

Abstract
NMDA receptors (NMDARs) expressed by cerebellar molecular layer interneurons (MLIs) are not activated by single exocytotic events but can respond to glutamate spillover following coactivation of adjacent parallel fibers (PFs), indicating that NMDARs are perisynaptic. Several types of synaptic plasticity rely on these receptors but whether they are activated at isolated synapses is not known. Using a combination of electrophysiological and optical recording techniques in acute slices of rat cerebellum, along with modeling, we find that repetitive activation of single PF–MLI synapses can activate NMDARs in MLIs. High-frequency stimulation, multivesicular release (MVR), or asynchronous release can each activate NMDARs. Frequency facilitation was found at all PF–MLI synapses but, while some showed robust MVR with increased release probability, most were limited to univesicular release. Together, these results reveal a functional diversity of PF synapses, which use different mechanisms to activate NMDARs.
Introduction
Spillover of glutamate from the synaptic cleft to distant receptors may impair synaptic specificity. However, activation of extrasynaptic receptors is essential for some forms of synaptic transmission and plasticity (Shin and Linden, 2005; Beierlein and Regehr, 2006; Szapiro and Barbour, 2007; Sun and June Liu, 2007). Investigation of spillover-activated receptors is hampered in many studies by simultaneous release from many neighboring synapses leading to pooling of glutamate and greater activation of distant receptors than would occur with spillover from a single, isolated synapse. In addition, spillover/pooling is usually monitored with synaptic receptors, which can be activated either directly or by spillover, leading to ambiguity in the source of transmitter. In the cerebellar cortex, glutamatergic granule cell axons form excitatory synapses on molecular layer interneurons (MLIs) that can be activated individually by stimulation in the granule cell layer (GCL) (Foster et al., 2005; Marcaggi and Attwell, 2007). In addition, MLIs express both 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid and N-methyl-d-aspartate receptors (AMPARs and NMDARs, respectively) but single exocytotic release events activate only AMPARs at parallel fiber (PF)–MLI synapses (Clark and Cull-Candy, 2002), similar to quantal events at retinal bipolar to ganglion cell synapses (Taylor et al., 1995). Repetitive stimulation of the PFs results in activation of MLI NMDARs, suggesting that NMDARs are located adjacent to the postsynaptic density and can be used to study spillover and pooling of extracellular glutamate from densely packed PF terminals (Carter and Regehr, 2000; Clark and Cull-Candy, 2002; Abrahamsson et al., 2012). Whether extrasynaptic NMDARs require glutamate release from multiple neighboring PFs or can respond to spillover of glutamate from a single synapse during physiological activity is not known. Due to their involvement in plastic events (Shin and Linden, 2005; Beierlein and Regehr, 2006; Sun and June Liu, 2007), determining the conditions under which MLI NMDARs are activated is critical to understanding how synaptic plasticity affects cerebellar function.
Granule cells can respond to mossy fiber input with bursts of action potentials (APs) at frequencies of several hundred hertz (Chadderton et al., 2004; Jörntell and Ekerot, 2006) and their synapses onto both Purkinje cells (PCs) and MLIs exhibit frequency facilitation (Konnerth et al., 1990; Perkel et al., 1990; Atluri and Regehr, 1996). PF boutons have an average of seven vesicles docked at each PF–PC active zone (Xu-Friedman et al., 2001), the presumed source for multivesicular release (MVR) (Foster et al., 2005; Rancillac and Barbara, 2005; Bender et al., 2009; Valera et al., 2012). In addition to MVR, bursts of PF APs cause asynchronous vesicular release (Atluri and Regehr, 1998; Chen and Regehr, 1999). Here, we determine that extrasynaptic NMDARs can be activated at single PF–MLI synapses by physiological stimulation rates. We find that asynchronous release is primarily responsible for activating extrasynaptic NMDARs at single PF–MLI synapses at moderate stimulation frequencies though MVR and high-frequency synchronous release can also activate NMDARs. In addition, synchronous release at the majority of PF–MLI synapses is limited to a single vesicle per AP even at high probability (Pr).
Materials and Methods
Slice preparation and electrophysiology.
Coronal cerebellar slices were prepared from postnatal day 14–19 rats of either sex in accordance with Oregon Health and Science University Institutional Animal Care and Use Committee guidelines. Rats were deeply anesthetized with isoflurane, decapitated, and the cerebellum submerged in ice-cold, oxygenated cutting solution that contained the following (in mm): 110 choline chloride, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 10 glucose, 1.25 NaH2PO4, 25 NaHCO3, 1.3 sodium ascorbate, and 3 sodium pyruvate. Slices (250 μm) were cut from the vermis using a vibroslicer (VT1200S; Leica Instruments) and transferred to oxygenated artificial CSF (ACSF) at 34°C for 30–40 min then allowed to equilibrate at room temperature. ACSF consisted of the following (in mm): 124 NaCl, 2.5 KCl, 1.5 CaCl2, 1 MgCl2, 1 NaH2PO4, 10 glucose, 26 NaHCO3, 1.3 sodium ascorbate, and 3 sodium pyruvate. During whole-cell recording, slices were submerged in oxygenated ACSF (31−32°C) and superfused at a rate of 2–3 ml/min. The GABAAR antagonist picrotoxin (100 μm) was added to the ACSF for all recordings and the NMDAR antagonist R-CPP (5 μm) or d-AP5 (50 μm) was included in the experiments in Figures 7–10.
Figure 7.
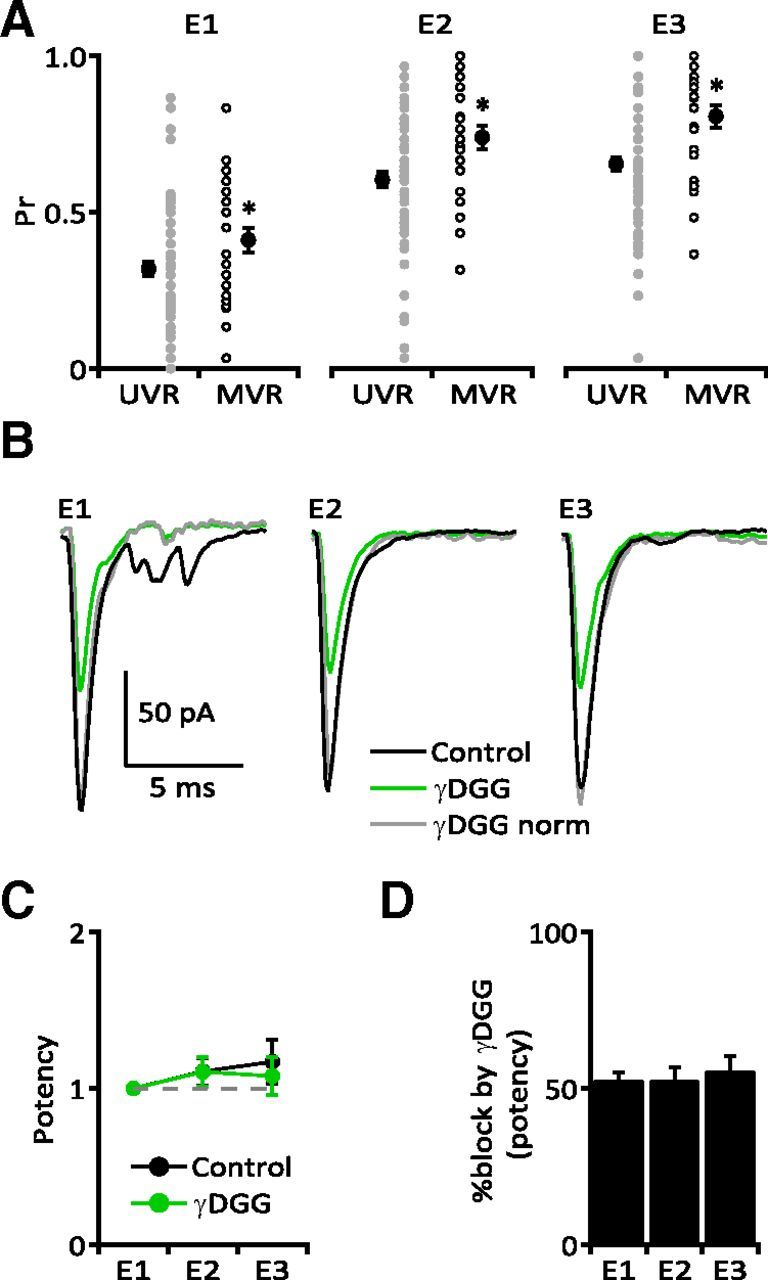
UVR is not an artifact of presynaptic release probability or postsynaptic AMPAR saturation. A, Both UVR (n = 76) and MVR (n = 26) synapses showed significant facilitation of Pr following each stimulus in the train (p < 0.0001 for both) but Pr at MVR synapses was significantly higher than UVR synapses for each stimulus response (E1: UVR 0.32 ± 0.024 vs MVR 0.41 ± 0.040, p = 0.0419; E2: UVR 0.60 ± 0.025 vs MVR 0.74 ± 0.037, p = 0.0078; E3: UVR 0.65 ± 0.021 vs MVR 0.81 ± 0.035, p = 0.0004). However, the distributions of Pr overlapped for each response at UVR and MVR synapses (E1: UVR 0–0.87 vs MVR 0.03–0.83; E2: UVR 0.03–0.97 vs MVR 0.32–1; E3: UVR 0.03–1 vs MVR 0.37–1). B, Sample UVR synapse treated with γDGG. The competitive antagonist reduced potency equally across all three responses. At five UVR synapses γDGG did not alter the potency ratio within the stimulus train (C; control vs γDGG: E2: 1.11 ± 0.090 vs 1.11 ± 0.097; E3: 1.17 ± 0.139 vs 1.08 ± 0.120; n = 5, p > 0.05) and inhibited all three eEPSCs equally (D; E1: 52.9 ± 2.06% block; E2: 52.7 ± 3.79%; E3: 55.7 ± 5.43%; n = 5, p > 0.05).
