

Briso et al. find that c-fos expression in the mouse epidermis is sufficient to promote inflammation-mediated epidermal hyperplasia. c-Fos transcriptionally controls mmp10 and s100a7a15 expression in keratinocytes, promoting CD4 T-cell recruitment to the skin. Combining c-fos expression with the carcinogen DMBA leads to the development of highly invasive SCCs, which was prevented by the anti-inflammatory drug sulindac. Human SCCs display a correlation between c-FOS and MMP10 and S100A15 proteins as well as CD4 T-cell infiltration. This work reveals promising therapeutic strategies to treat SCCs.
Keywords: c-Fos, AP-1, inflammation, cancer, skin, CD4 T cell
Abstract
Skin squamous cell carcinomas (SCCs) are the second most prevalent skin cancers. Chronic skin inflammation has been associated with the development of SCCs, but the contribution of skin inflammation to SCC development remains largely unknown. In this study, we demonstrate that inducible expression of c-fos in the epidermis of adult mice is sufficient to promote inflammation-mediated epidermal hyperplasia, leading to the development of preneoplastic lesions. Interestingly, c-Fos transcriptionally controls mmp10 and s100a7a15 expression in keratinocytes, subsequently leading to CD4 T-cell recruitment to the skin, thereby promoting epidermal hyperplasia that is likely induced by CD4 T-cell-derived IL-22. Combining inducible c-fos expression in the epidermis with a single dose of the carcinogen 7,12-dimethylbenz(a)anthracene (DMBA) leads to the development of highly invasive SCCs, which are prevented by using the anti-inflammatory drug sulindac. Moreover, human SCCs display a correlation between c-FOS expression and elevated levels of MMP10 and S100A15 proteins as well as CD4 T-cell infiltration. Our studies demonstrate a bidirectional cross-talk between premalignant keratinocytes and infiltrating CD4 T cells in SCC development. Therefore, targeting inflammation along with the newly identified targets, such as MMP10 and S100A15, represents promising therapeutic strategies to treat SCCs.
Skin squamous cell carcinomas (SCCs) are the second most common type of human nonmelanoma skin cancers (NMSK), with an incidence of 16 out of 100,000 people in Europe (Brantsch et al. 2008). Unlike basal cell carcinomas (BCCs), SCCs are characterized by an increased risk of metastasis (Ratushny et al. 2012). SCCs arise from keratinocytes of the epidermis and oral mucosa and are most commonly found in sun-exposed areas. Besides ultraviolet (UV) light, other risk factors have been associated with skin SCCs, such as arsenic exposure, tobacco, and human papilloma virus infection. SCCs typically manifest as a spectrum of progressively advanced malignancies, ranging from a precursor lesion like actinic keratosis to SCCs to, eventually, invasive SCCs.
Inflammatory processes often facilitate cancer development by fostering infiltration of immune cells and promoting stromal remodeling (Hanahan and Coussens 2012). In humans, inflammatory skin diseases such as lupus vulgaris or chronic skin ulcers predispose patients to develop cutaneous SCCs (Baldursson et al. 1993; Motswaledi and Doman 2007). In addition, deregulated expression of oncogenes or tumor suppressors in cancer cells can promote immune cell recruitment to the tumor microenvironment by activating the expression of cytokines or metalloproteases that control proinflammatory pathways (Kerkela and Saarialho-Kere 2003; Zenz et al. 2005; Soucek et al. 2007). Interestingly, CD4 T-cell depletion delayed the appearance of tumors in a skin SCC-prone genetic mouse model (Daniel et al. 2003).
A number of signaling pathways have been described as important in the development of SCCs, such as RAS, p53, Notch, and AP-1 (Guinea-Viniegra et al. 2012; Ratushny et al. 2012). Expression of a dominant-negative AP-1 transgene in the skin protects mice from UV-induced, chemically induced, and papillomavirus-induced tumor formation, indicating that AP-1 activity is essential for tumor development (Cooper et al. 2003). In skin physiology, c-Fos is required for the development of RAS-induced malignant papilloma or squamous cell lesions in the background of an epidermal-specific or complete c-fos knockout mouse, while it is dispensable for mouse skin development and homeostasis (Saez et al. 1995; Guinea-Viniegra et al. 2012). Moreover, keratinocyte-specific c-Fos deficiency promotes skin tumor suppression in a p53/TACE-dependent cell-autonomous manner (Guinea-Viniegra et al. 2012). Importantly, increased levels of c-Fos have been found in human SCCs (Sachdev et al. 2008).
In inflammatory diseases such as arthritis, c-Fos was shown to promote disease development (Aikawa et al. 2008; Shiozawa and Tsumiyama 2009). In contrast, c-Fos can act as a negative regulator of proinflammatory responses in myeloid and lymphoid cell lineages (Ray et al. 2006). It was reported that the AP-1 transcription factor regulates a number of genes affecting the microenvironment in the epidermis, including matrix proteins and other secreted factors (Eferl and Wagner 2003; Wagner and Eferl 2005). Whether c-Fos plays a role in epithelial inflammation and in an epithelial/immune cross-talk remains unknown.
Using inducible and regulatable mouse models, we show that c-Fos expression in the epidermis is sufficient to promote inflammation-dependent development of preneoplastic lesions. This phenotype seems to be induced by recruitment of proinflammatory CD4 T cells to the skin through the induction of novel transcriptional c-Fos target genes mmp10 and s100a7a15 in epithelial cells. We further show that this cross-talk between c-Fos expression in the epidermis and CD4 T-cell recruitment may also be conserved in human SCCs.
Results
Inducible epidermal c-fos expression in adult mice leads to epidermal hyperplasia
Increased c-Fos levels have been observed in SCCs arising from different stratified squamous epithelia (Sachdev et al. 2008; Guinea-Viniegra et al. 2012). To investigate whether c-Fos expression is sufficient to promote SCC development in the skin, we generated a doxycycline (Dox)-inducible mouse model. To achieve inducible, keratinocyte-specific expression of c-fos in vivo, keratin 5 (K5)-rtTA mice were crossed to colTetO-Fos mice (Supplemental Fig. 1A). Double-transgenic mice are referred to as c-FosEp-tetON mice, and wild-type or single-transgenic littermates are referred to as controls. Following 4 wk of Dox treatment, adult c-FosEp-tetON mice showed reduced body size, hair loss, and limited life span compared with controls (Fig. 1A,B; Supplemental Fig. 1B). The mutant mice did not show enlarged spleen size indicative of a systemic inflammatory disease (Supplemental Fig. 1C). The affected skin was thickened, with cornified epidermis appearing as dry, scaly lesions (Fig. 1B). Expression of inducible c-fos was detected in the epidermis 2 wk after Dox induction by RT-qPCR and immunohistochemistry (IHC) (Fig. 1C,D). At 2 wk, c-Fos was mainly localized to the hair follicles, while, at 4 wk, it was clearly visible in the basal layer of the epidermis and outer root sheath of the hair follicles (Fig. 1D, arrows). Other K5-expressing epithelial tissues, such as esophagus, tongue, and forestomach, expressed inducible c-fos, showing a mild phenotype only in the forestomach (Supplemental Fig. 1D), while K5-negative tissues, such as spleen or liver, did not express exogenous c-fos (data not shown). Histological analyses of c-FosEp-tetON back skin revealed a complete disruption of skin architecture, leading to the development of preneoplastic lesions characterized by massive epidermal hyperplasia, hyperkeratosis, parakeratosis, and cell/nuclear atypia (Fig. 1E). Strikingly, most hair follicles were absent, and foci of central keratinization within concentric layers of abnormal squamous cells were often observed (Fig. 1E). Proliferation and survival pathways were increased, as assessed by Ki67 and p-STAT3 stainings, in the basal layer of the epidermis upon inducible c-fos expression as early as after 2 wk of Dox treatment (Fig. 1F,G). Interestingly, the presence of a pronounced leukocyte infiltrate, as compared with resident leukocyte infiltrates in wild-type skin, was observed at 4 wk upon c-fos expression, as assessed by CD45 immunostainings (Fig. 1H). Furthermore, epidermal differentiation marker analyses revealed a dramatic expansion of the different epidermal layers in mutant mice. Multiple cell layers were positive for K5 and K14 (basal compartment), and the K1 (spinous layer) and the loricrin (granular layer) were also expanded (Supplemental Fig. 1E). Furthermore, calcium-induced keratinocyte differentiation, as measured by impaired Keratin10, Notch1, and Hes1 expression, was slightly impaired upon inducible c-fos expression (Supplemental Fig. 1F–K). These results show that inducible expression of c-fos in the epidermis is sufficient to trigger skin hyperplasia.
Figure 1.

