

Programmed cell death in C. elegans requires caspase CED-3 activation. The CED-4 apoptosome facilitates CED-3 activation, yet how CED-4 recognizes CED-3 is unknown. Huang et al. now show that CED-3 directly binds CED-4 and plays a pivotal role in forming an active CED-4–CED-3 holoenzyme. Structural and biochemical analyses suggest a model whereby two CED-3 molecules are forced into the CED-4 apoptosome, which then undergoes dimerization and autocatalytic maturation. This work provides a major revision of the prevailing model for initiator caspase activation.
Keywords: programmed cell death (PCD), apoptosis, caspase activation, CED-4, CED-3
Abstract
Programmed cell death in Caenorhabditis elegans requires activation of the caspase CED-3, which strictly depends on CED-4. CED-4 forms an octameric apoptosome, which binds the CED-3 zymogen and facilitates its autocatalytic maturation. Despite recent advances, major questions remain unanswered. Importantly, how CED-4 recognizes CED-3 and how such binding facilitates CED-3 activation remain completely unknown. Here we demonstrate that the L2′ loop of CED-3 directly binds CED-4 and plays a major role in the formation of an active CED-4–CED-3 holoenzyme. The crystal structure of the CED-4 apoptosome bound to the L2′ loop fragment of CED-3, determined at 3.2 Å resolution, reveals specific interactions between a stretch of five hydrophobic amino acids from CED-3 and a shallow surface pocket within the hutch of the funnel-shaped CED-4 apoptosome. Structure-guided biochemical analysis confirms the functional importance of the observed CED-4–CED-3 interface. Structural analysis together with published evidence strongly suggest a working model in which two molecules of CED-3 zymogen, through specific recognition, are forced into the hutch of the CED-4 apoptosome, consequently undergoing dimerization and autocatalytic maturation. The mechanism of CED-3 activation represents a major revision of the prevailing model for initiator caspase activation.
Many concepts of programmed cell death (PCD), also known as apoptosis, were derived from genetic investigation of cell deaths in the nematode Caenorhabditis elegans (Horvitz et al. 1994). Initiation of PCD in C. elegans is controlled by four genes—ced-3, ced-4, ced-9, and egl-1—that act in a linear pathway (Fig. 1A; Horvitz 2003). The cell-killing protease CED-3 belongs to the caspase family (Yuan et al. 1993; Xue et al. 1996) and is synthesized as an inactive zymogen in cells. Activation of CED-3 occurs in three sequential steps (Fig. 1B). First, in response to cell death stimuli, the proapoptotic, BH3-only protein EGL-1 is activated and negatively regulates the anti-apoptotic protein CED-9 through direct interaction (Conradt and Horvitz 1998; del Peso et al. 1998, 2000; Parrish et al. 2000). Second, binding of EGL-1 to CED-9 leads to allosteric disruption of the interactions between CED-9 and CED-4, resulting in the release of CED-4 from the inhibitory CED-9–CED-4 complex (Hengartner and Horvitz 1994; Chinnaiyan et al. 1997; James et al. 1997; Spector et al. 1997; Wu et al. 1997; Chen et al. 2000). Third and finally, the released proapoptotic protein CED-4 facilitates autocataltytic activation of the CED-3 zymogen (Yuan and Horvitz 1992; Chinnaiyan et al. 1997; Irmler et al. 1997; Seshagiri and Miller 1997; Wu et al. 1997; Yang et al. 1998). Elucidating the molecular mechanisms by which CED-3, CED-4, CED-9, and EGL-1 control the onset of PCD is central to understanding the regulation of PCD.
Figure 1.

CED-4-mediated CED-3 activation is a central step to initiate PCD in C. elegans. (A) Four genes—egl-1, ced-9, ced-4, and ced-3—act in a linear fashion to control the onset of PCD in C. elegans. (B) Current model of the linear PCD pathway. In normal cells, CED-4 is sequestered by CED-9, unable to activate the cell-killing caspase CED-3. Upon cell death stimuli, EGL-1 is activated and binds to CED-9. Binding by EGL-1 causes marked conformational changes in CED-9, resulting in the release of CED-4. The released CED-4 dimer oligomerizes to form the CED-4 apoptosome, which in turn facilitates the activation of CED-3. How CED-4 binds CED-3 remains unclear, hindering a mechanistic understanding of CED-3 activation. (C) Structure of the two-chain CED-3 (residues 198–374, 389–503, and C358S). Despite being a monomer in solution, CED-3 was crystallized as an asymmetric dimer, likely due to the high concentrations required for crystallization.
Rigorous investigations in the past decade have revealed mechanistic insights into the first two steps of the linear PCD pathway in C. elegans (Fig. 1B). First, EGL-1 uses an extended BH3 helix to recognize a hydrophobic groove on the surface of CED-9 (Yan et al. 2004). Second, binding of EGL-1 to CED-9 induces pronounced conformational changes in CED-9, which, through allostery, destabilize the interactions between CED-9 and CED-4, ultimately leading to the dissociation of CED-9 from an asymmetric dimer of CED-4 (Yan et al. 2005; Yan et al. 2006b). The freed CED-4 dimer further oligomerizes to form the CED-4 apoptosome, which is responsible for the activation of CED-3. In sharp contrast to the mechanistic elucidation of the first two steps in the linear PCD pathway, the third and arguably most important step—how CED-4 mediates the activation of CED-3—remains largely enigmatic.
