentiation by sevoflurane of the gamma-aminobutyric acid-induced chloride current in acutely dissociated CA1 pyramidal neurones from rat hippocampus. British J Pharmacol. 1996;119:1013–1021. doi: 10.1111/j.1476-5381.1996.tb15772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakura T, Borghese C, Harris RA. A transmembrane site determines sensitivity of neuronal nicotinic acetylcholine receptors to general anesthetics. J Biol Chem. 2000;275:40879–40886. doi: 10.1074/jbc.M005771200. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Mori T, Nagata K, Yeh JZ, Narahashi T. Isoflurane modulation of neuronal nicotinic acetylcholine receptors expressed in human embryonic kidney cells. Anesthesiology. 2005;102:76–84. doi: 10.1097/00000542-200501000-00015. [DOI] [PubMed] [Google Scholar]