Inducible expression of c-fos in the epidermis of adult mice promotes epidermal hyperplasia with increased proliferation. (A) Scheme of the experimental design: Six-week-old (6w) mice were fed with Dox (0.5 g/L) in the drinking water, and the analyses were performed after 2 and 4 wk (2w and 4w). (B) Picture of a representative control (n = 11) (left) and a c-FosEp-tetON (n = 17) (right) mouse is shown at 4 wk of inducible c-fos expression with Dox in the drinking water. (C) Total c-Fos and exogenous c-Fos-Flag mRNA expression analyses in the back skin of control and c-FosEp-tetON mice at 4 wk of inducible c-fos expression (n = 3). Mean ± SD. (D, left) c-Fos immunostainings of back skin of control (littermate) and c-FosEp-tetON mice at 2 and 4 wk (n = 3) of inducible c-fos expression. (Right panel) Epidermal c-Fos positive nuclei IHC quantification. (E, left) H&E staining of the back skin of c-FosEp-tetON and control mice at 2 and 4 wk of inducible c-fos expression. (Right panel) Epidermal thickness quantification. (F, left) Ki67 immunostainings of the back skin of control and c-FosEp-tetON mice at 2 wk (n = 3) and 4 wk (n = 3) of inducible c-fos expression. (Right panel) Epidermal Ki67-positive nucleus IHC quantification. Mean ± SD. (G, left) p-STAT3 immunostainings of the back skin of control and c-FosEp-tetON mice at 2 wk (n = 3) and 4 wk (n = 3) of inducible c-fos expression. (Right panel) Epidermal p-STAT3-positive nucleus IHC quantification. Mean ± SD. (H) CD45 immunostainings of the back skin of control and c-FosEp-tetON mice at 2 wk (n = 3) and 4 wk (n = 3) of inducible c-fos expression.
Inducible c-Fos expression promotes skin inflammation through chronic CD4 T-cell recruitment
To analyze whether the observed skin phenotype is a direct consequence of c-Fos-induced keratinocyte proliferation and whether c-Fos can promote proliferation cell-autonomously, primary FostetON keratinocytes were employed. Inducible expression of c-Fos was detected in vitro upon Dox treatment (Supplemental Fig. 2A). However, no increased proliferation—as assessed by keratinocyte numbers (Supplemental Fig. 2B), EdU incorporation (Supplemental Fig. 2C), colony formation assays (Supplemental Fig. 2D), and enhanced CyclinD1/2 or CyclinE2 expression—was observed (Supplemental Fig. 2E–G). Furthermore, no changes in the Fos-dependent p53/TACE differentiation pathway were observed upon c-Fos expression (Supplemental Fig. 2I,J). These data demonstrate that c-Fos does not promote proliferation in a cell-autonomous manner.
In addition to the epidermal hyperplasia, histological analysis of c-FosEp-tetON mouse skin revealed an accumulation of inflammatory cells, as shown in Figure 1H. To confirm the infiltration of immune cells in the skin of c-FosEp-tetON mice and determine the nature of these cells, we performed flow cytometry (FACS) analyses in skin homogenates from mice 2 wk after inducible expression of c-fos, when there is no microscopic skin phenotype. Although there were no significant changes in the relative presence of total leukocyte infiltration, as determined by CD45 cells, there was a pronounced accumulation of CD4 T cells as well as Gr1+ granulocytes in the c-FosEp-tetON mouse skin (Fig. 2A). Four weeks after inducible c-fos expression, when the epidermis shows a pronounced hyperplasia, there were increased leukocyte infiltrates in the skin of c-FosEp-tetON mice (Fig. 2B). Moreover, there was a clear accumulation of CD4 T cells in c-FosEp-tetON mice (Fig. 2B), while the number of Gr1+ cells was restored to normal (Fig. 2B). No changes were observed in the infiltration of CD8 T cells or B cells at either 2 wk or 4 wk, but macrophages were significantly reduced at 4 wk (Supplemental Fig. 3A,B). Thus, inducible c-fos expression in epidermis caused selective recruitment of CD4 T cells to the skin. Most CD4 T cells present in the skin of c-FosEp-tetON mutant mice were CD44high (data not shown) according to the expected activated status of the cells. Analyses of the CD4 T cells in the skin draining lymph nodes also revealed increased CD4+CD44high cells in c-FosEp-tetON mice compared with control mice (Fig. 2C), suggesting that accumulation of CD4 T cells in the skin of mutant mice could potentially be the result of the recruitment of activated CD4 T cells from the draining lymph nodes.
Figure 2.

c-Fos expression induces skin inflammation characterized by chronic CD4 T-cell recruitment. (A) FACS (flow cytometry) analyses of the skin immune cell infiltrate (CD45+, CD45+CD4+, and CD45+Gr1+ populations) of control (n = 6) and c-FosEp-tetON (n = 9) mice after 2 wk of inducible c-fos expression. (B) FACS analyses of the skin immune cell infiltrate of control (n = 6) and c-FosEp-tetON (n = 9) mice at 4 wk of inducible c-fos expression. (C) FACS analyses of the immune cell infiltrate in the skin draining lymph nodes of CD4+/CD44+ cells from control and c-FosEp-tetON (n = 3) mice at 2 wk of inducible c-fos expression. (D) FACS analyses of the immune cell infiltrate (CD45+CD4+ population) in the skin of control and c-FosEp-tetON mice at 4 wk of inducible c-fos expression followed by 4 wk and 16 wk without Dox treatment. (E) Chemotaxis assay. CD4 T lymphocytes isolated from murine wild-type lymph nodes were activated for 5 h with anti-CD28 and anti-CD3 antibodies. Activated CD4 T lymphocytes were subjected to chemotaxis in a transwell assay using either medium alone, SDF-1 (10 ng/mL), or conditioned medium (CM) from control or c-FostetON keratinocytes cultured ±Dox and ±the recombinant protein S100a7a15. (F, right) Immunohistochemical analyses depicting H&E stainings of the back skin of control (Col-Fos+/+; K5rtTA+/+; Rag−/−) and c-FosEp-tetON [Col-Fos (+/KI); K5rtTA (+/T); Rag+/−] and seven c-FosEp-tetON Rag−/− mice [Col-Fos (+/KI); K5rtTA (+/T); Rag−/−] mice on Dox for 4 wk (n = 7, 4, or 10 males). Right: Epidermal thickness quantification of control, RAG1+/−, and RAG−/− c-FosEp-tetON mice. (G, right) Immunohistochemical analyses depicting Ki67 stainings of the back skin of control, c-FosEp-tetON; Rag+/−, and c-FosEp-tetON Rag−/− mice on Dox for 4 wk (n = 7, 4, or 10 males). (Left) Epidermal Ki67-positive nucleus quantification of control, c-FosEp-tetON; Rag+/−, and c-FosEp-tetON Rag−/− mice. (H) RT-qPCR expression analyses of mRNA of sorted CD4 T cells from the back skin of control and c-FosEp-tetON mice after 4 wk of Dox treatment (n = 3).
To demonstrate that the presence of these cells in the skin was dependent on c-fos expression in keratinocytes, Dox was removed after 4 wk of induction, and the mutant mice were analyzed after an additional 4 or 16 wk, when c-fos expression was shut off. The accumulation of CD4 T cells in the skin was restored to almost normal levels compared with control mice (Fig. 2D), and c-Fos levels were normalized (data not shown). The selective presence of CD4 T cells in the skin is therefore dependent on continuous epidermal c-fos expression.
To assess whether specific factors secreted by c-Fos-expressing keratinocytes can promote migration of CD4 T cells, chemotaxis assays were performed in vitro using wild-type murine primary lymph node-derived CD4 T cells. Supernatants from Dox-treated c-FostetON primary keratinocytes induced a twofold chemotactic migration of CD4 T cells, similar to SDF-1α, which was used as a positive control (Fig. 2E). These results indicate that specific proteins secreted upon c-Fos expression into the supernatants of Dox-treated c-FostetON keratinocytes are able to act as chemotactic molecules for CD4 T cells.
To address whether the selective accumulation of CD4 T cells in the skin of c-FosEp-tetON mice plays a functional role in the development of skin hyperplasia, c-FosEp-tetON mice were crossed to RAG1−/−-deficient mice (Mombaerts et al. 1992). Unlike c-FosEp-tetON mice, double-transgenic c-FosEp-tetON RAG1−/− mice showed significantly smaller skin lesions (Fig. 2F). Moreover, keratinocyte hyperproliferation and activation of the STAT3 pathway detected in FosEp-tetON mice was restored to normal levels in FosEp-tetON RAG1−/− mice, as assessed by Ki67 and p-STAT3 staining (Fig. 2G; Supplemental Fig. 3D), whereas c-Fos levels remained increased (Supplemental Fig. 3E). Thus, CD4 T cells accumulate in the skin upon c-fos expression in keratinocytes and contribute to c-Fos-mediated development of epidermal hyperplasia.
