

The crystal structure of succinyl-CoA: 3-ketoacid CoA transferase from Drosophila melanogaster was determined at 2.64 Å resolution.
Keywords: succinyl-CoA:3-ketoacid CoA transferase, Drosophila melanogaster, class I CoA transferases
Abstract
Succinyl-CoA:3-ketoacid CoA transferase (SCOT) plays a crucial role in ketone-body metabolism. SCOT from Drosophila melanogaster (DmSCOT) was purified and crystallized. The crystal structure of DmSCOT was determined at 2.64 Å resolution and belonged to space group P212121, with unit-cell parameters a = 76.638, b = 101.921, c = 122.457 Å, α = β = γ = 90°. Sequence alignment and structural analysis identified DmSCOT as a class I CoA transferase. Compared with Acetobacter aceti succinyl-CoA:acetate CoA transferase, DmSCOT has a different substrate-binding pocket, which may explain the difference in their substrate specificities.
1. Introduction
Succinyl-CoA:3-ketoacid CoA transferase (SCOT; EC 2.8.3.5) is a key enzyme involved in ketone-body metabolism. It catalyzes the transfer of CoA from succinyl-CoA to acetoacetate to produce acetoacetyl-CoA and succinate via a ping-pong mechanism (Stern et al., 1956 ▶),
Acetoacetyl-CoA is further converted into acetyl-CoA, which enters the citric acid cycle to either provide energy or be stored as a fatty acid. SCOT belongs to the class I CoA transferases and can form a thioester intermediate during catalysis in which the CoA group from succinyl-CoA is covalently bound to a conserved glutamate residue at the active site (Solomon & Jencks, 1969 ▶; Rochet & Bridger, 1994 ▶). The glutamate-CoA thiolester replaces an anhydride intermediate which is formed by the succinyl group binding to the active-site Glu (Benson & Boyer, 1969 ▶).
Previous studies showed that eukaryotic SCOTs such as that from pig heart are homodimeric with one active site per subunit (Lloyd & Shoolingin-Jordan, 2001 ▶). In contrast to the eukaryotic SCOTs, the bacterial enzymes are heterotetrameric (α2β2), in which the α subunits of the prokaryotic proteins bind CoA and correspond to the N-terminal domains of eukaryotic SCOTs, whereas the β subunits provide the catalytic glutamates and correspond to the C-terminal domains (Parales & Harwood, 1992 ▶; Corthesy-Theulaz et al., 1997 ▶). The structures of CoA transferases from the bacterial source Acidaminococcus fermentans (GCT) and from pig heart show that these types of enzymes share a similar fold despite having little sequence homology (Jacob et al., 1997 ▶; Bateman et al., 2002 ▶). The formation of the pig heart SCOT–CoA intermediate triggers significant conformational changes in which the pocket between the N-terminal and C-terminal domains becomes narrower to bury the reactive intermediate inside and to protect it from reacting with the solvent (Fraser et al., 2010 ▶). Interestingly, the N-terminal and C-terminal domains of eukaryotic SCOT are usually connected by a highly hydrophilic linker which can be specifically digested at the helix–turn–helix motif without affecting the catalytic activity (Lin & Bridger, 1992 ▶). Patients with SCOT deficiency (OMIM 245050) suffer a metabolic disease leading to severe ketoacidosis. Under conditions of diabetes or limited availability of carbohydrates, ketolysis enables ketone bodies in the liver to produce fat-derived energy that can be used by extrahepatic tissues such as heart, kidney, skeletal muscle and brain (Mitchell et al., 1995 ▶). Diminution in the SCOT catalytic activity may lead to a rise in the concentration of ketone bodies (acetoacetate and 3-hydroxybutyrate) and cause severe ketoacidosis (Grinblat et al., 1986 ▶).
Here, we report the crystal structure of SCOT from Drosophila melanogaster (DmSCOT) at 2.64 Å resolution. The protein shares 63% sequence identity with both human SCOT and pig heart SCOT (Fig. 1 ▶). The overall structure of DmSCOT is similar to those of its homologues from other organisms, such as Homo sapiens and Sus scrofa (PDB entries 3dlx and 1ooy; Structural Genomics Consortium, unpublished work; Coros et al., 2004 ▶). Sequence alignment and structural analysis reveal that DmSCOT contains a conservative CoA-binding site and a potential acetoacetate-binding site.
