

Spermatogonial stem cells (SSCs) have the potential to acquire pluripotency under specific culture conditions. Takashima et al. report that global DNA hypomethylation triggered by Dnmt1 depletion induces pluripotent cell derivation. Dnmt1 depletion down-regulates Dmrt1, a gene involved in sexual differentiation. Dmrt1 depletion up-regulates Sox2, which in turn up-regulates Oct4 and produces pluripotent cells. These results suggest that the Dmrt1–Sox2 axis plays a crucial role in repression of SSC pluripotency.
Keywords: DNA methylation, pluripotency, reprogramming, spermatogonia, teratoma
Abstract
Spermatogonial stem cells (SSCs) present the potential to acquire pluripotency under specific culture conditions. However, the frequency of pluripotent cell derivation is low, and the mechanism of SSC reprogramming remains unknown. In this study, we report that induction of global DNA hypomethylation in germline stem (GS) cells (cultured SSCs) induces pluripotent cell derivation. When DNA demethylation was triggered by Dnmt1 depletion, GS cells underwent apoptosis. However, GS cells were converted into embryonic stem (ES)-like cells by double knockdown of Dnmt1 and p53. This treatment down-regulated Dmrt1, a gene involved in sexual differentiation, meiosis, and pluripotency. Dmrt1 depletion caused apoptosis of GS cells, but a combination of Dmrt1 and p53 depletion also induced pluripotency. Functional screening of putative Dmrt1 target genes revealed that Dmrt1 depletion up-regulates Sox2. Sox2 transfection up-regulated Oct4 and produced pluripotent cells. This conversion was enhanced by Oct1 depletion, suggesting that the balance of Oct proteins maintains SSC identity. These results suggest that spontaneous SSC reprogramming is caused by unstable DNA methylation and that a Dmrt1–Sox2 cascade is critical for regulating pluripotency in SSCs.
Germ cells are thought to have pluripotency potential because they form teratomas. Teratomas are spontaneously formed in mice on a 129 background, and this strain was used to derive embryonic stem (ES) cells (Evans and Kaufman 1981; Stevens 1984). Although the frequency of spontaneous teratoma development was found to be limited to ∼2%, studies have shown that primordial germ cells (PGCs) in the fetus can be induced to form teratomas by transplanting the genital ridges of mid-gestational embryos into ectopic locations in other animals (Stevens 1984). In the most successful cases, teratomas were found in ∼80% of the PGC transplants. PGCs have pluripotency potential up until 12.5 d post-coitum (dpc). Although these classic experiments provided the foundation for teratoma studies, the mechanism of teratoma development has remained unknown. However, in 1992, two groups (Matsui et al. 1992; Resnick et al. 1992) demonstrated that PGCs can become ES-like pluripotent embryonic germ (EG) cells in vitro when they are cultured with Kit ligand (KitL), fibroblast growth factor2 (FGF2), and leukemia inhibitory factor (LIF). EG cells are similar to ES cells, except in their DNA methylation patterns, and formed chimeras when the cells were transferred into blastocysts (Matsui et al. 1992; Labosky et al. 1994). The frequency of EG cell formation decreases gradually during development, and such potential pluripotency is no longer found in the germline later than 12.5 dpc (Labosky et al. 1994).
In 2003, a long-term culture system for spermatogonial stem cells (SSCs), in which SSCs proliferated in vitro in the presence of FGF2 and glial cell line-derived neurotrophic factor (GDNF), was developed (Kanatsu-Shinohara et al. 2003). Cultured SSCs, designated as germline stem (GS) cells, form grape-like clusters of spermatogonia in vitro but reinitiate spermatogenesis when transplanted into seminiferous tubules of infertile testes. Although GS cells are unipotent and produce sperm, they were subsequently found to transform into ES-like cells (Kanatsu-Shinohara et al. 2004). These ES-like cells, called multipotent GS (mGS) cells, often appear as sheets of epiblast-like cells, which transformed into ES-like compact colonies upon passaging. Despite their spermatogonial origin, they proliferate without GDNF and produce teratomas in seminiferous tubules but are able to contribute to the blastocyst, thereby producing germline chimeras. Additional studies revealed that GS cells directly transform into mGS cells (Kanatsu-Shinohara et al. 2008), and similar cells were also derived from other species, including humans, although some of these results are being questioned (Ko et al. 2010b; Tapia et al. 2011).
While these results showed the pluripotency potential of SSCs, several critical issues remain to be answered. One is their low derivation frequency: mGS cells develop in only one out of every ∼30 testes during GS cell derivation (Kanatsu-Shinohara et al. 2004). Although several groups also reported the derivation of pluripotent cells from postnatal male germ cells, discrepancies exist among these studies in terms of the nature and efficiency of reprogramming (Geijsen and Hochedlinger 2009). For example, one study showed the usefulness of LIF and obtained four lines from 21 mice (Guan et al. 2006). However, multipotent adult GS (maGS) cells produced in this study were unique in that they contribute to not only spermatogenesis but also embryogenesis, but the result of this study is being questioned (Geijsen and Hochedlinger 2009). Although beneficial effects of coculturing on testicular CD34+ stromal cells in pluripotency induction were reported in another study (Seandel et al. 2007), even better results were obtained by low-cell-density culture on mouse embryonic fibroblasts (MEFs), and six ES-like germline-derived pluripotent stem (gPS) cells were derived from eight testes within 4 wk in the most successful case (Ko et al. 2012). However, the quality of MEFs is still a critical factor, and an additional 3–4 wk are required when ES-like cells do not appear by low-cell-density cultures (Ko et al. 2010a). In addition to the variation in induction procedure, the difference in DNA methylation patterns in the differentially methylated region (DMR) of H19 is also pointed out. While gPS cells show androgenetic DNA methylation patterns, maGS cells exhibit somatic cell DNA methylation patterns (Guan et al. 2006; Ko et al. 2009). It also should be noted here that many of these studies claimed derivation of ES-like cells from 129 or C57BL/6 (B6) mice, whose SSCs never proliferate without augmenting GDNF signal by GFRα1 supplementation (Kubota and Brinster 2008).
Such low or inconsistent derivation efficiency has made it difficult to study the molecular mechanism underlying pluripotency induction. We initially noticed that mGS cells often develop during initiation of GS cell cultures and that p53 deficiency improves their derivation (Kanatsu-Shinohara et al. 2004). We also found that mGS cells occasionally appear after freezing–thawing or electroporation (Kanatsu-Shinohara et al. 2005, 2008). Unexpectedly, GS cells were resistant to transfection of Yamanaka factors and did not become pluripotent (Morimoto et al. 2012). However, the mechanism of pluripotency regulation in SSCs has remained unknown. Thus, there is clearly a need to develop a fast and efficient system to induce SSC reprogramming, which will enable us to dissect the molecular mechanism involved in this process.