To understand how CD4 T cells could contribute to the development of the skin phenotype, CD4 T cells were FACS-sorted from the back skin of control and c-FosEp-tetON mice after 4 wk of Dox treatment. Interestingly, mRNA expression analyses of T-helper cell subpopulations revealed significant enhanced expression of IL-22 in CD4 T cells infiltrating the skin of c-FosEp-tetON mice when compared with controls (Fig. 2H). IL-22 has been shown to be involved in barrier defense and can promote keratinocyte survival by activating the STAT3 pathway (Boniface et al. 2005; Zhang et al. 2012).
c-Fos is necessary and sufficient to up-regulate the expression of mmp10 and s100a7a15 in keratinocytes
To understand the underlying molecular mechanisms and identify novel transcriptional targets of c-Fos, including putative secreted factors, we next employed genome-wide expression analyses using primary keratinocytes isolated from the c-FostetON mice. No major changes in the expression of genes involved in proliferation or differentiation were observed between c-FostetON keratinocytes upon inducible expression of c-fos (Supplemental Table 1). However, among a number of c-Fos-induced genes, two consistently up-regulated genes encoding secreted factors with a role in triggering inflammatory responses were identified: mmp10 and s100a7a15 (Fig. 3A). MMP10 is an MMP that facilitates the recruitment of leukocytes from the bloodstream by degrading components of the extracellular matrix and modulating cytokine and chemokine activity (Elkington et al. 2005). In addition, S100a7a15 is involved in the recruitment of CD4 T cells and granulocytes (Wolf et al. 2010).
Figure 3.

MMP10 and S100a7a15 are novel transcriptional targets of c-Fos. (A) Schematic representation of the two most up-regulated genes in a microarray expression analysis comparing in vitro cultured c-FostetON [coltetO-Fos (+/KI), Rosa-rtTA (+/KI)] keratinocytes treated ±Dox (1 mg/mL; n = 3) for 6, 12, 24, 48, and 72 h. (B) RT-qPCR expression analyses of c-fos, mmp10, and s100a7a15 mRNA in c-FostetON keratinocytes treated ±Dox (1 μg/mL; n = 3). (C) Immunoblot depicting S100a7a15 and MMP10 protein levels as well as vinculin and GAPDH as loading controls in c-FostetON keratinocytes treated ±Dox for 48 h. (D) RT-qPCR expression analyses of c-fos, mmp10, and s100a7a15 mRNA in control and c-FosEp-tetON back skin at 4 wk of inducible c-fos expression (n = 3). Mean ± SD. (E) MMP10 immunohistochemical analyses of the back skin of control and c-FosEp-tetON mice after 2 and 4 wk of inducible c-fos expression (n = 3). (F) S100a7a15 immunofluorescence analyses in the back skin of control and c-FosEp-tetON mice after 4 wk of inducible c-fos expression (n = 3). (G) ChIP of c-Fos at the mmp10 promoter. (Left, top) Scheme of the TRE element at the mmp10 promoter where c-Fos binds. (Right) Endpoint qPCR fragments are shown together with the representation of the percentage of binding of c-Fos to mmp10 promoter. (Left, bottom) Chromatin was immunoprecipitated using c-Fos antibody from c-FostetON keratinocytes treated ±Dox for 24 h. (H) ChIP of c-Fos at the s100a7a15 promoter. (Left, top) Scheme of the TRE element at the mmp10 promoter where c-Fos binds. (Right) Endpoint qPCR-fragments are shown together with the representation of the percentage of binding of c-Fos to s100a7a15 promoters. (Left, bottom) Chromatin was immunoprecipitated using c-Fos antibody from c-FostetON keratinocytes treated ±Dox for 24 h.
The expression of these novel target genes was validated at the RNA and protein levels in vitro and in vivo. A significant increase in mRNA (Fig. 3B) and protein (Fig. 3C) expression levels of mmp10 and of s100a7a15 was observed upon c-Fos induction in cultured keratinocytes. Moreover, a significant increase of MMP10 and S100a7a15 was detected by ELISA in the serum of c-FosEp-tetON mice (Supplemental Fig. 4B; data not shown) and in vitro in the cell supernatants of c-FostetON keratinocytes (Supplemental Fig. 4C; data not shown). Importantly, no other MMPs or TACE were differentially expressed upon c-fos expression, as assessed in the microarray analyses or by RT-qPCR (Supplemental Figs. 4A, 2I). Both mmp10 and s100a7a15 mRNA levels were increased in the skin of c-FosEp-tetON mice compared with controls, as assessed by RT-qPCR (Fig. 3D), IHC, and immunofluorescence (Fig. 3E,F). Furthermore, the chemotactic effect observed using supernatants from c-FosEp-tetON keratinocytes was abrogated when conditioned medium was preincubated with an s100a7a15-blocking antibody (Supplemental Fig. 3C). In addition, CD4 T-cell chemotaxis was also observed upon stimulation with recombinant s100a7a15 protein (Supplemental Fig. 3C). These results suggest that S100a7a15 protein promotes CD4 T-cell recruitment in c-FosEp-tetON mice.
To further assess whether these genes are putative c-Fos target genes, c-fos-deficient keratinocytes treated with TPA were analyzed for the expression of mmp10 and s100a7a15. c-fos levels were gradually increased 2 h after TPA treatment (data not shown), and, after 6 h, 12 h, and 24 h, a gradual increase of mmp10 and s100a7a15 expression was observed. However, this induction was significantly abrogated in the absence of c-Fos (Supplemental Fig. 4D,E).
The promoter regions of mmp10 and s100a7a15 contain TPA response elements (TREs) to which AP-1 transcription factors can bind (Fig. 3G,H, top panel). To evaluate direct binding of c-Fos to both mmp10 and s100a7a15 promoters, chromatin immunoprecipitation (ChIP) assays were performed. Twenty-four hours after c-fos expression in primary c-FosEp-tetON keratinocytes, mmp10 and s100a7a15 showed binding of c-Fos to their promoters (Fig. 3G,H). Recruitment of c-Fos to mmp10 and s100a7a15 promoters was confirmed by ChIP analyses in c-fos-deficient keratinocytes stimulated with TPA for 3 h. Upon in vitro c-fos deletion, no binding of c-Fos to mmp10 and s100a7a15 promoters was detected (Supplemental Fig. 4F,G). These results show that mmp10 and s100a7a15 are direct transcriptional targets of c-Fos in keratinocytes and are likely responsible for the recruitment of CD4 T cells and consequent preneoplastic lesion development. In addition, increased MMP10 expression was also present in other K5-expressing tissues, such as the esophagus, forestomach, or tongue, and leukocyte recruitment was observed specifically in the forestomach (Supplemental Fig. 5A,B).
In addition, continuous TPA application in mice lacking c-fos in the epidermis showed that c-fos is necessary for MMP10 expression and, consequently, the recruitment of CD45+ and CD3+ inflammatory cells (Supplemental Fig. 6A–E). In addition, impaired expression of mmp10 and s100a7a15 was observed in keratinocytes lacking c-fos and stimulated for 4 h and 16 h with TPA (Supplemental Fig. 6F–H).
MMP10 blockade impairs c-Fos-mediated epidermal hyperplasia
To address the question of whether MMP10 is an important contributor to the development of c-Fos-induced preneoplastic lesions, we blocked MMP10 activity by using a broad MMP inhibitor (TAPI-1) (Slack et al. 2001). The inhibitor was used in a preventive treatment and administered intraperitoneally (i.p.) three times per week to mice subjected to Dox treatment. As assessed by H&E, c-FosEp-tetON mice treated with the MMP inhibitor showed significant prevention of preneoplastic lesion development (Fig. 4A). However, even though TAPI-1-treated c-FosEp-tetON mice did not show a dramatic hyperplasia, the mice exhibited other features commonly developed by c-FosEp-tetON mice, such as hair follicle cysts (Fig. 4A). While c-FosEp-tetON mice still expressed MMP10 protein levels upon TAPI-1 treatment, the mutants showed no recruitment of T lymphocytes (Supplemental Fig. 7A,B). Keratinocyte proliferation and survival, as assessed by Ki67 and p-STAT3 immunostaining, were reduced compared with vehicle-treated mice (Fig. 4B; Supplemental Fig. 7C). Importantly, an MMP10 activity assay using skin protein lysates demonstrated that the drug reached the skin and inhibited MMP10 (Fig. 4C). mRNA levels of c-fos, s100a7a15, and mmp10 remained elevated upon TAPI treatment (Supplemental Fig. 7D). These results indicate that inhibition of broad MMP/ADAM signaling ameliorates the pathological features developed by the c-FosEp-tetON mice.
Figure 4.