Figure 8.

Presynaptic inhibition differentially affects UVR and MVR synapses. A, Overlaid averaged evoked responses showing potency for a sample UVR (top) and MVR (bottom) synapse. B, Diary plots for the same synapses shown in A. Baclofen increased the failure rate at both synapses but decreased eEPSC amplitude only at the MVR synapse. C, Summary data showing increased failure rate in the presence of baclofen for UVR (top; control vs baclofen: E1: 0.68 ± 0.062 vs 0.85 ± 0.058, p = 0.0313; E2: 0.37 ± 0.073 vs 0.61 ± 0.104, p = 0.0313; E3: 0.36 ± 0.057 vs 0.53 ± 0.101, p > 0.05; n = 6) and MVR synapses (bottom; control vs baclofen: E1: 0.74 ± 0.058 vs 0.96 ± 0.019; E2: 0.38 ± 0.079 vs 0.86 ± 0.050; E3: 0.26 ± 0.046 vs 0.65 ± 0.070; n = 6, p = 0.0313 for each). CGP (5 μm) reversed failure rate to near control levels for both UVR (E1: 0.70 ± 0.054; E2: 0.40 ± 0.094; E3: 0.27 ± 0.077; n = 5, p > 0.05) and MVR synapses (E1: 0.80 ± 0.094; E2: 0.56 ± 0.113; E3: 0.44 ± 0.111; n = 6, p > 0.05). D, Top, Baclofen and CGP did not alter potency at UVR synapses (ctrl vs bac vs cgp: E1: 1 vs 1.00 ± 0.115 vs 1.14 ± 0.111; E2: 1.07 ± 0.018 vs 1.04 ± 0.082 vs 1.10 ± 0.097; E3: 1.06 ± 0.022 vs 1.05 ± 0.046 vs 1.12 ± 0.076; n = 6,6,5, p > 0.05). Bottom, Baclofen reduced potency at MVR synapses to apparent UVR levels, with only a partial reversal by CGP (ctrl vs bac vs cgp: E1: 1 vs 0.88 ± 0.221 vs 0.85 ± 0.018, n = 6,4,4, p = 0.0364; E2: 1.54 ± 0.092 vs 0.97 ± 0.102 vs 1.07 ± 0.019, n = 6, p = 0.0031; E3: 1.63 ± 0.124 vs 1.01 ± 0.169 vs 1.14 ± 0.084, n = 6, p = 0.0133).
Figure 9.
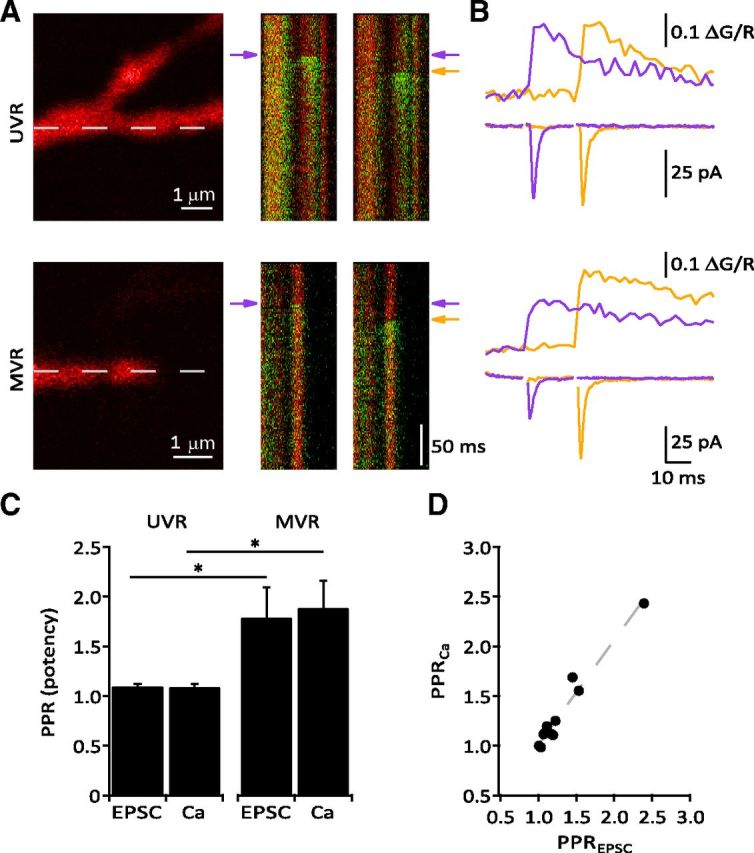
Fluorescence calcium analysis confirms different release modes at optically identified PF–MLI synapses. A Left, Reference images of MLI dendrites used for line scans of a UVR (top) and MVR (bottom) synapse. Right, Line scans with overlaid Alexa Flour 594 (red) and Fluo5F (green) signals. A rise in postsynaptic calcium can be clearly seen in response to either a single stimulus (purple arrow, left line scans) or the second of a pair of stimuli following a failure on the first (orange arrow, right line scans). B, Fluorescence intensity profiles and physiology traces, color-coded to match the indicator arrows in A. C, Group data comparing the PPR (of potency) at UVR (eEPSC: 1.10 ± 0.025, calcium: 1.09 ± 0.028, n = 7) and MVR synapses (eEPSC: 1.79 ± 0.302, calcium: 1.89 ± 0.273, n = 3). PPR at MVR synapses was significantly greater than at UVR synapses for both eEPSCs and Ca2+ transients (p = 0.0167 for both). D, A strong linear correlation existed between the PPR (of potency) for the eEPSCs and calcium transients (n = 11, Pearson's r = 0.981, p < 0.0001).
Figure 10.

MVA and synaptic modeling reveal two populations of synapses. A, MVA using data from the baclofen experiments. Normalized UVR responses were closely approximated by a binomial model for an idealized synapse with a single release site (N = 1; adjusted r2 = 0.949) and quantal content q = 1 (see Results, Eq. 4). The root of the UVR fit (adjusted r2 = 0.968) occurred at 1.14, consistent with primarily UVR release. MVR responses showed significantly more variability and were poorly fit by the same model (N = 1; adjusted r2 = −0.603). Models for N = 2 and N = 7 release sites are displayed for reference. B, A model of two independent synapses did not reproduce the facilitation observed at a MVR synapse. Inset, EPSC rising phase for model and actual E2. C, Comparison of the model predictions to peak amplitude and rise times for all 26 MVR synapses. The model consistently underestimated both the amplitude facilitation and slowing of the rising phase of the E2 response (ratio E2/E2 model: amplitude, 1.27 ± 0.030, p < 0.0001; rise time, 1.15 ± 0.044, p = 0.0009).
Interneurons in the outer third of the molecular layer were visualized using infrared gradient contrast optics (Dodt et al., 2002). Borosilicate patch pipettes were pulled on a Narishige PP-83 upright pipette puller, had a tip resistance of 2–4 MΩ, and were filled with an internal recording solution containing the following (in mm): 130 cesium gluconate, 4 MgCl2, 4 NaCl, 10 EGTA, 20 HEPES, 4 Mg-ATP, 0.4 Na-GTP, and 0.1 spermine, pH 7.3. For the experiments presented in Figures 2, 3A, and 4A, spermine was excluded to allow measurement of AMPAR-mediated currents recorded at positive holding potentials. For the experiments in Figures 3 and 4, the presence or absence of spermine did not affect the NMDAR current and so data were pooled. Currents were recorded with an Axopatch 1C or Multiclamp 700B amplifier (Molecular Devices), filtered at 2 kHz and digitized at 20–50 kHz. Data were collected using custom software (JS Diamond) written in IgorPro (WaveMetrics). Access resistance (Ra) was 12–20 MΩ (uncompensated) upon whole-cell break-in and was monitored throughout each experiment; any experiment in which Ra deviated by >20% from control was excluded from analysis.
Figure 2.
