

Abstract
Recent evidence indicates that inflammatory insults in neonates significantly influenced white matter development and caused behavioral deficits that manifest in young adulthood. The mechanisms underlying these developmental and behavioral complications, however, are not well understood. We hypothesize that acute brain inflammation caused by neonatal infection reduces the bioavailability of iron required for oligodendrocyte maturation and white matter development. Here, we confirm that peripheral Escherichia coli infection in neonates at postnatal day 3 (P3) caused acute brain inflammation that was resolved within 72 h. Nonetheless, transient early life infection (ELI) profoundly influenced behavior, white matter development, and iron homeostasis in the brain. For instance, mice exposed to E. coli as neonates had increased locomotor activity and impaired motor coordination as juveniles (P35) and young adults (P60). In addition, these behavioral deficits were associated with marked hypomyelination and a reduction of oligodendrocytes in subcortical white matter and motor cortex. Moreover, ELI altered transcripts related to cellular sequestration of iron in the brain including hepcidin, ferroportin, and L-ferritin. For example, ELI increased hepcidin mRNA and decreased ferroportin mRNA and protein in the brain at P4, which preceded increased L-ferritin mRNA at P12. Consistent with the mRNA results, L-ferritin protein was robustly increased at P12 specifically in neurons of E. coli infected mice. We interpret these data to indicate that neonatal infection causes significant neuronal sequestration of iron at a time point before myelination. Together, these data indicate a possible role for aberrant neuronal iron storage in neonatal infection-induced disturbances in myelination and behavior.
Introduction
Perinatal inflammatory insults, including bacterial infections, may influence normal brain development and lead to significant cognitive and behavioral impairments (Bilbo and Schwarz, 2009; Hagberg et al., 2012). For instance, maternal bacterial infection during pregnancy increases the risk for schizophrenia or autism in offspring (Sørensen et al., 2009; Atladóttir et al., 2010). Clinical reports indicate that neonatal infection increases susceptibility to impairments in cognition and motor ability (Wald et al., 1986; Jones et al., 2004; Libster et al., 2012) and to white matter injury associated with impaired neurobehavioral development (Stoll et al., 2004; Chau et al., 2012). Thus, an inflammatory insult during a crucial period of development could have profound consequences later in life.
Several experimental models using rodents indicate that early life inflammatory events are associated with disruptions in white matter development and neurobehavioral deficits. For example, a recent study showed that repeated IL-1β injections during neonatal development (P1–P5) impaired oligodendrocyte maturation, disrupted white matter development, and impaired cognition (Favrais et al., 2011). One plausible explanation for these phenomena is that early life infection reduces the availability of iron for oligodendrocyte maturation and subsequent white matter development. In support of this notion, systemic iron availability is critical for oligodendrocyte maturation, lipid metabolism, and subsequent myelination of the CNS (Yu et al., 1986; Morath and Mayer-Pröschel, 2002; Ortiz et al., 2004). Moreover, iron is dynamically reduced in response to inflammation (Nemeth et al., 2004b). Reduced iron availability during infection is mediated by hepatic production of the iron regulatory hormone hepcidin (Nemeth et al., 2004b). Hepcidin decreases serum iron by inhibiting the iron transport channel, ferroportin, which is responsible for the exocytosis of cellular iron stores (Nemeth et al., 2004a). Hepcidin is present in the brain (Zechel et al., 2006; Wang et al., 2010), is increased following a peripheral immune challenge with lipopolysaccharide (Wang et al., 2008), and can alter iron transport in the brain. For example, Li et al. (2011) reported that intracerebroventricular injection of hepcidin results in degradation of brain ferroportin. The consequence of this hepcidin response is increased intracellular iron concentrations that drive increases in iron storage proteins, such as ferritins (De Domenico et al., 2006). Thus altered iron homeostasis in the brain may link early life inflammatory events to subsequent myelin-related deficits in brain and behavior.
Therefore, the objectives of the current study were to: (1) characterize the long-term behavioral complications that manifested after neonatal exposure to Escherichia coli and (2) determine the degree to which neonatal infection altered iron homeostasis in the brain and impaired white matter development. Here, we show that transient neuroinflammation in neonates led to impaired motor coordination and increased locomotor activity as juveniles. These early life infection (ELI)-induced behavioral complications were associated with reduced oligodendrocytes in the white matter and a significant reduction in myelin. Furthermore, ELI altered the expression of several transcripts and proteins related to cellular sequestration of iron in the brain. Last, ELI resulted in increased iron sequestration specifically in neurons at a time in brain development before peak myelination.
Materials and Methods
Mice.
Neonatal BALB/c mice were obtained from a breeding colony kept in a specific pathogen-free facility at The Ohio State University. Naive female mice were used for breeding. Two naive females were housed together and conception was synchronized so that multiple cohorts of neonatal mice were available simultaneously. All procedures were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by The Ohio State University Institutional Animal Care and Use Committee.
Neonatal E. coli infection paradigm.
For exposure to neonatal infection, male and female mice were subcutaneously injected with sterile saline or 1 × 106 colony-forming units (CFU) of K-12 Escherichia coli (strain 10798; ATCC) at postnatal day 3 (P3). Both female and male mice were used in the experiments in this study, except for experiments that involved the collection of behavioral data at postnatal day 35 and 60, where only males were used. Female dams were removed during experimental injections, but were placed back in the home cage within 10 min. A separate cohort of neonatal mice (P3) received a subcutaneous injection of sterile saline or 1 × 106 CFU of GFP+ E. coli (strain LT004) derived from the UPEC strain CFT073 with the genotype: cobS::φ(PLtetO-1–gfp+)cmR [chloramphenicol (Cm, 20 μg/ml); Månsson et al., 2007]. All littermates received the same treatment to avoid cross contamination and to reduce extra manipulation of pups while marking pups with treatment group identifiers (e.g., toe clips or tattoos). Congruent with a report using a similar neonatal E. coli infection paradigm in rats (Bilbo et al., 2007), no gross differences in maternal care including grooming and nursing of the infected pups were observed. There was no mortality associated with injections of 1 × 106 CFU of K-12 E. coli. At P21, mice were weaned and placed with same-sex littermates (2–5 mice per cage). Mice were housed in polypropylene cages and maintained at 25°C with ad libitum access to water and rodent chow under a 12 h light/dark cycle from 600 to 1800 h until weaning and then 1200–2400 h after weaning. Each experimental group represents mice obtained from at least three different litters.
Experimental protocols.
In the first set of experiments, neonatal mice (P3) were injected subcutaneously with sterile saline or 1 × 106 CFU of K-12 E. coli. Neonates were killed by rapid decapitation 24, 48, or 72 h later. Brains were removed, flash frozen, and RNA was collected for analysis (n = 7). A separate cohort of neonatal mice (P3) were injected as above and killed by rapid decapitation 24 h later.
In a related set of experiments, neonatal mice (P3) were injected subcutaneously with sterile saline or 1 × 106 CFU of K-12 E. coli. Neonates were killed by rapid decapitation at P4 or P12. Blood was collected to determine iron concentration in the serum. Brains and livers were removed, flash frozen, and RNA was collected for analysis (n = 6).
In the second set of experiments, neonatal mice (P3) were injected subcutaneously with sterile saline or 1 × 106 CFU of the K-12 or the GFP+ strain of E. coli. Mice were injected as above and killed 1 or 7 d later. Brain, liver, and spleen tissues were aseptically removed, individually homogenized with a plunger through a filter mesh (0.22 μm), and rinsed through with 2 ml sterile PBS. Homogenates were cultured individually on nutrient dense brain–heart infusion agar overnight at 37°C. Bacterial colonies were counted 24 h later and the average number of colonies recovered was determined in each tissue.
In the third set of experiments, neonatal mice (P3) were injected subcutaneously with sterile saline or 1 × 106 CFU of K-12 E. coli. Locomotor activity (n = 7), motor coordination (n = 8), and novel object recognition (n = 14) were determined in the same male mice at P35 and P60.
In the fourth set of experiments, neonatal mice (P3) were injected subcutaneously with sterile saline or 1 × 106 CFU of K-12 E. coli. Mice were deeply anesthetized with ketamine/xylazine and transcardially perfused with PBS and 4% PFA at P4, P12, P35, and P60. Brains were postfixed in 4% formaldehyde for 24 h and incubated in 30% sucrose for an additional 24 h. Brains were sectioned, mounted on slides, and used for myelin, Iba-1, glial fibrillary acidic protein (GFAP), Olig2, NG2, L-ferritin, ferroportin, and NeuN analyses (n = 4–8).
RNA isolation and RT-PCR.
