

Abstract
A simple chemometrics-assisted spectrophotometric method for the simultaneous determination of lamivudine and stavudine in pharmaceutical tablets is described. The UV absorption spectra of the studied drugs, in the range of 200–310 nm, showed a considerable degree of spectral overlapping ([Di]0.5 = 94.9%). Resolution of the mixture has been accomplished by using classical least-squares regression analysis (CLS) and principle components regression analysis methods (PCR). Beer’s law was obeyed for both drugs in the general concentration ranges of 2–12 and 3–15 μg ml−1 for lamivudine and stavudine, respectively. The proposed methods were successfully applied for the determination of the two drugs in laboratory prepared mixtures. The overall recoveries percent were found 98.58 ± 1.53–101.30 ± 1.35 (CLS) and 98.62 ± 1.65–101.13 ± 1.04 (PCR) for lamivudine and 98.43 ± 1.62–99.42 ± 1.55 (CLS) and 98.23 ± 1.97–101.20 ± 1.79 (PCR) for stavudine, respectively. The commercial tablets percentage content was found 98.10 ± 2.5–102.47 ± 2.94 (CLS) and 99.12 ± 1.71–100.92 ± 1.54 (PCR) for lamivudine and 96.00 ± 2.94–98.17 ± 1.72 (CLS) and 97.40 ± 1.55–97.80 ± 1.92 (PCR) for stavudine, respectively. Good percentage recoveries and proper statistical data obtained with both the laboratory prepared mixtures and the commercial tablets proved the suitability and efficiency of the proposed procedures for routine analysis and quality control purposes with quite satisfactory precision. A comparison of the obtained results from CLS and PCR were also performed with those obtained from reported method. The obtained F- and t-values obtained indicating no significant differences between the results of the proposed and reported methods.
Keywords: Chemometrics, Pharmaceuticals, Spectrophotometry, HIV-inhibitors
1. Introduction
Simultaneous determination of commercial drug mixtures by using traditional spectrophotometric methods is a difficult problem in the field of analytical chemistry due to considerable spectral interference. Derivative spectrophotometry and the H-point standard addition methods have been frequently used to overcome the problems of interference due to the partial spectral overlapping (Sabry and Khamis, 2000; Tomsu et al., 2004; Catherine et al., 2004; Palabiyik et al., 2004).
The great advent and wide spread of the laboratory computer has been allowed analytical chemist to use chemometrics for facile and accurate analysis of drug mixtures. Among these techniques are the multivariate calibration methods which can be performed with easily accessible statistical software and they are currently widely used in pharmaceutical analysis (Palabiyik et al., 2004; Dinc et al., 2005; Mohamed and Salem, 2005; Salem et al., 2002).
Classical least squares (CLS), partial least-square regression (PLS) and principal component regression (PCR) are the most commonly used methods (Palabiyik et al., 2004; Dinc et al., 2005; Mohamed and Salem, 2005; Salem et al., 2002). On the other hand, derivative spectrometry is a fast, simple and rapid technique which is useful for determination of drugs in multicomponent systems as well as in single component formulations in presence of interfering excipients (Tomsu et al., 2004; Catherine et al., 2004; Palabiyik et al., 2004). It enhances the qualitative features and thus increases the fingerprinting utility for selective identification and quantification of pharmaceutical compounds.
HIV/AIDS requires lifelong treatment with potent life saving drugs that include nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors and protease inhibitors (Delgado and Reme, 1998; Williams and Lemke, 2002). Among these lamivudine (2′,3′-dideoxy-3′-thiacytidine) and stavudine (2′,3′-didehydro-3-doexythymidine) constitute first line therapy. Pharmaceutical preparations containing lamivudine together with stavudine are now commonly found on the market.
Lamivudine (Fig. 1a) is a nucleoside analog having potent in vitro and in vivo inhibitory activity against HIV reverse transcriptase. Lamivudine specifically refers to the (–) enantiomer of the cis-racemate and is marketed as tablets in different strengths. Stavudine (Fig. 1b) is chemically a thymidine nucleoside analog. It has a complete and less variable oral absorption as compared to other nucleoside analogs (Hardman and Limbird, 2001).
Figure 1.
Structural formulae of lamivudine and stavudine.