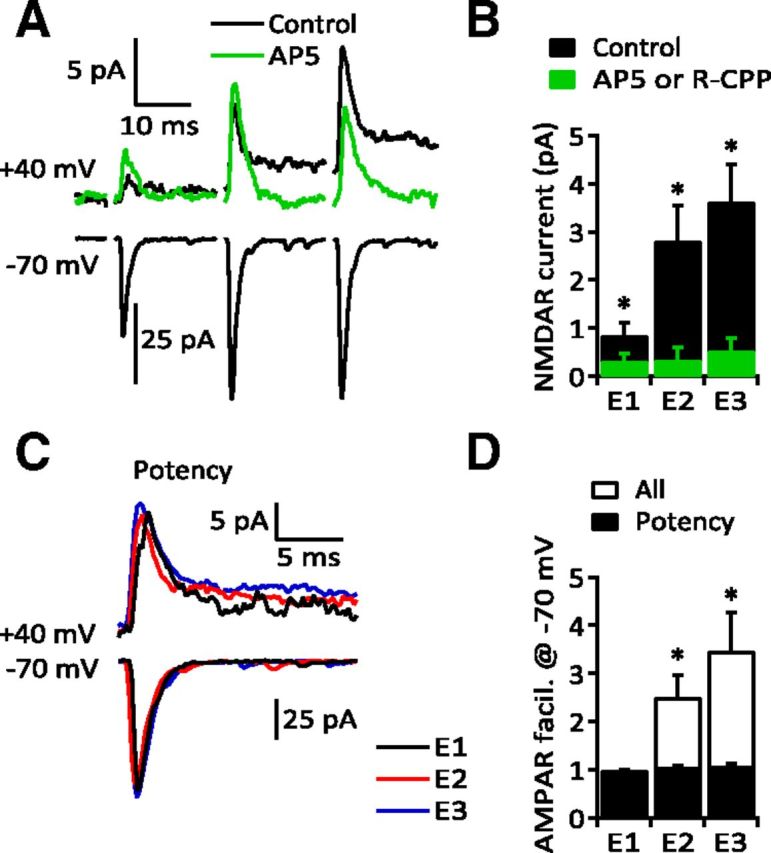
Single synapse activation of extrasynaptic NMDARs. A, Top, Average eEPSCs recorded from a MLI at +40 mV, evoked by a train of stimuli (3 × 50 Hz; black record). The late current visible after the second and third stimuli was blocked by d-AP5 (50 μm; green record). Bottom, Average AMPAR-mediated eEPSCs from the same cell recorded at −70 mV showed significant facilitation. B, Group data from six recordings for NMDAR currents measured in the absence (E1: 0.85 ± 0.246 pA; E2: 2.82 ± 0.719 pA; E3: 3.63 ± 0.764 pA; p < 0.0001) and presence of NMDAR antagonists (d-AP5, 50 μm or R-CPP, 5 μm) (E1: 0.32 ± 0.165 pA; E2: 0.35 ± 0.242 pA; E3: 0.52 ± 0.271 pA; p = 0.0313 for each). C, Synaptic potencies (successes only) of the AMPAR EPSCs recorded at +40 and −70 mV are the same for E1–E3 (same cell as in A). D, Group data comparing potency (black bars; E2: 1.06 ± 0.029; E3: 1.08 ± 0.038; n = 14, p > 0.05) and average of all responses (white bars; E2: 2.51 ± 0.451; E3: 3.47 ± 0.796; n = 14, p = 0.0002, both within group and compared to control).
Figure 3.
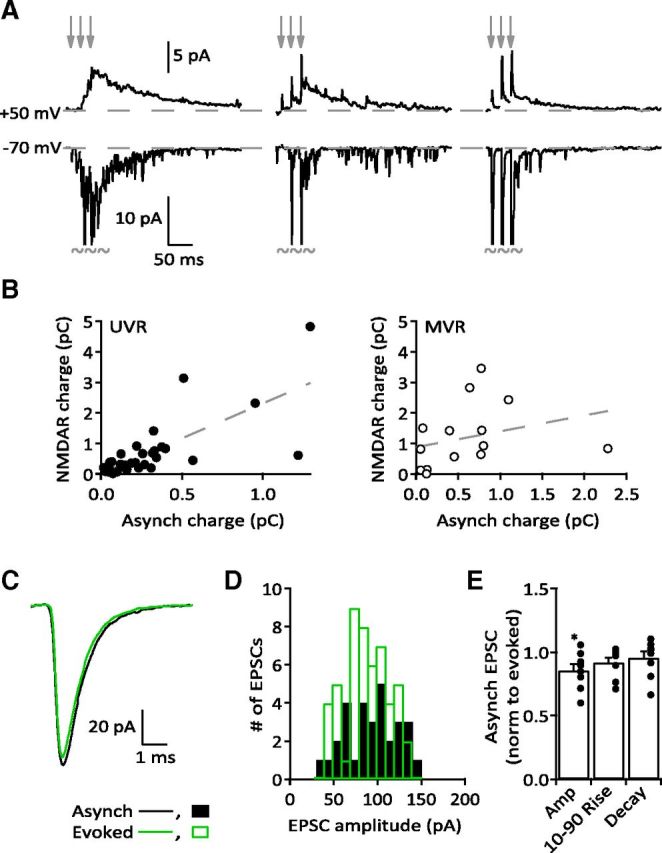
Asynchronous release correlates with NMDAR activity. A, Three sample cells in which the amount of asynchrony following stimulation (records at −70 mV) is correlated to the NMDAR current (records at +50 mV; within cell comparison). Arrows indicate stimulus timing. Each record is an average of 20–30 sweeps. Recordings at −70 mV have been truncated where shown to illustrate differences in asynchronous release. B, Comparisons of integrated NMDAR charge and total aEPSC charge (see Materials and Methods). Left, A strong correlation exists between asynchrony and NMDAR charge at UVR synapses (n = 34, Pearson's r = 0.732, p < 0.0001). Right, Lack of correlation at MVR synapses (n = 15, Pearson's r = 0.299, p > 0.05). C, Average evoked and asynchronous EPSCs from a single UVR synapse. D, Distribution of evoked and asynchronous EPSC amplitudes from the same recording as in C. E, Comparison of evoked and asynchronous EPSC parameters from nine cells with substantial asynchronous activity (>25 resolvable aEPSCs). aEPSCs were smaller than eEPSCs (ratio of aEPSC/eEPSC: 0.86 ± 0.046; p = 0.0117) but had nearly identical response kinetics (ratio of 10–90% rise time: 0.92 ± 0.038, p > 0.05; ratio of decay: 0.96 ± 0.047, p > 0.05).
Figure 4.

Asynchronous and NMDAR currents covary independent of synchronous release. A, EGTA-AM (1 μm) reduced asynchronous release and NMDAR currents in a sample cell with high levels of both in control. Arrows indicate stimulus timing. Each record is an average of 20–30 sweeps. Recordings at −70 mV have been truncated where shown to illustrate differences in asynchronous release. B, Summary data for the effect of EGTA-AM on eEPSCs (mix of UVR and MVR synapses) as well as the charge transfer from aEPSCs and NMDARs. EGTA-AM produced a decrease in eEPSC amplitude (left, E1: 67.7 ± 9.25% of control, n = 8, p = 0.01; E2: 77.9 ± 10.62%, n = 9, p > 0.05; E3: 73.3 ± 7.43%, n = 9, p = 0.007; one experiment had all failures for E1) but had a greater effect on the charge carried by asynchrony (right, 52.4 ± 9.94%, p = 0.0156, n = 8) and NMDARs (63.1 ± 10.52%, p = 0.0156, n = 8). C, Sample cell showing enhanced asynchronous release and NMDAR activation in the presence of 0.5 mm Ba2+. As in A, records at −70 mV have been truncated to illustrate changes in asynchronous release. D, Summary data for the effect of Ba2+ on eEPSCs (mix of UVR and MVR synapses) and charge transfer from aEPSCs and NMDARs. Left, Ba2+ significantly reduced eEPSC amplitudes for E1 and E2 (E1: 71.1 ± 9.24% of control, p = 0.0156; E2: 61.9 ± 8.56%, p = 0.0156; E3: 92.9 ± 18.26%, p > 0.05; n = 7). Right, Ba2+ increased both aEPSC and NMDAR charge transfer (aEPSC: 965 ± 378% of control; NMDAR: 401 ± 79.2%; n = 7, p = 0.0156 for both).
Monopolar stimulators were constructed from glass pipettes filled with ACSF and had a tip diameter ∼0.5–1.0 μm. Granule cells were activated by placing two stimulators (one anode, one cathode) in the granule cell layer within ∼20 μm of each other and 50–100 μm lateral to the recorded cell. This orientation was used to prevent spillover transmission from climbing fibers (Szapiro and Barbour, 2007), avoid synapses from ascending granule cell axons (Sims and Hartell, 2005), and decrease the likelihood of glutamate pooling and synaptic cross talk between PFs that can be induced with molecular layer stimulation (Carter and Regehr, 2000; Clark and Cull-Candy, 2002; Marcaggi and Attwell, 2007). While searching for a response, each electrode was tested as the cathode and anode repeatedly until a reliable response was found. No preference was given to one position of the cathode over another and therefore the consistent recruitment of multiple PFs just below the Purkinje cell layer is unlikely. Stimuli were delivered as a 50 Hz train of three pulses (100 μs, 5–90 V) every 10 s except for experiments in Figures 5 and 6. In Figure 5, a minimum of 30 trains at each frequency (50, 100, and 200 Hz) was delivered first while holding the postsynaptic cell at −70 mV then while holding at +50 mV. R-CPP (5 μm) was applied at the end of the 200 Hz train. Throughout the text, the eEPSC resulting from the first, second, or third stimulus of the train is referred to as E1, E2, or E3, respectively.
Figure 5.