RNA was isolated from the brain and liver using a Tri-reagent protocol (Sigma-Aldrich). RNA concentration was determined by spectrophotometry and reverse transcribed to cDNA using an RT-RETROscript kit (Ambion). Real-Time PCR (RT-PCR) was performed using the Applied Biosystems Assay-on-Demand Gene Expression protocol as previously described (Godbout et al., 2005; Wynne et al., 2010). In brief, cDNA was amplified by RT-PCR where a target cDNA (e.g., CD11b, IL-1β, IL-1R1, TNFα, hepcidin, ferroportin, transferrin receptor, and L-ferritin) and a reference cDNA (glyceraldehyde-3-phosphate dehydrogenase; GAPDH) were amplified simultaneously using an oligonucleotide probe with a 5′ fluorescent reporter dye (6-FAM) and a 3′ quencher dye (NFQ). Fluorescence was determined on an ABI PRISM 7300-sequence detection system (Applied Biosystems). Data were analyzed using the comparative threshold cycle (Ct) method and results are expressed as fold-difference from control.
Motor coordination.
Motor coordination was determined using the Accelerating Rotarod (Columbus Instruments, OH). All testing was performed in the light cycle ∼2 h before the onset of the dark cycle. For juvenile (P35) mice: Acclimation was completed on day 1 of testing. Mice were placed on a stationary bar for 45 s, and then the bar started at a constant speed of 5 rpm. When mice fell, they were placed back on the bar to facilitate learning. After 1 min of running at 5 rpm, mice were placed back into home cages. At least 15 min later, mice were placed on the bar that rotated at 5 rpm with an acceleration rate of 0.2 rpm/s. On day 2 (training phase) and day 3 (test phase), mice were subjected to three trials at 5 rpm with an acceleration rate of 0.2 rpm/s. Trials 1 and 2 were run back to back so mice would not associate falling off the bar with a reward to return to their home cage. Trial 3 was run at least 15 min following Trial 2. If mice fell off the bar at 3 s or less, that mouse was given a second chance for that trial. Trials were averaged for each day for each mouse. For young adult (P60) mice, rotarod testing was performed as described above except the start speed was 10 rpm.
Locomotor activity.
Overall locomotor activity was determined using a Versamax open-field activity box paradigm (AccuScan Instruments), as previously described (Wohleb et al., 2012). Before behavior testing, all mice were handled for 5 d, and all testing was completed during the light cycle during 800–1100 h, 2–3 h before onset of the dark cycle. To initiate testing, mice were placed into independent 40 cm × 40 cm chambers of the activity box. Movements were auto-recorded by a laser detection system for 5 min and data outputs were analyzed using VersaMap software (AccuScan Instruments).
Novel object recognition test.
Novel object recognition was used as a basic cognitive function test and was performed as previously described (Bevins and Besheer, 2006). In brief, all testing was completed during the dark cycle starting ∼1 h after onset (1300–1500 h). Mice were handled for 5 d before testing and allowed to acclimate to the bedding-free clean cage environment for 1–2 h. During pre-exposure, two identical objects were placed on opposite ends of the cage and mice were allowed to explore the objects for 10 min. Objects were removed and the mice were allowed to rest for 1.5 h. During testing, one familiar object and one novel object were then placed on opposite sides of the cage and mice were allowed to explore the objects for 5 min. All activity was recorded and later scored by a trained observer who was blind to the experimental treatments. The time mice actively investigated each object during the pre-exposure and novel object testing phases was determined.
Serum iron assay.
Blood was collected from P4 and P12 mice, allowed to clot at room temperature, and spun at 2000 × g for 20 min. Serum was then aspirated and stored at −80°C until analysis. Total iron [i.e., Fe(II) and Fe(III)] was determined in serum using a colorimetric iron assay kit according to the manufacturer's instructions (Abcam). Absorbance was read at 593 nm using a colorimetric plate reader. The assay was sensitive to >8 μm of iron.
Immunohistochemistry.
Brains were fixed (as described in Experimental protocols) and frozen with isopentane (−78°C), embedded in OCT compound and sectioned (12 μm) using a Microm HM550 cryostat. Brain regions were identified by reference markers in accordance with the stereotaxic mouse brain atlas (Paxinos and Franklin, 2008). Before incubation with primary antibodies, all sections were washed in PBS and incubated in a blocking solution containing PBS, 4% bovine serum albumin, 0.3% Triton X-100, and 3% normal serum from the same species as the host of the secondary antibody. After incubation with primary antibody, sections were washed and incubated in appropriate AlexaFluor secondary antibody solution (1:500 dilution in blocking solution). Double labeling was completed consecutively, with appropriate washing and blocking steps between the first secondary antibody (2°) and the second primary antibody.
To label for Iba-1, sections were incubated in Iba-1 primary (1:1000, Wako). To double label for Olig2 and GFAP, sections were first labeled for Olig2 (goat, 1:100, R&D Systems; donkey anti-goat 2°) and then labeled for GFAP (rabbit, 1:2000, Dako; goat anti-rabbit 2°). To label for Iba1 or Olig2 and L-ferritin, sections were first labeled for Olig2 (goat, 1:100, R&D Systems) or Iba-1 (goat, 1:500; Abcam) with donkey anti-goat secondary antibody and then labeled for L-ferritin (rabbit, 1:500; Abcam; goat anti-rabbit 2°). To label for GFAP or NeuN and L-ferritin, sections were first labeled for L-ferritin (rabbit, 1:500; Abcam; goat anti-rabbit 2°) and then for either GFAP (chicken, 1:500, Aves) or NeuN (chicken, 1:1000; Aves) with goat anti chicken secondary antibody. Rabbit anti-mouse ferroportin antibody (1:500; Abcam) and donkey anti-rabbit 596 secondary were used to label ferroportin. One-hundred × DAPI was used to stain for nuclear bodies (1:2000). Slides were coverslipped with Fluoromount-G slide mounting medium (Beckman Coulter) and stored at −20°C.
Fluorescence was visualized using an epi-fluorescent Leica DM5000B microscope. Images were captured using a Leica DFC300 FX camera and imaging software. A threshold for positive staining was determined for each image and was processed by densitometric scanning of the threshold targets using ImageJ software (NIH). Proportional area was reported as the average percentage area in the positive threshold for all representative pictures. L-ferritin localization within NeuN+ cells was confirmed using a Zeiss 510 LSM confocal microscope, and LSM software was used to generate orthogonal images.
Myelination.
Myelin was determined using an eriochrome cyanine (EC) stain as previously described (Kigerl et al., 2006; Donnelly et al., 2011). In brief, slides were placed in acetone for 5 min and then allowed to air dry at RT. Slides were stained in EC solution for 30 min and then briefly differentiated in 5% iron alum followed by a borax-ferricyanide solution. Slides were rinsed, dehydrated, and coverslipped using Permount (Fisher). Slides were analyzed by digital intensity and threshold analyses as previously described (Donnelly et al., 2009; Cate et al., 2010).
Statistical analysis.
To ensure a normal distribution, data were subjected to Shapiro–Wilk test using Statistical Analysis Systems (SAS) software). Observations greater than two interquartile ranges from the first and third quartile were considered outliers and were excluded from analysis. To determine significant main effects and interactions between main factors, data were analyzed using one- (infection) or two-way (infection × time) ANOVA using the General Linear Model procedures of SAS. When appropriate, differences between treatment means were evaluated by an F-protected t test using the Least-Significant Difference procedure of SAS. All data are expressed as treatment mean ± SEM.
Results
Neonatal (P3) E. coli infection caused transient brain inflammation and microglial activation
The objectives of this study were to (1) characterize the behavioral complications that manifested in young adulthood after neonatal exposure to E. coli and (2) determine the degree to which this neonatal infection altered iron bioavailability in the brain and impaired white matter development. To begin to address these objectives, we aimed to use a model of neonatal E. coli infection (ELI) that was acute and nonlethal. The E. coli (K-12) strain was selected for these experiments because it lacks the O and K antigens, which are involved in eluding host defenses and adhering to host cell surfaces (Liu and Reeves, 1994). A series of preliminary experiments were completed in neonatal (P3) BALB/c mice, and the dose of 1 × 106 CFU K-12 of E. coli was selected because the infection was transient and no mortalities were observed (data not shown).
Neonates (P3) were injected subcutaneously with saline (CON) or E. coli (ELI), and mRNA levels of several markers of inflammation (CD11b, IL-1β, IL-1R1, and TNFα) were determined in whole-brain homogenates collected 24, 48, and 72 h later. Figure 1A shows that CD11b was increased by E. coli infection (F(1,41) = 42.88, p < 0.0001) in a time-dependent manner (infection × time interaction; F(2,41) = 9.51, p < 0.001). For example, CD11b mRNA levels were increased 24 h (p < 0.001) and 48 h (p < 0.001) after E. coli infection but returned to baseline expression by 72 h. Similar to CD11b, Figure 1B shows that IL-1β mRNA was increased in the brain by E. coli infection (F(1,42) = 24.12, p < 0.0001) in a time-dependent manner (infection × time interaction; F(2,42) = 5.91, p < 0.007). Specifically, IL-1β mRNA was increased in the brain 24 h (p < 0.0001) and 48 h (p < 0.001) after infection, but returned to baseline levels by 72 h. Twenty-four hours after infection, IL-1R1 mRNA expression (Fig. 1C; p < 0.03) and TNFα mRNA expression (Fig. 1D; p < 0.002) were increased in the brain, and these returned to baseline by 48 h.
Figure 1.