Literature survey reveals several methods that have been used for quantitative determination of the two drugs individually, such as UV-spectrophotometry (Indian Pharmacopoeia, 1996a,b; Kapoor et al., 2006; Anbazhagan et al., 2005), HPLC (Kapoor et al., 2006; Anbazhagan et al., 2005; Sarasa et al., 2000; Kapoor et al., 2006; Pendela et al., 2009), HPLC with tandem mass spectrometric detection (Volosov et al., 2002; Hou et al., 2004; Xu et al., 2009; Mistri et al., 2007) and radio-immunoassay (Kaul et al., 1996).
Very few reports were found that involving simultaneous determination of the studied drugs in commercial combined formulations. These methods included derivative spectroscopy and HPLC (Kapoor et al., 2006) but there is no report involving the use of multivariate methods.
Lamivudine and stavudine show closely overlapping UV absorption bands in the range from 200 to 310 nm. Hence, it is not easily possible to analyze lamivudine and stavudine combinations by traditional UV-spectrophotometry. In this study two chemometric-assisted spectrophotometric methods are proposed for the simultaneous determination of lamivudine and stavudine in some laboratory prepared mixtures and in pharmaceutical preparations present in the Ethiopian market. Resolution of the mixtures under investigation has been accomplished mainly by using CLS and PCR methods but we use also the zero-crossing first derivative for the same results for the comparison proposes although it was previously reported (Kapoor et al., 2006). The proposed procedures are simple, rapid, non-expensive and provide a good resolution of both drugs with the minimal amount of errors and spectral and background interferences. In addition, the calibration data for the reference standards can be used for long time without change, making it a very suitable method for routine analysis and quality control purposes.
2. Experimental
2.1. Apparatus
Spectrophotometric measurements were carried out on a computerized Spectronic Genesys 2PC, UV/visible Spectrophotometer (Milton Roy, USA). The absorption spectra of test and reference solutions were recorded over the range 200–310 nm. The subsequent statistical manipulation was performed by transferring the spectral data to Microsoft excel 2003 program and processing them with the standard curve fit package and matrix calculations.
2.2. Chemicals
Pharmaceutical grade lamivudine (Glasko Smith-Kline, Durham, UK) and stavudine (Hetero Drugs Limited, Hyderabad, India) were used as working standards after confirming their purity and compliance with pharmaceutical requirements. All other reagents used were analytical grade.
2.3. Pharmaceutical preparation
The following pharmaceutical preparation was purchased from the local market and subjected to analysis by the proposed procedures; Coviro-LS™ tablets (Ranbaxy Laboratories Limited, India) labeled to contain 150 mg lamivudine and 30 mg stavudine for each film coated tablet.
2.4. Procedures
2.4.1. Preparation of standards
Into 100-ml volumetric flask an accurately weighed amount (50 mg) of either of the studies drugs is dissolved in about 80 ml of 0.1 M hydrochloric acid and diluted to volume with the same solution. The resulting solution is diluted quantitatively with 0.1 M hydrochloric acid to obtain the appropriate dilutions for each drug according to its linear calibration range or as specified under the analysis of the laboratory prepared mixtures.
2.4.2. Sample preparation
Twenty tablets are weighed and finely powdered. An accurately weighed amount of the powder equivalent to the averaged weight of one tablet is transferred to 100 ml volumetric flask and diluted to about 80 ml with 0.1 M hydrochloric acid. The mixture is shacked for about 10 min and then sonicated for additional 5 min, diluted to 100 ml with 0.1 M hydrochloric acid and filtered. The first portion of filtrate is discarded. The obtained clear solution is used as stock sample solution. The stock solution is diluted quantitatively with 0.1 M hydrochloric acid to obtain the suitable working sample solutions for the UV-measurements.
2.4.3. Standard solutions for multivariate calibration
In order to obtain the calibration matrixes for applying CLS and PCR analysis, 10 solutions of each of the pure components (lamivudine and stavudine) were prepared with concentrations ranged from: 2 to 12 and 3 to 15 μg ml−1 for lamivudine and stavudine respectively, as described in Section 2.4.1. These ranges were previously verified to obey Beer’s law for each of the studied drugs in the selected acid solutions. The absorption data in the range of 200–310 nm (digitized every 1.0 nm) were subjected to the least squares analysis in order to obtain the calibration matrix K for each drug (see Section 3). The laboratory prepared mixtures were then prepared by mixing known amounts of lamivudine with stavudine in different varied proportions as indicated in Table 3, in order to verify the precision of the method for analysis of such mixtures and matching the commercial tablets with those having the same or approximately comparable concentrations.