Dependence of NMDAR activation on stimulus frequency. A, Averaged NMDAR currents at three stimulus frequencies from a sample UVR (top) and MVR (bottom) synapse. B, Top, Increasing stimulus frequency at UVR synapses with few aEPSCs increased the NMDAR current (compared with 50 Hz; 100 Hz: 1.85 ± 0.293, p = 0.0313; 200 Hz: 1.92 ± 0.540, p > 0.05; n = 6,5) without changing asynchronous release (100 Hz: 1.16 ± 0.297; 200 Hz: 1.00 ± 0.216; n = 6,5, p > 0.05). Bottom, MVR synapses were not affected by different stimulus frequencies and showed no change in asynchronous release (100 Hz: 0.90 ± 0.271; 200 Hz: 1.49 ± 0.306; n = 5, p > 0.05) or NMDAR currents (100 Hz: 1.12 ± 0.275; 1.07 ± 0.235; n = 5, p > 0.05). NMDAR currents were blocked by R-CPP (p = 0.0007, n = 5). C, PPRpot did not vary with different stimulus frequencies at UVR (top; 50 Hz: 1.05 ± 0.039; 100 Hz: 1.11 ± 0.064; 200 Hz: 1.06 ± 0.068; n = 6,5,6, p > 0.05) or MVR synapses (bottom; 50 Hz: 1.39 ± 0.078; 100 Hz: 1.36 ± 0.84; 200 Hz: 1.39 ± 0.152; n = 5, 4, 4, p > 0.05).
Figure 6.

AMPAR-mediated EPSCs always precede NMDAR activation. A–D, Five trials from each of four MLIs during trains of five GCL stimuli at 100 Hz (A, B) or three stimuli at 50 Hz (C, D), all recorded at +50 mV. Arrows indicate timing of stimuli. Slow baseline shifts, indicative of NMDAR-mediated currents, were observed only following AMPAR EPSCs. Similar baseline shifts were not observed at −70 mV in the presence of external Mg2+.
Experiments in Figure 8 were performed in the presence of LY341495 (100 μm), a broad spectrum metabotropic glutamate antagonist, to prevent a switch in AMPAR subunit composition that occurs when both GABAB receptors and metabotropic glutamate receptors are activated (Kelly et al., 2009). LY341495 by itself did not alter Pr.
We cannot completely rule out simultaneous activation of multiple low probability synapses. After bifurcation in the molecular layer, PFs run in more or less a straight line (parallel to the pial surface) while MLI and PC dendritic arbors project orthogonally to the PFs. Although a single PF will form hundreds of synaptic contacts over the extent of its projection (Palay and Chan-Palay, 1974), these axons rarely form multiple contacts with an individual postsynaptic cell (Napper and Harvey, 1988). We defined a single synaptic response as meeting the following criteria: (1) E1 success amplitude and failure rate were stable with increasing stimulus intensity, (2) clear differentiation between successes and failures, (3) all-or-none stimulus threshold (Fig. 1A), (4) short response latency (<4 ms), and (5) consistent eEPSC kinetics throughout the experiment (Figs. 2C, 3C, 7B, 8A). Experiments in which two synaptic contacts were clearly detectable (i.e., distinct amplitudes, latencies or kinetics) were excluded from analysis.
Figure 1.
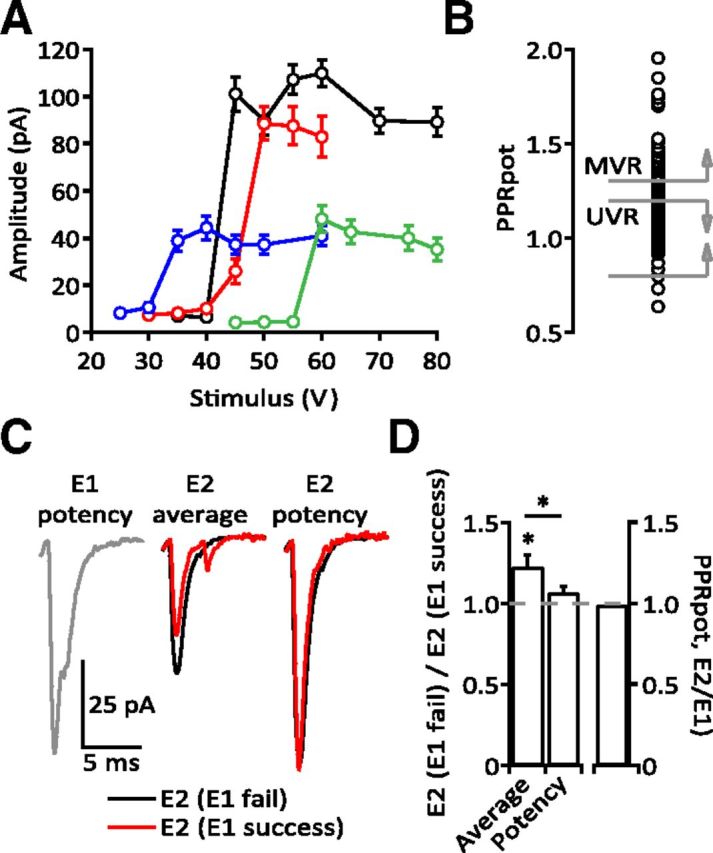
Single synapse isolation and classification. A, Average amplitudes of EPSCs from four different MLIs at varying stimulus voltages. The amplitudes of three EPSCs evoked at 50 Hz, E1–E3, were summed and then averaged across trials using the same stimulus voltage. This measurement included successes and failures and, by summing E1–E3, decreases variability across trials and results in a clearer delineation of threshold. All four examples are from UVR synapses, i.e., the potency did not change with stimulus number or stimulus voltage. The sharp threshold from failures to successes along with no additional increase in amplitude as stimulus voltage increased was used to identify single inputs to MLIs. The inflection point seen for the red trace is consistent with stimulation at threshold resulting in intermittent failures of AP generation. Stimulus voltages for experimentation were generally set ∼10 V above threshold. B, Plot of the extent of the PPRpot across synapses and the ranges assigned to UVR and MVR. Synapses classified as MVR (n = 26) have a PPRpot > 1.3 while synapses classified as UVR (n = 76) have a PPRpot between 0.8 and 1.2. Synapses with 1.2 < PPRpot < 1.3 were considered ambiguous and not analyzed further. C, Sample UVR synapse demonstrating the difference in Pr of E2 following either a failure or success on E1. The potency of E2 is independent of the success of E1. D, Group data for 14 UVR synapses (average PPRpot = 1.00 ± 0.007, right bar) comparing the ratio of the average amplitude of E2 following an E1 failure to E2 following an E1 success (1.23 ± 0.072, p = 0.0134, left bar) or the same ratio of the potency of E2 (1.07 ± 0.034, p > 0.05, middle bar). The slight change in potency is not sufficient to explain the much larger change in average amplitude (p = 0.0494).
Two-photon laser scanning microscopy.
For two-photon imaging of MLIs, cerebellar slices were cut in the parasagittal orientation (250 μm thick) and MLIs were patch clamped using an internal solution containing the following (in mm): 130 cesium gluconate, 4 MgCl2, 10 HEPES, 10 sodium phosphocreatine, 4 Na-ATP, 0.4 Na-GTP, 1 QX-314, 0.01 Alexa Fluor 594, and 0.2 Fluo5F. The internal solution was set to a pH of 7.30 using CsOH and volume adjusted to an osmolarity of 290 mOsm. Two-photon laser scanning microscopy was performed on a custom setup using an Olympus upright microscope and a Ti:sapphire laser (Chameleon; Coherent) tuned to 810 nm. Photomultipliers (H8224P-40 or H10770PA-40; Hamamatsu) collected red and green emissions in both epifluorescence and transfluorescence pathways (Christie and Jahr, 2008). For the experiments presented in Figure 9, data were acquired using ScanImage software (Pologruto et al., 2003). Line scans were performed at 500 Hz and fluorescence changes were calculated by subtracting the baseline green signal followed by normalization to the red signal (ΔG/R). Single PF synaptic contacts onto MLIs were found by axially scanning short segments of MLI dendrites in conjunction with granule cell layer stimulation. In some instances, recordings were performed at room temperature; however, no differences were observed with high temperature recordings and data were pooled.
Analysis.
Data were analyzed using custom routines written in IgorPro. EPSCs were considered synchronous (evoked, eEPSCs) if they occurred within 4 ms of the stimulus artifact and asynchronous (aEPSCs) if they occurred later. Synaptic potency was calculated as the average amplitude of successful eEPSCs following a given stimulus. Synapses were classified as univesicular release (UVR) if the paired-pulse ratio of the potency (PPRpot) was 0.8 < PPRpot < 1.2 or MVR if 1.3 < PPRpot (Fig. 1B). UVR synapses were further identified by comparing the average E2 amplitude (both successes and failures) following either a success or failure of E1. Although PF synapses facilitate, a UVR synapse by definition can release only a single vesicle at a time. Thus, if a vesicle is released on E1, the second stimulus may occur before vesicle replenishment takes place; this was observed as a smaller average amplitude of E2 following a success on E1 than when E2 followed a failure of E1 (Fig. 1C,D). Throughout our experiments, we observed a spectrum of PPRpot; to study clear examples of UVR and MVR, we restricted our analyses to the ranges presented above. Mean variance analysis (MVA) presented in Figure 10 was performed in IgorPro using a binomial model of synaptic release (see Results).