Here, we report a critical role of Dmrt1 (a gene involved in sex determination) (Raymond et al. 2000) in GS cell reprogramming. We found previously that mGS cells often exhibit abnormal DNA methylation in DMRs of imprinted genes (Kanatsu-Shinohara et al. 2004). Because Dnmt1 is responsible for maintaining genomic methylation, we depleted Dnmt1 and found that Dnmt1 knockdown induces H19 demethylation and mGS cell formation. Furthermore, Dnmt1 knockdown in GS cells was accompanied by the down-regulation of Dmrt1, a gene involved in sex differentiation (Raymond et al. 2000). Although this gene is thought to be responsible for meiotic induction in spermatogonia (Matson et al. 2010), Dmrt1 knockdown in GS cells up-regulates Sox2 and efficiently induces mGS cells, suggesting that Dmrt1 plays a crucial role in repression of pluripotency in SSCs. We also propose a model in which spermatogonial identity is regulated by the balance of Oct proteins.
Results
Reprogramming of GS cells by induction of DNA demethylation
Global methylation of genomic DNA in GS cells is significantly higher than those in mGS and ES cells (Fig. 1A). Because DNA demethylation is often found in DMRs of H19-imprinted genes after reprogramming into mGS cells, we directly examined the role of Dnmt1, which maintains DNA methylation. To induce DNA demethylation, we used a Dnmt1 knockdown vector. When we induced demethylation by Dnmt1 knockdown, the majority of cells underwent p53-dependent apoptosis (Fig. 1B), but no mGS cells were found. However, because Dnmt1 is a maintenance methylase and passive demethylation due to Dnmt1 depletion requires multiple cell divisions (Jackson-Grusby et al. 2001), poor proliferation of GS cells in these cultures probably prevented efficient global DNA demethylation at 7 d. Therefore, GS cells from p53 knockout mice were transduced with a Dnmt1 knockdown vector and kept for ∼1 mo. Although extensive apoptosis occurred after knockdown, some cells divided slowly and formed GS cell colonies. Using this protocol, mGS cells were obtained within 4 wk after Dnmt1 knockdown (Fig. 1C; Table 1; Supplemental Table S1). The morphology and the proliferation pattern of the Dnmt1 knockdown-induced mGS (Dnmt1-mGS) cells were indistinguishable from those of cells that developed spontaneously. Real-time PCR confirmed down-regulation of Dnmt1 expression after knockdown (Supplemental Fig. S1A). Examination of global DNA methylation showed 3.7% ± 0.6% reduction in total methylcytosine levels by Dnmt1 knockdown 2 wk after transfection (n = 3; P < 0.05 by t-test). Combined bisulfite restriction analysis (COBRA) showed progressive demethylation of H19 (Fig. 1D).
Figure 1.

Development of mGS cells after Dnmt1 knockdown (KD). (A) Global DNA methylation levels in GS, mGS, and ES cells (n = 3). (B) Suppression of Dnmt1 knockdown-induced apoptosis by p53 knockdown. For each cell type, at least 99 cells were counted 7 d after transfection (n = 5). pSicoR was used as a control. (C) Development of mGS cells from p53 knockout (KO) GS cells after Dnmt1 knockdown. (D) COBRA of cultured cells. Open arrowheads indicate the sizes of the unmethylated DNA fragments, and closed arrowheads denote the sizes of the methylated DNA fragments. PCR products were digested with the indicated enzymes. Percent methylation is shown below the gel. pSicoR was used as a control. (E) Development of mGS cells from wild-type (WT) ROSA26 GS cells after double knockdown of Dnmt1 and p53. (F) RT–PCR analysis. Bars: C,E, 100 μm.
Table 1.
Summary of mGS induction efficiency
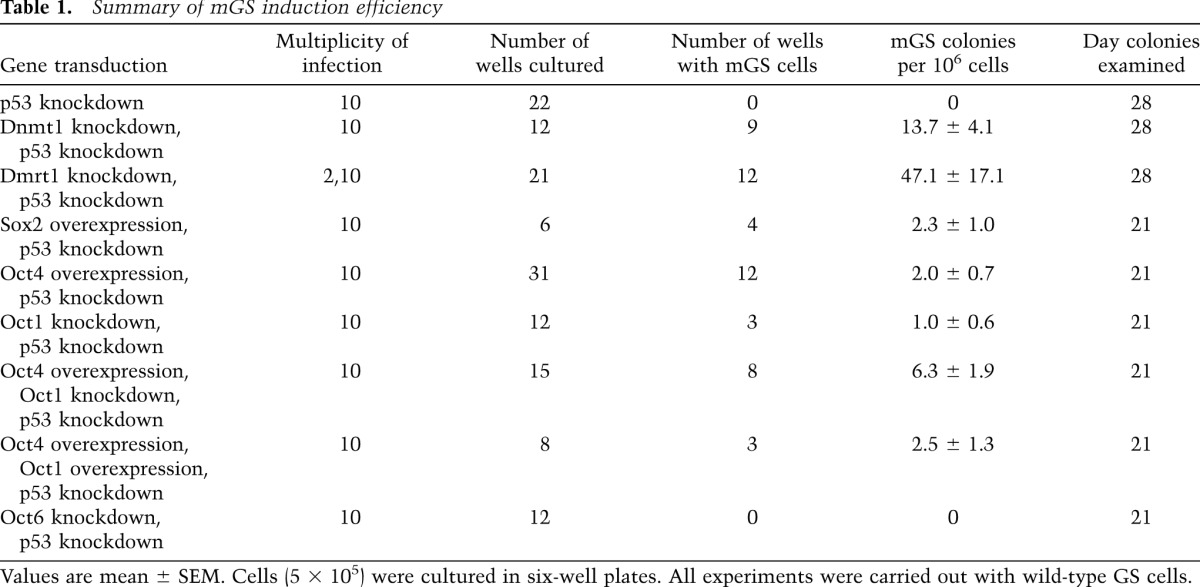
To examine whether the wild-type GS cells also transform into mGS cells, we established GS cells from transgenic pups carrying an enhanced green fluorescent protein (EGFP) gene (green mouse). Logarithmically growing GS cells were transduced with Dnmt1 and p53 knockdown vectors. Real-time PCR confirmed down-regulation of both p53 and Dnmt1 (Supplemental Fig. S1B,C). Wild-type GS cells produced mGS cells within 4 wk (Fig. 1E; Supplemental Table S1). RT–PCR confirmed Nanog expression in Dnmt1-mGS cells, which was accompanied by loss of Nanos3 expression, suggesting that GS cells lost their spermatogonial identity and became ES-like cells (Fig. 1F). We did not find mGS cells using 5-azacytidine treatment using both wild-type and p53 knockout GS cells.