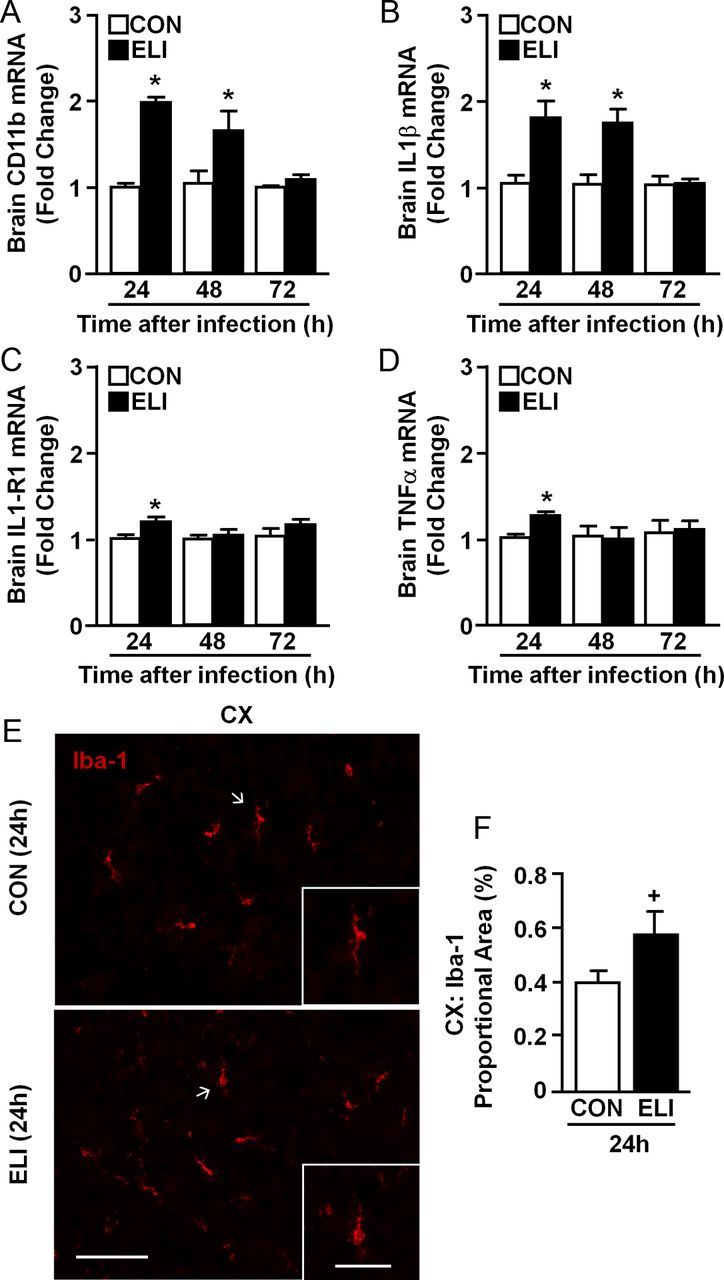
Neonatal (P3) E. coli infection caused transient brain inflammation and microglial activation. Neonatal BALB/c mice (P3) were injected subcutaneously with either sterile saline (CON) or E. coli (ELI). Brains were collected from male and female mice 24, 48, or 72 h later and mRNA levels of (A) CD11b, (B) IL-1β, (C) IL-1R1 and (D) TNFα were determined (n = 7). In a related study, male and female mice were infected as above. The brain was collected 24 h later and Iba-1 protein expression was determined. E, Representative images of Iba-1 labeling in the CX of both CON and ELI mice are shown (scale bar, 100 μm). Insets show enlarged image of microglia (scale bar, 50 μm). F, Digital image analysis of Iba-1 proportional area in the cortex (n = 4). Bars represent the mean ± SEM. Means with asterisk are significantly different from controls (p < 0.05). Means with plus sign denote a tendency (p ≤ 0.1).
Next, microglial activation was determined 24 h after neonatal E. coli infection. Figure 1E shows representative images of Iba-1 labeling in the cortex (CX) of control and E. coli-infected mice. The E. coli-infected mice had a more activated microglial phenotype indicated by increased deramified morphology (i.e., shorter and thicker processes) (Fig. 1E, insert). Digital image analysis of Iba-1 labeling indicated that ELI tended to increase the proportional area of Iba-1 in the cortex (Fig. 1F; F(1,7) = 3.0, p = 0.1).
These mRNA and histological data indicate that neonatal E. coli infection induced an inflammatory response in the brain that was resolved within 72 h. To confirm the transience of the E. coli infection, brain, liver, and spleen were collected 1 or 7 d after infection. K-12 E. coli was present in all tissue cultures 1 d after infection, but was no longer detected after 7 d (Table 1). Cultures from mice injected with a GFP+ strain of E. coli confirmed that the recovered bacteria were derived from the experimental injections and not from another contaminating source (data not shown). In addition, GFP+ bacteria were not detected in the brain parenchyma or meninges 24 h after injection (data not shown). Together, these data indicate E. coli infection in neonates caused a transient increase in brain inflammation and microglia activation that was resolved by 72 h. Moreover, this bacterial infection was cleared from neonates within 7 d (P10).
Table 1.
E. coli CFU recovered from tissue following a neonatal infection at P3
| 1 d after infection |
7 d after infection |
|||||
|---|---|---|---|---|---|---|
| Brain | Liver | Spleen | Brain | Liver | Spleen | |
| Control | — | — | — | — | — | — |
| K-12 E.coli | ++ | +++ | + | — | — | — |
Male and Female neonatal mice were injected subcutaneously with K-12 E. coli at P3. Brain, liver, and spleen tissues were collected 1 or 7 d later and cultured on nutrient-rich agar. Results expressed as average CFU recovered per specimen. —, No CFU recovered; +, 1–10 CFU; ++, 10–150 CFU; +++ ≥ 150 CFU (n = 6).
ELI reduced motor coordination and increased locomotor activity in juvenile and young adult mice
Inflammatory insults early in life, such as intracerebral administration of LPS in neonatal rodents (Fan et al., 2005) or neonatal bacterial infection in preterm infants (Stoll et al., 2004; Chau et al., 2012), can increase the susceptibility to motor and behavioral deficits in adulthood. Therefore, the extent to which neonatal E. coli infection resulted in impaired behavioral responses in juveniles (P35) and young adult mice (P60) was determined. In these experiments, motor coordination, locomotor activity, and novel object recognition were assessed in male mice 1 and 2 months after neonatal E. coli infection.
In these experiments, motor coordination was determined using the accelerating rotarod at P35 (Fig. 2A, juveniles) and again at P60 (Fig. 2B, young adulthood). Figure 2A shows that ELI mice spent less time on the rotarod as juveniles (P35) compared with noninfected controls (F(1,67) = 26.28, p < 0.0001) on day 1 (p < 0.002), day 2 (p < 0.008), and day 3 (p < 0.007) of testing. It is important to note that both groups of juvenile mice improved their times on the rotarod over the three testing days. Nonetheless, these data show that ELI mice had impaired motor coordination on each day of testing. The same cohort of mice was also tested 1 month later as young adults (P60). Figure 2B shows that ELI mice (P60) demonstrated decrements in motor coordination compared with control mice (main effect of ELI; F(1,67) = 6.00, p < 0.02). Post hoc analysis indicated that ELI mice tended to fall off the rotarod faster on day 2 (p = 0.1) and day 3 (p = 0.1) of testing compared with the control mice. These data indicate that neonatal infection (P3) resulted in impaired motor coordination that was evident in the mice as juveniles and young adults.
Figure 2.

Reduced motor coordination and hyperactivity were observed in juvenile and young adult after neonatal infection. Neonatal mice (P3) were injected subcutaneously with either saline (CON) or E. coli (ELI), and motor coordination in males was determined when mice were juveniles (P35) and again as young adults (P60). Time spent on the accelerating rotarod is shown at (A) P35 and (B) P60 (n = 8). In a separate study, locomotor activity was determined in male CON and ELI mice as juveniles (P35) and young adults (P60). C, Total distance traveled and (D) total time moving in an open field at P35 and P60 are shown (n = 7). In a separate study, object memory was determined in CON and ELI mice as (E) juveniles (P35) and (F) young adults (P60) (n = 14). Bars represent the mean ± SEM. Means with asterisk are significantly different from controls (p < 0.05), and means with plus sign tended to be different from controls (p ≤ 0.1).
In the next experiment, locomotor activity (i.e., total distance traveled and total time moving) was determined using an open-field paradigm when mice were juveniles (P35) and young adults (P60). Figure 2C shows that ELI mice traveled a greater distance in the open field compared with controls (F(1,96) = 10.99, p < 0.002). For instance, post hoc analysis confirmed that total distance traveled was increased in juvenile (P35) ELI mice (p < 0.002) and young adult (P60) ELI mice (p = 0.09) compared with their age-matched controls. Parallel to these results, Figure 2D shows that ELI mice spent more time moving (F(1,96) = 8.25, p < 0.002) in the open field than controls. Post hoc analysis indicated that total time moving was increased in juvenile (P35) ELI mice (p < 0.003) and young adult (P60) ELI mice (p < 0.05) compared with their respective age-matched controls. These data indicate that ELI mice had increased locomotor activity compared with their age matched controls.