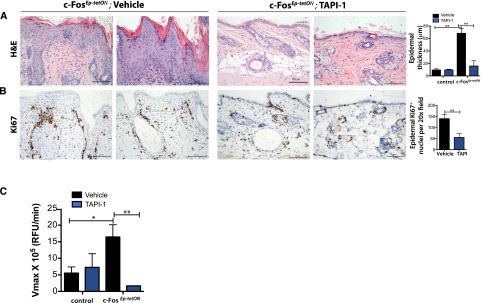
Blocking MMP10 signaling suppresses c-Fos-mediated epidermal hyperplasia. (A, left) Immunohistochemical analyses depicting H&E staining of c-FosEp-tetON mice [Col-Fos (+/KI); K5rtTA (+/T)] treated with vehicle or TAPI-1 (10 mg/kg, three times per week for 4 wk; n = 3 or 3). (Right panel) Epidermal thickness IHC quantification. Mean ± SD. (B, left) Immunohistochemical analyses depicting Ki67 stainings of the back skin of c-FosEp-tetON mice [Col-Fos (+/KI); K5rtTA (+/T)] treated with vehicle or TAPI-1 (10 mg/kg, three times per week for 4 wk), (n = 3 or 3). (Right panel) Ki67-positive nucleus IHC quantification. Mean ± SD. (C) MMP10 activity assay of the back skin of vehicle- and TAPI-treated control and c-FosEp-tetON mice.
7,12-dimethylbenz(a)anthracene (DMBA)-induced papilloma and SCC development is accelerated by c-Fos
Hras-activating mutations have been shown to render the epidermis susceptible to the development of skin cancers (Wong et al. 2013). To investigate whether c-Fos is sufficient to promote tumor development upon DMBA treatment, we applied an adapted version of the two-step skin carcinogenesis protocol (DMBA/TPA) (Bremner et al. 1994) to the c-FosEp-tetON mice. Hras mutations were induced by DMBA in the back skin of adult mice, and, instead of TPA, inducible c-fos expression was activated 1 wk later using a low dose of Dox (0.25 g/L) (Fig. 5A). Eight weeks after Dox treatment, c-FosEp-tetON mice developed papillomas, which became ulcerative at 11 wk (Fig. 5B–D). The tumor promoter TPA was included in a small set of animals as a positive control (Supplemental Fig. 8A–C). mRNA expression analyses of the whole tumor revealed increased levels of mmp10 as well as s100a7a15 compared with control skin (Fig. 5E). Histologically, tumors invaded the adjacent dermal and fat layers with features of SCCs, such as aberrant mitosis and atypia (Supplemental Fig. 8D; data not shown). IHC analyses revealed that expression of c-Fos was present throughout the tumor (Supplemental Fig. 8D). Furthermore, the tumors expressed high levels of MMP10 and Keratin 6, a keratin mainly expressed by proliferating keratinocytes (Supplemental Fig. 8D). Moreover, markers associated with cancer cell migration and invasiveness, such as podoplanin, a direct c-Fos target gene (Durchdewald et al. 2008), were up-regulated at the invading front of the tumor (Supplemental Fig. 8D).
Figure 5.

c-Fos expression accelerates DMBA-induced papilloma and SCC development. (A) Scheme of the experimental design: Six-week-old (6w) mice were given a topical single dose of DMBA (0.5% [w/v] in acetone), and Dox was supplied 1 wk later (1w) in the water at a concentration of 0.25 g/L. (B) Representative picture of control (littermate) and c-FosEp-tetON (n = 10). (C) Quantification of the papilloma number after 8 wk of inducible c-fos expression. (D) Tumor size in c-FosEp-tetON female and male mice after DMBA and 8 and 11 wk of inducible c-fos expression. Mean ± SEM. (E) mRNA expression analyses of c-fos, mmp10, and s100a7a15 in control back skin and c-FosEp-tetON SCCs. (F) Scheme of the experimental design: Six-week-old mice were given a topical single dose of DMBA (0.5% [w/v]), and Dox was supplied 1 wk later (7w) in the food pellets. Sulindac was supplied in the drinking water at a concentration of 180 mg/L. (G, top panels) Representative pictures of DMBA-treated c-FosEp-tetON mice ± sulindac treatment after 7 wk of inducible c-fos expression. (Bottom)Immunohistochemical analyses depicting H&E and cleaved caspase 3 (cl.caspase 3) immunostaining of the back skin of DMBA-treated c-FosEp-tetON mice ± sulindac treatment after 7 wk of inducible c-fos expression. (H) Lesion number quantified when the mice were sacrificed, comparing controls and c-FosEp-tetON mice treated with DMBA ± sulindac. (I) Lesion size quantified when the mice were sacrificed, comparing controls and c-FosEp-tetON mice treated with DMBA ± sulindac. (J) Schematic representation of changes in the epidermis upon induction of c-Fos, leading to increased MMP10 and S100a7a15 and, subsequently, the recruitment of CD4 T cells. As a consequence, paracrine cytokine signaling (IL-22) promotes keratinocyte proliferation and survival, leading to epidermal hyperplasia and, when combined with DMBA, SCC development.
To test whether c-Fos promotes epidermal proliferation and, subsequently, SCC development by recruitment of inflammatory cells, specifically CD4 T cells, we aimed to inhibit inflammation by using the COX1/COX2 inhibitor sulindac. The adapted version of the two-step carcinogenesis protocol comparing sulindac-treated versus untreated mice was performed (Fig. 5F). After one dose of topical DMBA and 7 wk of inducible c-fos expression, mutant mice developed ∼100-mm2 tumors (Fig. 5G–I). Interestingly, sulindac-treated mice developed fewer tumors, and the ones developed were significantly smaller and did not show increased apoptotic rates (Fig. 5G). Therefore, we conclude that c-Fos is sufficient to promote SCC development and that pharmacological blockade of inflammatory responses, likely preventing CD4 T-cell recruitment, diminishes SCC incidence in c-FosEp-tetON mice.
Human SCCs express high c-FOS levels, correlating with high MMP10 and S100A15 expression levels and CD4 T-cell recruitment
To address whether the findings from the c-FosEp-tetON mouse model were applicable to human skin tumors, expression of c-FOS, MMP10, and the corresponding human orthologs S100A7 (psoriasin) and S100A15 (koebnerisin) was assessed. Sections from 85 BCC, 96 SCC, and 20 perilesional skin samples were analyzed by IHC (Fig. 6A,B). In BCCs, none of the tumors expressed c-FOS protein, which correlated with negative MMP10 staining (Fig. 6A). In contrast, 75% of the SCCs expressed high c-FOS protein levels, correlating with high MMP10 expression (Fig. 6A), and the remaining 25% percent of SCCs that expressed low or undetectable c-FOS levels had low or undetectable MMP10 levels (Fig. 6A). Therefore, a high correlation between c-Fos and MMP10 protein expression in human SCCs was observed (Fig. 6A). However, no correlation was observed between c-FOS and S100A7 protein levels in the same set of samples (data not shown), while 89% (41 positive for S100A15 out of 46 positive for c-FOS) of the S100A15 immunostainings correlated with c-FOS expression in human SCCs (Fig. 6B). Human SCC cell lines treated with a c-FOS/AP-1 inhibitor were used to confirm previous findings. mRNA expression analyses revealed decreased expression of MMP10 and S100A15 when SCC cell lines were cultured in the presence of the c-FOS/AP-1 inhibitor (Supplemental Fig. 9A,B). In addition, lentiviral infection of human epidermal keratinocytes (obtained from American Type Culture Collection) with a plasmid overexpressing c-FOS showed a good correlation between the levels of c-FOS and MMP10 but not with S100A15 (Supplemental Fig. 9C).
Figure 6.

Correlating c-FOS levels and MMP10 and S100A15 in SCCs but not in BCCs or perilesional healthy skin. (A, top) c-FOS immunostainings on human “perilesional” skin (n = 20), BCCs (n = 85), and SCCs (n = 46). (Middle) MMP10 immunostainings on human “perilesional” skin (n = 20), BCCs (n = 85), and SCCs (n = 42). (Bottom) CD4 immunostainings on human “perilesional” skin (n = 20), BCCs (n = 85), and SCCs (n = 42). (Below) Correlation of c-FOS and MMP10 expression levels on human SCC immunostainings. (B) S100A15 immunostainings on human healthy skin (n = 20), BCCs (n = 85), and SCCs (n = 66).
Since c-FOS expression correlated with MMP10 and S100A15 in SCCs, the levels of CD4 T-cell infiltrates were assessed by immunostaining on human samples. Interestingly, 73% of the c-FOS-positive SCCs showed CD4 T-cell infiltrates, which was confirmed by CD3 immunohistochemical analyses (data not shown). In addition, a correlation between IL-22 expression and CD4 was observed in the tumor-infiltrating CD4 T lymphocytes (Supplemental Fig. 9D). Collectively, these data show a strong correlation between c-FOS expression in human SCCs, MMP10, S100A15, and CD4 T-cell infiltrates (Fig. 6A,B). These results suggest that the presence of CD4 T cells in human SCCs is likely the consequence of increased c-FOS, MMP10, and S100A15 protein levels.
