

Abstract
Tumor necrosis factor-alpha (TNF-α), a member of the TNF superfamily, was the first cytokine to be evaluated for cancer biotherapy. However, the clinical use of TNF-α is severely limited by its toxicity. Currently, TNF-α is administered only through locoregional drug delivery systems such as isolated limb perfusion and isolated hepatic perfusion. To reduce the systemic toxicity of TNF-α, various strategies have been explored over the last several decades. This review summarizes current state-of-the-art targeted cancer therapy using TNF-α. Passive targeting, cell-based therapy, gene therapy with inducible or tissue-specific promoters, targeted polymer-DNA complexes, tumor pre-targeting, antibody-TNF-α conjugate, scFv/TNF-α fusion proteins, and peptide/TNF-α fusion proteins have all been investigated to combat cancer. Many of these agents are already in advanced clinical trials. Molecular imaging, which can significantly speed up the drug development process, and nanomedicine, which can integrate both imaging and therapeutic components, has the potential to revolutionize future cancer patient management. Cooperative efforts from scientists within multiple disciplines, as well as close partnerships among many organizations/entities, are needed to quickly translate novel TNF-α-based therapeutics into clinical investigation.
Keywords: tumor necrosis factor-alpha (TNF-α), cancer therapy, fusion protein, gene therapy, TNF superfamily, targeted delivery
Introduction
Tumor necrosis factor-alpha (TNF-α) is a cytokine of the TNF superfamily, composed of at least 20 members (Mocellin et al. 2005). TNF-α is primarily produced as a 212 amino acid transmembrane protein which forms stable homotrimers. The soluble homotrimeric TNF-α (157 amino acid residues per monomer) can be released via proteolytic cleavage by a metalloprotease, the TNF-α converting enzyme. The C-terminus of TNF-α is embedded in the base of the trimer, while the N-terminus is relatively free of the base structure (Schottelius et al. 2004). Therefore, the N-terminal residues do not participate in the trimer interactions and are not crucial for the biological activities of TNF-α.
Two receptors, TNFR1 and TNFR2, bind to TNF-α (Zhang, 2004). TNFR1, constitutively expressed in most tissues, can be activated by both membrane-bound and soluble trimeric forms of TNF-α. TNFR2, generally found in cells of the immune system, responds only to the membrane-bound form of TNF-α homotrimer. Mounting experimental evidence has revealed that TNFR1 initiates the majority of TNF-α’s biological activities (Chen and Goeddel, 2002). Upon TNF-α binding, TNFRs also form homotrimers which cause conformational changes to the receptor. A series of intracellular events then occur which can lead to the activation of three major signalling cascades: the nuclear factor kappa B (NF-κB) pathway, the mitogen-activated protein kinase (MAPK) pathway, and the induction of death signaling (Fig. 1).
Figure 1.

The TNF signalling pathway. DD: death domain; FADD: Fas-associated death domain; FAN: factor associated with neutral-sphingomyelinase; FLICE: FADD-like interleukin-1β-converting enzyme; JNK/SAPK: c-Jun N-terminal kinase/stress-activated protein kinase; MADD: MAPK-activating death domain; NF-κB: nuclear factor kappa B; NIK: NFκB-inducing kinase; RIP: receptor interacting protein; SMase: sphingomyelinase; TRADD: TNFR-associated death domain; TRAF-2: TNFR-associated factor-2.
Mainly produced by macrophages, TNF-α can also be found in a wide variety of cell types including lymphoid cells, mast cells, endothelial cells, cardiac myocytes, and fibroblasts (Spriggs et al. 1992). When initially isolated from the serum of mice treated with bacterial endotoxin, TNF-α was found to be capable of replicating the ability of endotoxin in inducing hemorrhagic tumor necrosis (Carswell et al. 1975). Cloning of the TNF-α gene in the mid-1980s led to the era of its clinical investigation (Aggarwal et al. 1985; Aggarwal et al. 1984; Shirai et al. 1985). TNF-α was the first cytokine to be employed for cancer biotherapy. Although systemic administration can inhibit tumor progression in mice, the clinical use of TNF-α is hindered by its severe dose-limiting toxicity. To avoid such toxicity, regional administration was used either as a stand alone therapy or in combination with certain chemotherapeutic agents (Mocellin et al. 2005). Currently, TNF-α is administered into patients only through locoregional drug delivery systems such as isolated limb perfusion (ILP) and isolated hepatic perfusion (IHP) (Alexander et al. 2000; Grunhagen et al. 2004; Grunhagen et al. 2006). Since both of these procedures are technically demanding which require surgery, they are primarily used for the treatment of locally advanced solid tumors such as limb-threatening soft tissue sarcomas, in-transit melanoma metastases, and primary or metastatic unresectable liver tumors.
Another approach to reduce the systemic toxicity of TNF-α is through tumor targeted delivery, where the ultimate goal is to deliver high doses of TNF-α to the tumor tissue for cell killing while sparing the normal tissue/organs. Various strategies have been explored for tumor targeted delivery of TNF-α over the last two decades, such as the use of inducible or tissue-specific viral vectors for cancer gene therapy and the generation of fusion proteins specific for certain molecular cancer markers. In this review, we will summarize the progress to date on targeted cancer therapy using TNF-α.
Passive Targeting
Tumor growth depends on the formation of new blood vessels to provide oxygen and nutrients (Folkman, 1971). Typically the newly formed tumor blood vessels are abnormal in their form and architecture, which makes them leakier than normal blood vessels (Hashizume et al. 2000). Furthermore, tumor tissues usually lack effective lymphatic drainage. Both of these characteristics can lead to the accumulation of macromolecular drugs and long-circulating nanoparticles in the tumor, termed the “enhanced permeability and retention (EPR) effect” (Maeda et al. 2000). Since such tumor accumulation is not target specific, it is also called “passive targeting”.
Recombinant adenovirus (Ad) has been widely used as a gene delivery vehicle in mammalian cells because of its unparalleled gene transfer efficiency in vivo (Smith, 1995). However, the wide native tropism of Ad, which can lead to toxic side effects, is a major concern. Cell-specific gene delivery via Ad vectors has been achieved by both genetic and chemical approaches (Hedley et al. 2006; Kreppel and Kochanek, 2008; Niu et al. 2007; Noureddini and Curiel, 2005). Modification of an Ad vector with poly(ethylene glycol) (PEG) can prolong the circulation half-life, ablate its native tropism, and circumvent the inflammatory and humoral immune responses. PEGylated Ad (PEG-Ad) vectors at various modification ratios, encoding TNF-α, were injected intravenously (i.v.) into tumor-bearing mice to determine the blood kinetics, viral distribution, and gene expression patterns (Gao et al. 2007). As expected, the plasma half-life of PEG-Ad was longer than that of the unmodified Ad, and the accumulation of PEG-Ad in the tumor tissue increased with the PEGylation ratio. PEG-Ad encoding TNF-α exhibited stronger tumor-suppressive activity, as well as fewer hepatotoxic side effects, when compared with the unmodified Ad. Although this study suggested that systemic injection of PEG-Ad may have potential in cancer therapy, PEGylation of the Ad vectors also led to significant loss of infectivity due to the steric bulk of the PEG chains. In future studies, incorporation of tumor targeting moieties at the end of the PEG chain may partially restore the infectivity of the PEG-Ad vector.
CYT-6091 is a PEGylated colloidal gold-based nanotherapeutic that can be used for delivering TNF-α to solid tumors (Visaria et al. 2007). A few hours after i.v. injection of either TNF-α or CYT-6091, localized heating was performed and the therapeutic responses were assessed by growth delay and/or perfusion of the tumor. Although both TNF-α and CYT-6091 reduced tumor perfusion, TNF-α was toxic to the mice while CYT-6091 was not whether it was administered alone or in combination with hyperthermia. More importantly, combination of CYT-6091 with heat resulted in significant tumor regression when compared to either heating or CYT-6091 alone. Phase I clinical trials are currently ongoing to evaluate the safety, pharmacokinetics, and clinical efficacy of CYT-6091.
Although both these studies showed that PEGylation improved the therapeutic effect to a certain extent when compared with the non-PEGylated analog, the use of passive targeting alone is neither optimal nor sufficient for efficacious cancer therapy. Combination of both molecularly specific targeting and passive targeting will almost certainly give better clinical outcome.
Cell-Based Therapy
Cell-based therapy is an alternative to conventional cancer therapy regimes (Fazle Akbar et al. 2006; Lotze et al. 1997). Human tumor-infiltrating lymphocytes (TILs), retrovirally transduced with the TNF-α gene, have been explored for delivering TNF-α to the tumor site (Hwu et al. 1993). Disappointingly, the overall production of TNF-α was at least 30-fold lower than a transduced and highly selected tumor cell line control. To increase TNF-α production, the TILs were transduced with a mutated form of TNF-α (which lacks the trans-membrane region) to enhance its secretion into the endoplasmic reticulum. A modest increase in TNF-α production was observed. This early report demonstrated that TILs can be genetically modified to express and secrete certain proteins for potential cancer therapy applications. Subsequently, retinoic acid was used to upregulate TNF-α expression in retrovirally transduced TILs (Treisman et al. 1994). Again, only modest improvement was achieved.
In the clinical setting, patients with metastatic cancer could be vaccinated with genetically modified autologous tumor cells to stimulate the host immune response (Herr et al. 1994). Mimicking this situation, a poorly immunogenic murine sarcoma genetically engineered to secrete TNF-α was studied (Marincola et al. 1994). After implantation in animals bearing pulmonary metastases established with the parental cell line (without TNF-α transduction), a reduction in the number of pulmonary metastases was occasionally seen, however not reproducible. Significant growth inhibition of the modified cells was observed in animals bearing pulmonary metastases, and this effect was demonstrated to be tumor specific, TNF-α specific, and cellular immunity dependent. These findings suggested that genetically modified tumor cells may be used for the treatment of established cancer. However, much more future research and optimization will be needed before it can become a clinically useful therapeutic strategy.
