

Abstract
Prion diseases encompass a diverse group of neurodegenerative conditions characterized by the accumulation of misfolded prion protein (PrP) isoforms. Other conformational variants of PrP have also been proposed to contribute to neurotoxicity in prion diseases, including misfolded intermediates as well as cytosolic and transmembrane isoforms. To better understand PrP neurotoxicity, we analyzed the role of two highly conserved methionines in helix 3 on PrP biogenesis, folding and pathogenesis. Expression of the PrP-M205S and -M205,212S mutants in Drosophila led to hyperglycosylation, intracellular accumulation and widespread conformational changes due to failure of oxidative folding. Surprisingly, PrP-M205S and -M205,212S acquired a transmembrane topology (Ctm) previously linked to mutations in the signal peptide (SP) and the transmembrane domain (TMD). PrP-M205,212S also disrupted the accumulation of key neurodevelopmental proteins in lipid rafts, resulting in shortened axonal projections. These results uncover a new role for the hydrophobic domain in promoting oxidative folding and preventing the formation of neurotoxic Ctm PrP, mechanisms that may be relevant in the pathogenesis of both inherited and sporadic prion diseases.
INTRODUCTION
The prion protein (PrP) is a glycosylphosphatidylinosotol (GPI)-anchored glycoprotein with a central role in a group of neurodegenerative disorders characterized by spongiform vacuolation, collectively known as prion diseases (1). Creutzfeldt–Jakob disease, Gerstmann–Straussler–Scheinker (GSS) syndrome, kuru and fatal insomnia are examples of human prion diseases with varied clinical manifestations, including cognitive, behavioral and locomotor disturbances. The common link to these diverse diseases is the accumulation of abnormal conformers of PrP in the brain. The native folding of PrP, called cellular PrP (PrPC), contains a globular domain in the C-terminus with three α-helices (α1-3) and two short β-strands (β1-2). Conversion of PrP into its pathogenic conformations, including scrapie PrP (PrPSc), increases the β-strand content at the expense of helices, which favors aggregation. Despite the wealth of information on the PrP structure from biochemical and biophysical studies as well as molecular dynamics modeling in silico, the mechanisms mediating PrP conversion and neurotoxicity in vivo are still poorly understood.
In addition to the misfolded PrP conformations, cytoplasmic and transmembrane PrP isoforms have been described in human patients and animal models (2,3). These alternative isoforms have been ascribed to aberrant PrP biogenesis and seem to be favored by pathogenic mutations linked to inherited prion diseases (2,4,5). Two different transmembrane topologies have been described, one with the N-terminus in the ER lumen (Ntm) and another with the C-terminus in the ER (Ctm) (4,6). The Ntm and Ctm PrP topologies were first observed in cell-free translation systems exhibiting a single transmembrane pass that corresponds to hydrophobic stretch spanning residues 111–135, the so-called transmembrane domain (TMD) (4). While Ntm does not seem to play a role in PrP pathogenesis, mutations in the TMD, such as the GSS-linked A117V, promote the accumulation of Ctm topologies (2). Additional mutations in the TMD, including an artificial triple A–V substitution (3AV), result in a more prominent production of Ctm at the expense of both secreted and Ntm PrP (2,4). These results suggested that mutations that increase the helicity of the TMD promote its transmembrane insertion (4). The accumulation of Ctm topologies was also explained as a failure of nascent PrP chains to completely translocate into the ER lumen. To determine the role of the SP in the accumulation of Ctm PrP, the Harris’ laboratory introduced a mutation in the SP (L9R) that reduced the efficiency of translocation to the ER and also increased Ctm topology (5). Moreover, the combination of SP and TMD mutations (L9R, 3AV) resulted in a more efficient production of Ctm, with all PrP accumulating as Ctm in cell-free translation systems and 50% of the chains appearing as Ctm in transgenic mice (5,7). Interestingly, mice expressing PrP-L9R,3AV showed strong neuropathology in the absence of PrPSc and transmissibility, supporting the idea that PrPSc is not required for neurodegeneration (7). In those mice, the neurotoxicity of PrP-L9R,3AV required endogenous PrP-WT, suggesting that Ctm topologies may mediate the conversion of secreted PrP to induce neurotoxic conformations (7). These observations suggest that Ctm PrP plays a relevant role in disease and has even been postulated as the neurotoxic component in some inherited prion diseases (2). So far, only the SP and the TMD seem to be involved in the production of Ctm PrP. But if other PrP motifs were implicated in the formation of Ctm, that would contribute to explain neurodegeneration in other forms of inherited prion diseases and, possibly, some sporadic cases.