Figure 1.

Multiple alignment of DmSCOT, pig heart SCOT and human SCOT. The alignment was performed using MultAlin (Corpet, 1988 ▶) and ESPript (Gouet et al., 2003 ▶). The secondary-structural elements of human SCOT are displayed at the top of the alignment. α-Helices, η-helices, β-sheets and strict β-turns are denoted α, η, β and TT, respectively. The CoA-binding sites are marked by filled blue circles.
2. Materials and methods
2.1. Cloning, expression and purification of DmSCOT
DmSCOT devoid of the signal peptide (residues 34–516) was amplified by PCR from a D. melanogaster cDNA library using sense (5′-CCGGAATTCGGCAAGATCTACGAGTCGGCCATAG-3′) and antisense (5′-CCGCTCGAGTCAGACCGGAATTTGACCCATTTTC-3′) primers. The amplified fragments were then cloned into a modified pET28a vector (Novagen) with an additional 6×His coding sequence following a TEV protease cleavage site at the 5′ end of the gene. The recombinant DmSCOT expression plasmid was identified by restriction-endonuclease digestion and was further verified using DNA sequencing at Sangon Biotech (Shanghai, People’s Republic of China). 20 µl of a glycerol stock of transformed Escherichia coli cells was used to inoculate 4 ml Luria broth (LB) medium supplemented with 0.01 mg ml−1 kanamycin. This culture was grown overnight at 310 K and was then transferred into 300 ml LB supplemented with 0.01 mg ml−1 kanamycin. The culture was grown at 310 K to an A 600 nm of 0.66 and was induced at 290 K with 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 16 h. After harvesting, the cells were resuspended in 50 ml buffer consisting of 500 mM NaCl, 20 mM Tris–HCl pH 8.5, 10 mM imidazole, 1 mM DTT, 5% glycerol. After three freeze–thaw cycles followed by 3 min sonication, the lysed cells were centrifuged at 15 000g for 40 min. The supernatant was loaded onto an Ni2+–NTA column (GE Healthcare) equilibrated with binding buffer (500 mM NaCl, 20 mM Tris–HCl pH 8.5, 10 mM imidazole, 1 mM DTT, 10% glycerol). The column was eluted with a 20–500 mM imidazole gradient. Fractions containing the recombinant protein were pooled and desalted by dialysis against buffer consisting of 100 mM NaCl, 20 mM Tris–HCl pH 8.5, 1 mM DTT, 10% glycerol. Tag cleavage was performed at 303 K for 2 h with TEV protease (16 µg ml−1) and the protein was then applied onto an Ni2+–NTA column to remove the His tag. The sample was subsequently loaded onto a Superdex 200 column (Amersham Biosciences) equilibrated with 100 mM NaCl, 20 mM Tris–HCl pH 8.5, 1 mM DTT, 10% glycerol. The purity of the fractions was checked by SDS–PAGE.
2.2. Enzyme-activity assay
The DmSCOT activity was determined by measuring the formation of acetoacetyl-CoA, in which the increase in the A 310 nm was monitored every 20 s for 5 min. The reaction buffer consisted of 67 mM lithium acetoacetate, 300 mM succinyl CoA, 15 mM MgCl2 in 50 mM Tris–HCl pH 9.1 at 298 K (Williamson et al., 1971 ▶). The extinction coefficient for acetoacetyl-CoA at 310 nm under these conditions is 7.8 × 103 M −1 cm−1 (Howard et al., 1986 ▶). All experiments were performed in triplicate. The specific activity of DmSCOT was defined as the amount of enzyme required to convert 1 µmol of substrate to product in 1 min under the assay conditions.