Dmrt1 knockdown induces mGS cells
Because Dnmt1 knockdown causes tumors in somatic cells without p53 (Gaudet et al. 2003), we hypothesized that DNA demethylation might have changed the expression of genes responsible for germ cell tumor (GCT) development. We therefore examined the impact of 14 GCT candidate genes by deregulating their expression in a p53 knockout GS cell line. Dnmt1 knockdown down-regulated the expression of several genes, including Dnd1 and Dmrt1, both of which are implicated in the formation of teratomas from PGCs (Fig. 2A; Supplemental Fig. S2A,B; Gilbert et al. 2011). When we carried out knockdown experiments, knockdown of Dnd1 or Dmrt1 yielded mGS cell colonies within 4 wk (Supplemental Table S1). However, none of the other genes showed evidence of conversion.
Figure 2.

Development of mGS cells after Dmrt1 knockdown (KD). (A) Real-time PCR analysis of GCT candidate gene expression in p53 knockout (KO) GS cells after Dnmt1 knockdown (n = 3). pSicoR was used as a control. (B) Suppression of mGS cell development by Dmrt1 overexpression (OE) 28 d after Dnmt1 knockdown (n = 9). CSII-EF1α-IRES2-Venus was used as a control. (C) Suppression of Dmrt1-induced apoptosis by p53 or Bax knockdown. For each cell type, at least 68 cells were counted 7 d after transfection (n = 5).
To confirm the effects of Dnd1 and Dmrt1 knockdown on wild-type GS cells, we cotransfected each knockdown vector with a p53 knockdown vector. Although we obtained no mGS cells with Dnd1 knockdown, Dmrt1 knockdown successfully yielded mGS cells (Table 1). Compared with Dnmt1 knockdown, Dmrt1 knockdown induced mGS cells at a higher frequency with a shorter incubation time, and colony development was observed as early as 12 d. Concurrent overexpression of Dmrt1 cDNA with a Dnmt1 knockdown vector significantly reduced mGS cell development (Fig. 2B), which suggests that Dmrt1 down-regulation is responsible for Dnmt1 knockdown-induced pluripotency. As in Dnmt1-mGS cells, simultaneous p53 knockdown was necessary because cells underwent apoptosis due to Dmrt1 knockdown (Fig. 2C). Although Bax knockdown was able to rescue cells from Dmrt1 knockdown-induced apoptosis, we were unable to observe mGS cells (Fig. 2C), suggesting that p53 knockdown has an additional function to promote pluripotency.
Because of the relatively high efficiency of mGS cell formation, we used this system to characterize the reprogramming process. As previously noted for spontaneously developed mGS cells, Dmrt1-mGS cell development is often accompanied by sudden generation of an epiblast-like sheet, which has Nanog expression in the center (Supplemental Fig. S3A). All Dmrt1-mGS cell colonies expressed Nanog on the day when colonies were discovered. This protocol was useful in deriving mGS cells from all seven tested wild-type GS cell lines, and mGS cells were obtained from not only pups but also adults (Supplemental Fig. S3B). These results suggest that Dmrt1 regulates pluripotency in GS cells.
Characterization of Dnmt1- and Dmrt1-mGS cells
RT–PCR and flow cytometry indicated that both Dnmt1- and Dmrt1-mGS cells have typical features of ES cells (Supplemental Fig. S4A,B). They expressed high levels of Oct4 and Sox2 and were positive for Nanog in immunohistochemical analyses (Supplemental Fig. S4C). However, several lines of Dnmt1- or Dmrt1-mGS cells expressed Neurog3 or Sohlh1, which suggested that the cells retain a spermatogonial phenotype (Supplemental Fig. S4A). We carried out bisulfite sequencing analysis to check the DNA methylation patterns in imprinted genes. Consistent with previous studies (Kanatsu-Shinohara et al. 2004), GS cells showed typical androgenetic DNA methylation patterns: hypermethylation in H19 and hypomethylation in Igf2r (Supplemental Fig. S4D). In contrast, Dnmt1- and Dmrt1-mGS cells showed demethylation in H19. Although we occasionally observed Igf2r methylation in mGS cells, no apparent difference in Igf2r was found, regardless of the induction method.
Functional characterization of Dnmt1- and Dmrt1-mGS cells
To determine whether Dnmt1- and Dmrt1-mGS cells are functionally similar to ES cells, we examined their differentiation potential. In these experiments, Dnmt1-mGS cells were produced by shRNA transfection, and the shRNA was removed by cre treatment (Supplemental Fig. S4E,F). We first induced neuroectodermal differentiation in adherent monocultures (Ying and Smith 2003). The cultured cells successfully differentiated into cells expressing βIII-tubulin, a neuronal marker. We then generated embryoid bodies (EBs) in vitro and examined whether the cells could differentiate into other lineages (Fig. 3A). After 5 d of culture on gelatin-coated dishes, the EBs were stained with several antibodies. Some cells stained positively for α-smooth muscle actin, a mesodermal marker, while others expressed cells with α-fetoprotein expression, suggesting that they belong to the endodermal lineage. We found no obvious difference between Dnmt1- and Dmrt1-mGS cells in terms of differentiation patterns and efficiencies. We also transplanted both cell types under the subcutaneous tissues of nude mice to examine their teratoma-forming potential. All clones produced tumors within 4 wk after transplantation. Histological analyses showed that the transplanted cells produced teratomas with the ectoderm (neural tube), mesoderm (chondrocytes), and endoderm (gut epithelium) (Supplemental Fig. S4G).
Figure 3.

Differentiation potential of Dnmt1- and Dmrt1-mGS cells. (A) Neuronal or EB-mediated differentiation of mGS cells. (B,C) A live chimera formed by microinjection of Dnmt1-mGS cells (B) or Dmrt1-mGS cells (C) into ICR or B6 blastocyst, respectively. The cinnamon coat color represents the contribution of the Dnmt1- or Dmrt1-mGS cells on a DBA/2 background. (D) Contribution of Dmrt1-mGS cells to various organs, as indicated by EGFP fluorescence. Bar: A, 100 μm.