Control and ELI mice were also tested for basic cognitive function in a novel object recognition test as juvenile (P35) and young adult mice (P60). There was no effect of ELI on novel object recognition at either P35 or P60 (Fig. 2E,F). There was also no difference in total time exploring objects during the pre-exposure or testing at both time points between treatment groups (data not shown). Collectively, these results indicate that neonatal E. coli infection resulted in motor coordination deficits and hyperactive locomotion that were evident in young adulthood.
Hypomyelination in the brain of juvenile mice exposed to E. coli as neonates
Several clinical studies reported that hyperactive behavior and motor deficits are associated with impairments in myelination (Counsell et al., 2008; Nagel et al., 2011; Spittle et al., 2011) and elevated inflammatory cytokines during the early postnatal period (O'Shea et al., 2013). Therefore, myelin was assessed in the brain of control and ELI juvenile mice (P35), a time-point when motor impairment and increased locomotor activity were evident (Fig. 2). Figure 3A shows representative images of myelin staining from coronal sections of juvenile (P35) control and ELI mice. Figure 3B shows that there was markedly less myelin in the white matter (F(1,22) = 7.08, p < 0.01) and gray matter (F(1,22) = 5.98, p < 0.03) of juvenile ELI mice compared with controls. Based on these data, myelin was determined in white matter areas (corpus callosum) and areas associated with motor coordination (motor cortex and cerebellum). Representative images of myelin staining are also shown from the mid-section of the corpus callosum (Fig. 3C), cingulum bundle of the corpus callosum (Fig. 3E), motor cortex (Fig. 3G), and cerebellum (Fig. 3I) of control and ELI mice. Overall, the myelin content was reduced in the brains of mice exposed to E. coli as neonates compared with controls (F(1,17) = 17.86, p < 0.0001). Analysis of each of these sections for relative intensity of myelin staining confirmed that neonatal infection was associated with reduced myelin in the mid-section of the corpus callosum (Fig. 3D; p < 0.01), cingulum bundle of the corpus callosum (Fig. 3F; p < 0.05), and motor cortex (Fig. 3H; p < 0.01). In the cerebellum, however, there were no detectable differences in myelin between the two treatment groups. In addition, there tended to be less myelin in the hippocampus of ELI mice compared with controls (p = 0.08, data not shown). Together, there was significantly less myelin in the corpus callosum and motor cortex of juvenile mice exposed to E. coli as neonates.
Figure 3.

Hypomyelination was observed in the brains of juvenile mice exposed to E. coli as neonates. Neonatal mice (P3) were injected subcutaneously with either saline (CON) or E. coli (ELI), and brain tissue from male and female mice was collected and stained for myelin 1 month later (P35). Representative images and quantification of myelin staining are shown in: A, coronal section and B, quantification of total white and gray matter areas examined; C, D, the mid-section of the corpus callosum; E, F, the cingulum bundle of the corpus callosum; G, H, the motor cortex; and I, J, cerebellum. Bars represent the mean ± SEM (n = 6–9). Means with asterisk are significantly different from controls (p < 0.05), and means with plus sign tended to be different from controls (p ≤ 0.1). Rel, Relative; CC, Corpus callosum; Cing, cingulum bundle; MCX, motor cortex; CBM, cerebellum. Scale bars: A, C, 500 μm; E, G, I, 250 μm.
Reduced number of oligodendrocytes in the corpus callosum and motor cortex at P35 in ELI mice
Oligodendrocytes are responsible for the myelination of axons in the CNS, and myelin was reduced in the brain of young adult mice that were infected with E. coli as neonates (Fig. 3). Therefore, we investigated the extent to which ELI alters oligodendrocyte survival and maturation by determining the total number of oligodendrocytes (Olig2+/NG2−) and oligodendrocyte progenitors (Olig2+/NG2+) in the corpus callosum, motor cortex, and hippocampus. Figure 4A shows representative images of Olig2+ labeling in the corpus callosum of control and ELI mice at P35. Olig2 did not colocalize with GFAP+ astrocytes (Fig. 4B) confirming that the Olig2+ cells were of oligodendrocyte lineage. In addition, Figure 4C shows representative images of Olig2/NG2 double labeling of oligodendrocyte progenitors in the corpus callosum. Although ELI had no effect on the number of oligodendrocyte progenitors in the motor cortex (Fig. 4D), corpus callosum (Fig. 4E), or hippocampus (Fig. 4F), exposure to neonatal infection reduced both the total number of Olig2+ cells (F(1,19) = 12.28, p = 0.01) and the total number of Olig2+/NG2− cells (F(1,9) = 11.34, p < 0.01) across brain regions. ELI reduced Olig2+/NG2− cells in the corpus callosum (p = 0.0001; Fig. 4H) and motor cortex (tendency, p = 0.1; Fig. 4G) compared with controls, but not in the hippocampus (Fig. 4I). Together, these results indicate that early life infection with E. coli resulted in a reduction of oligodendrocytes (Olig2+/NG2−) in the corpus callosum and motor cortex of juvenile mice (P35), but did not affect the number of oligodendrocyte progenitors (Olig2+/NG2+).
Figure 4.
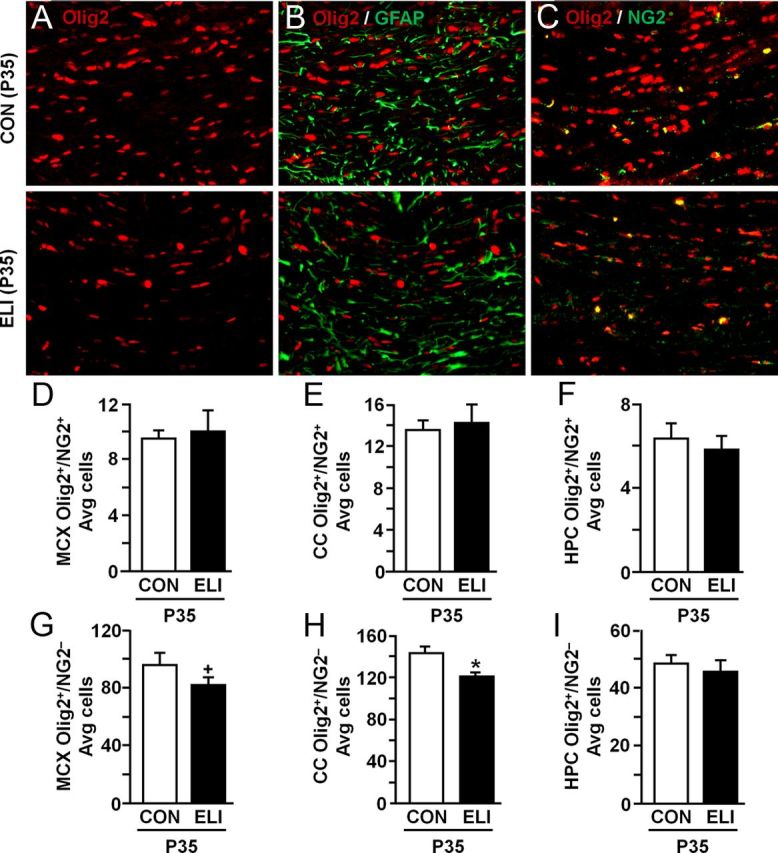
Reduced number of oligodendrocytes in the corpus callosum and motor cortex of juvenile mice exposed to E. coli as neonates. Neonatal mice (P3) were injected subcutaneously with either saline (CON) or E. coli (ELI), and brain tissue from male and female mice was collected at P35 and Olig2 (red) and GFAP (green) or Olig2 (red) and NG2 (green) labeling was determined. Representative images of (A) Olig2, (B) Olig2/GFAP, (C) Olig2/NG2 from the CC of CON and ELI mice are shown. Average number of Olig2+/NG2+ cells in the (D) MCX, (E) CC, and (F) hippocampus (HPC). Average number of Olig2+/NG2− cells in the (G) MCX, (H) CC, and (I) HPC. Bars represent the mean ± SEM (n = 6). Means with asterisk are significantly different from controls (p < 0.05), and means with plus sign tend to be different from controls (p ≤ 0.1).