Gene Therapy with Inducible or Tissue-Specific Promoters
The safety of gene therapy has always been a concern for scientists, clinicians, and the general public (McCormick, 2001). Therefore, gene therapy is generally performed through local administration. Additionally, various strategies have been investigated to improve the safety profile of gene therapy for clinical applications (Yi et al. 2005). For example, transcriptional targeting using cell type-specific promoters and enhancers can minimize the expression of transduced genes in other types of cells, thereby reducing potential side effects; Inducible expression systems (by radiation, chemotherapy, hormone, etc.) can be used to increase safety and efficacy; Co-transduction of a suicidal gene under the control of an inducible promoter can also serve as a safety feature, so that destruction of the transduced cells can be triggered if abnormal growth is observed. Cancer gene therapy using TNF-α has been explored using various approaches to avoid the dose-limiting toxicity. TNFerade, an Ad vector with a radiation-inducible promoter, is the most advanced in clinical development.
TNFerade
The discovery of radiation-inducible genes led to the concept and development of radiation targeted gene therapy, in which promoters of the radiation-inducible genes are used to drive the transcription of transgenes in response to radiation (Vilaboa and Voellmy, 2006). Since transcriptional regulation of the promoter/enhancer region of the early growth response gene 1 (egr-1) is activated by ionizing radiation (Datta et al. 1992), the DNA sequence from the promoter region of egr-1 was linked to a human TNF-α encoding sequence (Weichselbaum et al. 1994). Upon transfection into a human cell line, it was found that the cells have radiation-inducible TNF-α secretion. Animals treated with radiation and intratumoral injection of the transfected cells exhibited an increase in tumor ablation when compared with those treated with either radiation or the cells alone. Importantly, no increase in local or systemic toxicity was observed, which makes this approach a promising new paradigm for cancer gene therapy.
Preclinical studies carried out to characterize the toxicity and pharmacokinetics of the Ad vector, subsequently termed “TNFerade”, in conjunction with radiation in nude and immunocompetent mice revealed that TNFerade was well-tolerated (Rasmussen et al. 2002). High, sustained levels of TNF-α were found in the tumor homogenate with no “spillover” to the plasma. Radiation increased the intratumoral level of TNF-α by more than ten fold. After confirming that gene therapy with TNFerade, in combination with radiation, can lead to anti-cancer efficacy without systemic toxicity, clinical trials in many solid tumor types were carried out (Mezhir et al. 2006).
Weekly intratumoral administration of TNFerade for six weeks with concomitant radiation in patients with solid tumors did not cause any dose-limiting toxicity in a Phase I trial, although certain adverse effects (e.g. acute delirium, nausea, anemia, and pain) were observed (Senzer et al. 2004). Another Phase I study of TNFerade in patients with soft tissue sarcoma in the extremities was also considered to be safe (Mundt et al. 2004). Eleven out of thirteen patients showed objective or pathological tumor response, which clearly warranted additional studies in larger controlled prospective trials. In one report, patients with advanced solid tumors were subjected to postoperative and long-term follow-up after TNFerade treatment (McLoughlin et al. 2005). Therapeutic efficacy, as well as short-term and long-term safety, was observed.
Data from a Phase I study in a small number of melanoma patients suggested the potential of TNFerade to impact distal metastases following intratumoral injection. In a “bedside-to-bench” transition, this hypothesis was evaluated in a spontaneously metastatic, syngeneic murine melanoma model (MacGill et al. 2007). Comparison of the metastatic burden with the control groups indicated that combination of TNFerade and radiation suppressed metastasis formation in the lymph nodes. Experiments in TNFR knockout mice further suggested that the local and distal activities of TNFerade are largely dependent on a host-mediated response. Taken together, local therapy of a solid tumor with TNFerade can lead to reduced tumor metastasis, in addition to therapeutic effects on the treated lesion. TNFerade is currently in Phase II/III clinical testing.
Other conditionally active vectors
A few other vectors with inducible or tissue-specific promoters have been reported for TNF-α-based cancer therapy (Table 1). The multidrug resistance (MDR) gene (mdr1) can be induced by MDR-associated drugs (Labialle et al. 2002; Scotto and Johnson, 2001). The mdr1 promoter sequence has been linked to human TNF-α cDNA in a retroviral vector and transduced into human cancer cell lines (Walther et al. 1997). Upon treatment with various MDR-associated drugs, it was found that the mdr1 promoter-driven TNF-α expression is inducible which is dependent on the drug concentration and exposure time. In a later study, single doxorubicin and vincristine (both chemotherapeutic drugs) treatment of nude mice, inoculated with this inducible vector transduced tumors, led to drug-induced and time-dependent elevation of intratumoral TNF-α expression at both the mRNA and protein level (Walther et al. 2000). The drug-induced TNF-α expression also appeared to be more effective in inhibiting tumor growth than the combination of chemotherapy with constitutively TNF-α-expressing vectors.
Table 1.
The various strategies used for TNF-α-based cancer gene therapy
| Vector | Promoter | Specificity | Stage | References |
|---|---|---|---|---|
| Adenoviral | cytomegalovirus (CMV) |
none | preclinical | (Gao et al. 2007) |
| Adenoviral | early growth response gene 1 (egr-1) |
radiation inducible | Phase II/III trials | (MacGill et al. 2007; McLoughlin et al. 2005; Mezhir et al. 2006; Mundt et al. 2004; Rasmussen et al. 2002; Senzer et al. 2004; Weichselbaum et al. 1994) |
| Retroviral | multidrug resistance gene (mdr1) |
drug and heat inducible | preclinical | (Walther et al. 2007; Walther et al. 2000; Walther et al. 2002; Walther et al. 1997) |
| Adenoviral | uroplakinII (UPII) | urothelium-specific | preclinical | (Zhu et al. 2004) |
| Adenoviral | mucin 1 (MUC1) | integrin targeting and tumor selective |
preclinical | (Murugesan et al. 2007) |
| Adenoviral | CMV | tumor-associated glycoprotein targeting |
preclinical | (Wright et al. 1998) |
| Polymer- DNA complex |
none | transferrin receptor targeting |
preclinical | (Jiang et al. 2007; Kircheis et al. 2002a; Kircheis et al. 2002b; Kursa et al. 2003) |
Another report revealed that the same construct can also be activated by hyperthermia in a temperature- and time-dependent manner (Walther et al. 2002). However, the increase in TNF-α expression upon activation was only modest. When combined with doxorubicin, the heat-induced TNF-α expression was able to inhibit tumor growth in animals (Walther et al. 2007). These studies demonstrated the feasibility of using inducible expression of a chemotherapy-sensitizing cytokine to enhance the cytotoxicity of drugs in cancer therapy, which may be potentially useful in the clinic.
Driving therapeutic gene expression with tissue-specific promoters can potentially result in enhanced therapeutic effect with better specificity. The DNA fragment upstream of the uroplakinII (UPII, a urothelium-specific membrane protein) gene was investigated for targeted gene therapy of bladder cancer (Zhu et al. 2004). After identifying the specific fragment responsible for the tissue-specificity of the UPII promoter, and validating the tissue-specific expression of a reporter gene using this promoter, a recombinant Ad encoding the TNF-α gene driven by the UPII promoter was prepared. Intravesical inoculation of the vector inhibited tumor growth in an orthotopic human bladder cancer model. This strategy, potentially useful as a new therapeutic approach for bladder cancer, may also provide information on the molecular regulation of urothelial growth, differentiation, and pathological processes.
Combination of two modifications to the Ad vectors, capsid modification and expression control, was employed to selectively deliver TNF-α to the tumor tissue (Murugesan et al. 2007). Integrin αvβ3, a cell adhesion molecule, is overexpressed on activated endothelial cells and tumor cells but is not readily detectable in resting endothelial cells and most normal organ systems (Cai and Chen, 2006). The capsid fiber and the penton base of the Ad were modified to ablate their native receptor binding and enable integrin αvβ3 targeting, via incorporation of an arginineglycine-aspartic acid (RGD) motif (potent integrin αvβ3 antagonist (Cai et al. 2008)). To further increase the tumor selectivity, different promoters were incorporated into the capsid-modified Ad vector to drive the expression of a TNF-α gene (Murugesan et al. 2007). Both constitutive and potentially tumor selective promoters were evaluated in terms of the therapeutic efficacy, tumor selectivity, and safety. It was found that the RGD-modified Ad vectors containing the mucin 1 (MUC1, a transmembrane glycoprotein overexpressed in many tumor types (Gendler, 2001)) promoter exhibited anti-tumor activity in ovarian cancer xenograft models with little evidence of systemic TNF-α toxicity.
Although tissue-specific promoters have been used in many clinical trials of cancer gene therapy, the overall transcription efficiency achieved with these promoters is significantly weaker than the generally used viral promoters (Aguilar and Aguilar-Cordova, 2003; Takahashi et al. 2006). Improvement in gene expression efficiency, tumor specificity, and safety profile are three major tasks that have to be tackled in future preclinical/clinical studies. Combination of tumor-specific targeting and the use of inducible/tissue-specific promoters will likely lead to safer, more efficient future cancer therapeutics. Many molecular targets have been explored for target-specific delivery of TNF-α in cancer therapy (Table 2). The remainder of this review article will focus on molecularly specific delivery of TNF-α to the tumor.
Table 2.