2.3. Crystallization and data collection
DmSCOT was crystallized at 289 K using sitting-drop vapour diffusion. The crystals of DmSCOT were grown in a drop consisting of 20 mg ml−1 protein in 100 mM NaCl, 20 mM Tris–HCl pH 8.5, 1 mM DTT, 10% glycerol and an equal volume of reservoir solution consisting of 0.2 M lithium sulfate, 0.1 M bis-tris pH 5.5, 25% PEG 3350 within 7 d. The crystals were transferred into cryoprotectant (reservoir solution supplemented with 10% glycerol) and flash-cooled in liquid nitrogen. The X-ray diffraction data set was collected on beamline BL17U at Shanghai Synchrotron Radiation Facility (SSRF), People’s Republic of China and processed with HKL-2000 (Otwinowski & Minor, 1997 ▶).
2.4. Structure determination and refinement
The structure of DmSCOT was solved by the molecular-replacement method with Phaser using pig SCOT (PDB code 1ooy; Coros et al., 2004 ▶) as the model. One dimer was located in the crystallographic asymmetric unit. The initial model was refined using the maximum-likelihood method implemented in REFMAC5 as part of the CCP4 program suite (Murshudov et al., 2011 ▶) and model building was performed in Coot (Emsley & Cowtan, 2004 ▶). 5% of reflections were set side to calculate R free. The structure was refined with an R work of 0.204 and an R free of 0.261. The final models were evaluated with MolProbity (Chen et al., 2010 ▶) and PROCHECK (Winn et al., 2011 ▶). The final coordinates and structure factors were deposited in the Protein Data Bank (http://www.rcsb.org/pdb) under accession code 4kgb. The data-collection and structure-refinement statistics are listed in Table 1 ▶. All structure figures were prepared with PyMOL (http://www.pymol.org).
Table 1. Crystal parameters, data collection and structure refinement for DmSCOT.
Values in parentheses are for the highest resolution bin.
| Data processing | |
| Space group | P212121 |
| Unit-cell parameters (Å, °) | a = 76.638, b = 101.921, c = 122.457, α = β = γ = 90 |
| Resolution range (Å) | 50.0–2.65 (2.70–2.65) |
| Unique reflections | 28630 (1380) |
| Completeness (%) | 99.9 (100.0) |
| 〈I/σ(I)〉 | 21.7 (7.0) |
| R merge † | 0.108 (0.338) |
| Multiplicity | 5.7 (6.0) |
| Refinement statistics | |
| Resolution range (Å) | 43.34–2.64 |
| R factor‡/R free § | 0.204/0.261 |
| No. of atoms | |
| Protein | 6867 |
| Ligand | 5 |
| Water | 59 |
| R.m.s.d.¶ | |
| Bond lengths (Å) | 0.013 |
| Bond angles (°) | 1.680 |
| Average B factor (Å2) | 34.6 |
| Ramachandran plot†† | |
| Residues in favoured region (%) | 96.0 |
| Residues in allowed region (%) | 3.34 |
| Outliers (%) | 0.66 |
| PDB code | 4kgb |
R
merge = 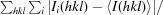
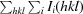 ∑hkl, where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean value for a unique reflection; summations are over all reflections.
∑hkl, where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean value for a unique reflection; summations are over all reflections.
R factor = 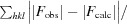
 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
R free was calculated using 5% of the data excluded from refinement.
Root-mean-square deviation from ideal values.
Categories were defined by MolProbity.
3. Results and discussion
3.1. Overall structure of DmSCOT and its comparison with other SCOTs
The crystal of DmSCOT diffracted to 2.64 Å resolution and belonged to space group P212121, with two molecules in the asymmetric unit (Fig. 2 ▶). DmSCOT preserves the typical SCOT tertiary structure, which adopts an open α/β fold. The DmSCOT monomer consists of N-terminal and C-terminal domains (residues 1–247 and 259–484, respectively), each displaying an α/β/α motif with a central seven-stranded β-sheet. Structural analysis using the PISA web server (Krissinel & Henrick, 2007 ▶) indicated that the dimer interface involves 63 residues from each monomer and buries an interface area of approximately 2200 Å2. The biologically relevant DmSCOT dimer interface involves hydrophobic interactions and some β-sheet hydrogen bonding. There are 12 hydrogen bonds (involving residues Glu104, Arg107, Gly110, Ala111, Lys186, Arg189, Asp218, His221, Arg353 and His356 from both monomers), numerous hydrophobic interactions and some salt bridges stabilizing the dimerization interface.