ELI altered the expression of iron-related transcripts in the brain
ELI resulted in hypomyelination (Fig. 3) and fewer oligodendrocytes (Olig2+/NG2−) at P35 (Fig. 4). Because iron is regulated by inflammation (Nemeth et al., 2004a) and is also critical for oligodendrocyte development and subsequent myelination (Yu et al., 1986; Morath and Mayer-Pröschel, 2002; Ortiz et al., 2004), we sought to determine whether peripheral and central iron homeostasis was altered by ELI. In the first study, blood was collected at P4 and P12 after neonatal E. coli infection and total serum iron was determined. Figure 5A shows that total serum iron was unaffected by the neonatal E. coli infection at P4 or P12. Next, mRNA expression of several transcripts associated with the cellular sequestration of iron (hepcidin, ferroportin, transferrin receptor, L-ferritin) was determined in whole brain homogenates collected at P4 and P12. In addition, hepcidin mRNA was determined in the liver collected at P4 and P12. Figure 5B shows that hepcidin mRNA was unchanged in the liver at P4 and P12. Analysis of hepcidin in the brain, however, revealed a robust increase (40-fold) in mRNA expression 24 h after neonatal E. coli infection compared with controls (Fig. 5C; p < 0.02). This ELI-induced increase in hepcidin mRNA was no longer detected in the brain by P12. Figure 5D shows that brain ferroportin mRNA tended to be decreased at P4 (p = 0.1) and elevated at P12 (p < 0.05). Transferrin receptor expression was unchanged at P4 and P12 (Fig. 5E). Figure 5F shows that L-ferritin mRNA was increased at P12 after E. coli infection (p < 0.05). These data indicate that neonatal infection significantly alters gene expression of important regulatory proteins that are associated with cellular sequestration of iron.
Figure 5.

ELI altered the expression of iron storage related genes. Neonatal mice (P3) were injected subcutaneously with saline (CON) or E. coli (ELI), and blood, liver, and brain from male and female mice were collected at P4 and P12. A, Total iron was determined from serum at P4 and P12. B, Liver mRNA levels of hepcidin were determined (n = 6). Brain mRNA levels of (C) hepcidin, (D) ferroportin, (E) transferrin receptor and (F) L-ferritin were determined at P4 and P12 (n = 6). Bars represent the mean ± SEM (n = 6). Means with asterisk are significantly different from controls (p < 0.05), and means with plus sign tended to be different from controls (p ≤ 0.1).
ELI decreased ferroportin in motor cortex at P4 and increased L-ferritin in the hippocampus and motor cortex at P12
Our data indicate that neonatal infection is associated with transient alterations in mRNA associated with increased cellular sequestration of iron. Therefore, ferroportin and L-ferritin protein levels were determined following ELI. Figure 6 shows representative images and quantification of ferroportin labeling in the hippocampus, motor cortex, and corpus callosum at P4 in control and ELI mice. Neonatal E. coli infection reduced ferroportin in the motor cortex at P4 (Fig. 6C,D; p < 0.01) and the hippocampus at P12 (Fig. 6G; p = 0.1). The motor cortex (P4) and the hippocampus (P12) were the only regions examined that had detectable reduction in ferroportin protein. Next, we analyzed protein levels of L-ferritin in control and ELI mice at P12, a time when increased mRNA expression of L-ferritin was observed (Fig. 5E). Representative images of L-ferritin labeling are shown in the CA1 of the hippocampus (Fig. 7A), motor cortex (Fig. 7B), and corpus callosum (Fig. 7C). ELI markedly increased L-ferritin labeling in the hippocampus (Fig. 7D; F(1,8) = 67.69, p < 0.0001) and cortex (Fig. 7E; F(1,8) = 7.83, p < 0.03). Levels of L-ferritin were not increased by ELI in the corpus callosum (Fig. 7F). Collectively, these data indicate that there was a reduction of protein in motor cortex at P4 and hippocampus at P12, and a marked elevation in L-ferritin in the brains of ELI mice at P12.
Figure 6.

ELI reduced ferroportin in the motor cortex at P4. Neonatal mice (P3) were injected subcutaneously with either saline (CON) or E. coli (ELI), and brain tissue from male and female mice was collected at P4 or P12. Protein levels of ferroportin (Ferro) were determined. Representative images of ferroportin labeling in the (A) CA1 of the HPC, (C) MCX, and (E) CC. Proportional area analyses of ferroportin labeling at P4 and P12 is provided for the (B, G) HPC, (D, H) MCX, and (F, I) CC. Hippocampus proportional area analysis represents average proportional area observed in the CA1, CA3, and dentate gyrus. Bars represent the mean ± SEM (n = 4). Means with asterisk are significantly different from controls (p < 0.05), and means with plus sign tended to be different from controls (p ≤ 0.1).
Figure 7.

ELI robustly increased L-ferritin in the hippocampus and cortex before myelination. Neonatal mice (P3) were injected subcutaneously with either saline (CON) or E. coli (ELI), and brain tissue from male and female mice was collected at P12 and the protein levels of L-ferritin (L-Fer) were determined. Representative images of L-ferritin labeling are shown in the (A) CA1 of the HPC, (C) MCX, and (E) CC. Proportional area analyses of L-ferritin labeling for the (B) HPC, (D) MCX, and (F) CC are provided. HPC proportional area analysis represents average proportional area observed in the CA1, CA3, and dentate gyrus. Bars represent the mean ± SEM (n = 4). Means with asterisk are significantly different from controls (p < 0.05).
Increased L-ferritin labeling after ELI did not colocalize with microglia, astrocytes, or oligodendrocytes
Microglia supply iron to oligodendrocytes during the early postnatal period concomitant with the production of myelin (Cheepsunthorn et al., 1998; Zhang et al., 2006). Inflammatory insults in neonatal rodents, including hypoxia/ischemia, cause hypomyelination that is correlated with increased ferritin storage in microglia (Cheepsunthorn et al., 2001). Therefore, the degree to which active microglia store iron in L-ferritin before onset of peak myelination (P12) in ELI mice was assessed. Microglial activation was determined by Iba-1 immunoreactivity. Figure 8A shows representative images of Iba-1 labeling in the corpus callosum and adjacent CA1 of the hippocampus. Microglia in ELI mice tended to have higher Iba-1 immunoreactivity in the corpus callosum compared with controls (Fig. 8B; F(1,8) = 4.28, p = 0.08). Increased Iba-1 immunoreactivity was not detected in either the hippocampus or cortex of ELI mice (Fig. 8B). Iba-1 and L-ferritin labeling showed that although there was higher L-ferritin labeling in ELI mice, the L-ferritin was not associated with microglia (Iba-1+; Fig. 8C).
Figure 8.
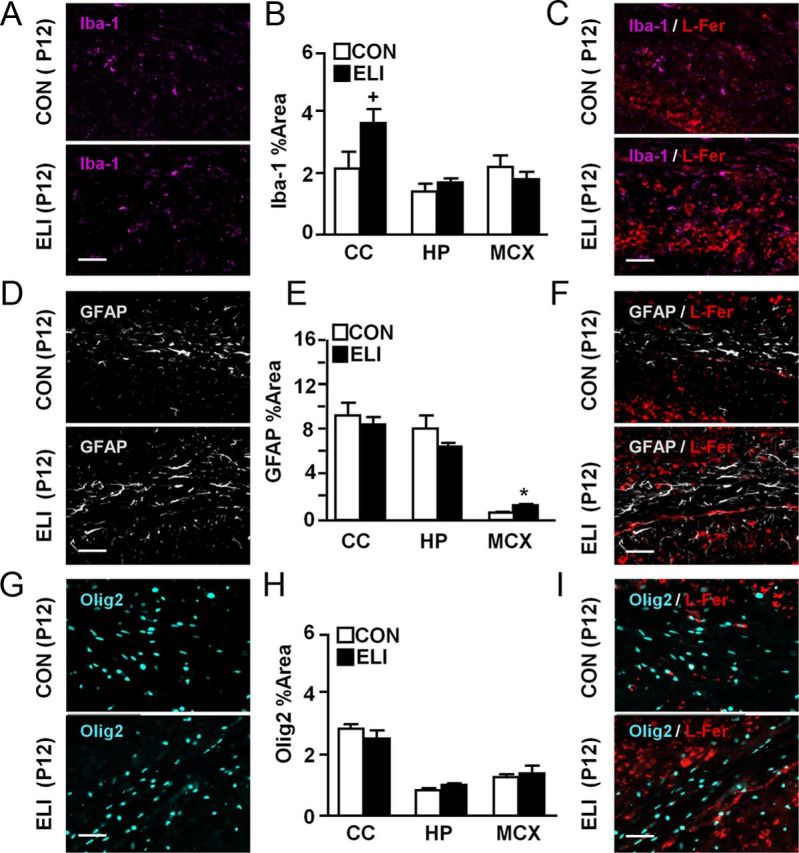
ELI-associated increase in L-ferritin did not colocalize with microglia, astrocytes, or oligodendrocytes. Neonatal mice (P3) were injected subcutaneously with either saline (CON) or E. coli (ELI), and brain tissue from male and female mice was collected at P12 and labeling for Iba-1 (pink), GFAP (white), Olig2 (light blue), and L-ferritin (L-Fer; red). Representative pictures of (A) microglia (Iba-1), (D) astrocytes (GFAP), and (G) oligodendrocytes (Olig2) in the corpus callosum (proximal to the hippocampal CA1 region) are shown. Quantification of immunopositive glia in the CC, HP, and CX for (B) Iba-1, (E) GFAP, and (H) Olig2 are provided. Representative images of colabeling with L-ferritin for (C) Iba-1, (F) GFAP, and (I) Olig2 are also provided. Bars represent the mean ± SEM (n = 4). Means with asterisk are significantly different from controls (p < 0.05). Means with plus sign denote a tendency (p ≤ 0.1). Scale bar, 100 μm.