Molecular targets that have been explored for TNF-α-based cancer therapy
| Target | Targeting ligand | Stage | Selected references |
|---|---|---|---|
| Transferrin receptor | transferrin | preclinical | (Kircheis et al. 2002a; Kircheis et al. 2002b) |
| Carcinoembryonic antigen (CEA) |
bispecific antibody, scFv |
preclinical | (Cooke et al. 2002; Robert et al. 1996) |
| Thy 1.1 | antibody | preclinical | (Gasparri et al. 1999; Moro et al. 1997) |
| Melanoma gp240 antigen |
antibody, scFv | preclinical | (Liu et al. 2004; Rosenblum et al. 1995; Rosenblum et al. 1991) |
| Tumor-associated glycoprotein (TAG-72) |
(Fab’)2 | preclinical | (Wright et al. 1998; Xiang et al. 1997) |
| Carbohydrate structure Lewis Y (LeY) |
scFv | preclinical | (Scherf et al. 1996) |
| Extradomain B of fibronectin |
scFv | preclinical | (Borsi et al. 2003; Halin et al. 2003) |
| Human epidermal growth factor receptor 2 (HER2) |
scFv | preclinical | (Lyu et al. 2008; Lyu and Rosenblum 2005; Rosenblum et al. 2000) |
| Integrin αvβ3 | peptide | preclinical | (Curnis et al. 2004; Wang et al. 2008) |
| CD13 | peptide | Phase I trial | (Crippa et al. 2008; Curnis et al. 2000; Curnis et al. 2002b) |
Targeted Polymer-DNA Complexes
Polymer-based conjugates have been explored for delivery of TNF-α to tumor sites. In one study, shielding of the polyethylenimine (PEI)-DNA complexes was achieved by PEGylation which prevents the complexes from non-specific interactions with blood components or normal cells (Kircheis et al. 2002b). Incorporation of transferrin at high densities can function not only as shielding but also as targeting ligands. Following systemic administration, the surface-shielded polymer-DNA complex caused hemorrhagic tumor necrosis and inhibition of tumor growth. The activity of TNF-α was confined to the tumor without systemic toxicity, suggesting that targeted gene delivery using such polymer-DNA complexes may be an attractive strategy for cancer therapy. A subsequent report expanded the therapeutic efficacy of these complexes to three murine tumor models of different tissue origins (Kircheis et al. 2002a). To further optimize the complex, PEG chains of varying size, as well as linear or branched PEI, were investigated for DNA conjugation at various ratios (Kursa et al. 2003). Transferrin was again incorporated as a targeting ligand for receptor-mediated cellular uptake. The optimized formulation, which contained TNF-α encoding plasmid DNA, was able to inhibit tumor growth in mouse models. Recently, the tumor inhibitory effect of a similar construct was also reported by another research group (Jiang et al. 2007). One intriguing phenomenon is that all these polymer-DNA complexes used transferrin as the targeting ligand. Other targeting ligands should be investigated in future studies, to find the optimal ligand/receptor pair for TNF-α-based gene therapy of various tumor types.
Tumor Pre-Targeting
A pre-targeting strategy can significantly increase tumor-to-background contrast over direct-targeting approaches. Typically, either the avidin/streptavidin-biotin pair or a bispecific antibody (BAb) is used (Gasparri et al. 1999; Sharkey et al. 2005). A certain waiting period is needed for the first agent to clear from the circulation and reach the tumor site. The second agent is then administered which will target and bind to the first agent, thus giving excellent tumor contrast if the system is designed properly.
A BAb directed against the carcinoembryonic antigen (CEA) and human TNF-α was constructed for tumor targeting (Robert et al. 1996). It was constructed by coupling the Fab’ fragment of an anti-CEA antibody to the Fab’ fragment of an anti-TNF-α antibody via a stable thioether linkage. Animal studies confirmed that the antigen specificity of the BAb was comparable to the parental antibodies, where the 125I-labeled BAb had greater than 20 percentage injected dose per gram of tissue (%ID/g) of tumor uptake in xenograft models. To direct TNF-α to the tumor, a two-step protocol was used. A variable dose of the 125I-labeled BAb was first injected, followed by injection of a constant dose of 131I-labeled TNF-α at a few days later. A modest increase in tumor TNF-α concentration, as well as its tumor retention time, was achieved when compared to direct TNF-α injection.
Tumor pre-targeting with biotinylated antibodies and avidin, followed by a delayed delivery of radiolabeled biotin, is currently used for cancer diagnosis and therapy in patients (Grana et al. 2002). A three-step approach to increase the local concentration and persistence of biotinylated human TNF-α was initially investigated in mouse lymphoma cells, transfected with the Thy 1.1 allele which serves as a tumor-associated antigen (Moro et al. 1997). Subsequently, intraperitoneal injection of a biotinylated anti-Thy 1.1 antibody and avidin in tumor-bearing mice increased the anti-tumor activity of systemically administered biotinylated TNF-α by at least five fold, without increasing the toxicity (Gasparri et al. 1999). Ex vivo analysis of tumor cells 24 h after treatment showed that biotinylated TNF-α persisted for several hours on the surface of pre-targeted tumors. The anti-tumor effect was related primarily to indirect mechanisms which involved a host-mediated response. These findings inspired the study of the structure-activity relationship of biotinylated TNF-α and the mechanism of its interaction with avidin and TNFRs on the tumor cells (Corti et al. 1998). It was suggested that biotinylated TNF-α trimers can slowly dissociate from the targeted cells in a bioactive form, through the trimer-monomer-trimer transition. The released TNF-α can also interact with TNFRs expressed on bystander cells, therefore cells other than those reached by the targeting antibody may also be affected in vivo.
Compared to these pre-targeting strategies, which can be quite technically challenging and hence not widely-used, direct tumor targeting using either chemical conjugates or fusion proteins has been extensively documented in the literature. A wide variety of targets (both tumor cell and tumor vasculature related) have been explored for tumor specific delivery of TNF-α. The targeting moieties span a wide range from intact antibodies (IgG, ~150 kDa) to small peptides (<1 kDa) (Fig. 2). Some of the fusion proteins are already being tested in clinical trials or are poised to enter clinical investigation.
Figure 2.

A variety of targeting ligands have been tested for tumor targeted delivery of TNF-α.
Antibody-TNF-α Conjugate
With the maturation of antibody production technologies over the last several decades, scientists can now raise antibodies against virtually any antigen of interest (Wu and Senter, 2005). Therefore, it is not surprising that tumor targeted delivery of TNF-α was first achieved using an antibody as the targeting moiety. A pioneering report of tumor targeted delivery of TNF-α appeared almost 2 decades ago (Rosenblum et al. 1991). A murine antibody ZME-018, which recognizes the melanoma gp240 antigen (a 240 kDa glycoprotein) present on melanoma cells, was chemically conjugated to TNF-α via a hetero-bifunctional cross-linking reagent. The ZME-018-TNF-α conjugate remained biologically active to antigen-expressing cells, more potent than TNF-α. Subsequent pharmacokinetic studies of the conjugate revealed that it cleared from the plasma biphasically, with half-lives similar to that of ZME-018, indicating that the low molecular weight TNF-α had little effect on the overall clearance rate (Rosenblum et al. 1995). Tissue distribution studies in tumor-bearing mice showed similar tumor localization pattern of the conjugate when compared to ZME-018. Treatment of tumor-bearing mice with ZME-018-TNF-α resulted in a statistically significant reduction in tumor growth rate over that of saline treated control animals.
These early studies demonstrated the feasibility of tumor targeted delivery of TNF-α using an antibody, which opened up new perspectives on TNF-α-based cancer therapy. However, the use of intact antibodies has several disadvantages. The long circulation half-life of antibody conjugates can cause a prolonged TNF-α exposure to normal organs. The field of antibody engineering has evolved rapidly in the past decade, fueled by novel technologies for the in vitro isolation of antibodies from combinatorial libraries and their functional expression in bacteria (Kim et al. 2005; Maynard and Georgiou, 2000). Many antibody fragments have since been investigated for tumor targeted delivery of TNF-α.
(Fab’)2/TNF-α Fusion Protein
The most straightforward approach to reduce the molecular weight of an antibody (and thereby speeding up its clearance from the circulation), without reducing its antigen binding affinity, is to remove the Fc region of the antibody. Recombinant antibody techniques were used to construct a fusion protein which contains TNF-α and a chimeric F(ab’)2 fragment, specific for a tumor-associated glycoprotein TAG-72 (Xiang et al. 1997). After purification, the fusion construct RM4/TNF-α retained both antigen binding affinity and TNF-α activity.
Interestingly, rather than systemically injecting the fusion protein for targeted cancer therapy, Ad-mediated gene therapy was investigated (Wright et al. 1998). A murine myeloma cell line was transfected with the light chain of the fusion protein, RM4/TNF-α. The Ad vector which contains a fused gene encoding the heavy chain of RM4/TNF-α was also constructed. In vitro transfection of the cells with the Ad resulted in significant level of RM4/TNF-α secretion. Intratumoral injection of the Ad vector with a repeated booster caused significant tumor regression in immunocompetent mice inoculated with the myeloma cell line. Besides tumor regression, the mice also developed protective immunity against a second challenge with the myeloma cells.
The molecular weight of the F(ab’)2 fragment is about 110 kDa, which still has a fairly long circulation half-life. The single chain variable fragment (scFv) is a fusion of the variable regions of the heavy and light chains of an antibody, linked together with a short peptide linker. Typically the scFv fragment retains the specificity of the original antibody, although it is monovalent instead of bivalent. With a molecular weight of about 28 kDa, the circulation half-life of a scFv fragment (less than a few hours) is much shorter than that of the intact antibody (several days) or the F(ab’)2 fragment (typically about a day), making it suitable for rapid tumor targeted delivery of TNF-α.
ScFv/TNF-α Fusion Proteins
A variety of antigens have been targeted for cancer therapy using scFv/TNF-α fusion proteins, including the carbohydrate structure Lewis Y (LeY), the extradomain B of fibronectin, the human epidermal growth factor receptor 2 (HER2), CEA, and the gp240 antigen.
Targeting the LeY antigen
A fusion protein composed of TNF-α fused at its C-terminus to the scFv fragment of an antibody, specific for the LeY antigen present on many human cancer cells, was reported (Scherf et al. 1996). The monomeric fusion protein has similar antigen binding affinity as the scFv itself. Since the C-terminus of TNF-α is involved in receptor binding, the fusion significantly diminished the binding affinity of the construct to TNFRs which reduced its toxicity. Specific targeting with the scFv fragment was able to compensate for this effect and exhibited selective killing of TNF-α sensitive, antigen-expressing cancer cells. The antigen specific and TNF-α specific toxicity of the fusion protein was demonstrated both in cell culture and in mouse tumor models, which caused significant tumor regression at doses that are not systemically toxic to the mice. Fusion of TNF-α to the targeting moieties is almost always at the N-terminus in other literature reports. Although this study demonstrated the feasibility of C-terminal fusion, no follow-up studies has since been reported.
Targeting angiogenesis
Angiogenesis, the sprouting of new blood vessels from preexisting ones, is a characteristic feature of many aggressive solid tumors (Cai and Chen, 2008). One established marker of angiogenesis is the extradomain B of fibronectin, present almost exclusively in the modified extracellular matrix that surrounds the newly-formed tumor blood vessels (Ebbinghaus et al. 2004). A fusion protein composed of murine TNF-α and a scFv fragment (L19) which recognizes the extradomain B of fibronectin was constructed (Borsi et al. 2003). L19/TNF-α, forms a homotrimer which is both immunoreactive and biologically active. Radiolabeled L19/TNF-α selectively targeted the tumor neovasculature in tumor-bearing mice, with an impressive tumor-to-blood ratio of about 700 at 48 h post-injection. L19/TNF-α had greater anti-cancer efficacy in mouse models than both TNF-α and a control fusion protein. The therapeutic efficacy of L19/TNF-α could also be enhanced when used in combination with melphalan (a chemotherapeutic drug) or another fusion protein, L19/IL-2.