At present, there is little information on how the CED-4 apoptosome specifically recognizes the CED-3 zymogen. There is no experimental identification of any specific sequence or structural motifs that are responsible for the interactions between CED-4 and CED-3. The unorthodox experimental revelation that the octameric CED-4 apoptosome only binds two molecules of CED-3 has engendered much excitement as well as doubt in the field, both of which ask for mechanistic explanation. In this study, we address these questions by deciphering a central piece of the puzzle: how CED-3 is specifically recognized by CED-4. The crystal structure unambiguously shows that CED-3 is bound within the hutch of the funnel-shaped CED-4 apoptosome, whereas our structural analysis indicates that only two CED-3 molecules can be accommodated within the hutch. These structural findings have important ramifications for understanding initiator caspase activation.
Results
Structure of processed CED-3
Despite the advent of the structural information on a number of caspases, CED-3 remains structurally uncharacterized. To gain insights into CED-3 activation, we crystallized the two-chain caspase domain of CED-3 (residues 198–374, 389–503, and C358S) and determined its structure at 2.65 Å resolution (Fig. 1C; Supplemental Table S1). Mutation of the catalytic residue Cys358 to Ser was essential for the generation of a stable CED-3 for crystallization. Although CED-3 exists mainly as a monomer in solution (Qi et al. 2010), it was crystallized as an asymmetric homodimer in the crystals, likely due to the high protein concentrations required for crystallization. As anticipated, the overall structure is very similar to that of other caspases. Notably, the two molecules of CED-3 exhibit contrasting features at the active site. The active site in one CED-3 molecule exists in a productive conformation, with loops L2 and L4 interacting with the L2′ loop of the adjacent molecule to form a loop bundle that is characteristic of activated caspases (Fig. 1C; Supplemental Fig. S1A). In contrast, the active site in the adjacent CED-3 molecule does not have a well-defined conformation, with the L2 loop largely disordered in the crystals. Asymmetric features of the active site have been previously observed in other initiator caspases, such as caspase-9 (Renatus et al. 2001) and Dronc (Yan et al. 2006a). This structural feature might be physiologically important; however, we cannot rule out the possibility that this feature is a consequence of crystal-packing interactions because the disordered active site is in close contact with another CED-3 molecule in the crystal lattice.
Requirement of the L2′ loop for CED-3 association with CED-4
The structural information of CED-3 empowered us to identify the surface elements that are responsible for binding to the CED-4 apoptosome. Guided by the structure, we carefully designed 21 CED-3 mutants, each containing one or more missense mutations on surface-exposed amino acids (Supplemental Fig. S1B). These targeted amino acids cover ∼50% of the exposed surface area on CED-3 and include most polar, charged, or long chain hydrophobic amino acids on the surface. Ten CED-3 mutant proteins exhibited poor solubility and were unfit for biochemical characterization; the rest were purified to homogeneity and assayed for their ability to interact with CED-4. Surprisingly, none of the remaining 11 CED-3 mutants lost the ability to associate with the CED-4 apoptosome (Supplemental Fig. S1B). This result suggests that the primary CED-4-interacting epitopes might be located on flexible surface loops. To examine this scenario, we generated a number of deletion variants, each involving removal of a stretch of surface loop sequences from CED-3. The two-chain CED-3 (residues 198–364 and 407–503), which has partial deletion of the L2 and L2′ loops, failed to form a stable complex with the CED-4 apoptosome (Fig. 2A, top two panels). Partial deletion of the L2′ loop (residues 389–406), but not the L2 loop (residues 365–374), led to abrogation of the interaction between CED-3 and CED-4 (Fig. 2A, bottom two panels). Thus, the L2′ loop of CED-3 is required for binding to the CED-4 apoptosome.
Figure 2.

The L2′ loop of CED-3 is required for stable association between CED-3 and the CED-4 apoptosome. (A) The L2′ loop is required for CED-3 to associate with the CED-4 apoptosome. Gel filtration analysis reveals that removal of the L2′ loop, but not the L2 loop, in the two-chain CED-3 (residues 198–374, 389–503, and C358A) resulted in loss of binding to the CED-4 apoptosome. (B) Deletion of the L2′ loop in CED-3 results in loss of CED-4-mediated stimulation of CED protease activity. (C) Missense mutations in the L2′ loop of CED-3 leads to abrogation of CED-4-mediated stimulation of CED protease activity. The three missense mutations L391D, F392D, and N393D target three consecutive amino acids in the L2′ loop of CED-3.
Next, we examined the functional consequence of deletion of the L2′ loop using an in vitro CED-3 protease activity assay. As previously reported (Qi et al. 2010), the protease activity of the two-chain CED-3 (residues 198–374 and 389–503) toward the peptide substrate DEVD-AMC was markedly stimulated by the presence of the CED-4 apoptosome (Fig. 2B). Strikingly, partial deletion of the L2′ loop in CED-3 (residues 198–374 and 410–503) nearly abolished CED-4-mediated stimulation of the CED-3 protease activity (Fig. 2B). Serendipitously, a CED-3 variant (198–503, C358S, L391D, F392D, and N393D) that contains three missense mutations in the L2′ loop was found to cripple interactions with the CED-4 apoptosome (Supplemental Fig. S2). Consequently, the protease activity of the wild-type CED-3 (residues 198–503), but not the CED-3 variant (residues 198–503, L391D, F392D, and N393D), was markedly stimulated by the presence of the CED-4 apoptosome (Fig. 2C). These results unambiguously show that the L2′ loop is required for CED-4-mediated stimulation of CED-3 protease activity.