Discussion
Here we show that inducible expression of c-fos in the basal layer of the epidermis leads to non-cell-autonomous keratinocyte proliferation and development of preneoplastic lesions caused by chronic recruitment of CD4 T cells (which secrete IL-22) through s100a7a15 and MMP10 signaling. Moreover, we show that upon a single dose of DMBA, c-Fos is sufficient to promote SCC development, which is ameliorated by anti-inflammatory treatment (Fig. 5J).
The expression of a number of proto-oncogenes in keratinocytes has been shown to favor the development of SCCs (Ratushny et al. 2012). However, in most cases, it is unclear whether this phenotype is solely due to an intrinsic effect on proliferation and/or survival of the targeted keratinocytes or an indirect effect on the microenvironment that feeds back to the keratinocytes. In this study, we show that c-Fos expression in adult epidermis is sufficient to cause skin hyperplasia. Nevertheless, it does not affect cell proliferation and/or survival of isolated keratinocytes, indicating that the effect of c-FOS on the development of skin hyperplasia and SCCs is non-cell-autonomous. Previous loss-of-function studies employed conditional targeted mice and keratinocytes lacking c-Fos to demonstrate a differentiation-induced inhibition of SCC development in a cell-autonomous manner (Guinea-Viniegra et al. 2012). Our study is the first one showing that the expression of c-Fos in keratinocyte requires additional signals from the microenvironment to cause the early signs of skin malignancy. In addition, we also show that paracrine external signals are provided by inflammatory cells that are recruited to the epidermis upon c-fos expression in keratinocytes.
The association of chronic inflammation and cancer initiation/progression has now been reported for a number of cancers (Colotta et al. 2009; DiDonato et al. 2012). In skin, certain inflammatory diseases such as lupus vulgaris (Motswaledi and Doman 2007) or chronic skin ulcers (Baldursson et al. 1993) predispose patients to NMSK (SCC). We show here that expression of c-Fos in keratinocytes is sufficient to trigger chronic accumulation of CD4 T cells in the epidermis with no effect on CD8 T cells and B cells. Since RAG deficiency prevents c-Fos-mediated skin hyperplasia, we propose that selective accumulation of CD4 T cells in the skin is directly promoting the development of the preneoplastic lesions in the epidermis. Mechanistically, increased intralesional/intratumoral IL-22-producing CD4 T cells could explain the epidermal hyperplasia or SCC development observed upon inducible c-Fos expression. The overall role of CD4 T cells in the incidence of preneoplastic lesions and SCCs in the skin has also been supported by other studies using the K14-HPV tumor-prone mice (Kemp et al. 1999; Daniel et al. 2003; de Visser et al. 2005). Unlike CD4 T cells, CD8 T cells seem to protect against skin carcinogenesis (Di Piazza et al. 2012), in agreement with the current view in cancer and inflammation assigning a protective effect to cytotoxic CD8 T cells and a tumor-promoting effect to helper CD4 T cells (Shiao et al. 2011; Brown et al. 2012). Nevertheless, there are some specific cases where CD4 T cells may have a tumor-protective role (Demehri et al. 2012). Although the presence of inflammatory cells in tumors has been previously reported in different types of cancer, it remains unknown when during development of the malignancy the inflammatory response occurs and what the extracellular signals that trigger this accumulation of inflammatory cells are. In this study, genome-wide expression analyses revealed two genes (s100a7a15 and mmp10) consistently up-regulated in c-Fos-expressing keratinocytes that could mediate the recruitment of CD4 T cells to the epidermis. We show here that mouse S100a7a15 is able to induce CD4 T-cell recruitment. Furthermore, S100a7a15, also named psoriasin in humans because of its involvement in psoriasis, is known to act as a chemoattractant molecule for granulocytes and CD4 T cells but not for CD8 T cells, B cells, or macrophages (Wolf et al. 2008, 2010). Altered expression of S100A7 and S100A15 has also been found during human epithelial tumorigenesis (Alowami et al. 2003; Kesting et al. 2009; Tiveron et al. 2012; Hattinger et al. 2013), although it is not known what is inducing their expression. Inducible expression of s100a7a15 in the mammary gland has also been shown to enhance tumor growth and metastasis (Nasser et al. 2012). MMP10, similar to other metalloproteases, can facilitate the recruitment of infiltrating cells by remodeling the extracellular matrix at the proximity of the c-Fos-expressing keratinocytes. Increased levels of MMP10 have been associated with cancer progression/initiation (Liu et al. 2012). MMP10 deficiency in a K-Ras-mediated lung cancer model leads to a decrease in tumor burden compared with controls (Regala et al. 2011). In addition, MMP10 has also been associated with inflammatory disease, such as arthritis (Barksby et al. 2006). Here we show that pharmacological blockers of MMPs in vivo delay the development of the preneoplastic lesions caused by c-fos expression, likely due to inhibition of MMP10, the only MMP/ADAM up-regulated upon c-Fos expression. Importantly, our studies also identify S100a7a15 and MMP10 genes as two novel direct targets of c-Fos in keratinocytes. Following DMBA-induced Hras mutations, inducible c-fos expression in the epidermis is sufficient to promote SCC development in mice with high levels of S100a7a15 as well as MMP10 proteins, further supporting the role of these proteins in mediating c-Fos functions in skin cancer.
Thus, our study in mouse models has uncovered a non-cell-autonomous function of c-Fos in the epidermis, linking proinflammatory responses to preneoplastic and neoplastic lesion development with a major contribution of CD4 T cells, likely due to the induction of newly uncovered target genes like s100a7a15 and MMP10. Importantly, our results also support that this novel pathway is relevant for human SCCs. In humans, there are two genes—S100A7 (psoriasin) and S100A15 (koebnerisin)—that correspond to the single s100a7a15 ortholog in mice. Psoriasin and koebnerisin have been previously shown to be up-regulated in human epithelial skin tumors (Moubayed et al. 2007; Hattinger et al. 2013). Here we show that S100A7 and S100A15 expression is increased in cutaneous SCCs compared with normal skin and epithelial BCCs. However, while c-FOS and S100A15 proteins are coexpressed in the SCCs, the c-FOS and S100A7 expression pattern are not overlapping. Thus, functionally, S100A15 in humans may resemble mouse s100a7a15 in SCCs. Similar to S100A15 and S100A17, increased MMP10 protein levels have also been reported in human epithelial tumors such as esophageal or oral SCCs (Stott-Miller et al. 2011; Liu et al. 2012). Indeed, tumoral and salivary MMP10 levels are being used as markers of oral SCCs (Stott-Miller et al. 2011). Our study shows a strong correlation between the expression of c-FOS and MMP10 in human SCCs. Interestingly, our results also revealed CD4 T-cell infiltrates in human SCCs, but not BCCs, and a positive correlation between high levels of c-FOS, MMP10, and CD4 T cells, and these cells secrete IL-22, which induces keratinocyte proliferation. Therefore, induction of MMP10 and S10015 expression by c-Fos in human SCCs could promote the selective accumulation of CD4 T cells and cancer progression. S100A15 and MMP10 may emerge as novel targets in human skin cancer progression as an alternative to intervene with CD4 T-cell responses in the skin.
Materials and methods
Generation of c-FosEp-tetON mice
ColTetO-Fos mice were generated by knocking in a Flag-tagged c-Fos cDNA controlled by a tetracycline operator (tetO) downstream from the col1a1 gene (L Bakiri and EF Wagner, unpubl.). K5-rtTA mice were obtained from S. Gutkind. Control and mutants were identified by PCR genotyping on tail DNA. Mice were maintained in a C57BL/6x129sv background. For the carcinogenesis treatment, F1 FvB mice were used, since this background has been shown to accelerate tumor formation (Naito and DiGiovanni 1989). Dox (Harlan) was supplied ad libitum at 0.25–0.5g/L (Sigma-Aldrich) in 2.5%–5% (w/v) -sucrose-supplemented drinking water or in the food pellets. Double-transgenic mice are referred to as c-FosEp-tetON mice, and wild-type or single-transgenic littermates were used as controls. Mice were treated with Dox between 6 and 8 wk of age.
Study approval
Mice were kept in the animal facility in accordance with institutional policies and federal guidelines for animals used in biomedical research following the recommendations of the Ethics Committee and Animal Welfare of the Insituto de Salud Carlos III (Madrid, Spain), following the Royal Decree 10th of October 1201/2005. Animal experiments were approved by the Animal Experimental Ethics Committee of the Instituto de Salud Carlos III.