To gain insight into the formation of toxic PrP conformations, we have focused on the role of hydrophobic residues in α3. Long-range hydrophobic interactions play a key role in stabilizing the secondary structures of the globular domain. In fact, α2 and α3 form the rigid core of PrP, which is stabilized by a short loop, a disulfide bond (C179–C214, human numbering) and hydrophobic interactions that include distant residues between α1 and the other loops. Interestingly, several disease-causing mutations are located in the extensive hydrophobic network between α2 and α3, including the conservative substitutions V180I and V210I, suggesting that even small structural perturbations in this domain promote the accumulation of neurotoxic conformations. Molecular dynamics simulations predict that the pathogenic mutations V180I and V210I can disrupt distant residues within the hydrophobic core, including the displacement of M206 (8). M206 and M213 are highly conserved residues in α3 (α3M hereafter), suggesting that they are critical for maintaining the native folding of PrP. α3M have received special attention because they appeared to be oxidized in PrPSc in human and rodent brains (6,9,10). In fact, α3M sulfoxidation preceded PrPSc formation in vitro, arguing that this covalent modification can promote PrP misfolding and disease (11), although other studies reported the opposite results (12). Molecular dynamics simulation predicted that the increase in polarity at α3M would promote PrP misfolding (13). Moreover, in vitro incorporation of polar alternatives (methoxinine) increased the aggregation propensity of PrP (14), while PrP oxidation disturbed α3M along with other nearby hydrophobic residues (15). In addition, replacement of α3M by Ser (α3M>S) in recPrP(23–231) revealed the population of partially unstructured states and increased aggregation by thioflavin-T binding (16). Furthermore, expression of hamster PrP-M206S and -M213S in human cell lines revealed aberrant PrP biogenesis linked to failure of oxidative folding (17). Overall, these observations indicate that increased polarity at α3M promotes PrP misfolding and aggregation, suggesting that it may play a physiological role in prion diseases. Despite these interesting results, the consequence of these polar substitutions has not been tested in animal models. Doing so may provide insight about the role of disease-causing mutations in α3.
In this study, we describe the generation of transgenic flies expressing mouse PrP-α3M>S substitutions to investigate the role of key hydrophobic residues in proper PrP folding, biogenesis and pathogenesis. We found that PrP-α3M>S mutants (M205 and M212, mouse numbering) were hyperglycosylated, accumulated intracellularly and displayed widespread conformational changes, including the disruption of the disulfide bond. Moreover, PrP-α3M>S mutants induced neurodevelopmental phenotypes that were only accompanied with progressive neurodegeneration in the presence of PrP-WT. All these new and intriguing features of the PrP-α3M>S mutants correlated with the accumulation of the transmembrane variant Ctm PrP. Interestingly, Ctm PrP perturbed vesicular trafficking and prevented the accumulation of key neurodevelopmental proteins in the lipid raft. These surprising results show, for the first time, that mutations outside the SP and TMD can produce Ctm PrP in vivo, and argue for the critical role of the oxidative folding of the globular domain in hiding the TMD and preventing its membrane integration. Our studies identify a new mechanism driving the formation of toxic Ctm PrP that may explain the pathogenesis of several disease-causing mutations in α3 and in sporadic forms of prion diseases.
RESULTS
PrP-α3M>S alters PrP maturation
To investigate the consequence of expressing the PrP-α3M>S substitutions in vivo, we generated transgenic flies expressing PrP-WT, -M205S and -M205,212S under the control of the UAS/Gal4 transcriptional system (18). We first studied the effects of the α3M>S mutants on PrP biogenesis by examining their electrophoretic mobility by western blot. For this, we produced homogenates with whole flies and resolved PrP by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) using the N-terminal anti-PrP antibody 6D11. PrP-WT produced the three characteristic glycoforms, with the un- and monoglycosylated isoforms being the most abundant (Fig. 1A). However, both PrP-α3M>S mutants run with a higher molecular weight than PrP-WT, with the M205,212S mutant running even higher than the single mutant (Fig. 1A). To determine whether the different electrophoretic mobility of the PrP-α3M>S substitutions was due to aberrant glycosylation, we eliminated all the sugar chains using PNGase F and detected PrP in western blot. Complete deglycosylation of PrP-WT and the PrP-α3M>S mutants resulted in single bands, although both PrP-α3M>S mutants exhibited a slightly higher molecular weight than the WT (Fig. 1B). Since M205S and M205,212S showed the same molecular weight after deglycosylation, the slower mobility of M205,212S could be explained by hyperglycosylation, which suggested that its maturation was incomplete. PrP is first glycosylated after the new protein chains translocate to the ER lumen, where they are sensitive to Endo H. Then, PrP moves to the Golgi apparatus, where sugar chains are modified to form the mature glycoprotein, which is resistant to Endo H. To determine whether the PrP-α3M>S substitutions led to immature PrP glycoforms, we treated the fly homogenates with Endo H. As expected, PrP-WT was not susceptible to Endo H, supporting its mature glycosylation (Fig. 1C). Endo H treatment had no effect on M205S either; however, it reduced the size of the M205,212S mutant (Fig. 1C), supporting the immature, hyperglycosylated state of the double mutant.