Next, L-ferritin was determined in astrocytes (GFAP) and oligodendrocyte lineage cells (Olig2). Representative pictures of GFAP+ astrocytes and Olig2+ oligodendrocytes lineage cells in the corpus callosum of ELI and control mice at P12 are shown in Figure 8D and G, respectively. GFAP immunoreactivity was increased in the cortex of ELI mice compared with controls (p < 0.007), but not in the hippocampus or corpus callosum (Fig. 8E). Olig2 immunoreactivity was not significantly different between treatment groups at P12 (Fig. 8H). L-ferritin did not localize with either GFAP+ astrocytes (Fig. 8F) or Olig2+ cells (Fig. 8I). Collectively, these data indicate that L-ferritin was increased in the ELI brain, but was not sequestered in glia.
ELI-induced L-ferritin labeling colocalized with neurons
The data provided in Figures 5 and 7 indicate that there was a marked increase in the amount of L-ferritin mRNA and protein in the brain of ELI mice at P12. This increase in L-ferritin, however, was not associated with microglia (Iba-1+), astrocytes (GFAP+), or oligodendrocytes lineage cells (Olig2+; Fig. 8). Because L-ferritin appeared to localize within cells in the pyramidal and granule cell layers of the hippocampus, the expression of L-ferritin by neurons (NeuN+) was determined in the brain at P12. Consistent with observations in Figures 7 and 8, ELI was associated with higher levels of L-ferritin in the CA1 (Fig. 9E; F(1,10) = 5.49, p < 0.05), CA3 (Fig. 9F; F(1,7) = 37.80, p < 0.0009), dentate gyrus (Fig. 9G; F(1,9) = 25.72, p < 0.002), and motor cortex (Fig. 9H; F(1,9) = 5.88, p < 0.05). Moreover, the representative images shown in Figure 9A–D indicate a close association between NeuN+ and L-ferritin+ labeling in the motor cortex and the CA1, CA3, and dentate gyrus of the hippocampus. Confocal analysis was used to further assess the proximity of NeuN and L-ferritin labeling on a cellular level. Orthogonal projections demonstrate that L-ferritin was localized to the cytoplasm surrounding the neuronal nuclei (Fig. 9I). This association between NeuN and L-ferritin was detected in both the control and ELI mice. Furthermore, these images confirmed that there was increased cytoplasmic L-ferritin labeling in the neurons of ELI mice. Together, ELI caused a robust increase in neuronal levels of L-ferritin in the hippocampus and motor cortex.
Figure 9.
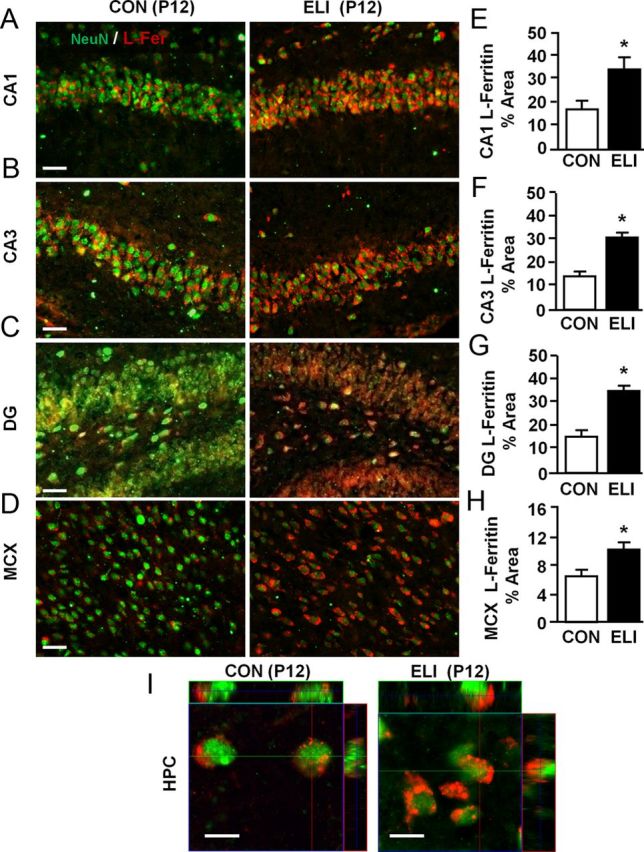
Neonatal infection leads to iron sequestration in neurons before peak myelination. Neonatal mice (P3) were injected subcutaneously with either saline (CON) or E. coli (ELI), and brain tissue from male and female mice was collected at P12 and labeled for NeuN and L-Fer. Representative images of NeuN+ and L-ferritin+ are shown in the (A) CA1, (B) CA3, and (C) dentate gyrus (DG) regions of the HPC, and (D) the CX. Proportional area analyses of L-ferritin labeling in the (E) CA1, (F) CA3, (G) dentate gyrus, and (H) CX are shown. I, Orthogonal projections of interneurons from the CA1 region of the hippocampus for control and ELI mice are provided. Bars represent the mean ± SEM (n = 4). Means with asterisk are significantly different from controls (p < 0.05). Scale bars: A–D, 100 μm; I, 5 μm.
Discussion
Perinatal infection can cause long-lasting behavioral impairments in humans and rodents (Bilbo and Schwarz, 2009; Hagberg et al., 2012). Here, we show that a transient infection during a critical period of neurodevelopment increased neuronal iron sequestration and impaired oligodendrogenesis, white matter development, and adult motor coordination. For instance, ELI significantly altered transcripts and proteins related to neuronal iron metabolism. Moreover, ELI caused a stark reduction in myelin content at P35, which was directly associated with decreased oligodendrocytes (Olig2+/GFAP−/NG2−) in subcortical white matter and motor cortex. Furthermore, these white matter deficits corresponded to deficits in motor coordination and locomotor activity evident at P35 and P60. Notably the time point used in this study (P3) is consistent with a prenatal time point in human gestation that is comparable with the third trimester (Dobbing and Sands, 1979; Rodier, 1980). Our findings are relevant because peripheral inflammatory events during neonatal development are associated with white matter deficits in humans (Chau et al., 2012) and rodents (Favrais et al., 2011). Moreover, clinical studies have correlated deficits in white matter integrity to neurobehavioral deficits including autism (Billeci et al., 2012), attention-deficit/hyperactivity disorder (Nagel et al., 2011), and motor impairments (Spittle et al., 2011).
One key element of this study was that a peripheral E. coli infection led to a transient inflammatory response in the brain. For example, ELI increased brain inflammatory markers including mRNA expression of IL-1β, IL-1R1, TNFα, and CD11b and increased Iba-1 immunoreactivity which were all only detectable for <72 h postinfection. These data are consistent with other studies using models of bacterial infection in neonatal rats (Bilbo et al., 2005a,b, 2006; Williamson et al., 2011). Parallel to these results, E. coli was present in brain, liver, and spleen 24 h after infection, but was no longer detectable 7 d later. Although E. coli colonies could be cultured from brain homogenates 24 h after infection, it is unlikely that E. coli colonized the brain. The K12 E. coli strain used in this study lacks O and K antigens that are involved in evading the immune system (Liu and Reeves, 1994). In support of this notion, GFP+ E. coli were undetected in brain sections collected 24 h after infection (data not shown). In addition, mice in these experiments were not perfused, so the presence of bacteria in circulation may explain the detection of E. coli in brain homogenates. Together, this model of neonatal E. coli infection likely causes acute bacteremia that does not lead to a persistent peripheral or CNS infection.
An important finding from this study was that neonatal infection altered the expression of transcripts and proteins associated with the cellular sequestration of iron in the brain. For example, brain mRNA expression of the iron regulatory protein, hepcidin, was robustly elevated 24 h after infection. Moreover, at this same time point, mRNA expression of the iron export channel, ferroportin, was also reduced. These data are consistent with previous reports showing substantial brain hepcidin induction after peripheral LPS challenge (Wang et al., 2008) and degradation of brain ferroportin following intracerebroventricular injection of hepcidin (Li et al., 2011). Neuronal ferroportin expression was reduced in the motor cortex 24 h after infection. These data support the notion that increased hepcidin expression in the brain following neonatal infection results in the preferential inhibition of neuronal iron export. Congruent with this notion, expression of the iron storage protein, L-ferritin, was robustly increased in neuronal cell bodies at P12. Because ferritin is rapidly mono-ubiquinated and degraded following depletion of iron from its subunits (De Domenico et al., 2006), we interpret these data to indicate that iron is being sequestered and stored into neuronal L-ferritin deposits. In addition, L-ferritin bound iron is not involved in intracellular iron exchange (Fisher et al., 2007), thus increased L-ferritin deposits result in reduced iron availability for other CNS cell types including oligodendrocytes. This is important because iron is critical for oligodendrocyte maturation, lipid metabolism, and myelination of the CNS (Yu et al., 1986; Morath and Mayer-Pröschel, 2002; Ortiz et al., 2004).