Table 3.
Results obtained by applying CLS and PCR calibration methods to laboratory prepared mixtures of lamivudine and stavudine in 0.1 M hydrochloric acid ± relative errors.
| Compounds | CLS |
PCR |
|||
|---|---|---|---|---|---|
| Actual (μg ml−1) | Found (μg ml−1) | % ±Relative errors⁎ | Found (μg ml−1) | % ±Relative errors⁎ | |
| LAM | 4.00 | 3.94 | 98.58 ± 1.53 | 3.95 | 98.80 ± 1.77 |
| STA | 10.00 | 9.88 | 98.77 ± 1.42 | 10.12 | 101.201 ± 1.79 |
| LAM | 6.00 | 5.98 | 99.72 ± 1.63 | 5.92 | 98.62 ± 1.65 |
| STA | 8.00 | 7.95 | 99.42 ± 1.55 | 8.07 | 100.91 ± 1.93 |
| LAM | 7.00 | 6.96 | 99.43 ± 1.50 | 7.09 | 101.31 ± 1.22 |
| STA | 7.00 | 6.89 | 98.43 ± 1.62 | 6.99 | 99.80 ± 1.58 |
| LAM | 8.00 | 8.05 | 100.62 ± 1.45 | 8.10 | 101.30 ± 1.34 |
| STA | 6.00 | 5.94 | 99.00 ± 1.10 | 6.02 | 100.03 ± 1.88 |
| LAM | 10.00 | 10.09 | 100.90 ± 1.14 | 10.11 | 101.13 ± 1.04 |
| STA | 4.00 | 3.87 | 98.75 ± 1.73 | 3.93 | 98.23 ± 1.97 |
| LAM | 10.00 | 10.13 | 101.30 ± 1.35 | 9.95 | 99.46 ± 1.08 |
| STA | 2.00 | 1.93 | 98.50 ± 2.00 | 1.97 | 98.35 ± 1.84 |
Relative errors as calculated from the corresponding binary mixtures results (n = 3).
PCR multivariate analysis follows the same procedure described for CLS except that the K matrix must be replaced by F matrix, which is calculated from the factorized data instead of absorbance values in case of CLS and A matrix must be replaced by Aproj. For details of the calculations see Section 3.
2.4.4. Data processing
Data were processed on an Intel Pentium IV-Veriton 7200D, PC-compatible computer equipped with the essential statistical programs for CLS and PCR calculations.
3. Results and discussion
Full spectrum methods usually provide significant improvement in precision over methods restricted to a small number of wavelengths. A convenient method for resolving the mixtures, which can in principle be applied to the present case, is the least square analysis. The simplest of them is the classical least squares (CLS). It should certainly be preferred when the selection of variables is simple. In such cases, the regression coefficients for different selected collinear wavelengths may have relatively little meaning for interpretation purposes, but the model performs well, both in the calibration and prediction stages, provided that the model having linearity between responses and concentrations and the prediction is performed within the calibration domain. In addition, the baseline effects and noise are probably non-significant or of very low significance. Under these conditions CLS is probably the method to be recommended.
The absorption spectra for the studied drugs in 0.1 M HCl is shown in Fig. 2. As can be seen, a considerable degree of spectral overlapping occurs in the region from 200 to 310 nm. The degree of spectral overlapping can be conveniently given by (Di)0.5, where Di is the magnitude of dependency that can be calculated for a two components mixture from the equation:
where k1 and k2 are the l × n matrices of regression coefficients for the compounds 1 and 2, respectively.
Figure 2.
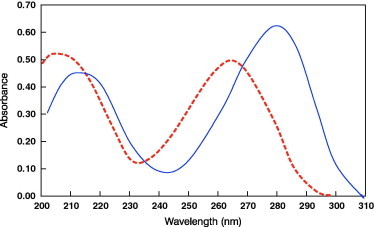
Zero-order spectra for 10 μg of lamivudine (—) and 10 μg/ml of stavudine (- - - - -) indicating the marked overlap between bands.
In case of the presently studied compounds, the spectra shown in Fig. 2 lead to Di = 0.90 implying a 94.9% of spectral overlapping for the studied mixtures.