To examine whether the L2′ loop alone is sufficient for binding to the CED-4 apoptosome, we engineered a caspase-9 variant in which its L2′ sequences (residues 316–331) were replaced by the corresponding CED-3 sequences (residues 389–404). We purified this caspase-9 variant and examined its potential interaction with the CED-4 apoptosome by gel filtration. The wild-type caspase-9 did not interact with the CED-4 apoptosome (Fig. 3A, top two panels). In contrast, a small fraction of the caspase-9 variant was shifted to earlier fractions upon incubation with CED-4, suggesting stable association (Fig. 3A, bottom two panels). We further speculated that, due to enhanced local concentrations that favor caspase-9 homodimerization, the CED-4 apoptosome might also be able to moderately stimulate the protease activity of the caspase-9 variant. Confirming this conjecture, this caspase-9 variant exhibited a markedly higher level of protease activity toward the peptide substrate LEHD-AFC in the presence of the CED-4 apoptosome (Fig. 3B).
Figure 3.

The L2′ loop of CED-3 is sufficient for stable association with the CED-4 apoptosome. (A) Replacement of the L2′ loop (residues 316–331) in caspase-9 by that in CED-3 (residues 389–404) allowed weak interaction between the caspase-9 variant and the CED-4 apoptosome. Contiguous fractions from gel filtration were visualized on Coomassie-stained SDS-PAGE gels. Red arrows denote a small fraction of the caspase-9 variant that had been shifted to earlier fractions. The wild-type (WT) caspase-9 controls are shown in the top two panels. (B) Replacement of the L2′ loop (residues 316–331) in caspase-9 by that in CED-3 (residues 389–404) resulted in higher protease activity in the presence of CED-4. LEHD-AFC was used as the substrate.
Recognition of the CED-3 L2′ loop by CED-4
Our experimental observations demonstrate that the processed CED-3, via its L2′ loop, interacts with the CED-4 apoptosome, ultimately leading to stimulation of the CED-3 protease activity. A pivotal point in the mechanistic understanding of CED-3 activation is identification of the CED-3-binding site in the CED-4 apoptosome. The importance of addressing this point is further necessitated by the need to understand the fourfold symmetry of the CED-4 octamer and the 8:2 stoichiometry between CED-4 and CED-3. We investigated this point by two parallel approaches. First, we generated a large number of CED-4 missense mutants, each targeting a surface-exposed amino acid. Unfortunately, the majority of these CED-4 mutants remained insoluble and defied purification; the remaining CED-4 variants appeared to retain association with CED-3 (data not shown). Second, we sought to crystallize a complex between CED-4 and the CED-3 L2′ loop fragment. After numerous trials, we succeeded in the crystallization of a CED-4 protein that is fused at its C terminus to the CED-3 L2′ loop fragment (residues 389–409) via a flexible 11-residue linker sequence. This CED-4–CED-3 fusion protein formed a stable complex, which presumably represents the CED-4 apoptosome bound to the L2′ loop fragment of CED-3.
We determined the crystal structure at 3.2 Å resolution (Supplemental Table S2). The CED-4 apoptosome bound to the L2′ loop of CED-3 exhibits a funnel-shaped architecture similar to that of the free CED-4 apoptosome (Fig. 4A; Qi et al. 2010). Eight CED-3 peptide fragments are clearly identified in the hutch of the CED-4 apoptosome, each binding to a shallow surface pocket on CED-4. Seven contiguous amino acids of the CED-3 L2′ fragment (Pro390–Leu391–Phe392–Asn393–Phe394–Leu395–Gly396) were unambiguously assigned (Fig. 4B). Correct assignment of these amino acids was further confirmed by three related structures in which Leu391 and Leu395 were both or individually replaced by selenomethionine to allow observation of the selenium anomalous signals in the L2′ loop fragments (Fig. 4C; Supplemental Table S2).
Figure 4.

Crystal structure of the CED-4 apoptosome bound to a L2′ loop fragment of CED-3. (A) Overall structure of the CED-4 apoptosome bound to a L2′ loop fragment of CED-3, viewed from the wide opening side of the funnel-shaped CED-4 apoptosome. The CED-4 apoptosome binds eight molecules of the L2′ loop fragment. (B) A close-up view of the CED-3 L2′ loop fragment bound to CED-4. Seven contiguous amino acids (residues 390–396) were unambiguously assigned. (C) Confirmation of the L2′ sequence assignment by selenium anomalous signals. The two Leu residues in the L2′ loop fragment, Leu391 and Leu395, were singly or doubly mutated to Met for generation of selenium anomalous signals (colored magenta and shown at 3σ). 2Fo − Fc electron density (light blue) for the L2′ loop fragment is displayed at 1.0 sec. (D) A cut-through section of the side view of the CED-4 apoptosome bound to the CED-3 L2′ fragment. (E) The neighboring L2′ loop fragments within the CED-4 apoptosome are separated by distances ranging from 30 to 83 Å. This and all other structural figures were prepared using PyMol (http://www.pymol.org).