To test whether these mGS cells contribute to embryonic development, we microinjected them into blastocysts (Supplemental Table S2). In this experiment, GS cells from ROSA26 mice were used to derive Dnmt1-mGS cells, which were treated with cre to remove shRNA for Dnmt1 and p53 before the chimera experiment. On the other hand, we used green GS cells and transfected siRNAs for Dmrt1 and p53 to avoid vector integration for Dmrt1-mGS cell production. Both Dnmt1- and Dmrt1-mGS cells could produce chimeric offspring, albeit at low efficiency (Fig. 3B–D; Supplemental Table S2). Low efficiency of chimera production could be due to residual expression of Neurog3 in these cells (Supplemental Fig. S4H). Nevertheless, both offspring were apparently normal, and Dmrt1-mGS-derived F1 offspring were born by natural mating. No abnormalities in H19 were found by COBRA in these offspring (Supplemental Fig. S4I).
Identification of Dmrt1 target genes
In the next set of experiments, we sought to identify target genes regulated by Dmrt1. A previous study showed that Dmrt1 regulates >1400 genes in spermatogonia (Murphy et al. 2010). Among the diverse types of genes, we focused on those associated with cell cycle regulation (p18 and p19) and pluripotency (Nr5a1, Utf1, Sox2, and Zic3) because we reasoned that deregulation of these genes can potentially stimulate GCT formation. We analyzed the expression levels of these candidate genes in p53 knockout GS cells by real-time PCR after Dmrt1 knockdown (Fig. 4A). The analyses revealed that while Sox2 and Utf1 were up-regulated by Dmrt1 knockdown, Nr5a1 and Zic3 were down-regulated, which suggests that Nr5a1 and Zic3 are dispensable for reprogramming. Both p18 and p19 cyclin-dependent kinase inhibitors (CDKIs) were down-regulated, which may facilitate teratoma formation. Although similar results were obtained using wild-type GS cells with p53 knockdown, we failed to find statistically significant down-regulation of p19 (Supplemental Fig. S5).
Figure 4.

Development of mGS cells after Sox2 or Oct4 overexpression (OE). (A) Real-time PCR analysis of Dmrt1 target genes in p53 knockout (KO) GS cells after Dmrt1 knockdown (KD) (n = 3). pLKO.1 EGFP was used as a control. (B) Suppression of mGS cell development by Sox2 knockdown 28 d after Dmrt1 knockdown (n = 13). pLKO.1 EGFP was used as a control. (C) Real-time PCR analysis of Dnmt1, Dmrt1, Sox2, and Oct1/4 expression in p53 knockout GS cells 14 d after Dmrt1 knockdown (n = 4). pSicoR was used as a control. For concurrent Dmrt1 overexpression, CSII-EF1α-IRES2-Venus was a control. (D) Real-time PCR analysis of Oct4 expression in p53 knockout GS cells 14 d after Sox2 overexpression (n = 3). CSII-EF1α-IRES2-hKO1 was used as a control. (E) Reduced Sox2-mGS cell development 28 d after Oct4 knockdown (n = 9).
Based on this observation, we screened for genes that trigger pluripotency in GS cells. We overexpressed pluripotency-related genes or depleted the expression of CDKIs in wild-type GS cells. All transfection experiments were carried out by cotransfection with a p53 knockdown vector. Although Utf1 transfection did not result in mGS cell development, transfection of Sox2 yielded mGS cells using three different wild-type GS cell lines (Table 1; Supplemental Table S1; Supplemental Fig. S2C). Many cells underwent p53-dependent apoptosis after Sox2 overexpression (Supplemental Fig. S6A), but the remaining cells transformed into mGS cells, a pattern similar to that for Dmrt1 knockdown. Sox2 knockdown inhibited Dmrt1-mGS cell development (Fig. 4B; Supplemental Fig. S2A). Neither p18 nor p19 knockdown resulted in mGS cell production. Because concurrent Dmrt1 overexpression suppressed Sox2 expression induced by Dnmt1 depletion, the Dmrt1–Sox2 axis also appears to operate in Dnmt1-mGS cell development (Fig. 4C; Supplemental Fig. S2C).
Sox2 is necessary to regulate transcription factors that affect Oct4 expression in ES cells (Masui et al. 2007). Although GS cells express Oct4, its level is significantly lower than that found in ES cells (Imamura et al. 2006). However, we noted that Oct4 mRNA and protein expression were up-regulated in Sox2 transfected GS cells (Fig. 4D; Supplemental Fig. S7). Moreover, Oct4 knockdown prevented mGS cell development after Sox2 overexpression (Fig. 4E). Although apparent changes in Oct4 expression were not observed after Dnmt1 knockdown (Fig. 4C), we reasoned that this could be due to the relatively variable timing and small number of pluripotent colonies by Dnmt1 knockdown. Because Sox2 maintains the requisite level of Oct4 in ES cells and Oct4 overexpression can sustain Sox2-null ES cells (Masui et al. 2007), we tested the possibility that Oct4 overexpression may abolish the need for Sox2 transfection. Transfection of Oct4 induced mGS cell formation using three different wild-type GS cell lines (Table 1; Supplemental Table S1). Unlike Sox2 overexpression, Oct4 overexpression did not induce GS cell apoptosis (Supplemental Fig. S6B) but did require p53 knockdown for mGS cell formation. These results suggest that Sox2 is responsible for Dmrt1-mGS cell generation and that increased Sox2 expression up-regulates Oct4, thereby inducing the formation of pluripotent cells.
Increased mGS cell development following Oct1 knockdown
Although these results show the importance of Oct4 regulation, the role of this protein in SSCs has been controversial. One study reported that Oct4 down-regulation induces GS cell apoptosis (Dann et al. 2008), while another study showed that Oct6, but not Oct4, is indispensable for SSCs (Wu et al. 2010). However, we noted that GS cells express Oct1 (Fig. 5A). Oct1 mRNA was down-regulated upon FGF2 or GDNF treatment, and Western blot analysis also confirmed this result (Fig. 5B,C). Although we did not observe apparent changes in Oct4 and Oct6 expression at mRNA levels, Western blotting showed that both Oct4 and Oct6 protein levels were slightly up-regulated by cytokine treatment. These results suggested that regulation of Oct1 and Oct4/6 expression is different. Because Oct1 often has the same targets as Oct4 (Kang et al. 2009), we reasoned that the relative balance of Oct proteins is important for regulating pluripotency in SSCs.
Figure 5.

Development of mGS cells after Oct1 knockdown (KD). (A) RT–PCR analysis of Oct gene expression in GS cells during logarithmic growth phase. (B) Real-time PCR analysis of Oct expression after cytokine stimulation. GS cells were starved for 3 d and restimulated with the indicated cytokines for 24 h before sample collection (n = 6). (C) Western blot analysis of Oct expression after cytokine stimulation. GS cells were starved for 3 d and restimulated with the indicated cytokines for 24 h before sample collection. (D–G) Real-time PCR analysis of Oct1/4 and Sox2 expression 14 d after Oct4 overexpression (OE) (D), Oct1 knockdown (E), Oct4 knockdown (F), or Oct1 overexpression (G) (n = 3). CSII-EF1α-IRES2-Venus was used as a control for Oct1 and Oct4 overexpression. pLKO.1 was a control for Oct1 and Oct4 knockdown. (H) Real-time PCR analysis of Oct1 and Sox2 expression 14 d after Sox2 overexpression (n = 3). CSII-EF1α-IRES2-hKO1 was used as a control.