3.1. CLS method
In CLS a linear relationship between absorbance and the component concentration at each wavelength is assumed. When calibration set of m samples of known content with n spectral variables are measured the relationship is given by the equation:
where A is the m × n matrix of calibration spectra, C the m × l matrix of component concentrations, K the l × n matrix of regression coefficient, and E the m × n matrix of spectral errors residuals not fit by the model. By means of the calibration sample set, estimation of absorptivities is possible by solving the matrix K according to the general least-squares solution indicated by the equation:
Analysis is then based on the spectrum (Aun) of the unknown samples by
where Cun is the vector of sought for concentrations.
3.2. PCR method
As a first step of PCR method, the proper number of components in the PCA model must be determined by one of the well known methods (Henrion and Henrion, 1994). The data from lamivudine and stavudine mixtures (Fig. 3) indicate that the residual variance approximately levels off after the second component and the first and second components accounts for about 99.37% of the total data variance fit by the whole components.
Figure 3.
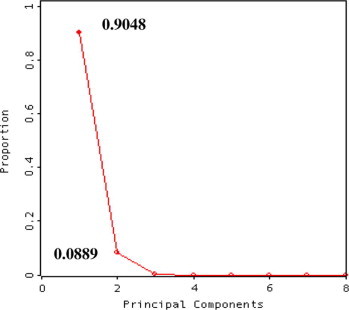
Screen-plot for principal component model of lamivudine and stavudine mixtures data.
The main equation for calculating the concentration can be written as:
where C is the concentration components and F is the regression matrix of the new coordinates, thus;
Then the calculated F-values are used to predict the concentrations in an unknown sample from its measured spectrum as follows:
where Fcal = the quantity pre-calculated at calibration time.
3.3. Comparison of the results form the proposed methods
Tables 1 and 2 show the actual and predicted amounts ± the relative errors (%) of the studied drugs as given by the least-squares regression analysis (CLS) and principle components regression (PCR) of the spectral data that obtained experimentally in the calibration range of each drug at the wavelength range from 200 to 310 nm. The results confirm the considerable degree of agreement between CLS and PCR techniques and indicate that these methods are suitable for this analysis in the given calibration domain for each drug if compared with results given by the first-derivative method especially for lamivudine.
Table 1.
Predicted concentrations of pure lamivudine samples in 0.1 M hydrochloric acid ± relative errors as calculated from CLS and PCR calibration methods.
| Concentrations (μg ml−1) | CLS |
PCR |
||
|---|---|---|---|---|
| Found (μg ml−1) | % ±Relative errorsa | Found (μg ml−1) | % ±Relative errorsa | |
| 2.00 | 2.06 | 101.00 ± 1.88 | 1.98 | 98.80 ± 2.11 |
| 3.00 | 3.06 | 98.67 ± 2.21 | 2.99 | 99.73 ± 2.23 |
| 4.00 | 3.92 | 98.00 ± 1.88 | 3.99 | 99.68 ± 1.96 |
| 5.00 | 4.93 | 98.80 ± 2.00 | 5.07 | 101.40 ± 1.77 |
| 6.00 | 5.87 | 99.17 ± 2.37 | 6.04 | 100.67 ± 1.98 |
| 7.00 | 6.96 | 99.43 ± 1.42 | 6.95 | 99.21 ± 1.45 |
| 8.00 | 8.03 | 100.40 ± 1.80 | 7.98 | 99.70 ± 2.00 |
| 9.00 | 9.04 | 100.41 ± 1.98 | 9.08 | 100.89 ± 1.14 |
| 10.00 | 10.00 | 100.00 ± 1.85 | 10.13 | 101.30 ± 1.56 |
| 11.00 | 10.93 | 99.40 ± 0.89 | 10.87 | 98.82 ± 1.78 |
Relative errors as calculated from the corresponding calibration model (n = 3).
Table 2.