Most notably, the L2′ loop of CED-3 is bound inside the hutch of the CED-4 apoptosome (Fig. 4D). The eight fragments of the CED-3 L2′ loop are separated from each other with distances ranging from 30 to 80 Å (Fig. 4E). Obviously, placement of eight L2′ fragments into the hutch of the octameric CED-4 apoptosome is facilitated by the small size of the peptide fragment. The processed caspase domains of the CED-3 homodimer measure ∼62 Å in length, 57 Å in height, and 48 Å in thickness (Supplemental Fig. S3). Taking into account the actual size of the CED-3 caspase domain, only one CED-3 homodimer (or two CED-3 molecules) can be accommodated into the hutch of the CED-4 apoptosome without steric clash. This conclusion is fully consistent with previous biochemical analysis, which revealed that only two molecules of CED-3 bind to each CED-4 apoptosome (Qi et al. 2010). Because the binding element comes from the flexible L2′ loop, there is more than one possibility of placing the two CED-3 molecules into the contacts of the eight CED-4 molecules.
Five of the seven amino acids in the bound CED-3 fragment are hydrophobic and make van der Waals contacts to surrounding structural elements of CED-4. In particular, the aromatic side chain of Phe392 in CED-3 mediates a number of interactions with the side chains of Val382, Leu393, Phe463, and Leu464 in CED-4 (Fig. 5A). Leu391 and Leu395, located on either side of Phe392, also contribute to the interactions. To corroborate the structural findings, we generated mutations on CED-3 and CED-4, each designed to weaken their interactions. First, we created a CED-3 variant (residue 198–503, C358S, L391D, F392D, and N393D), involving mutation of three residues in the L2′ loop. Replacement of the catalytic residue Cys358 by Ser was required for the generation of stable CED-3 for biochemical assays. As discussed previously, this mutant CED-3 nearly crippled its interaction with CED-4, while CED-3 without mutations in the L2′ loop retained stable association with CED-4 (Supplemental Fig. S2). Next, we generated a mutant CED-4 (A394W) that had a small amino acid, Ala394, in the hydrophobic CED-3-binding pocket replaced by a bulky residue, Trp. Presumably, Trp at this location may destabilize the CED-3–CED-4 interactions by pushing out the CED-3 L2′ element (Supplemental Fig. S4). Supporting this analysis, the interaction between CED-3 and CED-4 is markedly weakened by the mutation A394W in CED-4 (Fig. 5B). We also generated a number of other CED-4 mutants, targeting Val382, Leu393, Phe463, or Leu464; unfortunately, none of these CED-4 mutants was soluble, defying further biochemical characterization (data not shown).
Figure 5.

Recognition of the CED-3 L2′ loop by CED-4 is required for the stable association of the CED-3–CED-4 complex. (A) A close-up view on the predominantly hydrophobic interface between CED-4 and the CED-3 L2′ loop fragment. The interface involves four hydrophobic residues from CED-4 (Val382, Leu393, Phe463, and Leu464) and two from CED-3 (Phe392 and Leu391). (B) The mutation A394W in CED-4 led to weakened interaction between CED-3 and the CED-4 apoptosome, as judged by gel filtration. Compared with the wild-type (WT) CED-4 (left panels), the CED-4 A394W variant exhibited a markedly diminished ability to interact with CED-3 (residues 198–503 and C358S) (right panels).
Requirement of specific CED-4 association for CED-3 activation and activity
The CED-4 apoptosome can stimulate both autoproteolytic processing of the CED-3 zymogen and the protease activity of mature CED-3 caspase (Yan et al. 2005; Qi et al. 2010). We predicted that both aspects may depend on the specific association between the L2′ loop of CED-3 and the CED-4 apoptosome. First, we investigated the impact of loss of this specific association on the autocatalytic processing of the CED-3 zymogen. Under conditions in which CED-3 alone undergoes little autocatalytic processing, increasing concentrations of wild-type CED-4 stimulated the autocatalytic processing of CED-3 in a concentration-dependent manner (Fig. 6A, lanes 1–5). In contrast, the mutant CED-4 (A394W), which exhibited nearly crippled interaction with CED-3, failed to stimulate the autocatalytic processing of CED-3 (Fig. 6A, lanes 6–9).
Figure 6.
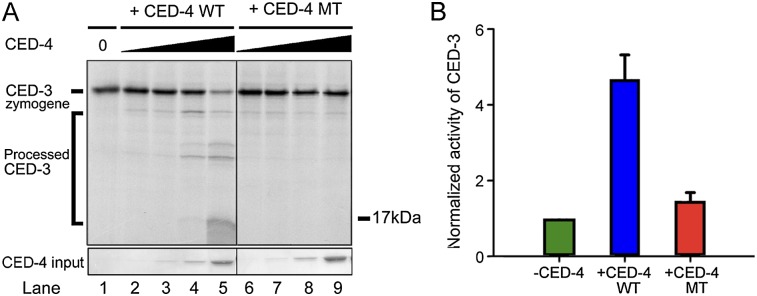
Recognition of the CED-3 L2′ loop by CED-4 is required for both efficient CED-3 activation and enhanced protease activity. (A) Autocatalytic processing of the CED-3 zymogen is greatly facilitated by the wild-type (WT) CED-4 but not the CED-4 A394W variant. Shown here is a representative autoradiograph. The full-length wild-type CED-3 protein was in vitro translated in the presence of 35S-Met. (B) The mutation A394W in the CED-4 hydrophobic pocket nearly abrogated the ability of the CED-4 apoptosome to stimulate the protease activity of the processed CED-3. DEVD-AMC was used as the substrate.