To test this hypothesis, we examined the impact of Oct knockdown in GS cells. Knockdown of Oct1 or Oct4 induced GS cell apoptosis in a p53-dependent manner (Supplemental Fig. S6C). Oct6 knockdown also caused apoptosis, which was not rescued by p53 deficiency. We transfected wild-type GS cells with Oct1 or Oct6 knockdown vectors together with a p53 knockdown vector. Knockdown of Oct1, but not Oct6, yielded mGS cells using two different wild-type GS cell lines (Table 1). The efficiencies of Oct4 overexpression and Oct1 knockdown were comparable, but combining Oct4 overexpression and Oct1 knockdown synergistically improved mGS cell derivation efficiency (P < 0.05 by ANOVA), and mGS cells were obtained (Table 1). While Oct4 overexpression increased Sox2 expression (Fig. 5D), Oct1 knockdown increased not only Sox2 expression but also Oct4 expression (Fig. 5E). On the other hand, Oct4 knockdown decreased Sox2 expression, suggesting that Oct4 is positively regulating Sox2 (Fig. 5F). Although Oct1 overexpression decreased Oct4 and Sox2 expression (Fig. 5G), it was not possible to suppress Oct4-mGS cell development, suggesting that Oct1 is necessary but not sufficient for suppressing pluripotency. Because neither Oct4 nor Sox2 overexpression influenced Oct1 expression (Fig. 5D,H), it is likely that Oct1 is regulated independently of Oct4 and Sox2. When we examined whether changes in Oct1/4 expression in GS cells can influence genes that are regulated by Oct4 in ES cells (van den Berg et al. 2010), several genes changed expression by Oct1 overexpression or knockdown (Supplemental Fig. S8). In particular, Utf1, one of the Dmrt1 target genes, was down-regulated by Oct1 overexpression, while it was up-regulated by Oct1 knockdown. There results suggest that Oct1 suppresses pluripotency in GS cells by competing with Oct4 for several pluripotency-related target genes.
Characterization of Sox2- and Oct4-mGS cells
Both Sox2- and Oct4-mGS cells were phenotypically similar to Dnmt1- and Dmrt1-mGS cells and were able to differentiate into cells of the three germ layers in vitro (Supplemental Fig. S9A–E). However, their global gene expression profiles were slightly different from those of ES cells (Fig. 6A; Supplemental Fig. S10). To confirm their pluripotency, we introduced Sox2 or Oct4 cDNA into a floxed vector that was transfected with a p53 knockdown vector. After transformation into mGS cells, the transgenes were removed by cre-mediated deletion (Supplemental Fig. S9F,G). The ability of the resulting cells to form chimeras by blastocyst injection was then tested (Supplemental Table S2). Chimeric offspring were obtained from both Sox2- and Oct4-mGS cells (Fig. 6B,C). Both types of mGS cells showed germline transmission by natural mating. These results suggest that Sox2- and Oct-4 mGS cells are pluripotent.
Figure 6.

Development of normal offspring from Sox2- or Oct4-mGS cells. (A) Hierarchical clustering of gene expression by microarray. (B,C) A live chimera formed by microinjection of Sox2-mGS cells (B) and Oct4-mGS cells (C) into B6 and ICR blastocysts, respectively. (Inset) Offspring produced after germline transmission. While Sox2-mGS cell origin was identified by the EGFP expression (B), Oct4-mGS cell origin was confirmed by the cinnamon coat color (C). (D) A model to explain the mechanism of pluripotency regulation in SSCs.
Discussion
The molecular mechanism of culture-induced pluripotency is not well understood. While DNA methylation patterns in ES cells often change within several months, GS cells maintain a stable karyotype and DNA methylation patterns in imprinted genes for >2 yr (Kanatsu-Shinohara et al. 2005). Because demethylation of H19 DMRs is almost always found in spontaneously developed mGS cells, we reasoned that DNA demethylation might have contributed to the reprogramming of GS cells. To test our hypothesis directly, we used Dnmt1 knockdown to induce hypomethylation in GS cells. Unlike ES cells, which can proliferate without Dnmt genes (Tsumura et al. 2006), GS cell survival depends critically on Dnmt1 expression (Takashima et al. 2009). Although Dnmt1 knockdown resulted in apoptosis of GS cells, simultaneous p53 knockdown successfully induced mGS cell formation, suggesting that DNA demethylation and suppression of p53 function are necessary for dedifferentiation of GS cells into pluripotent cells.
In our previous study, we did not observe mGS cells by Dnmt1 knockdown, probably because of the higher virus titer and shorter incubation time (Takashima et al. 2009), and H19 DMR demethylation was not confirmed. The higher virus titer induced extensive apoptosis of GS cells, and the shorter incubation time (∼3 wk) was probably insufficient to induce passive demethylation by cell replication because the current study shows that most of the mGS cell colonies developed after 3 wk. Because deficiency of Dnmt1 causes somatic cell tumors (Gaudet et al. 2003), acquisition of pluripotency in GS cells is suggested to share similarities with tumorigenesis. Consistent with this notion, H19 is often demethylated in human GCTs (Looijenga et al. 1998). In particular, seminomas and nonseminomas generally show biallelic expression of the H19 gene. Moreover, we also recently showed H19 demethylation in ES-like colonies produced by in vitro transformation of mouse SSCs with activated Ras, c-myc, and p53 dominant-negative constructs (Morimoto et al. 2012). Although the roles of imprinted genes in GCTs are unknown, similarities between GCT formation and GS cell reprogramming led us to examine whether GCT candidate genes are deregulated as a result of global DNA hypomethylation.