Figure 2.

Cartoon representation of the DmSCOT dimer.
Using the DALI server (Holm & Rosenström, 2010 ▶), we confirmed that the DmSCOT structure is very similar to that of pig heart SCOT as well as those of class I CoA transferases from other species such as A. fermentans glutaconate CoA transferase (PDB entry 1poi; Jacob et al., 1997 ▶), E. coli YdiF (PDB entry 2ahu; Rangarajan et al., 2005 ▶) and human SCOT (PDB entry 3dlx; Structural Genomics Consortium, unpublished work). Glu307 of DmSCOT can be superposed onto Glu305 of pig heart SCOT, which is known to be the conserved glutamate residue in the active site of SCOTs (Fig. 3 ▶ a). Residues 250–257 in the hydrophilic linker, which is absent in the bacterial homologues, are invisible in the electron-density maps. The region consisting of residues 373–383 is also disordered in the DmSCOT structure. There are conformational differences between DmSCOT and pig heart SCOT in several loops. The most obvious difference lies in residues 125–157 of DmSCOT, which contains three extra twisted β-hairpins.
Figure 3.

(a) Superposition of the monomers of DmSCOT (yellow) and pig heart SCOT (cyan). The active-site glutamate residues are shown as stick representations. (b) Superposition of the CoA-binding sites of pig heart SCOT bound to CoA (green) and DmSCOT (yellow). The CoA-binding site residues are shown as sticks.
3.2. The substrate-binding site of DmSCOT
Previous study showed that CoA binding results in a conformational change of pig heart SCOT to allow the closure of the substrate-binding pocket, thus fully shielding the reactive intermediates from the solvent (Fraser et al., 2010 ▶). The enzyme forms stabilizing interactions with both the nucleotide and pantoic acid portions of CoA in the binding pocket. CoA is covalently bound to the active-site Glu305 and forms hydrogen bonds to Tyr76, Ile284, Ile286, Met363, Asn373, Gly386 and Lys407 of pig heart SCOT. Superposition of the DmSCOT structure onto that of pig heart SCOT with bound CoA shows that the residues in the CoA-binding site are highly conserved and that the pocket occupied by the bound CoA in the pig heart SCOT structure is also present in DmSCOT (Fig. 3 ▶ b). By determining the SCOT activity, we found that the specific activity of DmSCOT is 3.69 ± 0.008 µmol min−1 mg−1, which is lower than that of pig heart SCOT (25.2 ± 1.2 µmol min−1 mg−1; Tammam et al., 2007 ▶). Given the highly conserved active sites and overall structures, we speculate that the single-residue difference in the CoA-binding pocket (Arg409 in DmSCOT versus Lys407 in pig heart SCOT) may account for the lowered catalytic activity of DmSCOT.
In previous studies, it has been shown that Asn51, Asn52 and Gln99 in the structure of pig heart SCOT interact with a glycerol molecule, which mimics the substrate acetoacetate (Coker et al., 2010 ▶). The conservation of these residues in DmSCOT suggests that a similar acetoacetate-binding site is present in DmSCOT. In the structure of Acetobacter aceti succinyl-CoA:acetate CoA transferase the substrate acetate binds to Ser71, Thr94 and Arg228, which are different from the acetoacetate-binding site in SCOTs (Mullins & Kappock, 2012 ▶). These differences may bring about the diverse substrate specificity and distinct functions of CoA transferases.
Supplementary Material
PDB reference: DmSCOT, 4kgb
Acknowledgments
We thank Professors Congzhao Zhou and Yuxing Chen at USTC for their generous assistance. We also thank Dr Yongliang Jiang at USTC for helpful discussions. The authors also thank beamline BL17U of the Shanghai Synchrotron Radiation Facility (SSRF) for providing the beam time. This work was supported by grants from the National Natural Science Foundation of China (grants 30970565, 31170688 and 31270808), the National Basic Research Program of China (grant 2009CB825502) and the Science and Technological Fund of Anhui Province for Outstanding Youth (grant 10040606Y15).