Predicted concentrations of pure stavudine samples in 0.1 M hydrochloric acid ± relative errors as calculated from CLS and PCR calibration methods.
| Concentrations (μg ml−1) | CLS |
PCR |
||
|---|---|---|---|---|
| Found (μg ml−1) | % ±Relative errorsa | Found (μg ml−1) | % ±Relative errorsa | |
| 3.00 | 2.89 | 98.33 ± 1.96 | 2.95 | 98.40 ± 1.96 |
| 4.00 | 3.83 | 98.33 ± 1.41 | 3.95 | 98.65 ± 1.88 |
| 5.00 | 4.96 | 99.10 ± 1.10 | 4.93 | 98.66 ± 1.86 |
| 6.00 | 5.91 | 97.94 ± 1.48 | 5.88 | 98.07 ± 1.23 |
| 7.00 | 6.91 | 98.76 ± 1.48 | 6.91 | 98.73 ± 1.67 |
| 8.00 | 7.99 | 99.88 ± 0.48 | 7.92 | 99.01 ± 1.33 |
| 9.00 | 9.04 | 100.44 ± 0.63 | 9.05 | 100.58 ± 1.74 |
| 10.00 | 10.19 | 101.91 ± 1.24 | 10.02 | 100.23 ± 2.05 |
| 11.00 | 11.09 | 101.82 ± 1.21 | 10.96 | 99.68 ± 0.98 |
| 12.00 | 12.08 | 100.70 ± 0.71 | 12.11 | 100.92 ± 0.92 |
Relative errors as calculated from the corresponding calibration model (n = 3).
Several laboratory prepared mixtures of the studied drugs were subjected to analysis by the two techniques in order to confirm the suitability of the calibration models for determination of the studied drugs in the pharmaceutical sample solutions and to verify the precision of the methods for analysis of such mixtures and matching the commercial dosage forms with those having comparable concentrations. Table 3 summarizes the results obtained for the suggested binary mixtures. As can be seen, the concentrations predicted by the models are considerably close to the real concentrations, the recoveries in most cases were satisfactory and the deviation range between the estimated and true concentrations were found between 0.28% and 1.57% with relative errors between 1.10% and 2.00% for CLS and 0.03% and 1.77% with relative errors between 1.04% and 1.97% for PCR, respectively. It can be observed from this set of results that the drug mixtures determination is feasible for the multivariate procedures (CLS and PCR), but the factorized multivariate calibration models (PCR) allows a more significant reduction of errors in relation to the determination by both the CLS procedure, especially in the mixtures containing low concentrations of stavudine (10:4 and 10:2 lamivudine:stavudine, respectively).
3.4. Analysis of commercial tablets
The proposed methods were used for the simultaneous determination of lamivudine and stavudine in pharmaceuticals without prior separation of the excipients and additives present in the commercial tablets. Table 4 shows the results obtained form the analysis of Coviro-LS tablets that contain 150 mg of lamivudine and 30 mg of stavudine at three concentration levels. The results indicated that the proposed techniques could be used for simultaneous quantitation and routine quality control analysis of this binary mixture in pharmaceuticals.
Table 4.
Results from analysis of the studied drugs in Coviro-LS™ tablets (150 mg lamivudine and 30 mg of stavudine) with the 1D (reported method), CLS and PCR methods.
| Drugs | Claimed analyzed concentrations (μg ml−1) | Founda |
Calculated t-valuesb | Calculated F-valuesb | ||
|---|---|---|---|---|---|---|
| 1D (% ±SD) | CLS (% ±SD) | PCR (% ±SD) | ||||
| Lamivudine | 5 | 100.21 ± 2.05 | 102.47 ± 2.94 | 99.12 ± 1.71 | t1 = 0.594 | F1 = 2.060 |
| t2 = 0.385 | F2 = 1.440 | |||||
| Stavudine | 1 | 95.00 ± 2.65 | 96 ± 2.94 | 97.65 ± 2.24 | t1 = 0.238 | F1 = 1.230 |
| t2 = 0.720 | F2 = 1.400 | |||||
| Lamivudine | 10 | 102.4 ± 2.47 | 98.10 ± 2.5 | 100.92 ± 1.54 | t1 = 1.153 | F1 = 1.020 |
| t2 = 0.479 | F2 = 2571 | |||||
| Stavudine | 2 | 97.00 ± 2.22 | 96.25 ± 2.17 | 97.80 ± 1.92 | t1 = 0.228 | F1 = 1.050 |
| t2 = 0.257 | F2 = 1.342 | |||||
| Lamivudine | 15 | 98.50 ± 1.63 | 100.18 ± 0.92 | 100.44 ± 1.38 | t1 = 0.661 | F1 = 3.139 |
| t2 = 0.702 | F2 = 1.395 | |||||
| Stavudine | 3 | 96.76 ± 1.27 | 98.17 ± 1.72 | 97.40 ± 1.55 | t1 = 0.927 | F1 = 1.834 |
| t2 = 0.736 | F2 = 1.489 | |||||
Theoretical values for t- and F-tests at 95% confidence limit are 2.31 and 6.39, respectively.