Dmrt1 was one of the several candidate genes that were influenced by Dnmt1 knockdown, and Dmrt1 knockdown successfully induced mGS cell formation. Dmrt1 is one of a group of conserved transcriptional regulators of sexual differentiation that share a Doublesex/Mab-3 (DM) domain DNA-binding motif and is required for testicular development in vertebrates (Raymond et al. 2000). In mice, this gene is expressed only in the gonad and is essential for differentiation of germ cells and Sertoli cells. Strikingly, testes without Dmrt1 show ovarian differentiation even at the adult stage (Matson et al. 2011). Humans lacking one copy of Dmrt1 exhibit testicular dysgenesis and in some cases are feminized (Krentz et al. 2009). In germ cells, this gene is responsible for the formation of teratomas from PGCs, but it is limited to the 129 background (Krentz et al. 2009). In the postnatal testis, Dmrt1 has been considered as a transcriptional gatekeeper that controls mitosis versus meiosis in germ cells (Matson et al. 2010). Undifferentiated spermatogonia without Dmrt1 showed precocious entry into meiosis and reached meiotic prophase by skipping amplifying divisions of the differentiating spermatogonia population, but no tumor formation was reported in postnatal animals without Dmrt1. In contrast, Dmrt1 overexpression is thought to cause spermatocytic seminomas in human adults by increasing Ret expression (Krentz et al. 2009).
Although regulation of pluripotency by Dmrt1 in spermatogonia has not been reported, we speculate that the discrepancy between our results and previous studies is due to two factors. One is the suppression of apoptosis by p53 knockdown. Dmrt1 is involved in germ cell survival, and while Dmrt1 deficiency in PGCs caused teratomas on a 129 background, germ cell loss was observed in non-129 strains (Krentz et al. 2009). Apoptosis of germ cells is probably caused by reduced GDNF signaling because analyses of mutant testes showed that both GFRα1 and Ret are down-regulated, which suggests that Dmrt1 influences the responsiveness to GDNF. In the present study, we also observed that simple Dmrt1 knockdown induced apoptosis of GS cells, but simultaneous p53 knockdown rescued them from apoptosis, resulting in mGS cell formation. The other factor is the differentiation status of the target cells. In the previous study, analysis of Dmrt1 function in spermatogonia was carried out using cre driven by a Neurog3 promoter (Matson et al. 2010). However, Neurog3 is thought to be expressed in a subpopulation of SSCs in a reversible manner (Yoshida 2010). Therefore, some SSCs may not have undergone Neurog3-cre-mediated Dmrt1 deletion, and the function of Dmrt1 in the remaining SSC population is not known. The differentiation level of the target cell population is important for germ cell transformation because cells enriched for SSCs showed a higher frequency of development into transformed cells than committed spermatogonia after transfection of oncogenes or Yamanaka factors (Morimoto et al. 2012).
Knockdown of two GCT candidate genes, Dnd1 and Dmrt1, induced pluripotency in p53 knockout GS cells. Dnd1 is an RNA-binding protein that regulates PGC viability and suppresses teratoma formation (Zhu et al. 2007). It is similar to Dmrt1 in that it only causes teratomas on a 129 background. However, the mechanism of pluripotency regulation seems to differ between these two genes. Although we were able to obtain mGS cells by Dnd1 or Dmr1 knockdown using p53 knockout GS cells, only Dmrt1 was useful for deriving mGS cells from wild-type cells. In fact, studies on PGCs also suggest that they have independent functions (Krentz et al. 2009). For example, Dnd1 mutants undergo a severe loss of germ cells before 11.5 dpc, whereas those in Dmrt1 mutants survive until birth. Although Pten deficiency induces teratomas in PGCs on non-129 backgrounds, Dmrt1 deficiency did not change levels of Pten or Akt phosphorylation (Kimura et al. 2003; Krentz et al. 2009). These results suggested that Dmrt1 acts either independently of Pten or downstream from the Pten pathway in PGCs. Although p53 deficiency also causes teratomas on non-129 backgrounds, it is distinct from Pten in that Pten-deficient gonocytes and spermatogonia do not show pluripotency (Goertz et al. 2011). Therefore, p53 and Dmrt1 seem to be guardians of pluripotency at both embryonic and postnatal stages.
Of the Dmrt1 target genes, our functional screening suggested that Sox2 acts downstream from Dmrt1 to induce reprogramming. Sox2 mRNA is expressed from the early stages of PGC development, but Sox2 protein decreases from 13.5 dpc to 17.5 dpc in fetal gonocytes and is absent in spermatogonia (Imamura et al. 2006; Campolo et al. 2013). Dmrt1 protein expression is absent by 15.5 dpc but is re-expressed in spermatogonia (Lei et al. 2007). This suggests that Dmrt1 protein expression is discrepant with Sox2 protein expression, which apparently involves additional molecules. On the other hand, although the Oct4 protein is expressed in GS cells, its level is only 10% of that found in ES cells; Oct4/Sox2 sites in GS cells are not occupied by Oct4 and Sox2 despite their hypomethylated status, possibly due to the smaller amount of Oct4 and the absence of Sox2 (Imamura et al. 2006). Nevertheless, our results showed that Sox2 and Oct4 expression levels are closely correlated in GS cells. Although the role of Oct4 in SSCs is still under debate (Wu et al. 2010), high expression of Sox2 appears to overcome Sox2 repression at the translational level and tip the balance toward pluripotency by up-regulating Oct4. Although changes in Oct4 levels were modest, this may have a large impact on pluripotency regulation, considering that the threshold for inducing differentiation in ES cells is set at 50% above or below the normal Oct4 expression (Niwa et al. 2000). Because Oct1 knockdown also induced up-regulation of Oct4, Sox2, and Utf1, relative reduction in Oct1 may perhaps induce pluripotency genes by increasing the access of Oct4 to target genes to regulate several pluripotency-related genes (Fig. 6C). However, given our production of mGS cells by Dnd1 knockdown in p53 knockout GS cells and the fact that Oct1 overexpression could not suppress Oct4-mGS cell development, suppression of pluripotency probably involves additional molecules. Identifying other genes that modulate the response of GS cells to Sox2 overexpression is warranted.
Because mGS cells can be derived without genetic manipulation, SSCs represent a unique resource for deriving pluripotent cells. However, conflicting reports on the induction method and nature of SSC-derived PS cells need to be reconciled. In particular, it still remains unknown why mGS cell development is often accompanied by epiblast-like colony formation, while similar colonies were not reported in other studies (Seandel et al. 2007; Geijsen and Hochedlinger 2009). Such discrepancies could be due to the differences in the cell of origin and/or mode of reprogramming. This may also explain why mGS cell formation occurred at relatively lower frequency compared with gPS cells (0.0047% vs. 0.01%), which gradually form ES-like colonies over several weeks. We also do not know how p53 is involved in pluripotency regulation. Because mGS cell development often occurred after freezing–thawing or electroporation, we think that such changes in culture conditions induce cellular stress, which may influence p53 levels. However, because Bax knockdown could not induce mGS cells, the role of p53 knockdown is not simply rescuing apoptosis but has an additional role in pluripotency regulation. Because we can reproducibly obtain mGS cells with our new protocol, analysis of reprogramming mechanisms is now possible, which will improve our understanding of how pluripotency is suppressed in germ cells despite their similarity to ES cells.