Since L19/TNF-α was not able to cause complete remission of established tumors in immunocompetent murine models, combinations of L19/TNF-α and L19/IL-12, as well as a triple fusion protein of IL-12, L19, and TNF-α, were investigated (Halin et al. 2003). Disappointingly, the triple fusion protein failed to target the tumor in mouse models. However, the combination of L19/IL-12 and L19/TNF-α displayed potent synergistic anti-cancer activity, eradicating the F9 teratocarcinomas grafted in immunocompetent mice. In another report, a single systemic administration of L19/TNF-α in combination with melphalan induced complete tumor eradication in mice, as well as triggering a specific T cell-based immune response (Balza et al. 2006). Such an immune response was able to protect the animals from a second tumor challenge, and from challenges with syngeneic tumor cells of a different histologic origin.
Targeting HER2
Overexpression of the HER family members has been implicated in many cancer types because of their pivotal roles in signaling pathways that regulate cellular proliferation, differentiation, motility, and survival (Cai et al. 2007d; Niu et al. 2008). The HER family consists of four members: HER1 (also known as the epidermal growth factor receptor), HER2, HER3, and HER4 (Casalini et al. 2004; Mass, 2004).
HER2 overexpression in breast cancer and certain other tumor types has been well documented (Ferretti et al. 2007; Hicks and Kulkarni, 2008). A fusion protein, scFv23/TNF-α which is composed of TNF-α and an anti-HER2 scFv fragment (scFv23), was more cytotoxic in vitro than TNF-α (Rosenblum et al. 2000). Intriguingly, it was found that it may only be effective against tumor cells expressing intermediate, but not high, levels of HER2. It was proposed that scFv23/TNF-α can overcome the cellular resistance mechanisms to TNF-α induced by intermediate levels of HER2, but not by extremely high cellular levels of HER2 when the critical signaling pathways for TNF-α mediated cytotoxicity is blocked beyond the ability of scFv23/TNF-α to salvage. A subsequent investigation revealed that scFv23/TNF-α sensitizes HER2-overexpressing cells to TNF-α induced apoptosis via TNFR1 (Lyu and Rosenblum, 2005). Treatment of cells with scFv23/TNF-α caused up-regulation of TNFR1 expression, down-regulation of Akt phosphorylation, and TNF-α induced apoptosis through caspase-3 and caspase-8.
HER2 expression has recently been proposed as a negative prognostic marker in pancreatic intraepithelial neoplasia (Burtness, 2007). Therefore, scFv23/TNF-α was tested in a panel of human pancreatic cell lines (Lyu et al. 2008). It was found that all pancreatic cancer cell lines were resistant to the cytotoxic effects of TNF-α. However, scFv23/TNF-α was highly cytotoxic to TNF-α resistant HER2-expressing cells, comparable to conventional chemotherapeutic agents. A synergistic cytotoxic effect of scFv23/TNF-α with 5-fluorouracil (5-FU) in TNF-α resistant pancreatic cancer cell lines was observed, which induced caspase-3/8-dependent apoptosis. In vivo pharmacokinetic, tissue distribution, and therapeutic studies of scFv23/TNF-α are currently ongoing using mouse models to evaluate whether this strategy could be considered for clinical development.
Targeting CEA
CEA is a 180 kDa cell-surface glycoprotein expressed during the development of the fetal gut (Krupey et al. 1967). Subsequently, expression of CEA is minimal and limited to the lumen of the colon in adult humans. However, CEA expression is elevated in a substantial proportion of carcinomas of various tissue origins (Hammarstrom, 1999). Recombinant antibody technology was used to generate high affinity anti-CEA scFv fragments from filamentous bacteriophage libraries (Verhaar et al. 1995). One of these scFvs, MFE-23, has been tested in clinical trials after radioiodine labeling (Chester et al. 2000; Mayer et al. 2000). Subsequently, fusion protein of MFE-23 and TNF-α was also constructed, aiming to reduce the sequestration and increase the tumor concentration of TNF-α (Cooke et al. 2002). The 144 kDa homotrimer of the fusion protein retained the antigen binding activity of the scFv and the cytotoxicity of TNF-α. Radiolabeled MFE-23/TNF-α bound to both human and mouse TNFR1, and it was able to target human LS174T colorectal (high CEA expression) tumor in nude mice.
Targeting the gp240 antigen
In recognition of the many disadvantages of the initial strategy, in which an intact antibody against the gp240 antigen was used to deliver TNF-α (Rosenblum et al. 1995; Rosenblum et al. 1991), the fusion construct of TNF-α and a scFv fragment was generated (Liu et al. 2004). ScFvMEL/TNF-α was active against melanoma cells which were resistant to TNF-α. Radiolabeled scFvMEL/TNF-α localized to human melanoma xenografts in nude mice with a tumor-to-blood ratio of about 8 at 72 h post-injection. Pharmacokinetic studies of 125I-scFvMEL/TNF-α in mice showed that it has a terminal-phase half-life of about 18 h after i.v. injection (Liu et al. 2006). The maximum tolerated dose of scFvMEL/TNF-α in nude mice was determined to be 4 mg/kg. ScFvMEL/TNF-α treatment of mice bearing established xenograft tumors, at a dose of 2.5 mg/kg, resulted in complete tumor regression of all lesions. Further, no subsequent tumor growth was observed from mice that were rendered tumor-free. This study indicated the relative safety of scFvMEL/TNF-α at doses that are therapeutically effective, thus providing a guidance to the starting dose of a projected Phase I clinical trial.
Drug development is typically a very long process which takes about 10–15 years (Cai et al. 2006b). Almost all scFv/TNF-α fusion proteins were developed during the last decade, therefore no clinical reports have appeared to date regarding the use of these fusions proteins in treating cancer patients. However, a few promising candidates are probably already under clinical evaluation. With a molecular weight of about 28 kDa, only a very small portion of the amino acid residues in the scFv fragment are actually involved in antigen binding. Newly emerged antibody fragments, such as affibodies which have molecular weights of only a few kDa (Tolmachev et al. 2007), may be more advantageous in tumor targeting than scFv. It is possible that more than one affibody molecule can be fused to TNF-α, thus taking advantage of the multivalency effect to improve the tumor targeting efficacy (Mammen et al. 1998).
Peptide/TNF-α Fusion Protein
Commonly used targeting ligands include antibodies, antibody fragments, non-antibody proteins, peptides, and small molecules. Antibody-antigen recognition involves a complementary surface of the two entities. In some cases, peptides can bind to their receptors with comparable affinity to antibodies. One classic example is the RGD-integrin αvβ3 pair which was initially discovered more than two decades ago (Pierschbacher and Ruoslahti, 1984). Since then, numerous reports have explored the use of RGD-based agents in multiple facets of cancer intervention such as noninvasive imaging and anti-angiogenic therapy (Cai and Chen, 2006; Cai and Chen, 2008; Cai et al. 2005; Cai et al. 2008).
Targeting integrin αvβ3
TNF-α fused with the ACDCRGDCFCG peptide, a ligand of αv integrins, has been evaluated (Curnis et al. 2004). Sub-nanogram doses of this conjugate, when used in combination with melphalan, were found to be sufficient for inducing anti-cancer effects in mouse tumor models. Intramuscular administration of plasmid DNA encoding RGD/TNF-α or NGR/TNF-α (NGR denotes the CNGRC peptide which binds to CD13 in activated tumor blood vessels (Arap et al. 1998)) inhibited the growth of subcutaneous murine tumors implanted at positions distant from the injection site (Zarovni et al. 2004). Combination of RGD/TNF-α or NGR/TNF-α with doxorubicin or melphalan were able to confer better therapeutic effects than any of the agents administered alone. Tumstatin, a 28 kDa fragment of the type IV collagen that binds to integrin αvβ3 (Maeshima et al. 2002), was also fused with human TNF-α and tested for targeted cancer therapy (Luo et al. 2006).
Molecular imaging, “the visualization, characterization and measurement of biological processes at the molecular and cellular levels in humans and other living systems” (Mankoff, 2007), has flourished over the last decade. Continued development and wider availability of scanners dedicated to small animal imaging studies, which can provide a similar in vivo imaging capability in mice, primates, and humans, can enable smooth transfer of knowledge and molecular measurements between species thereby facilitating clinical translation. Molecular imaging can give a whole body readout in an intact system which is much more relevant and reliable than in vitro/ex vivo assays; help decrease the workload and speed up the drug development process; provide more statistically accurate results since longitudinal studies can be performed in the same animal which serves as its own control; aid in lesion detection in cancer patients and patient stratification; and help in treatment monitoring and/or dose optimization of individualized anti-cancer therapies (Cai et al. 2006b; Rudin and Weissleder, 2003). Many pharmaceutical companies already have a molecular imaging department/branch. It is expected that in the near future, molecular imaging will be routinely applied in many stages of the drug development process.
In a recent study, molecular imaging techniques were employed to quantitatively evaluate the tumor targeting efficacy of a RGD/TNF-α fusion protein (Wang et al. 2008). 64Cu (half-life: 12.8 h) has recently become increasingly more popular for positron emission tomography (PET) imaging applications (Wadas et al. 2007). One of the most extensively used chelators for 64Cu is 1,4,7,10-tetraazacyclododecane-N,N’,N’’,N’’’-tetraacetic acid (DOTA) (Cai et al. 2007a; Cai et al. 2007b; Cai et al. 2006a; Cai et al. 2007c; Liu et al. 2007), which was also employed for 64Cu-labeling of RGD/TNF-α or TNF-α in this study. Fusion of the RGD peptide to the N-terminus of TNF-α had little effect on its cytolytic toxicity effect in cell culture. Using non-invasive PET imaging, it was found that 64Cu-RGD/TNF-α had significantly higher accumulation in integrin-positive tumors, but not in integrin-negative tumors, than that of 64Cu-TNF-α (Fig. 3). The tumor accumulation of RGD/TNF-α was attributed to both integrin and TNFR targeting. Although fusion of the RGD moiety to TNF-α had little effect on the bioactivity and cytotoxicity in cell culture, RGD/TNF-α was significantly more potent than TNF-α in inhibiting integrin αvβ3-positive, orthotopic tumor growth in mouse models. The significance of this study is that non-invasive molecular imaging was employed to enable a direct, quantitative evaluation of the pharmacokinetics as well as the tumor targeting efficacy of the fusion protein, which was further confirmed by ex vivo histology of the tumor tissue (Fig. 3).