Chemical carcinogenesis
For DMBA/TPA-induced skin carcinogenesis, cohorts of c-FosEp-tetON and control mice at 5 wk of age received a single application of 100 μL of an acetone solution containing 0.5% DMBA (Sigma-Aldrich) applied to the dorsal surface. TPA (10−4 M; Sigma-Aldrich) in acetone was applied twice per week to the dorsal surface. Dox treatment was used either at 0.25 g/L in the drinking water or ad libitum in the food pellets. DMBA was combined with Dox or with Dox and TPA. A small cohort of mice also received 180 mg/L sulindac (Sigma-Aldrich) in the water at the same time as Dox in the food pellets.
MMP inhibitory treatment
Six-week-old to 8-wk-old control and c-FosEp-tetON mice were treated with Dox for 4 wk. TAPI-1 (MMP inhibitor; Peptides International) was injected i.p. every other day three times per week for 4 wk. TAPI was diluted in 5% DMSO in saline. The final concentration used was 10 mg/kg.
Histological analysis
Histology and IHC were performed according to Zenz et al. (2003). Briefly, tissues were fixed in PBS-buffered 3.7% formalin or frozen in OCT at −80°C. Formalin-fixed tissues were dehydrated, and 4-μm sections were used; 7-μm cryosections were cut from OCT-fixed and frozen tissues. Human tissue arrays from paraffin blocks were produced as 0.5-mm punches taken out of the paraffin-embedded material by parallel viewing of H&E-stained sections. H&E staining was performed according to standard procedures (Sigma-Aldrich). The following primary antibodies were used: rabbit monoclonal to Ki-67 (SP6 Master Diagnostica); c-Fos (Calbiochem); Flag-DYKDDDDK tag (Cell Signaling); K6, K1, K5, and loricrin (Covance); podoplanin (Developmental Studies Hybridoma Bank); S100a7a15 and S100A15 (donated by R. Wolf, Munich, Germany); MMP10 (Leica); c-FOS (Santa Cruz Biotechnology, D1); and CD4 (DAKO, 4B12). Alexa Fluor 488 dye-labeled secondary antibodies (Invitrogen) were used at concentrations from 1 μg/mL to 50 μg/mL. Sections were counterstained with Carazzi's hematoxylin (Panreac) and analyzed by light microscopy (Leica, DM2500).
Human skin SCC samples obtained after informed consent (ethics permission no. 125/10/2012 EK 405/2006) and approval were provided and evaluated by Dr. Peter Petzelbauer (Medical University of Vienna, Austria). Production of tissue arrays from paraffin blocks from BCC and SCC lesions (from the dermato-pathological data bank) was performed according to ethics committee permission 405/2006 and extension 125/10/2012 (Medical University of Vienna, Austria). In brief, by this technique, 0.5-mm punches are taken out of the paraffin-embedded material by parallel viewing of H&E-stained sections.
Flow cytometry
Total skin from control and c-FosEp-tetON mice was subjected to mild digestion with liberase and DNase and mechanically disrupted using GentleMACS Dissociator (Miltenyi). The isolated cells were then stained for 30 min with NK1.1 PerCPCy5.5 (1:200; BD Pharmingen), CD25 APC (1:200; BD Pharmingen), CD45.2 FITC (1:400; BD Pharmingen), g/d TCR Brilliant Violet 421 (1:200; Biolegend), CD3 (1:200; BD Pharmingen), CD4 PECy7 (1:100; BD Pharmingen), CD8 FITC (1:200; BD Pharmingen), Gr1 FITC (1:100; BD Pharmingen), CD45 (1:500; BD Pharmingen), CD11b PerCPCy5.5 (1:200; BD Pharmingen), B220 PE (1:200; BD Pharmingen), CD11c PE (1:100; BD Pharmingen), CD69 PE (1:200; BD Pharmingen), and CD44 and APC (1:200; BD Pharmingen). The cells were then fixed using BD Cytofix buffer. Samples were collected in a FACS CANTO II (BD Pharmingen) equipped with 488-nm, 640-nm, and 405-nm lines. We used pulse processing to exclude cell aggregates and live/dead fixable dye Aqua to exclude dead cells. At least 10,000 live single CD45+ events were collected; all data were analyzed using FlowJo 9.5.3 (Treestar). Skin draining lymph nodes were removed from the mouse and kept in 5% FBS RPMI. Lymph nodes were smashed with a plunger against a 40-mm cell strainer. The strainer was washed with 5% FBS RPMI. Cells were spun down at 300g for 10 min. Finally, leukocytes were resuspended in PB buffer with low BSA, and staining was carried out.
Cell culture and adenoviral infection
Isolation and culture of mouse primary tail keratinocytes was performed as described elsewhere (Zenz et al. 2003). Twenty-four hours after plating, keratinocyte medium was changed to KC-SFM (Gibco). Keratinocytes were cultured at 32°C. The medium was changed every other day and, if needed, supplemented with Dox at 1 mg/L. Ca2+ (0.05 mM or 2 mM CaCl2) stimulation of keratinocytes was performed in equally semiconfluent cultures for proper comparison. Cells were collected at different time points after Dox treatment. Primary c-Fosfl/fl keratinocytes were cultured in the same conditions and treated with adenoviruses. Adeno-Cre (AdCre) or Adeno-green fluorescent protein (AdGFP) adenoviruses were purchased from the University of Iowa. Primary keratinocytes were infected with 300 particles per cell in KC-SFM medium. Cells were treated with TPA for 3 h and collected at different times post-infection. The epidermal-derived SCC9 cell line was kindly provided by M. Sibilia (Institute for Cancer Research, Vienna, Austria) (Lichtenberger et al. 2010), and SCC12 and SCC22 were provided by G.P. Dotto (University of Lausanne, Lausanne, Switzerland) (Lefort et al. 2007). All of the SCC cells were grown in DMEM and 10% FBS. The SCC cells were cultured with vehicle or with a c-Fos/AP-1 inhibitor (Aikawa et al. 2008).
Packaging of retroviral constructs and retroviral transduction
For production of viruses, 293T cells were transfected with Lipofectamine 2000 (Gibco) and the indicated plasmids according to manufacturer's instructions. After 48 h, supernatants were filtered through a 0.45-μm sterile filter and used directly or stored at −80°C until further use. Plasmids used were LeGO-iG2 (Weber et al. 2008) and Precision LentiORF human FOS (Thermo Scientific, OHS5897-202616290). Viruses were pseudotyped with VSV-G. Human keratinocytes were infected with the different lentiviral particles.
Fos activity assay
TAPI- and vehicle-treated control and c-FosEp-tetON mouse skin extracts or keratinocyte lysates were used in c-Fos activity assays (Active Motif, TransAm AP-1 c-Fos) following the manufacturer's instructions.
CD4 T-cell chemotaxis assay
CD4 T cells were purified from wild-type mouse spleens by positive selection using MACS kits as recommended by the manufacturer (Miltenyi Biotec). CD4 T cells were activated with plate-bound anti-CD3 mouse antibody (3 μg/mL; BD Pharmingen, OKT3) and a soluble anti-CD28 mouse antibody (2 μg/mL; BD Pharmingen) for 5 h. SDF-1α (Vitro) was used at 10 ng/mL. Chemotaxis assay was performed using a plate with a transwell containing a 5-μm polycarbonate membrane. CD4 T cells were diluted in complete RPMI medium (5% fetal calf serum, 2.5 mg/L glucose, 2 mM glutamine, 10 μg/mL folate, 1 mM pyruvate, 50 μM β−mercaptoethanol). In the lower chamber, the condition medium was added (±SDF-1α). Two hours after plating CD4 T cells, the insert was lifted, and cells that migrated to the lower chamber were analyzed using a BD FACS Canto II flow cytometer (BD Biosciences).
MMP10 enzymatic assay
An enzymatic assay using back skin protein lysates was performed following the manufacturer's instructions (AnaSpec).
Protein isolation
Back skin from mice was shaved and snap-frozen in dry ice, or cells were scraped from the plate with a cell scraper. Protein isolation for Western blot was performed in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% deoxycholate, 1% NP-40) containing a protease inhibitor cocktail (0.1 mM Na3VO4, 40 mM b-glycerophospate, 40 mM NaPPi, 1 mM NaF) (Sigma-Aldrich), and tissues were homogenized using Precellys 24 (Bertin Technologies). For protein lysates from cultured keratinocytes, cells were scraped (100 μL for each well in a six-well plate) with a cell scraper and kept for 15 min in ice. The cell lysate was spun down at maximum speed, and the supernatant was collected. Protein lysates were quantified by using Pierce BCA protein assay reagent (Thermo Scientific). Western blot analysis was performed according to standard procedures using the following antibodies: c-Fos (BD-Pharmingen), Flag (Sigma-Aldrich), vinculin (Sigma-Aldrich), MMP10 (DAKO), S100a7a15 (provided by Dr. Ronald Wolf), b-Actin (Sigma-Aldrich), and GAPDH (Sigma-Aldrich).