Materials and methods
Cell culture
GS cells were derived from the transgenic mouse lines C57BL6/Tg14(act-EGFP-OsbY01) and B6-TgR(ROSA26)26Sor, as previously described (Kanatsu-Shinohara et al. 2003, 2004, 2011). GS cells were also established from the transgenic mouse line Tg(Nanog-GFP, Puro)1Yam (Okita et al. 2007). GS cells from p53 knockout mice were previously described (Kanatsu-Shinohara et al. 2004). Culture medium was based on StemPro-34 SFM (Invitrogen) as previously described (Kanatsu-Shinohara et al. 2003). We also used mGS cells on a DBA/2 background that spontaneously developed during green GS cell derivation (Kanatsu-Shinohara et al. 2004). Growth factors used were 10 ng/mL human FGF2 and 15 ng/mL recombinant rat GDNF (both from Peprotech). ES cells (R1) were a generous gift from Dr. M. Ikawa (Osaka University, Suita, Japan). ES cells (BRC1 and B6-6) were provided by RIKEN BRC.
All pluripotent cell lines were maintained on MEFs in Dulbecco's modified Eagle's medium (DMEM) supplemented with 15% fetal bovine serum (FBS), 1000 U/mL LIF (ESGRO; Merck Millipore), nonessential amino acid mixture (Invitrogen), and 0.1 mM 2-mercaptoethanol (2-ME). We also used 2 μM PD0325901 (Selleck Chemicals), 3 μM CHIR99021 (Biovision, Inc.), and 1000 U/mL LIF in N2B27 medium to maintain these cells for chimera production (Ying and Smith 2003).
For differentiation into neuronal lineages, cells were cultured on gelatin-coated plates for 8 d in N2B27 medium. Neural cell differentiation was induced by replating on a LAB-TEK chamber slide (Thermo Fisher Scientific) coated with 0.2 mg/mL fibronectin (Invitrogen) and maintenance in N2B27 medium supplemented with 20 ng/mL FGF2 for 10 d (Ying and Smith 2003). For EB formation, cells were suspended in DMEM supplemented with 20% FBS, 10 μM 2-ME, and nonessential amino acid mixture and plated on a low-cell-binding plate at a density of 7.5 × 104 cells per 9.6 cm2. Two days after culture, the serum concentration was reduced to 15% FBS, and the cells were cultured for 8 d. EBs were then transferred to a LAB-TEK chamber slide coated with 0.1% gelatin and cultured for 5 d.
Statistical analyses
The results are presented as the mean ± SEM. Significant differences between means for single comparisons were identified using Student's t-test. Multiple comparison analyses were performed using ANOVA followed by Tukey's HSD test.
Accession number
Raw data sets have been submitted to the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo) and are available under the accession number GSE43850.
Acknowledgments
We thank Ms. Y. Ogata for technical assistance. This research was supported by Japan Science and Technology Agency (CREST), and the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.220194.113.
References
- Campolo F, Gori M, Favaro R, Nicolis S, Pellegrini M, Botti F, Rossi P, Jannini EA, Dolci S 2013. Essential role of Sox2 for the establishment and maintenance of the germ cell line. Stem Cells 31: 1408–1421 [DOI] [PubMed] [Google Scholar]
- Dann CT, Alvarado AL, Molyneux LA, Denard BS, Garbers DL, Porteus MH 2008. Spermatogonial stem cell self-renewal requires OCT4, a factor downregulated during retinoic acid-induced differentiation. Stem Cells 26: 2928–2937 [DOI] [PubMed] [Google Scholar]
- Evans MJ, Kaufman MH 1981. Establishment in culture of pluripotential cells from mouse embryos. Nature 292: 154–156 [DOI] [PubMed] [Google Scholar]
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R 2003. Induction of tumors in mice by genomic hypomethylation. Science 300: 489–492 [DOI] [PubMed] [Google Scholar]
- Geijsen N, Hochedlinger K 2009. gPS navigates germ cells to pluripotency. Cell Stem Cell 5: 3–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert D, Rapley E, Shipley J 2011. Testicular germ cell tumours: Predisposition genes and the male germ cell niche. Nat Rev Cancer 11: 278–288 [DOI] [PubMed] [Google Scholar]
- Goertz MJ, Wu Z, Gallardo TD, Hamra FK, Castrillon DH 2011. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J Clin Invest 121: 3456–3466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan K, Nayernia K, Maier LS, Wagner S, Dressel R, Lee JH, Nolte J, Wolf F, Li M, Engel W, et al. 2006. Pluripotency of spermatogonial stem cells from adult mouse testis. Nature 440: 1199–1203 [DOI] [PubMed] [Google Scholar]
- Imamura M, Miura K, Iwabuchi K, Ichisaka T, Nakagawa M, Lee J, Kanatsu-Shinohara M, Shinohara T, Yamanaka S 2006. Transcriptional repression and DNA hypermethylation of a small set of ES cell marker genes in male germline stem cells. BMC Dev Biol 6: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, Dausman J, Lee P, Wilson C, Lander E, et al. 2001. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet 27: 31–39 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, Toyokuni S, Shinohara T 2003. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod 69: 612–616 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Inoue K, Lee J, Yoshimoto M, Ogonuki N, Miki H, Baba S, Kato T, Kazuki Y, Toyokuni S, et al. 2004. Generation of pluripotent stem cells from neonatal mouse testis. Cell 119: 1001–1012 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Ogonuki N, Iwano T, Lee J, Kazuki Y, Inoue K, Miki H, Takehashi M, Toyokuni S, Shinkai Y, et al. 2005. Genetic and epigenetic properties of mouse male germline stem cells during long-term culture. Development 132: 4155–4163 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Lee J, Inoue K, Ogonuki N, Miki H, Toyokuni S, Ikawa M, Nakamura T, Ogura A, Shinohara T 2008. Pluripotency of a single spermatogonial stem cell in mice. Biol Reprod 78: 681–687 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Inoue K, Ogonuki N, Morimoto H, Ogura A, Shinohara T 2011. Serum- and feeder-free culture of mouse germline stem cells. Biol Reprod 84: 97–105 [DOI] [PubMed] [Google Scholar]
- Kang J, Shakya A, Tantin D 2009. Stem cells, stress, metabolism and cancer: A drama in two Octs. Trends Biochem Sci 34: 491–499 [DOI] [PubMed] [Google Scholar]
- Kimura T, Suzuki A, Fujita Y, Yomogida K, Lomeli H, Asada N, Ikeuchi M, Nagy A, Mak TW, Nakano T 2003. Conditional loss of PTEN leads to testicular teratoma and enhances embryonic germ cell production. Development 130: 1691–1700 [DOI] [PubMed] [Google Scholar]