For experiments in Figure 2, the initial NMDAR current was calculated as the average current from the 5 ms window 15–20 ms after each stimulus. For experiments in Figures 3–5, total charge transfer was determined as follows: (1) an average trace was constructed from a minimum of 30 consecutive baseline-adjusted sweeps in each condition. The baseline for individual sweeps was defined as the steady-state current from a quiet (i.e., lacking spontaneous events) 10 ms window preceding the first stimulus. (2) A 5 ms window including the third stimulus artifact and accompanying eEPSC was blanked to allow measurement of current due to asynchronous release only. (3) Integration of this adjusted average current from 5 ms after the second stimulus to 310 ms after the third stimulus (325 ms window in total, which was generally well after asynchronous release had ceased). For the experiments in Figure 5, the asynchronous and NMDAR currents elicited by a 50 Hz train were measured as described while for the 100 and 200 Hz trains the measurement window began 5 ms after the last stimulus artifact. This adjustment was necessary to prevent contamination by the decay of the second AMPAR eEPSC.
To compare aEPSC and eEPSC kinetics and amplitude distributions for a given synapse (Fig. 3C–E), we used a template-based automated event detection (IgorPro) to identify clearly distinguishable aEPSCs. Following event detection, all aEPSCs were manually checked to remove false positives, aligned to the midpoint of their rising phases, and averaged together to create a representative aEPSC. aEPSCs were considered to be a result of stimulation if they occurred within the same measurement window as the current integration described above (i.e., 310 ms after the third stimulus). The analysis in Figure 3, C–E, was performed on UVR synapses only; therefore, the evoked responses to all three stimuli were used to create an average eEPSC. eEPSCs were aligned in the same manner as aEPSCs before averaging. The 10–90% rise time was measured from the averaged eEPSC and the decay time constant was calculated by fitting the decay phase with a single exponential.
Nonparametric statistical tests, including Wilcoxon signed-rank, Wilcoxon matched-pairs, Mann–Whitney U, Friedman repeated-measures ANOVA, and Kruskal–Wallis one-way ANOVA with Dunn post hoc, were performed using Instat3 (GraphPad). Significance for linear fits was determined using a look-up table for Pearson's r values; r2 values for polynomial fits presented in Figure 10 were calculated manually using Excel (Microsoft). A value of p ≤ 0.05 was considered significant. Data are reported as mean ± SEM. Stimulus artifacts in illustrated traces have been blanked.
Results
Single synapse activation of NMDARs
Consistent with previous reports (Konnerth et al., 1990; Perkel et al., 1990; Atluri and Regehr, 1996), single-synapse PF–MLI eEPSCs exhibited frequency facilitation (averages of all trials; Fig. 2A,D). At +40 mV, a slowly decaying current built up during the stimulus train that was blocked by the NMDAR antagonists R-CPP (5 μm) or d-AP5 (50 μm) (Fig. 2A,B). NMDAR antagonists did not alter the potency of AMPAR EPSCs (E1: 100.7 ± 10.17%, E2: 96.8 ± 8.51%, E3: 89.3 ± 7.57% of control; n = 6, p > 0.05). Since MVR increases with facilitation (Foster et al., 2005; Bender et al., 2009), we initially hypothesized that MVR was responsible for elevating glutamate and activating NMDARs. However, in these experiments the synaptic potency (average amplitude of successes only) was constant across the stimulus train (Fig. 2C,D; n = 13/14). These results suggest that glutamate released from single PF–MLI synapses is capable of activating extrasynaptic NMDARs but MVR is not necessary to achieve this activation.
Asynchronous glutamate release is sufficient to activate NMDARs
Trains of PF action potentials can dramatically increase the frequency of aEPSCs. This asynchronous release can last hundreds of milliseconds following high-frequency stimulation and results from residual calcium in the PF terminal (Atluri and Regehr, 1998; Chen and Regehr, 1999). The amount of asynchronous release varied greatly from synapse to synapse (Fig. 3A), which afforded us an opportunity to determine whether asynchronous release from a single synapse could activate NMDARs. At 34 UVR synapses (defined as potency second eEPSC/potency first eEPSC < 1.2; see Materials and Methods), we observed a strong correlation between the total charge transfer of aEPSCs at −70 mV and the total charge transfer through NMDARs at +50 mV (Fig. 3B, left). In contrast, asynchronous release was not a good predictor of the NMDAR charge transfer at 15 MVR synapses (potency second eEPSC/potency first eEPSC > 1.3; Fig. 3B, right), although we did observe a positive trend in the relationship, which would be expected if asynchronous release is partially responsible for activating NMDARs at these synapses. Some MVR synapses were capable of inducing an NMDAR-mediated current even with very little asynchronous release. We compared the amplitude and response kinetics of the aEPSCs and eEPSCs to determine the source of asynchronous release. In nine cells with sufficient asynchronous activity (>25 clearly resolvable aEPSCs), asynchronous and evoked responses had similar 10–90% rise times and decay kinetics (Fig. 3C,E). Furthermore, the amplitude distributions showed considerable overlap (Fig. 3D) with eEPSCs being slightly larger than the associated aEPSCs (Fig. 3E). These results suggest that aEPSCs and eEPSCs originate from the same presynaptic release site.
If the correlation between the occurrence of aEPSCs and NMDAR currents represents a causal relationship, selective modulation of asynchronous release should similarly alter the associated NMDAR current. We used the acetoxymethyl (AM) ester of the calcium chelator EGTA (EGTA-AM, 1 μm) to reduce residual calcium in the presynaptic PF terminal (Atluri and Regehr, 1998). This produced a modest inhibition of average eEPSC amplitudes and a larger reduction of both the asynchronous and NMDAR currents (Fig. 4A,B; n = 8, UVR and MVR combined), again indicating a correlation between asynchronous release and NMDAR activation. In a corollary experiment, we added 0.5 mm barium (Ba2+) to the ACSF. Ba2+ also reduced eEPSC amplitudes for E1 and E2 but enhanced both asynchronous release and NMDAR currents as expected from previous studies (Silinsky, 1978; Fig. 4C,D; n = 7, UVR and MVR combined). In both experimental conditions, asynchronous release and NMDAR currents covaried, suggesting that extracellular glutamate levels at NMDARs were primarily dependent on the rate of asynchronous release.
Many synapses, particularly UVR contacts, lacked both asynchronous activity and NMDAR currents. The lack of NMDAR currents could result from low expression of nearby NMDARs. Alternatively, without asynchronous activity, evoked release events 20 ms apart (50 Hz train) may be inadequate to activate extrasynaptic NMDARs because clearance is too fast for successive glutamate quanta to summate. At six UVR synapses with little asynchrony or NMDAR activity, increasing the stimulus frequency (from 50 to 200 Hz) increased the NMDAR current without changing asynchronous release (Fig. 5A,B, top). In contrast, at five MVR synapses with low levels of asynchrony and small NMDAR currents, we found no significant increase in asynchronous release or NMDAR currents (Fig. 5A,B, bottom). It is possible that there were few NMDARs adjacent to the MVR synapses and they were already maximally activated at 50 Hz. At both UVR and MVR synapses, the change in stimulus frequency did not convert UVR synapses to MVR or vice versa, as the PPR of the potency (PPRpot) for each remained unchanged (Fig. 5C). Together with the previous results, these data indicate that activation of extrasynaptic NMDARs requires high-frequency release, whether it is synchronous or asynchronous.
A complication in interpreting our data is the possibility that transmitter released from synapses onto neighboring MLIs is responsible for NMDAR activation on the recorded MLI. Because of the relative affinities of AMPARs and NMDARs, spillover from neighboring synapses may activate NMDARs without activating an observable AMPAR-mediated EPSC. To determine whether such spillover contributed to NMDAR activation, we recorded from 54 MLIs and stimulated the GCL with trains of either three stimuli at 50 Hz or five stimuli at 100 Hz. In all cases, which included 1500 trials, an AMPAR-mediated EPSC always occurred before an NMDAR current, in many cases following failures on the first few stimuli (Fig. 6). In trials resulting in no AMPAR EPSCs, no apparent NMDAR current was observed. Unless neighboring synapses all released in lock-step with the direct synaptic contact, a gradual, NMDAR-dependent current in the absence of AMPAR EPSCs should be detectable if spillover could activate NMDARs. That NMDAR currents were not observed without a preceding AMPAR EPSC suggests that release from neighboring synapses was insufficient to activate NMDARs on the recorded MLI.
Heterogeneity of release properties between synapses
Our finding that many PF–MLI synapses are UVR is surprising given previous reports that MVR is a general property of these synapses (Foster et al., 2005; Bender et al., 2009). We observed a wide range of PPRpot for >100 PF–MLI synapses tested (Fig. 1), yet the majority (n = 76, 65%) were classified as UVR (PPRpot = 1.06 ± 0.0089). In comparison, only 22% (n = 26) were clearly MVR (PPRpot = 1.49 ± 0.0368). One explanation for the low percentage of MVR synapses is that the present conditions did not allow most synapses to reach a Pr high enough to support MVR (Wadiche and Jahr, 2001). However, this does not appear to be the case as individual UVR synapses frequently reached Pr levels equal to or greater than some MVR synapses (Fig. 7A).
The stable potency across stimuli at UVR synapses could result from saturation of AMPARs by a single quantum of glutamate, thus masking the effect of MVR on potency. We tested for receptor saturation with the low-affinity competitive AMPAR antagonist γDGG (Wadiche and Jahr, 2001; Christie and Jahr, 2006; Chanda and Xu-Friedman, 2010; Sun and Beierlein, 2011). Since γDGG is a rapidly dissociating antagonist, its effectiveness is reduced as the glutamate concentration rises and therefore it should inhibit MVR eEPSCs less than UVR eEPSCs regardless of saturation. However, at five UVR synapses γDGG (0.5 mm) inhibited the potency of E1–E3 to the same degree (Fig. 7B–D), suggesting the glutamate concentration transient in the synaptic cleft did not vary with frequency facilitation.