Next, we examined the impact on the catalytic activity of mature CED-3 protease using the fluorogenic substrate DEVD-AMC. As reported previously (Qi et al. 2010), the processed CED-3 exhibited a basal level of protease activity that was markedly stimulated by the presence of the full-length wild-type CED-4 (Fig. 6B). In contrast, the protease activity of the processed CED-3 remained largely unchanged in the presence of the mutant CED-4 (A394W), which exhibited crippled interaction with CED-3. These results, together with the findings that mutations in the L2′ loop of CED-3 abolish both CED-4 association and CED-4-mediated stimulation of CED-3 protease activity (Fig. 2C; Supplemental Fig. S2), demonstrate that specific association between CED-4 and CED-3 is essential in the activation process of CED-3.
To further corroborate this conclusion, we examined whether the free L2′ peptide fragment could interfere with CED-4-mediated stimulation of CED-3 protease activity. In agreement with our prediction, increasing concentrations of the L2′ peptide resulted in progressively diminished stimulation of CED-3 protease activity in the presence of the CED-4 apoptosome (Supplemental Fig. S5). Consistent with this observation, the L2′ loop peptide at high concentration interfered with the binding of CED-3 to the CED-4 apoptosome (data not shown).
Discussion
The structural and biochemical findings reported in this study, together with previous knowledge (Yan et al. 2004; Yan et al. 2005; Qi et al. 2010), allow us to propose a molecular model on the activation of the cell-killing protease CED-3 in C. elegans (Fig. 7). During homeostasis, the CED-3 zymogen exists as a monomer, and an asymmetric homodimer of CED-4 is sequestered by one molecule of CED-9. Upon sensing cell death signals, EGL-1 is transcriptionally activated and binds to CED-9, causing allosteric changes in CED-9 that ultimately result in the release of CED-4. The freed CED-4 dimer oligomerizes to form an octameric CED-4 complex: the CED-4 apoptosome, which recruits two molecules of the CED-3 zymogen. Once bound in the hutch of the CED-4 apoptosome, the caspase domains of the two CED-3 zymogen molecules have a greatly enhanced opportunity to homodimerize, thus undergoing autoproteolytic activation. Importantly, the two processed CED-3 molecules remain bound to the CED-4 apoptosome together as a holoenzyme with markedly higher protease activity compared with that of the free, processed CED-3. Specific association between the L2′ loop of CED-3 and the hydrophobic pocket of CED-4 serves to enhance dimerization of the CED-3 caspase domains.
Figure 7.

A mechanistic model of the regulation of PCD initiation in C. elegans. In nonapoptotic cells, CED-4 is sequestered by CED-9, unable to form an oligomeric apoptosome for CED-3 activation. In dying cells, EGL-1 is transcriptionally activated, binds CED-9, and causes allosteric changes in CED-9 that no longer allow CED-4 association. The freed CED-4 forms an apoptosome, which recruits the CED-3 zymogen. The caspase domains of two CED-3 molecules are likely bound within the hutch of the funnel-shaped CED-4 apoptosome, where dimerization and subsequent autoprocessing of CED-3 are greatly facilitated. The processed CED-3 remains bound to the CED-4 apoptosome as a holoenzyme, mainly via interaction between the L2′ loop of CED-3 and a hydrophobic surface pocket within the hutch of the CED-4 apoptosome.
The available structural information on CED-3 and CED-4 also allows us to rationalize a large number of in vivo mutations that have been reported for ced-3 or ced-4 (Yuan and Horvitz 1990; Yuan et al. 1993; Shaham et al. 1999; Sarin et al. 2007). We chose 16 missense mutations in CED-3 and three missense mutations in CED-4 and mapped these mutations onto the structures of CED-3 and CED-4 (Supplemental Fig. 6). On the basis of CED-3 and CED-4 structures, these mutations can be classified into two classes: functional and structural. The structural mutations are predicted to be detrimental to the structural stability of the proteins; this class is exemplified by L2F, G162E, and S163F in CED-4 (Supplemental Fig. 6A). The functional mutations are thought to disrupt the functions of CED-3 or CED-4; this class includes many missense mutations that map to the vicinity of the protease active site of CED-3 (Supplemental Fig. 6B).
The L2′ loop of CED-3 plays a major role in association with the CED-4 apoptosome because deletion of the L2′ loop crippled this association (Fig. 2A). A caspase-9 variant with the engrafted L2′ loop from CED-3 was found to form a stable complex with the CED-4 apoptosome (Fig. 3A). Consequently, this caspase-9 variant exhibited higher protease activity in the presence of CED-4 (Fig. 3B). Despite the important contribution from the L2′ loop, we cannot rule out the possibility that additional surface elements of CED-3 might be involved in binding to the CED-4 apoptosome. For example, a surface element of CED-3 might weakly associate with CED-4 to strengthen the interaction. Consistent with this speculation, the mutation A394W in CED-4 reduced, but failed to abrogate, the association between CED-3 and CED-4 (Fig. 5B). The weakened interaction between the L2′ loop of CED-3 and the hydrophobic pocket of the CED-4 A394W variant might cooperate with the yet-to-be-identified weak interface to retain some association. A similar case was reported previously for the association between caspase-9 and the BIR3 domain of XIAP (Shiozaki et al. 2003), where a major interaction between the ATPF motif of caspase-9 and a conserved surface groove of XIAP-BIR3 is strengthened by a weak interaction involving the caspase-9 dimerization interface and a C-terminal helix of XIAP-BIR3. Mutation of the latter interaction had no obvious effect on the binding between caspase-9 and XIAP-BIR3.