Figure 1.
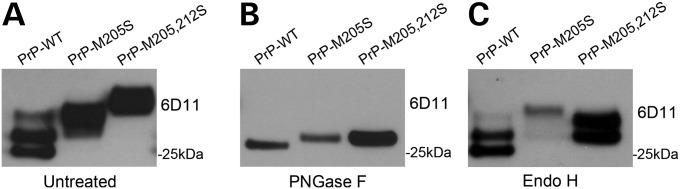
PrP-α3M>S mutants show incomplete maturation. (A–C) Tissue homogenates from flies expressing PrP-WT, -M205S or -M205,212S untreated (A) or incubated with PNGase F (B) or Endo H (C) and detected by 6D11 in western blot. (A) Both α3M>S mutants display a higher molecular weight than PrP-WT. (B) Complete deglycosylation with PNGase F produces a single band of unglycosylated PrP-WT at around 27 kDa. Both PrP-M205S and PrP-M205,212S exhibit a slightly higher molecular weight. (C) Treatment with Endo H has no effect on PrP-WT and -M205, but decreases the size of PrP-M205,212S.
PrP-α3M>S accumulates intracellularly
We next determined whether the aberrant post-translational modification of the PrP-α3M>S mutants resulted in abnormal PrP distribution in brain neurons. For this, we expressed PrP in interneurons of the larval ventral nerve cord under the control of OK107-Gal4. As described before, PrP-WT accumulated in the membrane, Golgi and secretory vesicles (Fig. 2A and B and Supplementary Material, Fig. S1E) (19). In contrast, PrP-M205,212S showed a punctate pattern throughout the cytoplasm, strikingly different from PrP-WT (Fig. 2A and B, and Supplementary Material, Fig. S1G). PrP-M205S showed an intermediate distribution between PrP-WT and PrP-M205,212S, with some accumulation in the Golgi and larger presence in smaller vesicles (Fig. 2A and B, and Supplementary Material, Fig. S1F). Since PrP-M205,212S is hyperglycosylated and may be stuck in the Golgi, we investigated whether the multivesicular distribution of PrP-M205,212S was due to Golgi fragmentation. To analyze the integrity of the Golgi apparatus, we used antibodies against p120 and Lava lamp (Lva), which label proximal and distal Golgi compartments, respectively (20,21). In neurons expressing PrP-WT, p120 and Lva partially co-localized in several puncta, indicating the presence of several Golgi per cell (Fig. 2C), the same distribution observed in control flies (Supplementary Material, Fig. S1A). Expression of PrP-α3M>S mutants did not change the number, size or distribution of the Golgi puncta, indicating that the integrity of the Golgi was not affected (Fig. 2C). To determine the identity of the puncta accumulating PrP-M205,212S, we used antibodies against several types of vesicles, including Rab11, Rab7 (not shown) and lysosomes. Whereas PrP-WT did not colocalize with Rab11, PrP-M205,212S partially co-localized with Rab11 vesicles (Fig. 2D and E), suggesting that mutant PrP is stuck in the secretory/endocytic pathway and cannot reach the membrane. However, we did not observe an increase of lysosomal vesicles or co-localization of PrP-M205,212S with the lysosomal marker Lamp1 (Fig. 2F and G) or the late endosome marker Rab7 (not shown). We observed no changes in the localization of Rab11 and Lamp1 in flies expressing PrP compared with control (LacZ) flies (Supplementary Material, Fig. S1B and C). These results indicated that PrP-M205,212S was not targeted for degradation through the autophagy/lysosomal pathway. Overall, these observations suggest that alterations in the post-translational modification of PrP-α3M>S mutants lead to abnormal PrP secretion and retention in recycling vesicles and other vesicles that do not reach the membrane.
Figure 2.