Figure 3.

Evaluation of a RGD/TNF-α fusion protein for targeted cancer therapy. A. The chemical structure of the RGD peptide used for tumor integrin αvβ3 targeting. B. Gel electrophoresis of the RGD/TNF-α fusion protein. Under non-reducing condition, the fusion protein runs as monomer, dimer, and trimer (middle lane). Under reducing condition, the fusion protein runs as a monomer (right lane). C. Non-invasive PET imaging demonstrated integrin αvβ3 specific tumor uptake of 64Cu-labeled RGD/TNF-α but not TNF-α. Decay-corrected coronal slices of tumor-bearing mice (arrowheads) at 4 h post-injection are shown. D. RGD/TNF-α caused statistically significant tumor growth inhibition comparedd to either TNF-α or PBS treated control. E. Immunofluorescence staining of the frozen tumor sections confirmed the anti-cancer effects observed in vivo. The fluorescence signal is colored coded red and the blue dots represent the nuclei. Adapted from (Wang et al. 2008).
Targeting CD13
In the first study to use peptides for tumor targeted TNF-α delivery, NGR/TNF-α was found to be ten fold more efficient than TNF-α in decreasing the tumor burden in lymphoma and melanoma animal models with comparable toxicity (Curnis et al. 2000). Compared to immunocytokines (e.g. scFv/TNF-α fusion proteins), peptide/TNF-α fusion proteins represent a new class of recombinant cytokines that have simpler structures and are potentially also less immunogenic. Examination of CD13 expression in normal and neoplastic human tissues and cells suggested that different forms of CD13 are expressed in myeloid cells, epithelia, and tumor-associated blood vessels (Curnis et al. 2002a), which may explain the tumor selective properties of NGR/TNF-α. The tumor specific expression of the particular form of CD13 may be exploited for future development of other vascular targeted cancer therapies based on the NGR/CD13 system.
Drug delivery and its penetration into neoplastic cells distant from the tumor vessels is critical for the effectiveness of solid tumor chemotherapy (Minchinton and Tannock, 2006). One study revealed that targeted delivery of TNF-α to tumor vessels using NGR/TNF-α was able to alter the endothelial barrier function and tumor interstitial pressure, thus enhancing the tumor penetration of doxorubicin and melphalan in mouse models (Curnis et al. 2002b). Such finding provided a rationale for the use of vascular targeting to increase the therapeutic index of chemotherapeutic drugs. Subsequently, the importance of endogenous interferon (IFN)-gamma on the anti-tumor activity of NGR/TNF-α was investigated (Sacchi et al. 2004). A crucial role for locally produced IFN-gamma was suggested and exogenous IFN-gamma can improve the therapeutic efficacy only in the cases of sub-optimal production of endogenous IFN-gamma.
The anti-cancer efficacy of NGR/TNF-α in combination with chemotherapeutic drugs acting via different mechanisms (e.g. cisplatin, paclitaxel, and gemcitabine) were then investigated (Sacchi et al. 2006). Pretreatment with NGR/TNF-α enhanced the tumor response to all these drugs, although to a different extent. Optimal administration schedule requires 2 h of pretreatment with NGR/TNF-α, independent of the mechanism of drug cytotoxicity, which provided important information for designing future clinical studies of NGR/TNF-α in combination with chemotherapy. In mouse models of hormone-dependent and hormone-independent prostate cancer, pretreatment with NGR/TNF-α also significantly expanded the therapeutic index of doxorubicin (Bertilaccio et al. 2008). In murine lymphoma models, the effect of NGR/TNF-α on the tumor microenvironment at 2 h after treatment was found to be transient and was subsequently overruled by other long-lasting effects (van Laarhoven et al. 2006). Taken together, the beneficial effect of NGR/TNF-α in combination with chemotherapy may critically depend on the sequence and timing of the treatment protocols.
Recently, it was reported that combining NGR/TNF-α with an ultra-low dose of endothelial monocyte activating polypeptide II (EMAP-II), a tumor-derived anti-angiogenic cytokine (van Horssen et al. 2006), can sensitize the tumor vasculature to TNF-α treatment with no evidence of systemic toxicity (Crippa et al. 2008). Ligand-directed targeting of TNF-α was critical because combination of non-targeted TNF-α with EMAP-II was inactive in the murine models tested. Interestingly, the synergistic anti-cancer effect was progressively lost when the dose of EMAP-II was increased from the nanogram to microgram level. NGR/TNF-α is currently being tested in clinical studies.
Summary
The potent anti-tumor activity of TNF-α and other cytokines is often achieved with the side effect of unacceptable toxicity. One avenue to improve the therapeutic index of TNF-α in cancer therapy is through specific targeting, as reviewed in this article. A diverse array of approaches has been explored for tumor targeted delivery of TNF-α in cancer therapy to avoid its non-specific toxicity. Passive targeting, cell-based therapy, gene therapy, pre-targeting, and fusion proteins have all been investigated (Fig. 4). Many of the agents are already in advanced clinical trials and new agents will continue to emerge. Another member of the TNF superfamily, TNF-related apoptosis-inducing ligand (TRAIL), has been reported to be able to specifically kill tumor cells without harming normal cells (Ashkenazi, 2002). Animal studies investigating the anti-cancer potential of TRAIL have been promising. Together with other TNF superfamily members, these agents possess enormous potential for future cancer therapy, if they can be rationally designed and specifically targeted.
Figure 4.
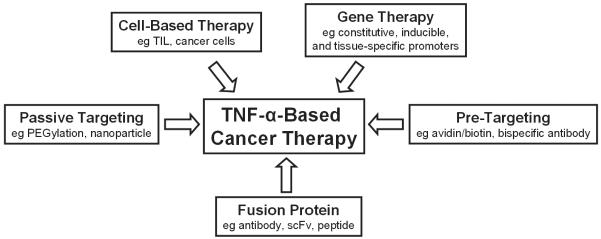
Many strategies have been investigated for tumor targeted delivery of TNF-α.
The TNF signalling pathway is a double-edged sword (Aggarwal, 2003). To accurately define the role of TNF-α in cancer therapy is challenging due to the pleiotropic nature of the cytokine. It is generally accepted that a single high dose of TNF-α can lead to tumor regression while chronic low dose of TNF-α may cause tumor progression. It is also possible that small amount of TNF-α released into the circulation, when a high dose is delivered for cancer therapy, may promote tumor metastasis. Further elucidation of the TNF superfamily biology, as well as conduction of novel clinical trials, is needed to explain some apparent inconsistencies in literature reports and define the exact role of TNF-α in cancer therapy.
Translating novel anti-cancer agents (e.g. TNF-α-based therapeutics) from bench to bedside is typically time-consuming and quite expensive (DiMasi et al. 2003). Multiple steps in preclinical development, especially the investigational new drug (IND)-directed toxicology, significantly slowed down this process. Development of protein-based therapeutics, such as those based on TNF-α, is even more difficult and expensive comparing to traditional small molecule-based drugs. Not only are proteins relatively difficult to manufacture in large quantities, good laboratory/manufacturing practice (GLP/GMP) compliance is also much more technically challenging. Currently, a vast number of new anti-cancer therapies are in pre-clinical and clinical testing. Whether TNF-α-based therapy is cost effective and whether it can be included in the clinical regimens for various cancers is still debatable. This is also an important aspect considered by various regulating agencies in approving new drugs.
Molecular imaging, as illustrated for the RGD/TNF-α fusion protein, will play an increasingly more significant role in drug development. By repeated imaging in preclinical models as well as in cancer patients, one can have several readouts of the tumor status pre- and post-treatment. The combination of molecular and anatomical/functional imaging techniques, many of which are already in routine clinical use (Townsend and Beyer, 2002), will be a powerful tool in assessing the anti-cancer efficacy of TNF-α-based agents.
Nanomedicine, briefly mentioned earlier in this review, may revolutionize future cancer patient management (Ferrari, 2005). Since most nanoparticles do not extravasate well, tumor vasculature targeted delivery of TNF-α using a nanoparticle carrier is probably the most feasible and appropriate strategy. The future of nanomedicine lies in multifunctional nanoplatforms which combine both therapeutic components and multimodality imaging (Cai and Chen, 2007). The ultimate goal is that nanoplatform-based agents can allow for efficient, specific in vivo delivery of drugs without systemic toxicity, and that the dose delivered as well as the therapeutic efficacy can be accurately measured non-invasively over time.
Despite the enormous potential of TNF-α in cancer therapy, most of the research efforts have so far come from academic/research institutions. To foster the continued discovery and development of TNF-α-based anti-cancer agents, cooperative efforts are needed from scientists within multiple disciplines (biology, chemistry, engineering, material science, among others). Close partnerships among academic researchers, clinicians, pharmaceutical industries, the National Cancer Institute, and the Food and Drug Administration are also needed to quickly apply novel TNF-α-based therapeutic agents to cancer patient management.