Relevant to the above findings, we show a reduction in myelin and oligodendrocytes (Olig2+/NG2−) in the corpus callosum and motor cortex in juvenile ELI mice (P35). Because oligodendrocytes myelinate axons, the reduction in Olig2+/NG2− cells is consistent with the reduction in myelin in these regions. Furthermore, these white matter impairments are congruent with the notion of reduced CNS iron availability following neonatal infection. For example, deficits in oligodendrogenesis (Morath and Mayer-Pröschel, 2002) and white matter development (Yu et al., 1986) are also observed following perinatal iron deficiency. Moreover, neurons are implicated in the extracellular export of iron into white matter regions via axonal trafficking. For instance, axonal expression of ferroportin is dynamically increased (Moos and Rosengren Nielsen, 2006) during developmental periods that correspond to peak myelination (Foran and Peterson, 1992). Thus, neuronal iron sequestration before myelination likely reduced the availability of iron for oligodendrocytes, resulting in impaired oligodendrogenesis and white matter development following neonatal infection.
A relevant finding from this study was that neonatal E. coli infection led to reduced motor coordination and increased locomotor activity in male juvenile (P35) and adult mice (P60). ELI, however, did not affect cognitive function in the novel-object recognition task. Some studies have linked poor cognitive performance following neonatal challenge with LPS (Harré et al., 2008) or IL-1β (Favrais et al., 2011), whereas other studies have not (Bilbo et al., 2005a,b, 2006). These discrepancies are likely related to methodological differences including the type (Schwarz and Bilbo, 2011) or timing (Favrais et al., 2011) of immune challenge. In addition, impairments in myelin and iron homeostasis were detected in both males and females, but behavior was only determined in male mice. This represents a limitation of the study. Nonetheless, our findings are consistent with previous studies showing that hyperactivity and impaired motor coordination are associated with disruptions in white matter. For example, severity of motor impairment was correlated with white matter abnormalities resulting from preterm birth (Counsell et al., 2008; Spittle et al., 2011) and postnatal infection (Chau et al., 2012). Furthermore, motor coordination deficits following perinatal iron deficiency (Lozoff et al., 2000, 2006; Shafir et al., 2008, 2009) are also associated with white matter impairments (Yu et al., 1986; Morath and Mayer-Pröschel, 2002). Collectively, ELI caused stark hypomyelination that was associated with motor impairment and hyperactivity observed at P35 and P60.
Similar to peripheral LPS challenge (Wang et al., 2008), we show that transient E. coli infection caused substantial brain hepcidin induction and neuronal iron sequestration. Thus, it is likely that a similar process occurs in other reports of neonatal inflammatory events. For example, it is plausible that reduced CNS iron availability mediates white matter impairments observed in mice following repeated neonatal IL-1β injections (Favrais et al., 2011) or in rats following neonatal intracerebroventricular LPS (Fan et al., 2005). Moreover, consistent with these results, Fan et al. (2005) observed a reduction in oligodendrocytes that was also associated with increased locomotion. Similarly, Favrais et al. (2011) reported impairments in oligodendrocyte maturation. Importantly, these data may have relevance to clinical reports of impaired white matter and motor coordination following early life inflammation. For example, inflammation and infection in human neonates is directly associated with neurobehavioral deficits, including impaired motor coordination that correlates with severity white matter abnormalities (Stoll et al., 2004; Chau et al., 2012; O'Shea et al., 2013). Thus, brain hepcidin expression, neuronal sequestration of iron, and reduced CNS iron availability may contribute to deficits in myelination and behavior following perinatal inflammatory events in both humans and rodents.
A final issue is that early life infection may have altered maternal care behaviors. Despite this, no gross changes in maternal care were observed. This is consistent with a report showing that maternal care was unaffected using a similar neonatal infection paradigm (Bilbo et al., 2007). Alterations in maternal care have been associated with behavioral and physiological changes later in life. In studies assessing maternal care, however, neither motor coordination deficits nor increases in locomotion were detected (Shalev and Kafkafi, 2002; Farkas et al., 2009). In addition, altered maternal care has not been linked to hypomyelination or altered iron homeostasis. Thus, we surmise that deficits reported here are caused by the neonatal infection, but future studies should closely examine maternal care behaviors.
In summary, activation of the innate immune system during neonatal development reduced motor coordination and caused hyperactivity that persisted into young adulthood. Moreover, neonatal infection induced brain hepcidin, reduced neuronal ferroportin, and increased neuronal iron sequestration. We provide data that supports the hypothesis that neuronal sequestration of iron reduced the bioavailability of iron for oligodendrocyte maturation and subsequent myelination. Notably, exposure to neonatal infection promoted aberrant neuronal iron storage at a critical window in brain development. Furthermore, preferential neuronal sequestration of iron may be a novel mechanism by which early life inflammatory insults induce white matter impairments and subsequent behavioral sequelae later in life.
Footnotes
This work was supported by an Abbott Nutrition Contract (ANUS1011) and NIH Grant R01-AG-033028 to J.P.G. We thank Todd Lash for his technical assistance on this project.
J.C. Lieblein-Boff is employed by Abbott Nutrition. J.P. Godbout received funding from Abbott Nutrition in support of this work.
References
- Atladóttir HO, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010;40:1423–1430. doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- Bevins RA, Besheer J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study “recognition memory.”. Nat Protoc. 2006;1:1306–1311. doi: 10.1038/nprot.2006.205. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Front Behav Neurosci. 2009;3:14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Levkoff LH, Mahoney JH, Watkins LR, Rudy JW, Maier SF. Neonatal infection induces memory impairments following an immune challenge in adulthood. Behav Neurosci. 2005a;119:293–301. doi: 10.1037/0735-7044.119.1.293. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci. 2005b;25:8000–8009. doi: 10.1523/JNEUROSCI.1748-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Rudy JW, Watkins LR, Maier SF. A behavioural characterization of neonatal infection-facilitated memory impairment in adult rats. Behav Brain Res. 2006;169:39–47. doi: 10.1016/j.bbr.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Newsum NJ, Sprunger DB, Watkins LR, Rudy JW, Maier SF. Differential effects of neonatal handling on early life infection-induced alterations in cognition in adulthood. Brain Behav Immun. 2007;21:332–342. doi: 10.1016/j.bbi.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Billeci L, Calderoni S, Tosetti M, Catani M, Muratori F. White matter connectivity in children with autism spectrum disorders: a tract-based spatial statistics study. BMC Neurol. 2012;12:148. doi: 10.1186/1471-2377-12-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cate HS, Wu QZ, Kemper D, Merlo D, Wang HX, Fang K, Egan GF, Kilpatrick TJ. Influence of methylprednisolone on magnetic resonance and histological measures during cuprizone-induced demyelination. Neurosci Lett. 2010;483:47–52. doi: 10.1016/j.neulet.2010.07.060. [DOI] [PubMed] [Google Scholar]
- Chau V, Brant R, Poskitt KJ, Tam EW, Synnes A, Miller SP. Postnatal infection is associated with widespread abnormalities of brain development in premature newborns. Pediatr Res. 2012;71:274–279. doi: 10.1038/pr.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheepsunthorn P, Palmer C, Connor JR. Cellular distribution of ferritin subunits in postnatal rat brain. J Comp Neurol. 1998;400:73–86. doi: 10.1002/(SICI)1096-9861(19981012)400:1<73::AID-CNE5>3.0.CO%3B2-Q. [DOI] [PubMed] [Google Scholar]
- Cheepsunthorn P, Palmer C, Menzies S, Roberts RL, Connor JR. Hypoxic/ischemic insult alters ferritin expression and myelination in neonatal rat brains. J Comp Neurol. 2001;431:382–396. doi: 10.1002/1096-9861(20010319)431:4<382::AID-CNE1077>3.0.CO%3B2-%23. [DOI] [PubMed] [Google Scholar]
- Counsell SJ, Edwards AD, Chew AT, Anjari M, Dyet LE, Srinivasan L, Boardman JP, Allsop JM, Hajnal JV, Rutherford MA, Cowan FM. Specific relations between neurodevelopmental abilities and white matter microstructure in children born preterm. Brain. 2008;131:3201–3208. doi: 10.1093/brain/awn268. [DOI] [PubMed] [Google Scholar]