Since PF–MLI synapses often have a large intrasynaptic variability of eEPSC amplitude (Fig. 3D; Crowley et al., 2007), we tested whether Pr at the PF–MLI synapse was sufficiently high to elicit MVR following the first stimulus in the train. GABAB receptor activation decreases Pr (Dittman and Regehr, 1996) and should lead to an increased failure rate at both UVR and MVR synapses but reduce synaptic potency only at MVR terminals. Application of the GABAB receptor agonist baclofen (1 μm) increased the failure rate at both UVR (n = 6) and MVR (n = 6) synapses (Fig. 8A–C). However, baclofen only decreased the potency at MVR, not UVR synapses (Fig. 8D). CGP55845 (5 μm, GABABR antagonist) returned the failure rate to control levels at all synapses but only partially reversed the effect on synaptic potency at MVR synapses. The synaptic potency of all three responses at MVR synapses in the presence of baclofen was close to that of the control E1, suggesting that in low Pr conditions MVR synapses reverted to UVR.
Activation of two (or more) nearly identical but spatially segregated UVR synapses could confound our interpretation of MVR data. We used optical quantal analysis to confirm that MVR represented exocytosis at individual synaptic contacts. MLIs were filled through the patch pipette with the morphological dye Alexa Fluor 594 (10 μm) and the Ca2+ indicator Fluo5F (200 μm). In the presence of R-CPP, synaptic contacts from PFs were located by sharp increases in Fluo5F fluorescence following GCL stimulation that were restricted to a small region (∼2 μm) of the dendrite (Soler-Llavina and Sabatini, 2006; Pugh and Jahr, 2011; Fig. 9). As MLIs were held at −70 mV in the presence of 1 mm Mg2+, these Ca2+ transients were mainly mediated by calcium-permeable AMPARs. Alternating single and pairs of stimuli were used to evoke EPSCs and Ca2+ transients in MLIs and only those recordings in which there was complete coincidence of successes and failures of the two measures were analyzed. Postsynaptic responses were separated into two categories: successes from the single stimulus (Fig. 9A,B, purple traces) and successes from the second stimulus following a failure on the first (Fig. 9A,B, orange traces). UVR synapses showed no facilitation in the eEPSC or Ca2+ transient while MVR synapses had robust facilitation of both (Fig. 9C). We also found a strong linear correlation between the PPR of the eEPSC potency and the PPR of the Ca2+ potency (Fig. 9D). Together with the observation that the rising phase of the Ca2+ transient lasted only as long as the charge transfer of the eEPSC (Fig. 9B), these data suggest that the Ca2+influx was mediated by the same Ca2+-permeable AMPARs that produced the eEPSC. The sum of Ca2+ transients resulting from a single success on either E1 or E2 (summed E2 peak) was indistinguishable from the Ca2+ transient when a success occurred with both stimuli (summed E2 peak: 0.23 ± 0.044 ΔG/R; double success E2 peak: 0.24 ± 0.042 ΔG/R, n = 10, p > 0.05) indicating that receptors at neither UVR nor MVR synapses were saturated following multiple release events (Fig. 7B–D).
MVA and synaptic modeling reveal two distinct synaptic populations
We used MVA to determine whether univesicular eEPSCs could be easily modeled by a simple, single release site synapse (Clements, 2003; Silver, 2003; Foster and Regehr, 2004; Saviane and Silver, 2007). To maximize the number of release conditions, we performed MVA on the three eEPSCs in the train in the presence and absence of baclofen.
The mean amplitude of a synaptic response (I), as given by Equation 1, depends on three factors: N, the number of release sites; p, the probability of release at each site; and q, the quantal size.
For a binomial model the variance of the response amplitude, σ2, is
Combining Equations 1 and 2 yields the relationship between the average response amplitude and its variance:
The large variability in potency between PF synapses prevented us from directly combining experiments from multiple cells. Therefore, the eEPSCs (E1, E2, and E3) from each recording were normalized to the mean of E1 from the same recording (which was assumed to represent a UVR response for that synapse) and then synapses were sorted into two groups–UVR and MVR–based on the PPRpot for each (see Materials and Methods). By creating a synapse with q = 1, Equation 3 reduces to the following:
and the two roots occur at 0 and N when Pr = 0 and 1, respectively. The normalized UVR data were well fit by the parabola N = 1 (Fig. 10A; the best fit of the UVR data had a second root at 1.14), suggesting that UVR synapses have a single release site. In contrast, the MVR data were poorly fit with this model and appeared to lie between the solutions for N = 2 and N = 7 (Fig. 10A), consistent with the average number of vesicles associated with PF active zones (Xu-Friedman et al., 2001). Under very low Pr conditions the UVR and MVR data overlap, suggesting that at MVR synapses a single action potential often causes the release of just one vesicle.
Finally, we assessed the possibility that MVR represents UVR release from two independent release sites. First, using Pr values measured from the UVR group, we calculated the predicted Pr for two independent synapses (E1, 0.54; E2, 0.84; E3, 0.88) which was significantly higher for the first two stimuli than the measured Pr for MVR synapses (Fig. 7A; E1, p = 0.0086; E2, p = 0.024; E3, p > 0.05) indicating that at least many of the synapses classified as MVR were indeed MVR. Second, we created a model of two independent synapses to simulate the facilitation observed at MVR synapses following the second stimulus. The average postsynaptic response from two independent release sites represents a mix of single and double release events. Based on the average failure rate for E2 at a single UVR synapse (Fr = 0.40), the probability of both synapses releasing a vesicle is 0.36 [(1 − Fr)*(1 − Fr)] and the probability that only one of the two synapses releases a vesicle is 0.48 [2*Fr*(1− Fr)]. The sum of these (0.84) represents the total probability (Pr) of recording a success given two independent synapses. Therefore, of all the postsynaptic successes, ∼43% should arise from “double” release events (0.36/0.84) with the remaining 57% due to single release events (0.48/0.84).
We modeled each MVR synapse by assuming the E1 potency for that synapse represented the average of single and double release events from two independent synapses. Similar to the situation described for E2, based on the Pr of E1 at UVR synapses, we expect E1 from two independent synapses to include ∼19% double events. Since we cannot easily distinguish these doubles from single release events, we estimated the average single release eEPSC as E1/1.19. We generated 10,000 replicates of which 5700 were identical to the adjusted E1 (i.e., 57% of the modeled EPSCs were single release events) and the remaining 4300 EPSCs were double release events. To generate realistic double events, we constructed a cumulative probability histogram of the response latencies from 10 UVR synapses (data not shown). We used a random selection from this distribution to add a second E1 to the first E1. The 10,000 replicates were then aligned by the midpoints of their rising phases and averaged to obtain the model E2 for each MVR synapse (Fig. 10B). For all MVR synapses, the actual E2 had a consistently larger amplitude and slower rising phase than the model E2 (Fig. 10B,C). These data suggest that MVR responses are not well described by release from two independent synapses and, together with the previous results, indicate at least two distinct populations of PF–MLI terminals exist with strikingly different release properties. This also suggests that AMPARs at MVR synapses are not saturated by single exocytotic events.
Discussion
We report that extrasynaptic NMDARs expressed by MLIs are activated by glutamate released from single PF terminals through asynchronous release, MVR, or high-frequency stimulus trains, all of which can result in multiple exocytotic events that occur in close temporal and spatial contiguity. We suggest that this results in two or more quanta of glutamate combining at extrasynaptic sites to produce a glutamate transient sufficient to activate NMDARs. In addition, we also find that the majority of PF terminals contacting MLIs are restricted to UVR, although MVR can occur at a smaller population.
Distinct populations of PF terminals
Our classification of synapses as UVR is based primarily on constant potency throughout a train of stimuli. Substantiation of this comes from MVA, invariant inhibition by γDGG, and uniform postsynaptic Ca2+ transients. Unambiguous classification of synapses as MVR is more problematic. PF–MLI synapses facilitate and therefore increase in potency during a train of stimuli could indicate either increasing MVR or the presence of two or more independent synapses. The classification of this second population of PF–MLI synapses as MVR synapses was justified by a number of considerations. First, all had increasing potency during the train of stimuli. Second, as the stimulus strength was increased and decreased, the first eEPSC was all or none, suggesting it was the result of activity in a single granule cell. Third, there was no correlation between the number of asynchronous events and the size of the NMDAR charge transfer at MVR synapses whereas the correlation was strong at UVR synapses. If synapses classified as MVR were actually two or more independent UVR synapses, the correlation between asynchronous release and NMDAR currents should be the same as at single UVR synapses. Fourth, postsynaptic Ca2+ transients and eEPSCs displayed nearly identical PPRs at individual MVR and UVR synapses, consistent with the imaged synapse representing the only source of receptor activation. Fifth, the Pr of the first and second eEPSCs at MVR synapses was lower than would be expected if two independent synapses contributed to these eEPSCs, based on the Pr of UVR synapses. Sixth, modeling synaptic release with two independent synapses underestimates the potency and rise time of E2 observed at MVR synapses. These two results, lower than expected Pr but higher potency of successes, are consistent if the Pr of a second vesicle is contingent on the release of the first. Seventh, several previous papers have reported that PF synapses, on both MLIs and PCs, can release multiple vesicles per AP (Foster et al., 2005; Rancillac and Barbara, 2005; Bender et al., 2009).