References
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 2003;3:745–56. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB, Kohr WJ, Hass PE, et al. Human tumor necrosis factor. Production, purification, and characterization. J. Biol. Chem. 1985;260:2345–54. [PubMed] [Google Scholar]
- Aggarwal BB, Moffat B, Harkins RN. Human lymphotoxin. Production by a lymphoblastoid cell line, purification, and initial characterization. J. Biol. Chem. 1984;259:686–91. [PubMed] [Google Scholar]
- Aguilar LK, Aguilar-Cordova E. Evolution of a gene therapy clinical trial. From bench to bedside and back. J. Neurooncol. 2003;65:307–15. doi: 10.1023/b:neon.0000003659.04633.6e. [DOI] [PubMed] [Google Scholar]
- Alexander HR, Jr., Bartlett DL, Libutti SK. Current status of isolated hepatic perfusion with or without tumor necrosis factor for the treatment of unresectable cancers confined to liver. Oncologist. 2000;5:416–24. doi: 10.1634/theoncologist.5-5-416. [DOI] [PubMed] [Google Scholar]
- Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–80. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer. 2002;2:420–30. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- Balza E, Mortara L, Sassi F, et al. Targeted delivery of tumor necrosis factor-alpha to tumor vessels induces a therapeutic T cell-mediated immune response that protects the host against syngeneic tumors of different histologic origin. Clin. Cancer Res. 2006;12:2575–82. doi: 10.1158/1078-0432.CCR-05-2448. [DOI] [PubMed] [Google Scholar]
- Bertilaccio MT, Grioni M, Sutherland BW, et al. Vasculature-targeted tumor necrosis factor-alpha increases the therapeutic index of doxorubicin against prostate cancer. Prostate. 2008;68:1105–15. doi: 10.1002/pros.20775. [DOI] [PubMed] [Google Scholar]
- Borsi L, Balza E, Carnemolla B, et al. Selective targeted delivery of TNFalpha to tumor blood vessels. Blood. 2003;102:4384–92. doi: 10.1182/blood-2003-04-1039. [DOI] [PubMed] [Google Scholar]
- Burtness B. Her signaling in pancreatic cancer. Expert. Opin. Biol. Ther. 2007;7:823–9. doi: 10.1517/14712598.7.6.823. [DOI] [PubMed] [Google Scholar]
- Cai W, Chen K, He L, et al. Quantitative PET of EGFR. expression in xenograft-bearing mice using 64Cu-labeled cetuximab, a chimeric anti-EGFR. monoclonal antibody. Eur. J. Nucl. Med. Mol. Imaging. 2007a;34:850–8. doi: 10.1007/s00259-006-0361-6. [DOI] [PubMed] [Google Scholar]
- Cai W, Chen K, Li ZB, et al. Dual-function probe for PET and near-infrared fluorescence imaging of tumor vasculature. J. Nucl. Med. 2007b;48:1862–70. doi: 10.2967/jnumed.107.043216. [DOI] [PubMed] [Google Scholar]
- Cai W, Chen K, Mohamedali KA, et al. PET of vascular endothelial growth factor receptor expression. J. Nucl. Med. 2006a;47:2048–56. [PubMed] [Google Scholar]
- Cai W, Chen X. Anti-angiogenic cancer therapy based on integrin αvβ3 antagonism. Anti-Cancer Agents Med. Chem. 2006;6:407–28. doi: 10.2174/187152006778226530. [DOI] [PubMed] [Google Scholar]
- Cai W, Chen X. Nanoplatforms for targeted molecular imaging in living subjects. Small. 2007;3:1840–54. doi: 10.1002/smll.200700351. [DOI] [PubMed] [Google Scholar]
- Cai W, Chen X. Multimodality imaging of tumor angiogenesis. J. Nucl. Med. 2008;49:113S–28S. doi: 10.2967/jnumed.107.045922. [DOI] [PubMed] [Google Scholar]
- Cai W, Ebrahimnejad A, Chen K, et al. Quantitative radioimmunoPET imaging of EphA2 in tumour-bearing mice. Eur. J. Nucl. Med. Mol. Imaging. 2007c;34:2024–36. doi: 10.1007/s00259-007-0503-5. [DOI] [PubMed] [Google Scholar]
- Cai W, Gambhir SS, Chen X. Multimodality tumor imaging targeting integrin αvβ3. Biotechniques. 2005;39:S6–S17. doi: 10.2144/000112091. [DOI] [PubMed] [Google Scholar]
- Cai W, Niu G, Chen X. Multimodality imaging of the HER-kinase axis in cancer. Eur. J. Nucl. Med. Mol. Imaging. 2007d;35:186–208. doi: 10.1007/s00259-007-0560-9. [DOI] [PubMed] [Google Scholar]
- Cai W, Niu G, Chen X. Imaging of integrins as biomarkers for tumor angiogenesis. Curr. Pharm Des. 2008 doi: 10.2174/138161208786404308. inpress. [DOI] [PubMed] [Google Scholar]
- Cai W, Rao J, Gambhir SS, et al. How molecular imaging is speeding up anti-angiogenic drug development. Mol. Cancer Ther. 2006b;5:2624–33. doi: 10.1158/1535-7163.MCT-06-0395. [DOI] [PubMed] [Google Scholar]
- Carswell EA, Old LJ, Kassel RL, et al. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. U.S.A. 1975;72:3666–70. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalini P, Iorio MV, Galmozzi E, et al. Role of HER. receptors family in development and differentiation. J. Cell Physiol. 2004;200:343–50. doi: 10.1002/jcp.20007. [DOI] [PubMed] [Google Scholar]
- Chen G, Goeddel DV. TNF-R.1 signaling: a beautiful pathway. Science. 2002;296:1634–5. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- Chester KA, Mayer A, Bhatia J, et al. Recombinant anti-carcinoembryonic antigen antibodies for targeting cancer. Cancer Chemother. Pharmacol. 2000;46(Suppl):S8–12. doi: 10.1007/pl00014055. [DOI] [PubMed] [Google Scholar]
- Cooke SP, Pedley RB, Boden R, et al. In vivo tumor delivery of a recombinant single chain Fv:tumor necrosis factor-alpha fusion [correction of factor: a fusion] protein. Bioconjug. Chem. 2002;13:7–15. doi: 10.1021/bc000178a. [DOI] [PubMed] [Google Scholar]
- Corti A, Gasparri A, Sacchi A, et al. Tumor targeting with biotinylated tumor necrosis factor alpha: structure-activity relationships and mechanism of action on avidin pretargeted tumor cells. Cancer Res. 1998;58:3866–72. [PubMed] [Google Scholar]
- Crippa L, Gasparri A, Sacchi A, et al. Synergistic damage of tumor vessels with ultra low-dose endothelial-monocyte activating poly-peptide-II and neovasculature-targeted tumor necrosis factor-alpha. Cancer Res. 2008;68:1154–61. doi: 10.1158/0008-5472.CAN-07-2085. [DOI] [PubMed] [Google Scholar]
- Curnis F, Arrigoni G, Sacchi A, et al. Differential binding of drugs containing the NGR. motif to CD13 isoforms in tumor vessels, epithelia, and myeloid cells. Cancer Res. 2002a;62:867–74. [PubMed] [Google Scholar]
- Curnis F, Gasparri A, Sacchi A, et al. Coupling tumor necrosis factor-alpha with alphaV integrin ligands improves its antineoplastic activity. Cancer Res. 2004;64:565–71. doi: 10.1158/0008-5472.can-03-1753. [DOI] [PubMed] [Google Scholar]
- Curnis F, Sacchi A, Borgna L, et al. Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13) Nat. Biotechnol. 2000;18:1185–90. doi: 10.1038/81183. [DOI] [PubMed] [Google Scholar]
- Curnis F, Sacchi A, Corti A. Improving chemotherapeutic drug penetration in tumors by vascular targeting and barrier alteration. J. Clin. Invest. 2002b;110:475–82. doi: 10.1172/JCI15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta R, Rubin E, Sukhatme V, et al. Ionizing radiation activates transcription of the EGR.1 gene via CArG elements. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10149–53. doi: 10.1073/pnas.89.21.10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J. Health Econ. 2003;22:151–85. doi: 10.1016/S0167-6296(02)00126-1. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus C, Scheuermann J, Neri D, et al. Diagnostic and therapeutic applications of recombinant antibodies: targeting the extra-domain B. of fibronectin, a marker of tumor angiogenesis. Curr. Pharm. Des. 2004;10:1537–49. doi: 10.2174/1381612043384808. [DOI] [PubMed] [Google Scholar]
- Fazle Akbar SM, Abe M, Yoshida O, et al. Dendritic cell-based therapy as a multidisciplinary approach to cancer treatment: present limitations and future scopes. Curr. Med. Chem. 2006;13:3113–9. doi: 10.2174/092986706778742882. [DOI] [PubMed] [Google Scholar]
- Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat. Rev. Cancer. 2005;5:161–71. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- Ferretti G, Felici A, Papaldo P, et al. HER2/neu role in breast cancer: from a prognostic foe to a predictive friend. Curr. Opin. Obstet. Gynecol. 2007;19:56–62. doi: 10.1097/GCO.0b013e328012980a. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Gao JQ, Eto Y, Yoshioka Y, et al. Effective tumor targeted gene transfer using PEGylated adenovirus vector via systemic administration. J. Control. Release. 2007;122:102–10. doi: 10.1016/j.jconrel.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Gasparri A, Moro M, Curnis F, et al. Tumor pretargeting with avidin improves the therapeutic index of biotinylated tumor necrosis factor alpha in mouse models. Cancer Res. 1999;59:2917–23. [PubMed] [Google Scholar]
- Gendler SJ. MUC1, the renaissance molecule. J. Mammary Gland Biol. Neoplasia. 2001;6:339–53. doi: 10.1023/a:1011379725811. [DOI] [PubMed] [Google Scholar]
- Grana C, Chinol M, Robertson C, et al. Pretargeted adjuvant radioimmunotherapy with yttrium-90-biotin in malignant glioma patients: a pilot study. Br. J. Cancer. 2002;86:207–12. doi: 10.1038/sj.bjc.6600047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunhagen DJ, Brunstein F, ten Hagen TL, et al. TNF-based isolated limb perfusion: a decade of experience with antivascular therapy in the management of locally advanced extremity soft tissue sarcomas. Cancer Treat. Res. 2004;120:65–79. doi: 10.1007/1-4020-7856-0_4. [DOI] [PubMed] [Google Scholar]