The CARD domain is known to play an important role in the activation of initiator caspases. The Apaf-1 CARD is responsible for the recruitment of caspase-9 through direct interaction with the CARD domain of caspase-9 (Qin et al. 1999). What is the function of the CARD domain in CED-3? Intriguingly, the free CED-3 CARD failed to form a stable complex with the isolated CED-4 CARD (WJ Huang, unpubl.), suggesting a variation of the Apaf-1–caspase-9 interaction. Nonetheless, CED-3 CARD formed a stable complex with the CED-4 apoptosome (Supplemental Fig. S7). We speculate that the CARD domain of CED-3 might play an important role in the recruitment of the CED-3 zymogen in vivo, perhaps through strengthened interactions. The role of the CED-3 CARD in CED-3 activation awaits further characterization.
One unanswered question is whether the Apaf-1-mediated apoptosome may employ a similar strategy to activate caspase-9. Specifically, would caspase-9 be bound within the hutch of the Apaf-1 apoptosome? The primary sequences of CED-4 and the Apaf-1 N-terminal half share ∼45% similarity (Supplemental Fig. S8), and the caspase domain of CED-3 and caspase-9 share ∼49% sequence similarity. Structural overlay and comparison (Supplemental Fig. S9) reveal a similarly conserved hydrophobic pocket in Apaf-1, which comprises hydrophobic amino acids Val372, Tyr383, Phe444, and Leu445. Except for Tyr383, the other three residues are identical between Apaf-1 and CED-4. This analysis raises the possibility that Apaf-1 might employ a similar interface to recruit caspase-9. However, given considerable sequence conservation between Apaf-1 and CED-4, the hydrophobic pocket in Apaf-1 could just be a pure coincidence and have nothing to do with the activation of caspase-9. Examination of this scenario requires further experimental investigation.
During apoptosis, the effector caspases (such as caspase-3 and caspase-7) are cleaved and activated by upstream initiator caspases (such as caspase-8 and caspase-9). Intrachain cleavage of the effector caspase allows free movement of the L2′ loop, which enables formation of a productive conformation at the active site and hence activation (Chai et al. 2001; Riedl et al. 2001). Activation of the initiator caspases, on the other hand, is mediated by upstream adaptor complexes. Caspase-8 and caspase-9 are activated by the membrane-associated death-inducing signaling complex (DISC) and the Apaf-1 apoptosome, respectively. Despite rigorous investigation in the past 15 years, how initiator caspases are activated by these adaptor complexes remains mechanistically unknown. The prevailing hypothesis is induced proximity (Salvesen and Dixit 1999) or the proximity-driven dimerization (Boatright et al. 2003). Unfortunately, this hypothesis merely states the fact that has been known for two decades; that is, active caspases are homodimers (Walker et al. 1994; Wilson et al. 1994), and overexpression of caspases (hence close proximity) readily causes their autoactivation. The crux of the problem—how close proximity drives caspase dimerization—remains unknown for any initiator caspase. In this study and other published work, we attempted to address this question by examining how, exactly, the CED-4 apoptosome facilitates the autoactivation of CED-3. Our finding argues against the conventional wisdom. First, the octameric CED-4 apoptosome recruits only two molecules of CED-3, not eight. Thus, close proximity is not a consequence of multiple molecules but only two being brought into the adaptor complexes. The conventional wisdom that an oligomeric adaptor complex recruits an equimolar amount of initiator caspases is clearly incorrect in the case of CED-3 activation. Second, the recognition between the adaptor complex and initiator caspase may not simply entail close proximity. Both the CARD domain and the caspase domain of CED-3 are capable of forming a stable complex with the CED-4 apoptosome. We suspect that additional interfaces are required for the recognition of the CED-3 caspase domain. The role of multiple interfaces is most likely to orient the initiator caspase for a specific conformation, allowing the highest probability of homodimerization. In this scenario, it is really induced conformation (Shi 2002a,b), not induced proximity, that may more faithfully represent the activation mechanism of initiator caspases.
Materials and methods
Protein preparation
All fragments and point mutations were constructed based on a standard PCR-based strategy. CED-4 was purified as described (Yan et al. 2005). All clones of CED-3 were expressed in Escherichia coli BL21 (DE3) and affinity-purified by a C-terminal 6xHis tag. Cells were grown at 37°C, and protein expression was optimized at 15°C. After overnight induction by IPTG, cells were collected and lysed in a buffer containing 500 mM NaCl and 25 mM HEPES (pH 7.5). Soluble protein was purified using Ni-NTA affinity resin followed by gel filtration (Superdex-200). To facilitate crystallization of the CED-4 apoptosome bound to the CED-3 L2′ loop fragment, we fused the L2′ loop sequence (residues 389–409) to the C terminus of CED-4 via a linker sequence (GSGSAGSAAGS). The CED-4–CED-3 fusion protein was expressed and purified similarly to CED-4.
Crystallization and structure determination of CED-3
Crystals of the processed CED-3 (198–374, 389–503, and C358S) were grown at 22°C using the hanging-drop vapor diffusion method. The well buffer contained 0.1 M HEPES (pH 7.5), 20% (w/v) PEG3350, and 0.2 M (NH4)2SO4. These crystals appeared immediately and grew to full size within 12 h, with a typical dimension of 0.2 × 0.2 × 0.4 mm3. The crystals belong to the space group I41 and contain two molecules of CED-3 per asymmetric unit. The unit cell has a dimension of a = b = 121 Å, c = 58 Å, and α = β = γ = 90°. Native data were collected on the A1 beamline of the Cornell High-Energy Synchrotron Source. The structure was determined by molecular replacement using residues 45–276 of caspase-3 from Protein Data Bank (PDB) entry 1CP3 (Mittl et al. 1997) using the program Phaser (Storoni et al. 2004). The two molecules formed a tightly associated dimer in the asymmetric unit. The structure was refined to a maximum resolution of 2.65 Å with CNS (Brunger et al. 1998) and Phenix (Adams et al. 2002) and rebuilt using O (Jones et al. 1991) and COOT (Emsley and Cowtan 2004). In the final stages of refinement, four TLS groups were used in conjunction with grouped B factor refinement with one group per residue. Refinement statistics are given in Supplemental Table S1. Ramachandran statistics were calculated by MolProbity.