What determines whether a given PF–MLI synapse can release multiple vesicles is not known. However, there is precedence for variation in the physiology of PF boutons. Adjacent PF boutons have heterogeneous sensitivities to different G-protein-coupled receptors that correlate with both bouton volume and the size of the calcium transient evoked by AP invasion (Zhang and Linden, 2009). Furthermore, different PF boutons have different calcium clearance rates (Zhang and Linden, 2012). Whether these differences correlate with UVR, MVR, or the frequency of asynchronous release remains to be determined. In PF and other systems the size of boutons correlates with active zone area, the number of docked vesicles, and, at least in hippocampal neurons, with Pr (Lisman and Harris, 1993; Schikorski and Stevens, 1997; Xu-Friedman et al., 2001). In addition, the likelihood of MVR increases as Pr increases (Tong and Jahr, 1994; Wadiche and Jahr, 2001; Foster et al., 2005; Biró et al., 2006; Christie and Jahr, 2006; Huang et al., 2010). However, in the present study, release probability was not dramatically different between UVR and MVR synapses. Indeed, UVR can occur at synapses with high Pr (Silver et al., 2003; Murphy et al., 2004).
Facilitating synapses that we have labeled MVR could instead be interpreted as resulting from UVR at synapses that transition during the stimulus train from “kiss and run” (KR) exocytosis to full-fusion (FF) release, given that KR occurs at lower release probabilities than FF (Gandhi and Stevens, 2003; Richards, 2009; Alabi and Tsien, 2013). However, UVR synapses, on average, have a lower Pr than the MVR synapses, though they attain high Pr by the end of the three stimulus train. If the first stimulated release event at such a UVR synapse is via KR, FF would become more likely following the second and third stimulus and result in facilitation of potency, which we do not see in UVR synapses. Alternatively, if UVR synapses remained FF throughout the train, the fast and complete release of glutamate would result in more perisynaptic NMDAR activation than at synapses that transition from KR to FF (MVR synapses in our interpretation). However, we find greater NMDAR activation at MVR than UVR synapses, excluding synapses with high rates of asynchrony. In addition, FF release is reported to be evenly distributed across the active zone while KR is located more centrally (Park et al., 2012); thus more glutamate would be released nearer the periphery at FF synapses than at KR-to-FF synapses and would lead to larger NMDAR currents, again contrary to our data. Though KR may well occur at PF synapses, we would suggest that, by itself, a KR-to-FF transient cannot account for our results.
NMDAR activation at single synapses
Glutamate diffusion is curtailed in the cerebellar molecular layer by glutamate transporters expressed by Bergmann glia and PCs (Furuta et al., 1997; Otis et al., 1997; Danbolt, 2001; Tzingounis and Wadiche, 2007; Satake et al., 2010). Excitatory synapses on PC dendritic spines are ensheathed by glial membranes (Yamada et al., 2000; Xu-Friedman et al., 2001) whereas shaft synapses on MLIs appear to have little or no accompanying glial coverage (Palay and Chan-Palay, 1974) allowing glutamate to diffuse more readily to receptors located adjacent to the active zone than at PC synapses. Using minimal stimulation of the granule cell layer to prevent pooling of glutamate released from neighboring synapses (Carter and Regehr, 2000; Foster et al., 2005; Marcaggi and Attwell, 2007), we find that release at single PF–MLI synapses can elevate extracellular glutamate sufficiently to activate extrasynaptic NMDARs. At synapses lacking either MVR or sufficient asynchrony, increasing the stimulation frequency to 100 Hz successfully elicited NMDAR currents where there were none at 50 Hz. These frequencies are well within the in vivo range of granule cell burst firing (Chadderton et al., 2004; Jörntell and Ekerot, 2006; Rancz et al., 2007). Nevertheless, the form of release that leads to elevated extracellular glutamate could affect the duration of the NMDAR current. MVR results in a large bolus of glutamate that rapidly dissipates while asynchronous release occurring over tens and hundreds of milliseconds could prolong elevated glutamate (albeit at a lower concentration than initially seen with MVR) and create sustained NMDAR activity. Additionally, we found no evidence for a contribution from neighboring synapses to NMDAR activation, consistent with the observation that neighboring granule cells bifurcate at variable levels within the molecular layer (Zong et al., 2005). We suggest that at synapses with high aEPSC frequency or with 100 Hz stimulus trains, glutamate from serial release events pools to reach an extrasynaptic concentration sufficient to activate NMDARs or that NMDARs singly bound by glutamate released by the first event remain bound long enough for a second binding to result from a subsequent release event.
Implications of NMDAR activation for synaptic plasticity
Extrasynaptic NMDARs on MLIs participate in several forms of synaptic plasticity. Synaptically evoked endocannabinoid release from stellate cells, which mediates a transient retrograde suppression of PF release (SSE), requires either NMDAR or mGluR1 activation (Beierlein and Regehr, 2006). This is effected by 10 PF stimuli at 50 Hz, a regime sufficient to activate stellate cell NMDARs (Carter and Regehr, 2000; Clark and Cull-Candy, 2002). Stellate cells also exhibit a unique form of synaptic depression in which AMPAR EPSCs are depressed at negative potentials but are either unaffected at positive potentials or enhanced; this results from a switch of calcium-permeable to calcium-impermeable AMPARs (Liu and Cull-Candy, 2000) or a change in the expression of transmembrane AMPAR regulatory proteins (TARPs; Jackson and Nicoll, 2011). Although this form of plasticity does not require NMDAR activation (Liu and Cull-Candy, 2000), NMDAR activation can induce it, again requiring bursts of PF stimulation (Sun and June Liu, 2007). The well studied form of long-term depression at PF–PC synapses also requires the activation of NMDARs (Casado et al., 2000, 2002; Shin and Linden, 2005). Despite controversy over the location of NMDAR expression, in one formulation they are expressed by stellate cells and, as above, require short bursts of PF stimulation (but see Casado et al., 2000, 2002; Shin and Linden, 2005; Bidoret et al., 2009). Activation of extrasynaptic NMDARs may also generate subthreshold depolarizations, which have been shown to modulate stellate cell release properties (Christie and Jahr, 2008). There appear to be at least four mechanisms of synaptic plasticity that depend on synaptic stimulation of MLI NMDARs. Despite differences in the magnitude of plasticity that depend on the site of stimulation (Sims and Hartell, 2006; Marcaggi and Attwell, 2007), the fact that release from single synapses can activate MLI NMDARs suggests that processes dependent on these receptors are physiological and do not necessarily require high density synaptic activity.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS040056 and NIH Training Grant NS007381 (B.N.). We thank the Jahr laboratory members for discussions and critical readings of this manuscript.
The authors declare no competing financial interests.