- Ko K, Tapia N, Wu G, Kim JB, Bravo MJA, Sasse P, Glaser T, Ruau D, Han DW, Greber B, et al. 2009. Induction of pluripotency in adult unipotent germline stem cells. Cell Stem Cell 5: 87–96 [DOI] [PubMed] [Google Scholar]
- Ko K, Araúzo-Bravo MJ, Kim J, Stehling M, Schöler HR 2010a. Conversion of adult mouse unipotent germline stem cells into pluripotent stem cells. Nat Protoc 5: 921–928 [DOI] [PubMed] [Google Scholar]
- Ko K, Araúzo-Bravo MJ, Tapia N, Kim J, Lin Q, Bernemann C, Han DW, Gentile L, Reinhardt P, Greber B, et al. 2010b. Human adult germline stem cells in question. Nature 465: E1. [DOI] [PubMed] [Google Scholar]
- Ko K, Wu G, Araúzo-Bravo MJ, Kim J, Francine J, Greber B, Mühlisch J, Joo JY, Sabour D, Fruhwald MC, et al. 2012. Autologous pluripotent stem cells generated from adult mouse testicular biopsy. Stem Cell Rev 8: 435–444 [DOI] [PubMed] [Google Scholar]
- Krentz AD, Murphy MW, Kim S, Cook MS, Capel B, Zhu R, Matin A, Sarver AL, Parker KL, Griswold MD, et al. 2009. The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. Proc Natl Acad Sci 106: 22323–22328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota H, Brinster RL 2008. Culture of rodent spermatogonial stem cells, male germline stem cells of the postnatal animal. Methods Cell Biol 86: 59–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labosky PA, Barlow DP, Hogan BLM 1994. Mouse embryonic germ (EG) cell lines: Transmission through the germline and differences in the methylation imprint of insulin-like growth factor 2 receptor (Igf2r) gene compared with embryonic stem (ES) cell lines. Development 120: 3197–3204 [DOI] [PubMed] [Google Scholar]
- Lei N, Hornbaker KI, Rice DA, Karpova T, Agbor VA, Heckert LL 2007. Sex-specific differences in mouse DMRT1 expression are both cell type- and stage-dependent during gonad development. Biol Reprod 77: 466–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looijenga LHJ, Verkerk AJMH, Dekker MC, van Gurp RJHLM, Gillis AJM, Oosterhuis JW 1998. Genomic imprinting in testicular germ cell tumours. APMIS 106: 187–197 [DOI] [PubMed] [Google Scholar]
- Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, Okochi H, Okuda A, Matoba R, Sharov AA, et al. 2007. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol 9: 625–635 [DOI] [PubMed] [Google Scholar]
- Matson CK, Murphy MW, Griswold MD, Yoshida S, Bardwell VJ, Zarkower D 2010. The mammalian Doublesex homologue DMRT1 is a transcriptional gatekeeper that controls the mitosis versus meiosis decision in male germ cells. Dev Cell 19: 612–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson CK, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D 2011. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 476: 101–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Zsebo K, Hogan BLM 1992. Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell 70: 841–847 [DOI] [PubMed] [Google Scholar]
- Morimoto H, Lee J, Tanaka T, Ishii K, Toyokuni S, Kanatsu-Shinohara M, Shinohara T 2012. In vitro transformation of mouse testis cells by oncogene transfection. Biol Reprod 86: 1–11 [DOI] [PubMed] [Google Scholar]
- Murphy MW, Sarver AL, Rice D, Hatzi K, Ye K, Melnick A, Heckert LL, Zarkower D, Bardwell VJ 2010. Genome-wide analysis of DNA binding and transcriptional regulation by the mammalian Doublesex homolog DMRT1 in the juvenile testis. Proc Natl Acad Sci 107: 13360–13365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Miyazaki J-I, Smith AG 2000. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet 24: 372–376 [DOI] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S 2007. Generation of germline-competent induced pluripotent stem cells. Nature 448: 313–317 [DOI] [PubMed] [Google Scholar]
- Raymond CS, Murphy MW, O'Sullivan MG, Bardwell VJ, Zarkower D 2000. Dmrt1, a gene related to worm and fly sexual regulators, is required for mammalian testis differentiation. Genes Dev 14: 2587–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick JL, Bixler LS, Cheng L, Donovan PJ 1992. Long-term proliferation of mouse primordial germ cells in culture. Nature 359: 550–551 [DOI] [PubMed] [Google Scholar]
- Seandel M, James D, Shmelkov SV, Falciatori I, Kim J, Chavala S, Scherr DS, Zhang F, Torres R, Gale NW, et al. 2007. Generation of functional mutipotent adult stem cells from GPR125+ germline progenitors. Nature 449: 346–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens LC 1984. Spontaneous and experimentally induced testicular teratomas in mice. Cell Differ 15: 69–74 [DOI] [PubMed] [Google Scholar]
- Takashima S, Takehashi M, Lee J, Chuma S, Okano M, Hata K, Suetake I, Nakatsuji N, Miyoshi H, Tajima S, et al. 2009. Abnormal DNA methyltransferase expression in mouse germline stem cells results in spermatogenic defects. Biol Reprod 81: 155–164 [DOI] [PubMed] [Google Scholar]
- Tapia N, Araúzo-Bravo MJ, Ko K, Schöler HR 2011. Concise review: Challenging the pluripotency of human testis-derived ESC-like cells. Stem Cells 29: 1165–1169 [DOI] [PubMed] [Google Scholar]
- Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S, Sakaue M, Matsuoka C, Shimotohno K, Ishikawa F, Li E, Ueda HR, et al. 2006. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 11: 805–814 [DOI] [PubMed] [Google Scholar]
- van den Berg DL, Snoek T, Mullin NP, Yates A, Bezstarosti K, Demmers J, Chambers I, Poot RA 2010. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 6: 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Oatley JM, Oatley MJ, Kaucher AV, Avarbock MR, Brinster RL 2010. The POU domain transcription factor POU3F1 is an important intrinsic regulator of GDNF-induced survival and self-renewal of mouse spermatogonial stem cells. Biol Reprod 82: 1103–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Smith AG 2003. Defined conditions for neural commitment and differentiation. Methods Enzymol 365: 327–341 [DOI] [PubMed] [Google Scholar]
- Yoshida S 2010. Stem cells in mammalian spermatogenesis. Dev Growth Differ 52: 311–317 [DOI] [PubMed] [Google Scholar]
- Zhu R, Bhattacharya C, Matin A 2007. The role of dead-end in germ-cell tumor development. Ann N Y Acad Sci 1120: 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]