The blots were incubated with the appropriate secondary horseradish peroxidase-coupled antibodies (GE Healthcare) and developed using the Amersham ECL Plus Western blotting detection reagent (GE Healthcare) and Amersham ECL Hyperfilms (GE Healthcare). Alternatively, blots were incubated with Alexa-Fluor 680-coupled secondary antibodies (Invitrogen) and visualized using the Odyssey imaging system (Li-Cor).
RT-qPCR
Total RNA was isolated using Trizol (Sigma-Aldrich) according to the manufacturer's instructions. Complementary DNA was synthesized using 1 μg of RNA, Ready-To-Go-You-Prime-First-Strand Beads (GE Healthcare), and random primers (Sigma-Aldrich) as described in the manufacturer's protocol. RT-qPCR reactions were performed using GoTaq RT-qPCR Master Mix (Promega) and Eppendorf fluorescence thermocyclers (Eppenddorf). The 2−ΔΔCT method was used to quantify amplified fragments. Expression levels were normalized using at least two housekeeping genes (gapdh and hprt). Primer sequences will be provided on request.
Genome-wide expression analyses
RNA of samples was hybridized to the Illumina MouseRef-8 V2 R3 BeadChip array according to the manufacturer's instructions (Illumina, Inc.). Microarray scanning was done using a Beadstation array scanner. Data preprocessing and quality control were conducted using packages of the Bioconductor project implemented at the DKFZ in-house-developed ChipYard framework for microarray data analysis (http://www.dkfz.de/genetics/ChipYard). In summary, microarray probes were annotated based on Ensembl (version 58) using an in-house BLAST-based pipeline. Before normalization with variance-stabilizing transformation (VST) and robust spline normalization (RSN) algorithms, beads with signals below the negative controls were removed, and positive, negative, and hybridization controls were excluded.
Normalized log2-transformed microarray data were used to calculate the gene fold change value (i.e., how many times the intensity of gene expression changed compared with the control). The fold change value has no measure of statistical significance but is biologically relevant and specifically usable for data that, other than the time series, have no other replication. Genes were ranked according to the Euclidian distance between the maximum expression within a probe set and the mean expression of a probe set over all time points (as shown in Busch et al. 2008), both scaled (normalized) to the maximum value measured in each data set.
ChIP
ChIP was performed using both the EZ-Zyme chromatin prep kit and EZ-Magna ChIP G ChIP kits (Millipore). Keratinocytes were isolated from the tails from adult c-Fosfl/fl or c-FostetON mice. Two confluent P100 plates were used per condition. c-FostetON keratinocytes were cultured as explained before and treated for 24 h with 1 μg/mL Dox. c-Fosfl/fl keratinocytes were infected with 300 particles of AdCre- or AdGFP-expressing viruses per cell. c-Fosfl/fl keratinocytes were cultured for 4 d after infection and serum-starved overnight. The next day, c-Fosfl/fl keratinocytes were treated for 3 h with 10 nM PMA (phorbol 12-myristate 13-acetate). Primers will be provided on request.
Flow cytometry and FACS sorting
Skin from c-FosEp-tetON mice and controls was shaved and removed from the mice. Dermal fat was scraped off using a scalpel. Tissues were finely minced with scissors and digested with 120 μL of 4 mg/mL DNase (Sigma-Aldrich) and 200 μL of 5 mg/mL liberase (Roche) in 4 mL of RPMI in MACs C tubes and incubated for 20 min at 37°C in a water bath. Tissue was homogenized using gentleMACS Dissociator. Enzymatic digestion was quenched by adding 10 mL of 10% Ch-FBS, 2 mM EDTA, and 250 μg/mL DNase RPMI. Released cells were filtered through a 70-μm cell strainer and spun down at 300g for 10 min at 4°C. Red blood cells were removed using 1 mL of Red Blood Cell Lysing buffer (Sigma-Aldrich) for 2 min at room temperature. Cells were washed in 5 mL of high PB buffer (2 %BSA, 2 mM EDTA, 250 μg/mL DNase in PBS) and resuspended in 1 mL of high PB buffer. FcBlock (1:200; BD Pharmingen) was used for 20 min at room temperature to avoid unspecific staining. Cells were washed and resuspended in low PB buffer (0.5 %BSA, 2 mM EDTA, 250 μg/mL DNase in PBS) and incubated with specific antibodies mixed previously in an antibody cocktail mix to avoid variability among samples (CD45-APC/PE [1:400], CD3-APC [1:200], Gr 1 FITC [1:100], B220 PE [1:200], CD44 APC [1:200], CD8 FITC [1:200], and F4/80 FITC [1:100] [BD-Pharmingen]). For FACS sorting, cells were used right after staining to avoid cell death. DAPI was used as a viability marker. Sorting was performed using a BD Influx cell sorter. For flow cytometry analyses, cells were fixed using BD cytofixed and Acqua live.
EdU proliferation
Sixty percent confluent c-FostetON keratinocyte cultures were treated ±Dox (1 mg/mL) for 1 d. An EdU proliferation assay was performed using the Click-iT EdU cell proliferation assay, and keratinocytes were treated following the manufacturer's instructions. Analyses were performed using a BD FACSCanto II flow cytometer. Analyses were done using FlowJo software.
Statistical analysis
Unless otherwise specified, results are presented as mean ± SD. P-value was calculated using Student's t-test.
Acknowledgments
We thank the members of the Wagner laboratory for critical reading of the manuscript and sharing reagents. We thank Marc Zapatka and Tobias Bauer and for normalization of expression profiling data and pathway analyses. We thank Guillermo Medrano and Gema Luque for taking care of the experimental mice. We thank Ultan Cronin for technical and analytical support in the cytometer and FACS sorter. E.F.W. is funded by the Banco Bilbao Vizcaya Argentaria (BBVA) Foundation and a European Research Council Advanced Grant (ERC FCK/2008/37). E.M.B. was funded by La Caixa International PhD Programme for 4 years. J.G.V. was funded by the Ramón y Cajal fellowship program (Spanish Ministry of Economy). P.A. and R.E. are funded by a grant from the German Ministry for Research and Education (BMBF) funding program AGENET.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.223339.113.
References
- Aikawa Y, Morimoto K, Yamamoto T, Chaki H, Hashiramoto A, Narita H, Hirono S, Shiozawa S 2008. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat Biotechnol 26: 817–823 [DOI] [PubMed] [Google Scholar]
- Alowami S, Qing G, Emberley E, Snell L, Watson PH 2003. Psoriasin (S100A7) expression is altered during skin tumorigenesis. BMC Dermatol 3: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldursson B, Sigurgeirsson B, Lindelof B 1993. Leg ulcers and squamous cell carcinoma. An epidemiological study and a review of the literature. Acta Derm Venereol 73: 171–174 [DOI] [PubMed] [Google Scholar]
- Barksby HE, Milner JM, Patterson AM, Peake NJ, Hui W, Robson T, Lakey R, Middleton J, Cawston TE, Richards CD, et al. 2006. Matrix metalloproteinase 10 promotion of collagenolysis via procollagenase activation: Implications for cartilage degradation in arthritis. Arthritis Rheum 54: 3244–3253 [DOI] [PubMed] [Google Scholar]
- Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F 2005. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol 174: 3695–3702 [DOI] [PubMed] [Google Scholar]
- Brantsch KD, Meisner C, Schonfisch B, Trilling B, Wehner-Caroli J, Rocken M, Breuninger H 2008. Analysis of risk factors determining prognosis of cutaneous squamous-cell carcinoma: A prospective study. Lancet Oncol 9: 713–720 [DOI] [PubMed] [Google Scholar]
- Bremner R, Kemp CJ, Balmain A 1994. Induction of different genetic changes by different classes of chemical carcinogens during progression of mouse skin tumors. Mol Carcinog 11: 90–97 [DOI] [PubMed] [Google Scholar]
- Brown MD, van der Most R, Vivian JB, Lake RA, Larma I, Robinson BW, Currie AJ 2012. Loss of antigen cross-presentation after complete tumor resection is associated with the generation of protective tumor-specific CD8+ T-cell immunity. OncoImmunology 1: 1084–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch H, Camacho-Trullio D, Rogon Z, Breuhahn K, Angel P, Eils R, Szabowski A 2008. Gene network dynamics controlling keratinocyte migration. Mol Syst Biol 4: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A 2009. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 30: 1073–1081 [DOI] [PubMed] [Google Scholar]
- Cooper SJ, MacGowan J, Ranger-Moore J, Young MR, Colburn NH, Bowden GT 2003. Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Mol Cancer Res 1: 848–854 [PubMed] [Google Scholar]