Average of three determinations.
F1, t1 for 1D/CLS methods and F2, t2 for 1D/PCR methods.
In order to provide the accuracy and precision of the proposed procedures a comparison between the used methods and 1D reported method (Kapoor et al., 2006) were done. According to the t-test and F-test, all the calculated t-values and F-values were smaller than the theoretical values at the 95% confidence level. This indicates that there is no significant difference between the performances of the used techniques and the reported method with regard to the mean values and their variances (Table 4).
Regarding these results and the recovery studies done for the pure compounds added to the commercial tablets which indicate the absence of the interferences due to the presence of the excipients and additives, we can conclude that the proposed methods are sufficiently accurate and precise in order to be applied to the pharmaceutical dosage forms.
4. Conclusion
The contents of several laboratory prepared mixtures and commercial tablets were simultaneously determined using UV-spectrophotometric measurements together with CLS and PCR calibration analysis. The good recoveries obtained in all cases as well as the reliable agreement with the reported procedures proved that, the proposed procedures could be applied efficiently for determination of studied drugs simultaneously in their binary mixtures as well as in the commercial dosage forms with satisfactory precision.
From the comparison of the results obtained with the proposed methods and the 1D reported method, they do not appear to have substantial differences, but a little superiority of the PCR technique over the 1D and CLS in the simultaneous determination of lamivudine and stavudine was observed especially in the low concentrations of stavudine and in presence of high background interferences.
The proposed procedures are rapid and sufficiently precise and suitable for quality control laboratories, where the economy and time are important factors.
References
- Anbazhagan S., Indumathy N., Shanmugapandiyan P., Krishnan S.S. Simultaneous quantification of stavudine, lamivudine and nevirapine by UV spectroscopy, reverse phase HPLC and HPTLC in tablets. J. Pharm. Biomed. Anal. 2005;39:801–804. doi: 10.1016/j.jpba.2005.04.044. [DOI] [PubMed] [Google Scholar]
- Catherine K.M., Eleftheria T.M., John E.K. Chemometric and derivative methods as flexible spectrophotometric approaches for dissolution and assaying tests in multicomponent tablets. Il Farmaco. 2004;59:627–636. doi: 10.1016/j.farmac.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Delgado J.N., Reme W.A. 10th ed. J.B. Lippincot Company; London/New York: 1998. Wilson and Gisvold’s Textbook Organic Medicinal and Pharmaceutical Chemistry. [Google Scholar]
- Dinc E., Ozdemir A., Baleanu D. Comparative study of the continuous wavelet transform, derivative and partial least squares methods applied to the overlapping spectra for the simultaneous quantitative resolution of ascorbic acid and acetylsalicylic acid in effervescent tablets. J. Pharm. Biomed. Anal. 2005;37:569–575. doi: 10.1016/j.jpba.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Hardman, J.G., Limbird, L.E. (Eds. in chief), 2001. Goodman and Gilman’s, The Pharmacological Basis of Therapeutics, 10th ed. McGraw-Hill, New York.
- Henrion R., Henrion G. Springer; Berlin: 1994. Multivariate Data Analysis. [Google Scholar]
- Hou W., Watters J.W., McLeod H.L. Simple and rapid docetaxel assay in plasma by protein precipitation and high-performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. B. 2004;804:263–267. doi: 10.1016/j.jchromb.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Indian Pharmacopoeia, 1996 (Addendum 2002). Govt. of India, Ministry of Health and Family Welfare, Controller of Publications, Delhi, pp. 930-938.
- Indian Pharmacopoeia, 1996 (Addendum 2002). Govt. of India, Ministry of Health and Family Welfare, Controller of Publications, Delhi, p. 913.