- De Domenico I, Vaughn MB, Li L, Bagley D, Musci G, Ward DM, Kaplan J. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 2006;25:5396–5404. doi: 10.1038/sj.emboj.7601409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Methods. 2009;181:36–44. doi: 10.1016/j.jneumeth.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. 2011;31:9910–9922. doi: 10.1523/JNEUROSCI.2114-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan LW, Pang Y, Lin S, Tien LT, Ma T, Rhodes PG, Cai Z. Minocycline reduces lipopolysaccharide-induced neurological dysfunction and brain injury in the neonatal rat. J Neurosci Res. 2005;82:71–82. doi: 10.1002/jnr.20623. [DOI] [PubMed] [Google Scholar]
- Farkas J, Reglodi D, Gaszner B, Szogyi D, Horvath G, Lubics A, Tamas A, Frank F, Besirevic D, Kiss P. Effects of maternal separation on the neurobehavioral development of newborn Wistar rats. Brain Res Bull. 2009;79:208–214. doi: 10.1016/j.brainresbull.2008.12.011. [DOI] [PubMed] [Google Scholar]
- Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, Lacaud A, Saliba E, Dammann O, Gallego J, Sizonenko S, Hagberg H, Lelièvre V, Gressens P. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol. 2011;70:550–565. doi: 10.1002/ana.22489. [DOI] [PubMed] [Google Scholar]
- Fisher J, Devraj K, Ingram J, Slagle-Webb B, Madhankumar AB, Liu X, Klinger M, Simpson IA, Connor JR. Ferritin: a novel mechanism for delivery of iron to the brain and other organs. Am J Physiol Cell Physiol. 2007;293:C641–C649. doi: 10.1152/ajpcell.00599.2006. [DOI] [PubMed] [Google Scholar]
- Foran DR, Peterson AC. Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac Z transgene. J Neurosci. 1992;12:4890–4897. doi: 10.1523/JNEUROSCI.12-12-04890.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005;19:1329–1331. doi: 10.1096/fj.05-3776fje. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol. 2012;71:444–457. doi: 10.1002/ana.22620. [DOI] [PubMed] [Google Scholar]
- Harré EM, Galic MA, Mouihate A, Noorbakhsh F, Pittman QJ. Neonatal inflammation produces selective behavioural deficits and alters N-methyl-d-aspartate receptor subunit mRNA in the adult rat brain. Eur J Neurosci. 2008;27:644–653. doi: 10.1111/j.1460-9568.2008.06031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B, Peake K, Morris AJ, McCowan LM, Battin MR. Escherichia coli: a growing problem in early onset neonatal sepsis. Aust N Z J Obstet Gynaecol. 2004;44:558–561. doi: 10.1111/j.1479-828X.2004.00304.x. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 2006;494:578–594. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Holscher C, Chen BB, Zhang ZF, Liu YZ. Hepcidin treatment modulates the expression of divalent metal transporter-1, ceruloplasmin, and ferroportin-1 in the rat cerebral cortex and hippocampus. Biol Trace Elem Res. 2011;143:1581–1593. doi: 10.1007/s12011-011-8967-3. [DOI] [PubMed] [Google Scholar]
- Libster R, Edwards KM, Levent F, Edwards MS, Rench MA, Castagnini LA, Cooper T, Sparks RC, Baker CJ, Shah PE. Long-term outcomes of group B streptococcal meningitis. Pediatrics. 2012;130:e8–e15. doi: 10.1542/peds.2011-3453. [DOI] [PubMed] [Google Scholar]
- Liu D, Reeves PR. Escherichia coli K12 regains its O antigen. Microbiology. 1994;140:49–57. doi: 10.1099/13500872-140-1-49. [DOI] [PubMed] [Google Scholar]
- Lozoff B, Jimenez E, Hagen J, Mollen E, Wolf AW. Poorer behavioral and developmental outcome more than 10 years after treatment for iron deficiency in infancy. Pediatrics. 2000;105:E51. doi: 10.1542/peds.105.4.e51. [DOI] [PubMed] [Google Scholar]
- Lozoff B, Beard J, Connor J, Barbara F, Georgieff M, Schallert T. Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutr Rev. 2006;64:S34–S43. doi: 10.1111/j.1753-4887.2006.tb00243.x. discussion S72–S91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Månsson LE, Melican K, Boekel J, Sandoval RM, Hautefort I, Tanner GA, Molitoris BA, Richter-Dahlfors A. Real-time studies of the progression of bacterial infections and immediate tissue responses in live animals. Cell Microbiol. 2007;9:413–424. doi: 10.1111/j.1462-5822.2006.00799.x. [DOI] [PubMed] [Google Scholar]
- Moos T, Rosengren Nielsen T. Ferroportin in the postnatal rat brain: implications for axonal transport and neuronal export of iron. Semin Pediatr Neurol. 2006;13:149–157. doi: 10.1016/j.spen.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Morath DJ, Mayer-Pröschel M. Iron deficiency during embryogenesis and consequences for oligodendrocyte generation in vivo. Dev Neurosci. 2002;24:197–207. doi: 10.1159/000065688. [DOI] [PubMed] [Google Scholar]
- Nagel BJ, Bathula D, Herting M, Schmitt C, Kroenke CD, Fair D, Nigg JT. Altered white matter microstructure in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2011;50:283–292. doi: 10.1016/j.jaac.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004a;113:1271–1276. doi: 10.1172/JCI200420945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004b;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Ortiz E, Pasquini JM, Thompson K, Felt B, Butkus G, Beard J, Connor JR. Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J Neurosci Res. 2004;77:681–689. doi: 10.1002/jnr.20207. [DOI] [PubMed] [Google Scholar]
- O'Shea TM, Shah B, Allred EN, Fichorova RN, Kuban KC, Dammann O, Leviton A. Inflammation-initiating illnesses, inflammation-related proteins, and cognitive impairment in extremely preterm infants. Brain Behav Immun. 2013. http://dx.doi.org/10.1016/j.bbi.2012.12.012. [DOI] [PMC free article] [PubMed]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. Ed 3. New York: Elsevier; 2008. [Google Scholar]
- Rodier PM. Chronology of neuron development: animal studies and their clinical implications. Dev Med Child Neurol. 1980;22:525–545. doi: 10.1111/j.1469-8749.1980.tb04363.x. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Bilbo SD. LPS elicits a much larger and broader inflammatory response than Escherichia coli infection within the hippocampus of neonatal rats. Neurosci Lett. 2011;497:110–115. doi: 10.1016/j.neulet.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafir T, Angulo-Barroso R, Jing Y, Angelilli ML, Jacobson SW, Lozoff B. Iron deficiency and infant motor development. Early Hum Dev. 2008;84:479–485. doi: 10.1016/j.earlhumdev.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafir T, Angulo-Barroso R, Su J, Jacobson SW, Lozoff B. Iron deficiency anemia in infancy and reach and grasp development. Infant Behav Dev. 2009;32:366–375. doi: 10.1016/j.infbeh.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalev U, Kafkafi N. Repeated maternal separation does not alter sucrose-reinforced and open-field behaviors. Pharmacol Biochem Behav. 2002;73:115–122. doi: 10.1016/S0091-3057(02)00756-6. [DOI] [PubMed] [Google Scholar]
- Sørensen HJ, Mortensen EL, Reinisch JM, Mednick SA. Association between prenatal exposure to bacterial infection and risk of schizophrenia. Schizophr Bull. 2009;35:631–637. doi: 10.1093/schbul/sbn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittle AJ, Cheong J, Doyle LW, Roberts G, Lee KJ, Lim J, Hunt RW, Inder TE, Anderson PJ. Neonatal white matter abnormality predicts childhood motor impairment in very preterm children. Dev Med Child Neurol. 2011;53:1000–1006. doi: 10.1111/j.1469-8749.2011.04095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll BJ, Hansen NI, Adams-Chapman I, Fanaroff AA, Hintz SR, Vohr B, Higgins RD. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. Jama. 2004;292:2357–2365. doi: 10.1001/jama.292.19.2357. [DOI] [PubMed] [Google Scholar]
- Wald ER, Bergman I, Taylor HG, Chiponis D, Porter C, Kubek K. Long-term outcome of group B streptococcal meningitis. Pediatrics. 1986;77:217–221. [PubMed] [Google Scholar]
- Wang Q, Du F, Qian ZM, Ge XH, Zhu L, Yung WH, Yang L, Ke Y. Lipopolysaccharide induces a significant increase in expression of iron regulatory hormone hepcidin in the cortex and substantia nigra in rat brain. Endocrinology. 2008;149:3920–3925. doi: 10.1210/en.2007-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SM, Fu LJ, Duan XL, Crooks DR, Yu P, Qian ZM, Di XJ, Li J, Rouault TA, Chang YZ. Role of hepcidin in murine brain iron metabolism. Cell Mol Life Sci. 2010;67:123–133. doi: 10.1007/s00018-009-0167-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. Microglia and memory: modulation by early-life infection. J Neurosci. 2011;31:15511–15521. doi: 10.1523/JNEUROSCI.3688-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology. 2012;37:1491–1505. doi: 10.1016/j.psyneuen.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynne AM, Henry CJ, Huang Y, Cleland A, Godbout JP. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav Immun. 2010;24:1190–1201. doi: 10.1016/j.bbi.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu GS, Steinkirchner TM, Rao GA, Larkin EC. Effect of prenatal iron deficiency on myelination in rat pups. Am J Pathol. 1986;125:620–624. [PMC free article] [PubMed] [Google Scholar]
- Zechel S, Huber-Wittmer K, von Bohlen und Halbach O. Distribution of the iron-regulating protein hepcidin in the murine central nervous system. J Neurosci Res. 2006;84:790–800. doi: 10.1002/jnr.20991. [DOI] [PubMed] [Google Scholar]
- Zhang X, Surguladze N, Slagle-Webb B, Cozzi A, Connor JR. Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia. 2006;54:795–804. doi: 10.1002/glia.20416. [DOI] [PubMed] [Google Scholar]