- Daniel D, Meyer-Morse N, Bergsland EK, Dehne K, Coussens LM, Hanahan D 2003. Immune enhancement of skin carcinogenesis by CD4+ T cells. J Exp Med 197: 1017–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demehri S, Turkoz A, Manivasagam S, Yockey LJ, Turkoz M, Kopan R 2012. Elevated epidermal thymic stromal lymphopoietin levels establish an antitumor environment in the skin. Cancer Cell 22: 494–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser KE, Korets LV, Coussens LM 2005. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 7: 411–423 [DOI] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Karin M 2012. NF-κB and the link between inflammation and cancer. Immunol Rev 246: 379–400 [DOI] [PubMed] [Google Scholar]
- Di Piazza M, Nowell CS, Koch U, Durham AD, Radtke F 2012. Loss of cutaneous TSLP-dependent immune responses skews the balance of inflammation from tumor protective to tumor promoting. Cancer Cell 22: 479–493 [DOI] [PubMed] [Google Scholar]
- Durchdewald M, Guinea-Viniegra J, Haag D, Riehl A, Lichter P, Hahn M, Wagner EF, Angel P, Hess J 2008. Podoplanin is a novel fos target gene in skin carcinogenesis. Cancer Res 68: 6877–6883 [DOI] [PubMed] [Google Scholar]
- Eferl R, Wagner EF 2003. AP-1: A double-edged sword in tumorigenesis. Nat Rev Cancer 3: 859–868 [DOI] [PubMed] [Google Scholar]
- Elkington PT, O'Kane CM, Friedland JS 2005. The paradox of matrix metalloproteinases in infectious disease. Clin Exp Immunol 142: 12–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinea-Viniegra J, Zenz R, Scheuch H, Jimenez M, Bakiri L, Petzelbauer P, Wagner EF 2012. Differentiation-induced skin cancer suppression by FOS, p53, and TACE/ADAM17. J Clin Invest 122: 2898–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Coussens LM 2012. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 21: 309–322 [DOI] [PubMed] [Google Scholar]
- Hattinger E, Zwicker S, Ruzicka T, Yuspa SH, Wolf R 2013. Opposing functions of psoriasin (S100A7) and koebnerisin (S100A15) in epithelial carcinogenesis. Curr Opin Pharmacol 13: 588–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CJ, Vo K, Gurley KE 1999. Resistance to skin tumorigenesis in DNAPK-deficient SCID mice is not due to immunodeficiency but results from hypersensitivity to TPA-induced apoptosis. Carcinogenesis 20: 2051–2056 [DOI] [PubMed] [Google Scholar]
- Kerkela E, Saarialho-Kere U 2003. Matrix metalloproteinases in tumor progression: Focus on basal and squamous cell skin cancer. Exp Dermatol 12: 109–125 [DOI] [PubMed] [Google Scholar]
- Kesting MR, Sudhoff H, Hasler RJ, Nieberler M, Pautke C, Wolff KD, Wagenpfeil S, Al-Benna S, Jacobsen F, Steinstraesser L 2009. Psoriasin (S100A7) up-regulation in oral squamous cell carcinoma and its relation to clinicopathologic features. Oral Oncol 45: 731–736 [DOI] [PubMed] [Google Scholar]
- Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, Devgan V, Lieb J, Raffoul W, Hohl D, et al. 2007. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKα kinases. Genes Dev 21: 562–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P, Sibilia M 2010. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 140: 268–279 [DOI] [PubMed] [Google Scholar]
- Liu H, Qin YR, Bi J, Guo A, Fu L, Guan XY 2012. Overexpression of matrix metalloproteinase 10 is associated with poor survival in patients with early stage of esophageal squamous cell carcinoma. Dis Esophagus 25: 656–663 [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68: 869–877 [DOI] [PubMed] [Google Scholar]
- Motswaledi MH, Doman C 2007. Lupus vulgaris with squamous cell carcinoma. J Cutan Pathol 34: 939–941 [DOI] [PubMed] [Google Scholar]
- Moubayed N, Weichenthal M, Harder J, Wandel E, Sticherling M, Glaser R 2007. Psoriasin (S100A7) is significantly up-regulated in human epithelial skin tumours. J Cancer Res Clin Oncol 133: 253–261 [DOI] [PubMed] [Google Scholar]
- Naito M, DiGiovanni J 1989. Genetic background and development of skin tumors. Carcinog Compr Surv 11: 187–212 [PubMed] [Google Scholar]
- Nasser MW, Qamri Z, Deol YS, Ravi J, Powell CA, Trikha P, Schwendener RA, Bai XF, Shilo K, Zou X, et al. 2012. S100A7 enhances mammary tumorigenesis through upregulation of inflammatory pathways. Cancer Res 72: 604–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratushny V, Gober MD, Hick R, Ridky TW, Seykora JT 2012. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J Clin Invest 122: 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray N, Kuwahara M, Takada Y, Maruyama K, Kawaguchi T, Tsubone H, Ishikawa H, Matsuo K 2006. c-Fos suppresses systemic inflammatory response to endotoxin. Int Immunol 18: 671–677 [DOI] [PubMed] [Google Scholar]
- Regala RP, Justilien V, Walsh MP, Weems C, Khoor A, Murray NR, Fields AP 2011. Matrix metalloproteinase-10 promotes Kras-mediated bronchio-alveolar stem cell expansion and lung cancer formation. PLoS ONE 6: e26439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdev R, Mandal AK, Singh I, Agarwal AK 2008. Progressive rise of c fos expression from premalignant to malignant lesions of oral cavity. Med Oral Patol Oral Cir Bucal 13: E683–E686 [PubMed] [Google Scholar]
- Saez E, Rutberg SE, Mueller E, Oppenheim H, Smoluk J, Yuspa SH, Spiegelman BM 1995. c-fos is required for malignant progression of skin tumors. Cell 82: 721–732 [DOI] [PubMed] [Google Scholar]
- Shiao SL, Ganesan AP, Rugo HS, Coussens LM 2011. Immune microenvironments in solid tumors: New targets for therapy. Genes Dev 25: 2559–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozawa S, Tsumiyama K 2009. Pathogenesis of rheumatoid arthritis and c-Fos/AP-1. Cell Cycle 8: 1539–1543 [DOI] [PubMed] [Google Scholar]
- Slack BE, Ma LK, Seah CC 2001. Constitutive shedding of the amyloid precursor protein ectodomain is up-regulated by tumour necrosis factor-α converting enzyme. Biochem J 357: 787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI 2007. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med 13: 1211–1218 [DOI] [PubMed] [Google Scholar]
- Stott-Miller M, Houck JR, Lohavanichbutr P, Mendez E, Upton MP, Futran ND, Schwartz SM, Chen C 2011. Tumor and salivary matrix metalloproteinase levels are strong diagnostic markers of oral squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 20: 2628–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiveron RC, de Freitas LC, Figueiredo DL, Serafini LN, Mamede RC, Zago MA 2012. Expression of calcium binding protein S100 A7 (psoriasin) in laryngeal carcinoma. Braz J Otorhinolaryngol 78: 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EF, Eferl R 2005. Fos/AP-1 proteins in bone and the immune system. Immunol Rev 208: 126–140 [DOI] [PubMed] [Google Scholar]
- Weber K, Bartsch U, Stocking C, Fehse B 2008. A multicolor panel of novel lentiviral ‘gene ontology’ (LeGO) vectors for functional gene analysis. Mol Ther 16: 698–706 [DOI] [PubMed] [Google Scholar]
- Wolf R, Howard OM, Dong HF, Voscopoulos C, Boeshans K, Winston J, Divi R, Gunsior M, Goldsmith P, Ahvazi B, et al. 2008. Chemotactic activity of S100A7 (psoriasin) is mediated by the receptor for advanced glycation end products and potentiates inflammation with highly homologous but functionally distinct S100A15. J Immunol 181: 1499–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf R, Mascia F, Dharamsi A, Howard OM, Cataisson C, Bliskovski V, Winston J, Feigenbaum L, Lichti U, Ruzicka T, et al. 2010. Gene from a psoriasis susceptibility locus primes the skin for inflammation. Sci Transl Med 2: 61ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CE, Yu JS, Quigley DA, To MD, Jen KY, Huang PY, Del Rosario R, Balmain A 2013. Inflammation and Hras signaling control epithelial–mesenchymal transition during skin tumor progression. Genes Dev 27: 670–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenz R, Scheuch H, Martin P, Frank C, Eferl R, Kenner L, Sibilia M, Wagner EF 2003. c-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Dev Cell 4: 879–889 [DOI] [PubMed] [Google Scholar]
- Zenz R, Eferl R, Kenner L, Florin L, Hummerich L, Mehic D, Scheuch H, Angel P, Tschachler E, Wagner EF 2005. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature 437: 369–375 [DOI] [PubMed] [Google Scholar]
- Zhang W, Dang E, Shi X, Jin L, Feng Z, Hu L, Wu Y, Wang G 2012. The pro-inflammatory cytokine IL-22 up-regulates keratin 17 expression in keratinocytes via STAT3 and ERK1/2. PLoS ONE 7: e40797. [DOI] [PMC free article] [PubMed] [Google Scholar]