- Kapoor N., Khandavilli S., Panchagnula R. Simultaneous determination of lamivudine and stavudine in antiretroviral fixed dose combinations by first derivative spectrophotometry and high performance liquid chromatography. J. Pharm. Biomed. Anal. 2006;41:761–765. doi: 10.1016/j.jpba.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Kapoor N., Khandavilli S., Panchagnula R. Simultaneous determination of lamivudine, stavudine and nevirapine in antiretroviral fixed dose combinations by high performance liquid chromatography. Anal. Chim. Acta. 2006;570(1):41–45. doi: 10.1016/j.jpba.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Kaul S., Stouffer B., Mummaneni V., Turabi N., Mantha S., Jayatilak P., Barbhaiya R. Specific radioimmuno-assays for the measurement of stavudine in human plasma and urine. J. Pharm. Biomed. Anal. 1996;15:165–174. doi: 10.1016/0731-7085(96)01839-0. [DOI] [PubMed] [Google Scholar]
- Mistri Hiren N., Jangid Arvind G., Pudage Ashutosh, Gomes Noel, Sanyal Mallika, Shrivastav Pranav. High throughput LC–MS/MS method for simultaneous quantification of lamivudine, stavudine and nevirapine in human plasma. J. Chromatogr. B. 2007;853:320–332. doi: 10.1016/j.jchromb.2007.03.047. [DOI] [PubMed] [Google Scholar]
- Mohamed A.E.-M.I., Salem H. Determination of certain antihypertensive mixtures using chemometrics-assisted spectrophotometric method. Anal. Bioanal. Chem. 2005;382:1066–1072. doi: 10.1007/s00216-005-3230-4. [DOI] [PubMed] [Google Scholar]
- Palabiyik M., Dinc E., Onur F. Simultaneous spectrophotometric determination of pseudoephedrine hydrochloride and ibuprofen in a pharmaceutical preparation using ratio spectra derivative spectrophotometry and multivariate calibration techniques. J. Pharm. Biomed. Anal. 2004;34:473–483. doi: 10.1016/s0731-7085(03)00578-8. [DOI] [PubMed] [Google Scholar]
- Pendela M., Van Gyseghem E., Van den Mooter G., Baert L., Rosier J., Hoogmartens J., Adams E. Development of a liquid chromatographic assay for an anti-HIV tablet containing lamivudine, zidovudine and TMC278 HCL. J. Pharm. Biomed. Anal. 2009;49:508–512. doi: 10.1016/j.jpba.2008.11.006. [DOI] [PubMed] [Google Scholar]
- Sabry S.M., Khamis E.F. Application of H-point standard additions method to spectrophotometric and spectrofluorometric determination of glafenine and glafenic acid in mixtures. Talanta. 2000;51:1219–1231. doi: 10.1016/s0039-9140(00)00310-6. [DOI] [PubMed] [Google Scholar]
- Salem M.Y., El-bardicy M.G., El-Tarras M.F., El-Zanfally E.S. Simultaneous determination of domperidone maleate and cinnarizine in a binary mixture using derivative ratio spectrophotometry and classical least squares calibration. J. Pharm. Biomed. Anal. 2002;30:21–33. doi: 10.1016/s0731-7085(02)00094-8. [DOI] [PubMed] [Google Scholar]
- Sarasa M., Riba N., Zamora L., Carné X. Determination of stavudine in human plasma and urine by high-performance liquid chromatography using a reduced sample volume. J. Chromatogr. B: Biomed. Sci. Appl. 2000;746:183–189. doi: 10.1016/s0378-4347(00)00324-8. [DOI] [PubMed] [Google Scholar]
- Tomsu D., Catala I.C., Catalayud J.M. Automated simultaneous triple dissolution profiles of two drugs, sulphamethoxazole–trimethoprim and hydrochlorothiazide–captopril in solid oral dosage forms by a multicommutation flow-assembly and derivative spectrophotometry. J. Pharm. Biomed. Anal. 2004;36:549–557. doi: 10.1016/j.jpba.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Volosov A., Alexander C., Ting L., Soldin S.J. Simple rapid method for quantification of antiretrovirals by liquid chromatography—tandem mass-spectrometry. Clin. Biochem. 2002;35(2):99–103. doi: 10.1016/s0009-9120(02)00286-2. [DOI] [PubMed] [Google Scholar]
- Williams D.A., Lemke T.L. fifth ed. Lea and Feiger; Philadelphia: 2002. Foye’s Principle of Medicinal Chemistry. [Google Scholar]
- Xu Meng, White Catherine A., Bartlett Michael G. Simultaneous determination of zalcitabine and stavudine in maternal plasma, amniotic fluid, placental, and fetal tissues using reversed phase on silica liquid chromatography/tandem mass spectrometry. J. Chromatogr. Relat. Technol. 2009;32:680–697. [Google Scholar]