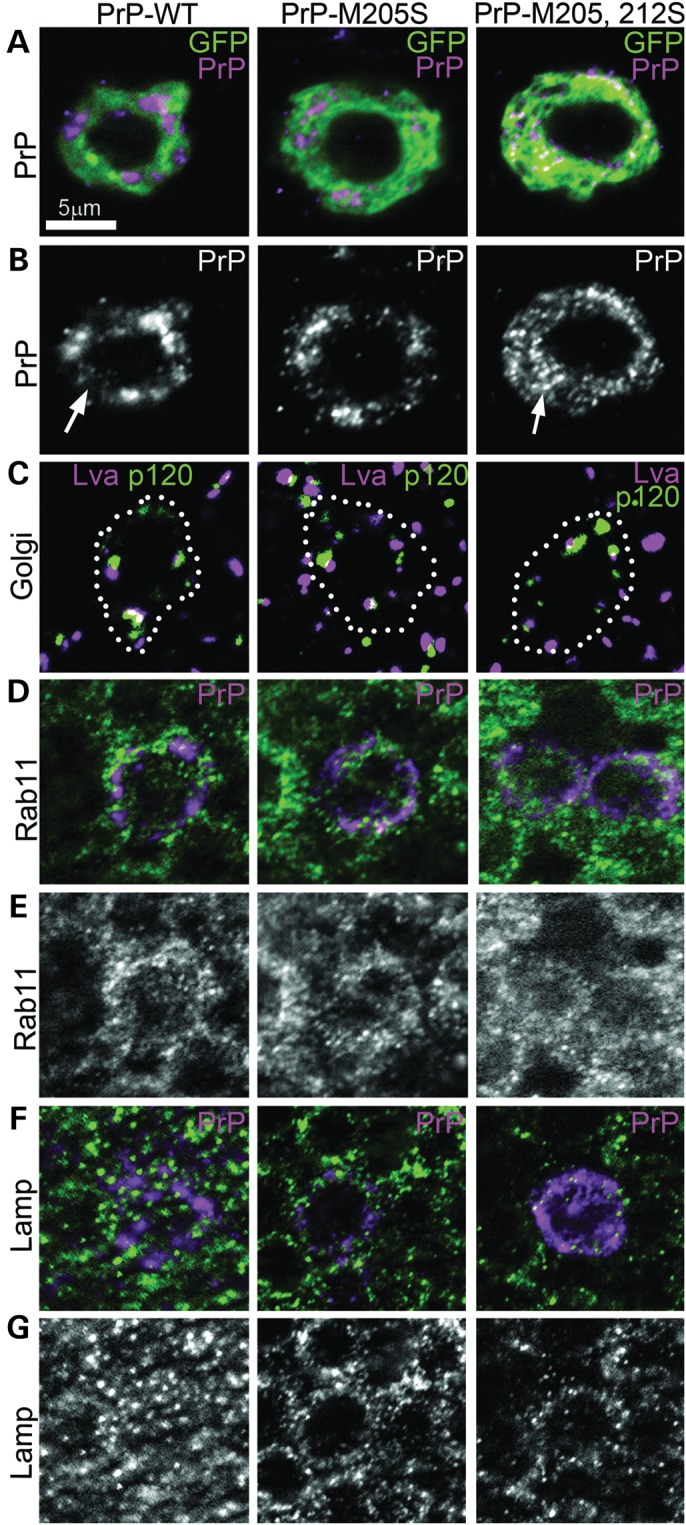
PrP-α3M>S mutants accumulate in intracellular vesicles. (A–E) Single focal plane micrographs of interneurons in the larval brain labeled with CD8-GFP showing the distribution of PrP-WT, -M205S, and -M205,212S. (A and B) PrP-WT accumulates in the Golgi and few secretory vesicles on its transition to the membrane. PrP-M205,212S accumulates in small puncta throughout the cytoplasm, while PrP-M205S shows a mixed distribution. (C) The α3M>S mutants do not disrupt the organization of the Golgi, as shown by the normal distribution of p120 and Lva. (D and E) PrP-WT and -M205S do not colocalize with endocytic vesicles containing Rab11, but PrP-M205,212S partially co-localizes with Rab11. (F and G) Neither PrP-WT nor the α3M>S mutants colocalize with the lysosome marker Lamp1. All images are shown at the same magnification.
PrP-α3M>S induces conformational changes
To determine whether the α3M>S mutations induced structural changes in PrP, we probed the flies expressing PrP with conformational antibodies. As control, we used the 6D11 antibody, which recognizes a linear epitope in the unstructured domain of PrP (amino acids 93–109) and results in positive immunoblotting of both WT and mutant PrP in western blot (Fig. 3A). In contrast, the 6H4 antibody detects an epitope in α1 (amino acids 144–152) that is perturbed when the disulfide bond is chemically reduced (22), thus resulting in sensitivity to the conformational states of PrP. 6H4 detected both PrP-WT and –M205S in immunoblot, but did not detect PrP-M205,212S (Fig. 3B). These results suggest that M205,212S induces significant conformational changes that can affect distant epitopes within the globular domain.