- Grunhagen DJ, de Wilt JH, ten Hagen TL, et al. Technology insight: Utility of TNF-alpha-based isolated limb perfusion to avoid amputation of irresectable tumors of the extremities. Nat. Clin. Pract. Oncol. 2006;3:94–103. doi: 10.1038/ncponc0426. [DOI] [PubMed] [Google Scholar]
- Halin C, Gafner V, Villani ME, et al. Synergistic therapeutic effects of a tumor targeting antibody fragment, fused to interleukin 12 and to tumor necrosis factor alpha. Cancer Res. 2003;63:3202–10. [PubMed] [Google Scholar]
- Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 1999;9:67–81. doi: 10.1006/scbi.1998.0119. [DOI] [PubMed] [Google Scholar]
- Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000;156:1363–80. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedley SJ, Chen J, Mountz JD, et al. Targeted and shielded adenovectors for cancer therapy. Cancer Immunol. Immunother. 2006;55:1412–9. doi: 10.1007/s00262-006-0158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr W, Wolfel T, Heike M, et al. Frequency analysis of tumor-reactive cytotoxic T lymphocytes in peripheral blood of a melanoma patient vaccinated with autologous tumor cells. Cancer Immunol. Immunother. 1994;39:93–9. doi: 10.1007/BF01525314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks DG, Kulkarni S. HER2+ breast cancer: review of biologic relevance and optimal use of diagnostic tools. Am. J. Clin. Pathol. 2008;129:263–73. doi: 10.1309/99AE032R9FM8WND1. [DOI] [PubMed] [Google Scholar]
- Hwu P, Yannelli J, Kriegler M, et al. Functional and molecular characterization of tumor-infiltrating lymphocytes transduced with tumor necrosis factor-alpha cDNA for the gene therapy of cancer in humans. J. Immunol. 1993;150:4104–15. [PubMed] [Google Scholar]
- Jiang YY, Liu C, Hong MH, et al. Tumor cell targeting of transferrin-PEG-TNF-alpha conjugate via a receptor-mediated delivery system: design, synthesis, and biological evaluation. Bioconjug. Chem. 2007;18:41–9. doi: 10.1021/bc060135f. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Park Y, Hong HJ. Antibody engineering for the development of therapeutic antibodies. Mol. Cells. 2005;20:17–29. [PubMed] [Google Scholar]
- Kircheis R, Ostermann E, Wolschek MF, et al. Tumor-targeted gene delivery of tumor necrosis factor-alpha induces tumor necrosis and tumor regression without systemic toxicity. Cancer Gene. Ther. 2002a;9:673–80. doi: 10.1038/sj.cgt.7700487. [DOI] [PubMed] [Google Scholar]
- Kircheis R, Wightman L, Kursa M, et al. Tumor-targeted gene delivery: an attractive strategy to use highly active effector molecules in cancer treatment. Gene. Ther. 2002b;9:731–5. doi: 10.1038/sj.gt.3301748. [DOI] [PubMed] [Google Scholar]
- Kreppel F, Kochanek S. Modification of adenovirus gene transfer vectors with synthetic polymers: a scientific review and technical guide. Mol. Ther. 2008;16:16–29. doi: 10.1038/sj.mt.6300321. [DOI] [PubMed] [Google Scholar]
- Krupey J, Gold P, Freedman SO. Purification and characterization of carcinoembryonic antigens of the human digestive system. Nature. 1967;215:67–8. doi: 10.1038/215067a0. [DOI] [PubMed] [Google Scholar]
- Kursa M, Walker GF, Roessler V, et al. Novel shielded transferrin-polyethylene glycol-polyethylenimine/DNA complexes for systemic tumor-targeted gene transfer. Bioconjug. Chem. 2003;14:222–31. doi: 10.1021/bc0256087. [DOI] [PubMed] [Google Scholar]
- Labialle S, Gayet L, Marthinet E, et al. Transcriptional regulators of the human multidrug resistance 1 gene: recent views. Biochem. Pharmacol. 2002;64:943–8. doi: 10.1016/s0006-2952(02)01156-5. [DOI] [PubMed] [Google Scholar]
- Liu Y, Cheung LH, Marks JW, et al. Recombinant single-chain antibody fusion construct targeting human melanoma cells and containing tumor necrosis factor. Int. J. Cancer. 2004;108:549–57. doi: 10.1002/ijc.11524. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhang W, Cheung LH, et al. The antimelanoma immunocytokine scFvMEL/TNF shows reduced toxicity and potent antitumor activity against human tumor xenografts. Neoplasia. 2006;8:384–93. doi: 10.1593/neo.06121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Cai W, He L, et al. In vivo biodistribution and highly efficient tumour targeting of carbon nanotubes in mice. Nat. Nanotechnol. 2007;2:47–52. doi: 10.1038/nnano.2006.170. [DOI] [PubMed] [Google Scholar]
- Lotze MT, Shurin M, Davis I, et al. Dendritic cell based therapy of cancer. Adv. Exp. Med. Biol. 1997;417:551–69. doi: 10.1007/978-1-4757-9966-8_91. [DOI] [PubMed] [Google Scholar]
- Luo YQ, Wang LH, Ma XL, Kong JX, Jiao BH. Construction, expression, and characterization of a new targeted bifunctional fusion protein: tumstatin45-132-TNF. IUBMB Life. 2006;58:647–53. doi: 10.1080/15216540600981743. [DOI] [PubMed] [Google Scholar]
- Lyu MA, Kurzrock R, Rosenblum MG. The immunocytokine scFv23/TNF targeting HER-2/neu induces synergistic cytotoxic effects with 5-fluorouracil in TNF-resistant pancreatic cancer cell lines. Biochem. Pharmacol. 2008;75:836–46. doi: 10.1016/j.bcp.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Lyu MA, Rosenblum MG. The immunocytokine scFv23/TNF sensitizes HER-2/neu-overexpressing SKBR-3 cells to tumor necrosis factor (TNF) via up-regulation of TNF receptor-1. Mol. Cancer Ther. 2005;4:1205–13. doi: 10.1158/1535-7163.MCT-05-0014. [DOI] [PubMed] [Google Scholar]
- MacGill RS, Davis TA, Macko J, et al. Local gene delivery of tumor necrosis factor alpha can impact primary tumor growth and metastases through a host-mediated response. Clin. Exp. Metastasis. 2007;24:521–31. doi: 10.1007/s10585-007-9089-3. [DOI] [PubMed] [Google Scholar]
- Maeda H, Wu J, Sawa T, et al. Tumor vascular permeability and the EPR. effect in macromolecular therapeutics. A review. J. Control Release. 2000;65:271–84. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- Maeshima Y, Sudhakar A, Lively JC, et al. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002;295:140–3. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- Mammen M, Chio S, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. Engl. 1998;37:2755–94. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Mankoff DA. A definition of molecular imaging. J. Nucl. Med. 2007;48:18N–21N. [PubMed] [Google Scholar]
- Marincola FM, Ettinghausen S, Cohen PA, et al. Treatment of established lung metastases with tumor-infiltrating lymphocytes derived from a poorly immunogenic tumor engineered to secrete human TNF-alpha. J. Immunol. 1994;152:3500–13. [PMC free article] [PubMed] [Google Scholar]
- Mass RD. The HER. receptor family: a rich target for therapeutic development. Int. J. Radiat. Oncol. Biol. Phys. 2004;58:932–40. doi: 10.1016/j.ijrobp.2003.09.093. [DOI] [PubMed] [Google Scholar]
- Mayer A, Tsiompanou E, O’Malley D, et al. Radioimmunoguided surgery in colorectal cancer using a genetically engineered anti-CEA single-chain Fv antibody. Clin. Cancer Res. 2000;6:1711–9. [PubMed] [Google Scholar]
- Maynard J, Georgiou G. Antibody engineering. Annu. Rev. Biomed. Eng. 2000;2:339–76. doi: 10.1146/annurev.bioeng.2.1.339. [DOI] [PubMed] [Google Scholar]
- McCormick F. Cancer gene therapy: fringe or cutting edge? Nat. Rev. Cancer. 2001;1:130–41. doi: 10.1038/35101008. [DOI] [PubMed] [Google Scholar]
- McLoughlin JM, McCarty TM, Cunningham C, et al. TNFerade, an adenovector carrying the transgene for human tumor necrosis factor alpha, for patients with advanced solid tumors: surgical experience and long-term follow-up. Ann. Surg. Oncol. 2005;12:825–30. doi: 10.1245/ASO.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Mezhir JJ, Smith KD, Posner MC, et al. Ionizing radiation: a genetic switch for cancer therapy. Cancer Gene. Ther. 2006;13:1–6. doi: 10.1038/sj.cgt.7700879. [DOI] [PubMed] [Google Scholar]
- Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat. Rev. Cancer. 2006;6:583–92. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- Mocellin S, Rossi CR, Pilati P, et al. Tumor necrosis factor, cancer and anticancer therapy. Cytokine Growth Factor Rev. 2005;16:35–53. doi: 10.1016/j.cytogfr.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Moro M, Pelagi M, Fulci G, et al. Tumor cell targeting with antibody-avidin complexes and biotinylated tumor necrosis factor alpha. Cancer Res. 1997;57:1922–8. [PubMed] [Google Scholar]
- Mundt AJ, Vijayakumar S, Nemunaitis J, et al. A Phase I trial of TNFerade biologic in patients with soft tissue sarcoma in the extremities. Clin. Cancer Res. 2004;10:5747–53. doi: 10.1158/1078-0432.CCR-04-0296. [DOI] [PubMed] [Google Scholar]
- Murugesan SR, Akiyama M, Einfeld DA, et al. Experimental treatment of ovarian cancers by adenovirus vectors combining receptor targeting and selective expression of tumor necrosis factor. Int. J. Oncol. 2007;31:813–22. [PubMed] [Google Scholar]
- Niu G, Cai W, Chen X. Molecular imaging of human epidermal growth factor receptor 2 (HER-2) expression. Front. Biosci. 2008;13:790–805. doi: 10.2741/2720. [DOI] [PubMed] [Google Scholar]
- Niu G, Xiong Z, Cheng Z, et al. In vivo bioluminescence tumor imaging of RGD peptide-modified adenoviral vector encoding firefly luciferase reporter gene. Mol. Imaging Biol. 2007;9:126–34. doi: 10.1007/s11307-007-0079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noureddini SC, Curiel DT. Genetic targeting strategies for adenovirus. Mol. Pharm. 2005;2:341–7. doi: 10.1021/mp050045c. [DOI] [PubMed] [Google Scholar]