References
- Abrahamsson T, Cathala L, Matsui K, Shigemoto R, Digregorio DA. Thin dendrites of cerebellar interneurons confer sublinear synaptic integration and a gradient of short-term plasticity. Neuron. 2012;73:1159–1172. doi: 10.1016/j.neuron.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Alabi AA, Tsien RW. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu Rev Physiol. 2013;75:393–422. doi: 10.1146/annurev-physiol-020911-153305. [DOI] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Regehr WG. Local interneurons regulate synaptic strength by retrograde release of endocannabinoids. J Neurosci. 2006;26:9935–9943. doi: 10.1523/JNEUROSCI.0958-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender VA, Pugh JR, Jahr CE. Presynaptically expressed long-term potentiation increases multivesicular release at parallel fiber synapses. J Neurosci. 2009;29:10974–10978. doi: 10.1523/JNEUROSCI.2123-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidoret C, Ayon A, Barbour B, Casado M. Presynaptic NR2A-containing NMDA receptors implement a high-pass filter synaptic plasticity rule. Proc Natl Acad Sci U S A. 2009;106:14126–14131. doi: 10.1073/pnas.0904284106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biró AA, Holderith NB, Nusser Z. Release probability-dependent scaling of the postsynaptic responses at single hippocampal GABAergic synapses. J Neurosci. 2006;26:12487–12496. doi: 10.1523/JNEUROSCI.3106-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Dieudonné S, Ascher P. Presynaptic N-methyl-D-aspartate receptors at the parallel fiber-Purkinje cell synapse. Proc Natl Acad Sci U S A. 2000;97:11593–11597. doi: 10.1073/pnas.200354297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Isope P, Ascher P. Involvement of presynaptic N-methyl-D-aspartate receptors in cerebellar long-term depression. Neuron. 2002;33:123–130. doi: 10.1016/S0896-6273(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Häusser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Chanda S, Xu-Friedman MA. A low-affinity antagonist reveals saturation and desensitization in mature synapses in the auditory brain stem. J Neurophysiol. 2010;103:1915–1926. doi: 10.1152/jn.00751.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Contributions of residual calcium to fast synaptic transmission. J Neurosci. 1999;19:6257–6266. doi: 10.1523/JNEUROSCI.19-15-06257.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Dendritic NMDA receptors activate axonal calcium channels. Neuron. 2008;60:298–307. doi: 10.1016/j.neuron.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG. Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci. 2002;22:4428–4436. doi: 10.1523/JNEUROSCI.22-11-04428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD. Variance-mean analysis: a simple and reliable approach for investigating synaptic transmission and modulation. J Neurosci Methods. 2003;130:115–125. doi: 10.1016/j.jneumeth.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Crowley JJ, Carter AG, Regehr WG. Fast vesicle replenishment and rapid recovery from desensitization at a single synaptic release site. J Neurosci. 2007;27:5448–5460. doi: 10.1523/JNEUROSCI.1186-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci. 1996;16:1623–1633. doi: 10.1523/JNEUROSCI.16-05-01623.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt HU, Eder M, Schierloh A, and Zieglgänsberger W. Infrared-guided laser stimulation of neurons in brain slices. Sci STKE. 2002;2002:pl2. doi: 10.1126/stke.2002.120.pl2. [DOI] [PubMed] [Google Scholar]
- Foster KA, Regehr WG. Variance-mean analysis in the presence of a rapid antagonist indicates vesicle depletion underlies depression at the climbing fiber synapse. Neuron. 2004;43:119–131. doi: 10.1016/j.neuron.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Foster KA, Crowley JJ, Regehr WG. The influence of multivesicular release and postsynaptic receptor saturation on transmission at granule cell to Purkinje cell synapses. J Neurosci. 2005;25:11655–11665. doi: 10.1523/JNEUROSCI.4029-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. 1997;17:8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi SP, Stevens CF. Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature. 2003;423:607–613. doi: 10.1038/nature01677. [DOI] [PubMed] [Google Scholar]
- Huang CH, Bao J, Sakaba T. Multivesicular release differentiates the reliability of synaptic transmission between the visual cortex and the somatosensory cortex. J Neurosci. 2010;30:11994–12004. doi: 10.1523/JNEUROSCI.2381-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Nicoll RA. Stargazin (TARP gamma-2) is required for compartment specific AMPA receptor trafficking and synaptic plasticity in cerebellar stellate cells. J Neurosci. 2011;31:3939–3952. doi: 10.1523/JNEUROSCI.5134-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörntell H, Ekerot CF. Properties of somatosensory synaptic integration in cerebellar granule cells in vivo. J Neurosci. 2006;26:11786–11797. doi: 10.1523/JNEUROSCI.2939-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly L, Farrant M, Cull-Candy SG. Synaptic mGluR activation drives plasticity of calcium-permeable AMPA receptors. Nat Neurosci. 2009;12:593–601. doi: 10.1038/nn.2309. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proc Natl Acad Sci U S A. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Harris KM. Quantal analysis and synaptic anatomy – integrating two views of hippocampal plasticity. Trends Neurosci. 1993;16:141–147. doi: 10.1016/0166-2236(93)90122-3. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Marcaggi P, Attwell D. Short- and long-term depression of rat cerebellar parallel fibre synaptic transmission mediated by synaptic crosstalk. J Physiol. 2007;578:545–550. doi: 10.1113/jphysiol.2006.115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GJ, Glickfeld LL, Balsen Z, Isaacson JS. Sensory neuron signaling to the brain: properties of transmitter release from olfactory nerve terminals. J Neurosci. 2004;24:3023–3030. doi: 10.1523/JNEUROSCI.5745-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napper RM, Harvey RJ. Number of parallel fiber synapses on an individual Purkinje cell in the cerebellum of the rat. J Comp Neurol. 1988;274:168–177. doi: 10.1002/cne.902740204. [DOI] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP, Jahr CE. Postsynaptic glutamate transport at the climbing fiber-Purkinje cell synapse. Science. 1997;277:1515–1518. doi: 10.1126/science.277.5331.1515. [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar cortex: cytology and organization. Berlin: Springer; 1974. [Google Scholar]
- Park H, Li Y, Tsien RW. Influence of synaptic vesicle position on release probability and exocytotic fusion mode. Science. 2012;335:1362–1366. doi: 10.1126/science.1216937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkel DJ, Hestrin S, Sah P, Nicoll RA. Excitatory synaptic currents in Purkinje cells. Proc Biol Sci. 1990;241:116–121. doi: 10.1098/rspb.1990.0074. [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh JR, Jahr CE. Axonal GABAA receptors increase cerebellar granule cell excitability and synaptic activity. J Neurosci. 2011;31:565–574. doi: 10.1523/JNEUROSCI.4506-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancillac A, Barbara JG. Frequency-dependent recruitment of inhibition mediated by stellate cells in the rat cerebellar cortex. J Neurosci Res. 2005;80:414–423. doi: 10.1002/jnr.20473. [DOI] [PubMed] [Google Scholar]
- Rancz EA, Ishikawa T, Duguid I, Chadderton P, Mahon S, Häusser M. High-fidelity transmission of sensory information by single cerebellar mossy fibre boutons. Nature. 2007;450:1245–1248. doi: 10.1038/nature05995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DA. Vesicular release mode shapes the postsynaptic response at hippocampal synapses. J Physiol. 2009;587:5073–5080. doi: 10.1113/jphysiol.2009.175315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake S, Song SY, Konishi S, Imoto K. Glutamate transporter EAAT4 in Purkinje cells controls intersynaptic diffusion of climbing fiber transmitter mediating inhibition of GABA release from interneurons. Eur J Neurosci. 2010;32:1843–1853. doi: 10.1111/j.1460-9568.2010.07469.x. [DOI] [PubMed] [Google Scholar]
- Saviane C, Silver RA. Estimation of quantal paramateres with multiple-probability fluctuation analysis. Methods Mol Biol. 2007;403:303–317. doi: 10.1007/978-1-59745-529-9_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Linden DJ. An NMDA receptor/nitric oxide cascade is involved in cerebellar LTD but is not localized to the parallel fiber terminal. J Neurophysiol. 2005;94:4281–4289. doi: 10.1152/jn.00661.2005. [DOI] [PubMed] [Google Scholar]
- Silinsky EM. On the role of barium in supporting the asynchronous release of acetylcholine quanta by motor nerve impulses. J Physiol. 1978;274:157–171. doi: 10.1113/jphysiol.1978.sp012141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA. Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J Neurosci Methods. 2003;130:127–141. doi: 10.1016/j.jneumeth.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Silver RA, Lubke J, Sakmann B, Feldmeyer D. High-probability uniquantal transmission at excitatory synapses in barrel cortex. Science. 2003;302:1981–1984. doi: 10.1126/science.1087160. [DOI] [PubMed] [Google Scholar]
- Sims RE, Hartell NA. Differences in transmission properties and susceptibility to long-term depression reveal functional specialization of ascending axon and parallel fiber synapses to Purkinje cells. J Neurosci. 2005;25:3246–3257. doi: 10.1523/JNEUROSCI.0073-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RE, Hartell NA. Differential susceptibility to synaptic plasticity reveals a functional specialization of ascending axon and parallel fiber synapses to cerebellar Purkinje cells. J Neurosci. 2006;26:5153–5159. doi: 10.1523/JNEUROSCI.4121-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Llavina GJ, Sabatini BL. Synapse-specific plasticity and compartmentalized signaling in cerebellar stellate cells. Nat Neurosci. 2006;9:798–806. doi: 10.1038/nn1698. [DOI] [PubMed] [Google Scholar]
- Sun L, June Liu S. Activation of extrasynaptic NMDA receptors induces a PKC-dependent switch in AMPA receptor subtypes in mouse cerebellar stellate cells. J Physiol. 2007;583:537–553. doi: 10.1113/jphysiol.2007.136788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YG, Beierlein M. Receptor saturation controls short-term synaptic plasticity at corticothalamic synapses. J Neurophysiol. 2011;105:2319–2329. doi: 10.1152/jn.00942.2010. [DOI] [PubMed] [Google Scholar]
- Szapiro G, Barbour B. Multiple climbing fibers signal to molecular layer interneurons exclusively via glutamate spillover. Nat Neurosci. 2007;10:735–742. doi: 10.1038/nn1907. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Chen E, Copenhagen DR. Characterization of spontaneous excitatory synaptic currents in salamander retinal ganglion cells. J Physiol. 1995;486:207–221. doi: 10.1113/jphysiol.1995.sp020803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8:935–947. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Valera AM, Doussau F, Poulain B, Barbour B, Isope P. Adaptation of granule cell to Purkinje cell synapses to high-frequency transmission. J Neurosci. 2012;32:3267–3280. doi: 10.1523/JNEUROSCI.3175-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/S0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Harris KM, Regehr WG. Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J Neurosci. 2001;21:6666–6672. doi: 10.1523/JNEUROSCI.21-17-06666.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Fukaya M, Shibata T, Kurihara H, Tanaka K, Inoue Y, Watanabe M. Dynamic transformation of Bergmann glial fibers proceeds in correlation with dendritic outgrowth and synapse formation of cerebellar Purkinje cells. J Comp Neurol. 2000;418:106–120. doi: 10.1002/(SICI)1096-9861(20000228)418:1<106::AID-CNE8>3.0.CO%3B2-N. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. Neuromodulation at single presynaptic boutons of cerebellar parallel fibers is determined by bouton size and basal action potential-evoked Ca transient amplitude. J Neurosci. 2009;29:15586–15594. doi: 10.1523/JNEUROSCI.3793-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. Calcium influx measured at single presynaptic boutons of cerebellar granule cell ascending axons and parallel fibers. Cerebellum. 2012;11:121–131. doi: 10.1007/s12311-009-0151-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic analysis with double markers in mice. Cell. 2005;121:479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]