References
- Bateman, K. S., Brownie, E. R., Wolodko, W. T. & Fraser, M. E. (2002). Biochemistry, 41, 14455–14462. [DOI] [PubMed]
- Benson, R. W. & Boyer, P. D. (1969). J. Biol. Chem. 244, 2366–2371. [PubMed]
- Coker, S.-F., Lloyd, A. J., Mitchell, E., Lewis, G. R., Coker, A. R. & Shoolingin-Jordan, P. M. (2010). Acta Cryst. D66, 797–805. [DOI] [PubMed]
- Coros, A. M., Swenson, L., Wolodko, W. T. & Fraser, M. E. (2004). Acta Cryst. D60, 1717–1725. [DOI] [PubMed]
- Corpet, F. (1988). Nucleic Acids Res. 16, 10881–10890. [DOI] [PMC free article] [PubMed]
- Corthesy-Theulaz, I. E., Bergonzelli, G. E., Henry, H., Bachmann, D., Schorderet, D. F., Blum, A. L. & Ornston, L. N. (1997). J. Biol. Chem. 272, 25659–25667. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fraser, M. E., Hayakawa, K. & Brown, W. D. (2010). Biochemistry, 49, 10319–10328. [DOI] [PubMed]
- Gouet, P., Robert, X. & Courcelle, E. (2003). Nucleic Acids Res. 31, 3320–3323. [DOI] [PMC free article] [PubMed]
- Grinblat, L., Pacheco Bolanos, L. F. & Stoppani, A. O. (1986). Biochem. J. 240, 49–56. [DOI] [PMC free article] [PubMed]
- Holm, L. & Rosenström, P. (2010). Nucleic Acids Res. 38, w545–W549. [DOI] [PMC free article] [PubMed]
- Howard, J. B., Zieske, L., Clarkson, J. & Rathe, L. (1986). J. Biol. Chem. 261, 60–65. [PubMed]
- Jacob, U., Mack, M., Clausen, T., Huber, R., Buckel, W. & Messerschmidt, A. (1997). Structure, 5, 415–426. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Lin, T. W. & Bridger, W. A. (1992). J. Biol. Chem. 267, 975–978. [PubMed]
- Lloyd, A. J. & Shoolingin-Jordan, P. M. (2001). Biochemistry, 40, 2455–2467. [DOI] [PubMed]
- Mitchell, G. A., Kassovska-Bratinova, S., Boukaftane, Y., Robert, M. F., Wang, S. P., Ashmarina, L., Lambert, M., Lapierre, P. & Potier, E. (1995). Clin. Invest. Med. 18, 193–216. [PubMed]
- Mullins, E. A. & Kappock, T. J. (2012). Biochemistry, 51, 8422–8434. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Parales, R. E. & Harwood, C. S. (1992). J. Bacteriol. 174, 4657–4666. [DOI] [PMC free article] [PubMed]
- Rangarajan, E. S., Li, Y., Ajamian, E., Iannuzzi, P., Kernaghan, S. D., Fraser, M. E., Cygler, M. & Matte, A. (2005). J. Biol. Chem. 280, 42919–42928. [DOI] [PubMed]
- Rochet, J. C. & Bridger, W. A. (1994). Protein Sci. 3, 975–981. [DOI] [PMC free article] [PubMed]
- Solomon, F. & Jencks, W. P. (1969). J. Biol. Chem. 244, 1079–1081. [PubMed]
- Stern, J. R., Coon, M. J. & Del Campillo, A. (1956). J. Biol. Chem. 221, 1–14. [PubMed]
- Tammam, S. D., Rochet, J. & Fraser, M. E. (2007). Biochemistry, 46, 10852–10863. [DOI] [PubMed]
- Williamson, D. H., Bates, M. W., Page, M. A. & Krebs, H. A. (1971). Biochem. J. 121, 41–47. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: DmSCOT, 4kgb