Crystallization of the CED-4–CED-3 complex
Crystals of the CED-4–CED-3 L2′ loop complex were grown at 18°C using the hanging-drop vapor diffusion method. The crystallization condition contained 0.6–0.8 M sodium acetate, 0.1 M HEPES (pH 7.5), and 0.1 M sodium fluoride. The crystals grew to full size overnight, with a typical dimension of 0.05 × 0.05 × 0.5 mm3. Fresh crystals were quickly transferred to a cryoprotectant buffer containing 25% (w/v) ethylene glycol before snap-freezing in liquid nitrogen prior to data collection.
Data collection and structure determination for the CED-4–CED-3 complex
The diffraction images were collected at beamline BL41XU (Spring-8) and BL17U (Shanghai Synchrotron Radiation Facility [SSRF]) and processed with the HKL2000 package (Otwinowski and Minor 1997). Further processing was carried out using programs from the CCP4 suite (Collaborative Computational Project, Number 4 1994). Data collection statistics are summarized in Supplemental Table S2. The atomic coordinates of CED-4 from the PDB entry 3LQQ (Qi et al. 2010) were used for molecular replacement with the program Phaser (McCoy et al. 2005). Manual model building was done using the program COOT (Emsley and Cowtan 2004). The model was refined in the program Phenix (Adams et al. 2002).
In vitro translation of CED-3
For in vitro translation of CED-3, the experiments were performed as described (Yan et al. 2005).
Caspase assay for CED-3 and caspase-9
The fluogenic peptides Ac-DEVD-AMC and Ac-LEHD-AFC were used as substrates to measure the protease activity of CED-3 variants and caspase-9 variants, respectively. For each assay, 250 μM substrate was used in the buffer containing 25 mM HEPES (pH 7.4) and 500 mM NaCl.
PDB deposition
The atomic coordinates are being deposited in the PDB with accession codes 4M9R, 4M9S, 4M9X, 4M9Y, and 4M9Z for CED-3, CED-4 bound to the CED-3 fragment (PMFNFMG), CED-4 bound to the CED-3 fragment (PLFNFLG), CED-4 bound to the CED-3 fragment (PLFNFMG), and CED-4 bound to the CED-3 fragment (PMFNFLG), respectively.
Acknowledgments
We thank J. He and S. Huang at SSRF for on-site assistance. This work was supported by funds from the Ministry of Science and Technology (grant no. 2009CB918801) and the National Natural Science Foundation (30888001, 31021002, and 31130002).
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.224428.113.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC 2002. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr 58: 1948–1954 [DOI] [PubMed] [Google Scholar]
- Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, et al. 2003. A unified model for apical caspase activation. Mol Cell 11: 529–541 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Chai J, Wu Q, Shiozaki E, Srinivasula SM, Alnemri ES, Shi Y 2001. Crystal structure of a procaspase-7 zymogen: Mechanisms of activation and substrate binding. Cell 107: 399–407 [DOI] [PubMed] [Google Scholar]
- Chen F, Hersh BM, Conradt B, Zhou Z, Riemer D, Gruenbaum Y, Horvitz HR 2000. Translocation of C. elegans CED-4 to nuclear membranes during programmed cell death. Science 287: 1485–1489 [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM 1997. Interaction of CED-4 with CED-3 and CED-9: A molecular framework for cell death. Science 275: 1122–1126 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Conradt B, Horvitz HR 1998. The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93: 519–529 [DOI] [PubMed] [Google Scholar]
- del Peso L, González VM, Nuñez G 1998. Caenorhabditis elegans EGL-1 disrupts the interaction of CED-9 with CED-4 and promotes CED-3 activation. J Biol Chem 273: 33495–33500 [DOI] [PubMed] [Google Scholar]
- del Peso L, Gonzalez VM, Inohara N, Ellis RE, Nunez G 2000. Disruption of the CED-9.CED-4 complex by EGL-1 is a critical step for programmed cell death in Caenorhabditis elegans. J Biol Chem 275: 27205–27211 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K 2004. Coot: Model-building tools for molecular graphics. Acta Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR 1994. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76: 665–676 [DOI] [PubMed] [Google Scholar]
- Horvitz HR 2003. Worms, life, and death (Nobel lecture). ChemBioChem 4: 697–711 [DOI] [PubMed] [Google Scholar]
- Horvitz HR, Shaham S, Hengartner MO 1994. The genetics of programmed cell death in the nematode Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol 59: 377–385 [DOI] [PubMed] [Google Scholar]
- Irmler M, Hofmann K, Vaux D, Tschopp J 1997. Direct physical interaction between the Caenorhabditis elegans ‘death proteins’ CED-3 and CED-4. FEBS Lett 406: 189–190 [DOI] [PubMed] [Google Scholar]
- James C, Gschmeissner S, Fraser A, Evan GI 1997. CED-4 induces chromatin condensation in Schizosaccharomyces pombe and is inhibited by direct physical association with CED-9. Curr Biol 7: 246–252 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ 2005. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr 61: 458–464 [DOI] [PubMed] [Google Scholar]