- Pierschbacher MD, Ruoslahti E. Cell. attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309:30–3. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- Rasmussen H, Rasmussen C, Lempicki M, et al. TNFerade Biologic: preclinical toxicology of a novel adenovector with a radiation-inducible promoter, carrying the human tumor necrosis factor alpha gene. Cancer Gene Ther. 2002;9:951–7. doi: 10.1038/sj.cgt.7700518. [DOI] [PubMed] [Google Scholar]
- Robert B, Mach JP, Mani JC, et al. Cytokine targeting in tumors using a bispecific antibody directed against carcinoembryonic antigen and tumor necrosis factor alpha. Cancer Res. 1996;56:4758–65. [PubMed] [Google Scholar]
- Rosenblum MG, Cheung L, Mujoo K, et al. An antimelanoma immunotoxin containing recombinant human tumor necrosis factor: tissue disposition, pharmacokinetic, and therapeutic studies in xeno-graft models. Cancer Immunol. Immunother. 1995;40:322–8. doi: 10.1007/BF01519633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum MG, Cheung L, Murray JL, et al. Antibody-mediated delivery of tumor necrosis factor (TNF-alpha): improvement of cytotoxicity and reduction of cellular resistance. Cancer Commun. 1991;3:21–7. [PubMed] [Google Scholar]
- Rosenblum MG, Horn SA, Cheung LH. A novel recombinant fusion toxin targeting HER-2/NEU-over-expressing cells and containing human tumor necrosis factor. Int. J. Cancer. 2000;88:267–73. doi: 10.1002/1097-0215(20001015)88:2<267::aid-ijc19>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Rudin M, Weissleder R. Molecular imaging in drug discovery and development. Nat. Rev. Drug Discov. 2003;2:123–31. doi: 10.1038/nrd1007. [DOI] [PubMed] [Google Scholar]
- Sacchi A, Gasparri A, Curnis F, et al. Crucial role for interferon gamma in the synergism between tumor vasculature-targeted tumor necrosis factor alpha (NGR-TNF) and doxorubicin. Cancer Res. 2004;64:7150–5. doi: 10.1158/0008-5472.CAN-04-1445. [DOI] [PubMed] [Google Scholar]
- Sacchi A, Gasparri A, Gallo-Stampino C, et al. Synergistic anti-tumor activity of cisplatin, paclitaxel, and gemcitabine with tumor vasculature-targeted tumor necrosis factor-alpha. Clin. Cancer Res. 2006;12:175–82. doi: 10.1158/1078-0432.CCR-05-1147. [DOI] [PubMed] [Google Scholar]
- Scherf U, Benhar I, Webber KO, et al. Cytotoxic and antitumor activity of a recombinant tumor necrosis factor-B1(Fv) fusion protein on LeY. antigen-expressing human cancer cells. Clin. Cancer Res. 1996;2:1523–31. [PubMed] [Google Scholar]
- Schottelius AJ, Moldawer LL, Dinarello CA, et al. Biology of tumor necrosis factor-alpha-implications for psoriasis. Exp. Dermatol. 2004;13:193–222. doi: 10.1111/j.0906-6705.2004.00205.x. [DOI] [PubMed] [Google Scholar]
- Scotto KW, Johnson RA. Transcription of the multidrug resistance gene MDR1: a therapeutic target. Mol. Interv. 2001;1:117–25. [PubMed] [Google Scholar]
- Senzer N, Mani S, Rosemurgy A, et al. TNFerade biologic, an adenovector with a radiation-inducible promoter, carrying the human tumor necrosis factor alpha gene: a phase I study in patients with solid tumors. J. Clin. Oncol. 2004;22:592–601. doi: 10.1200/JCO.2004.01.227. [DOI] [PubMed] [Google Scholar]
- Sharkey RM, Cardillo TM, Rossi EA, et al. Signal amplification in molecular imaging by pretargeting a multivalent, bispecific antibody. Nat. Med. 2005;11:1250–5. doi: 10.1038/nm1322. [DOI] [PubMed] [Google Scholar]
- Shirai T, Yamaguchi H, Ito H, et al. Cloning and expression in Escherichia coli of the gene for human tumour necrosis factor. Nature. 1985;313:803–6. doi: 10.1038/313803a0. [DOI] [PubMed] [Google Scholar]
- Smith AE. Viral vectors in gene therapy. Annu. Rev. Microbiol. 1995;49:807–38. doi: 10.1146/annurev.mi.49.100195.004111. [DOI] [PubMed] [Google Scholar]
- Spriggs DR, Deutsch S, Kufe DW. Genomic structure, induction, and production of TNF-alpha. Immunol. Ser. 1992;56:3–34. [PubMed] [Google Scholar]
- Takahashi S, Ito Y, Hatake K, et al. Gene therapy for breast cancer.—Review of clinical gene therapy trials for breast cancer and MDR.1 gene therapy trial in Cancer Institute Hospital. Breast Cancer. 2006;13:8–15. doi: 10.2325/jbcs.13.8. [DOI] [PubMed] [Google Scholar]
- Tolmachev V, Orlova A, Nilsson FY, et al. Affibody molecules: potential for in vivo imaging of molecular targets for cancer therapy. Expert. Opin. Biol. Ther. 2007;7:555–68. doi: 10.1517/14712598.7.4.555. [DOI] [PubMed] [Google Scholar]
- Townsend DW, Beyer T. A combined PET/CT scanner: the path to true image fusion. Br. J. Radiol. 2002;75(Spec(No)):S24–30. doi: 10.1259/bjr.75.suppl_9.750024. [DOI] [PubMed] [Google Scholar]
- Treisman J, Hwu P, Yannelli JR, et al. Upregulation of tumor necrosis factor-alpha production by retrovirally transduced human tumor-infiltrating lymphocytes using trans-retinoic acid. Cell. Immunol. 1994;156:448–57. doi: 10.1006/cimm.1994.1189. [DOI] [PubMed] [Google Scholar]
- van Horssen R, Eggermont AM, ten Hagen TL. Endothelial monocyte-activating polypeptide-II and its functions in (patho)physiological processes. Cytokine Growth Factor Rev. 2006;17:339–48. doi: 10.1016/j.cytogfr.2006.08.001. [DOI] [PubMed] [Google Scholar]
- van Laarhoven HW, Gambarota G, Heerschap A, et al. Effects of the tumor vasculature targeting agent NGR-TNF on the tumor microenvironment in murine lymphomas. Invest New Drugs. 2006;24:27–36. doi: 10.1007/s10637-005-4540-2. [DOI] [PubMed] [Google Scholar]
- Verhaar MJ, Chester KA, Keep PA, et al. A single chain Fv derived from a filamentous phage library has distinct tumor targeting advantages over one derived from a hybridoma. Int. J. Cancer. 1995;61:497–501. doi: 10.1002/ijc.2910610412. [DOI] [PubMed] [Google Scholar]
- Vilaboa N, Voellmy R. Regulatable gene expression systems for gene therapy. Curr. Gene. Ther. 2006;6:421–38. doi: 10.2174/156652306777934829. [DOI] [PubMed] [Google Scholar]
- Visaria R, Bischof JC, Loren M, et al. Nanotherapeutics for enhancing thermal therapy of cancer. Int. J. Hyperthermia. 2007;23:501–11. doi: 10.1080/02656730701611241. [DOI] [PubMed] [Google Scholar]
- Wadas TJ, Wong EH, Weisman GR, et al. Copper chelation chemistry and its role in copper radiopharmaceuticals. Curr. Pharm. Des. 2007;13:3–16. doi: 10.2174/138161207779313768. [DOI] [PubMed] [Google Scholar]
- Walther W, Arlt F, Fichtner I, et al. Heat-inducible in vivo gene therapy of colon carcinoma by human mdr1 promoter-regulated tumor necrosis factor-alpha expression. Mol. Cancer Ther. 2007;6:236–43. doi: 10.1158/1535-7163.MCT-06-0070. [DOI] [PubMed] [Google Scholar]
- Walther W, Stein U, Fichtner I, et al. Mdr1 promoter-driven tumor necrosis factor-alpha expression for a chemotherapy-controllable combined in vivo gene therapy and chemotherapy of tumors. Cancer Gene. Ther. 2000;7:893–900. doi: 10.1038/sj.cgt.7700196. [DOI] [PubMed] [Google Scholar]
- Walther W, Stein U, Schlag PM. Use of the human MDR.1 promoter for heat-inducible expression of therapeutic genes. Int. J. Cancer. 2002;98:291–6. doi: 10.1002/ijc.10174. [DOI] [PubMed] [Google Scholar]
- Walther W, Wendt J, Stein U. Employment of the mdr1 promoter for the chemotherapy-inducible expression of therapeutic genes in cancer gene therapy. Gene. Ther. 1997;4:544–52. doi: 10.1038/sj.gt.3300451. [DOI] [PubMed] [Google Scholar]
- Wang H, Chen K, Cai W, et al. Integrin-targeted imaging and therapy with RGD4C-TNF fusion protein. Mol. Cancer Ther. 2008;7:1044–53. doi: 10.1158/1535-7163.MCT-07-2084. [DOI] [PubMed] [Google Scholar]
- Weichselbaum RR, Hallahan DE, Beckett MA, et al. Gene therapy targeted by radiation preferentially radiosensitizes tumor cells. Cancer Res. 1994;54:4266–9. [PubMed] [Google Scholar]
- Wright P, Zheng C, Moyana T, et al. Intratumoral vaccination of adenoviruses expressing fusion protein RM4/tumor necrosis factor (TNF)-alpha induces significant tumor regression. Cancer Gene. Ther. 1998;5:371–9. [PubMed] [Google Scholar]
- Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat. Biotechnol. 2005;23:1137–46. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- Xiang J, Moyana T, Qi Y. Genetic engineering of a recombinant fusion possessing anti-tumor F(ab’)2 and tumor necrosis factor. J. Biotechnol. 1997;53:3–12. doi: 10.1016/s0168-1656(96)01639-2. [DOI] [PubMed] [Google Scholar]
- Yi Y, Hahm SH, Lee KH. Retroviral gene therapy: safety issues and possible solutions. Curr. Gene. Ther. 2005;5:25–35. doi: 10.2174/1566523052997514. [DOI] [PubMed] [Google Scholar]
- Zarovni N, Monaco L, Corti A. Inhibition of tumor growth by intramuscular injection of cDNA encoding tumor necrosis factor alpha coupled to NGR., and RGD tumor-homing peptides. Hum. Gene. Ther. 2004;15:373–82. doi: 10.1089/104303404322959524. [DOI] [PubMed] [Google Scholar]
- Zhang G. Tumor necrosis factor family ligand-receptor binding. Curr. Opin. Struct. Biol. 2004;14:154–60. doi: 10.1016/j.sbi.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Zhu HJ, Zhang ZQ, Zeng XF, et al. Cloning and analysis of human UroplakinII promoter and its application for gene therapy in bladder cancer. Cancer Gene. Ther. 2004;11:263–72. doi: 10.1038/sj.cgt.7700672. [DOI] [PubMed] [Google Scholar]