- Mittl PR, Di Marco S, Krebs JF, Bai X, Karanewsky DS, Priestle JP, Tomaselli KJ, Grutter MG 1997. Structure of recombinant human CPP32 in complex with the tetrapeptide acetyl-Asp–Val–Ala–Asp fluoromethyl ketone. J Biol Chem 272: 6539–6547 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Parrish J, Metters H, Chen L, Xue D 2000. Demonstration of the in vivo interaction of key cell death regulators by structure-based design of second-site suppressors. Proc Natl Acad Sci 97: 11916–11921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi S, Pang Y, Hu Q, Liu Q, Li H, Zhou Y, He T, Liang Q, Liu Y, Yuan X, et al. 2010. Crystal structure of the Caenorhabditis elegans apoptosome reveals an octameric assembly of CED-4. Cell 141: 446–457 [DOI] [PubMed] [Google Scholar]
- Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y 1999. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399: 549–557 [DOI] [PubMed] [Google Scholar]
- Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS 2001. Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci 98: 14250–14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Savesen GS, Bode W 2001. Structural basis for the activation of human procaspase-7. Proc Natl Acad Sci 98: 14790–14795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM 1999. Caspase activation: The induced-proximity model. Proc Natl Acad Sci 96: 10964–10967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S, O'Meara MM, Flowers EB, Antonio C, Poole RJ, Didiano D, Johnston RJ Jr, Chang S, Narula S, Hobert O 2007. Genetic screens for Caenorhabditis elegans mutants defective in left/right asymmetric neuronal fate specification. Genetics 176: 2109–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshagiri S, Miller LK 1997. Caenorhabditis elegans CED-4 stimulates CED-3 processing and CED-3-induced apoptosis. Curr Biol 7: 455–460 [DOI] [PubMed] [Google Scholar]
- Shaham S, Reddien PW, Davies B, Horvitz HR 1999. Mutational analysis of the Caenorhabditis elegans cell-death gene ced-3. Genetics 153: 1655–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y 2002a. Apoptosome: The cellular engine for the activation of caspase-9. Structure 10: 285–288 [DOI] [PubMed] [Google Scholar]
- Shi Y 2002b. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 9: 459–470 [DOI] [PubMed] [Google Scholar]
- Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y 2003. Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell 11: 519–527 [DOI] [PubMed] [Google Scholar]
- Spector MS, Desnoyers S, Hoeppner DJ, Hengartner MO 1997. Interaction between the C. elegans cell-death regulators CED-9 and CED-4. Nature 385: 653–656 [DOI] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ 2004. Likelihood-enhanced fast rotation functions. Acta Crystallogr 60: 432–438 [DOI] [PubMed] [Google Scholar]
- Walker NP, Talanian RV, Brady KD, Dang LC, Bump NJ, Ferenz CR, Franklin S, Ghayur T, Hackett MC, Hammill LD 1994. Crystal structure of the cysteine protease interleukin-1b-converting enzyme: A (p20/p10)2 homodimer. Cell 78: 343–352 [DOI] [PubMed] [Google Scholar]
- Wilson KP, Black J-A, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, et al. 1994. Structure and mechanism of interleukin-1b converting enzyme. Nature 370: 270–275 [DOI] [PubMed] [Google Scholar]
- Wu D, Wallen HD, Nunez G 1997. Interaction and regulation of subcellular localization of CED-4 by CED-9. Science 275: 1126–1129 [DOI] [PubMed] [Google Scholar]
- Xue D, Shaham S, Horvitz HR 1996. The Caenorhabditis elegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev 10: 1073–1083 [DOI] [PubMed] [Google Scholar]
- Yan N, Gu L, Kokel D, Chai J, Li W, Han A, Chen L, Xue D, Shi Y 2004. Structural, biochemical, and functional analyses of CED-9 recognition by the proapoptotic proteins EGL-1 and CED-4. Mol Cell 15: 999–1006 [DOI] [PubMed] [Google Scholar]
- Yan N, Chai J, Lee ES, Gu L, Liu Q, He J, Wu JW, Kokel D, Li H, Hao Q, et al. 2005. Structure of the CED-4–CED-9 complex provides insights into programmed cell death in Caenorhabditis elegans. Nature 437: 831–837 [DOI] [PubMed] [Google Scholar]
- Yan N, Huh JR, Schirf V, Demeler B, Hay BA, Shi Y 2006a. Structure and activation mechanism of the Drosophila initiator caspase Dronc. J Biol Chem 281: 8667–8674 [DOI] [PubMed] [Google Scholar]
- Yan N, Xu Y, Shi Y 2006b. 2:1 Stoichiometry of the CED-4–CED-9 complex and the tetrameric CED-4: Insights into the regulation of CED-3 activation. Cell cycle 5: 31–34 [DOI] [PubMed] [Google Scholar]
- Yang X, Chang HY, Baltimore D 1998. Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science 281: 1355–1357 [DOI] [PubMed] [Google Scholar]
- Yuan JY, Horvitz HR 1990. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol 138: 33–41 [DOI] [PubMed] [Google Scholar]
- Yuan J, Horvitz HR 1992. The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development 116: 309–320 [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR 1993. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell 75: 641–652 [DOI] [PubMed] [Google Scholar]