Figure 3.
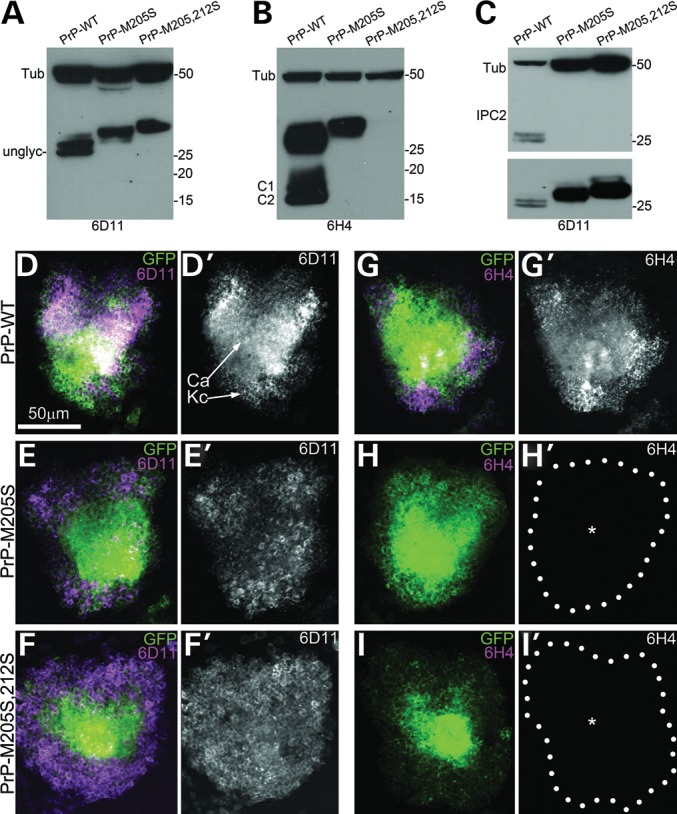
PrP-α3M>S prevents the formation of the disulfide bridge. (A–C) PrP immunoblots using 6D11 (A), 6H4 (B) and IPC2 (C). (A) 6D11 recognizes three bands in PrP-WT corresponding to its three glycoforms, while PrP-M205S and -M205,212S display higher molecular weights. (B) 6H4 recognizes the three glycoforms of PrP-WT plus the C1 and C2 proteolytic fragments. PrP-M205S runs slightly higher that WT, but C1 and C2 are not detected. PrP-M205,212S is not detected by 6H4. (C) IPC2 recognizes the un- and monoglycosylated forms of PrP. Neither PrP-M205S nor -M205,212S are recognized by IPC2 (top). The same membrane incubated with 6D11 reveals all PrPs (bottom). Tubulin is used as a loading control. (D–I) Micrographs show Kc clusters and the dendritic projections into the calix (Ca) by CD8-GFP in the adult brain. (D and D′) 6D11 recognizes PrP-WT, (E and E′) -M205S and (F and F′) -M205S,212. (G and G′) 6H4 immunoreacts with PrP-WT, but produces no signal in neurons expressing PrP-M205S (H and H′, asterisk) or -M205,212S (I and I′, asterisk).
Next, we examined the effect of the PrP-α3M>S substitutions on the 6D11 and 6H4 epitopes by immunofluorescence. For this, we expressed PrP along with CD8-GFP in the mushroom body neurons under the control of OK107-Gal4, and imaged the reactivity of the antibodies in the cell bodies (Kenyon cells, Kc, custom in field). The Kc form two clusters of ∼2500 tightly packed neurons involved in learning and memory (see Fig. 7A). 6D11 recognized PrP-WT and both mutants, labeling the cell bodies and calices (dendritic projections) of the Kc (Fig. 3D–F′). In contrast, 6H4 detected PrP-WT (Fig. 3G and G′), but did not recognize the M205S and M205,212S mutants (Fig. 3H and I′). These results supported the observation that the PrP-α3M>S substitutions induced structural perturbations that altered the availability of the 6H4 epitope in α1. Since 6H4 recognized M205S by western blot but not by immunofluorescence, natively (formaldehyde-fixed) folded PrP-M205S was more sensitive to the loss of the 6H4 epitope than its denatured (SDS, 95°C) version.
Figure 7.

PrP-M205,212S induces the abnormal distribution of other membrane proteins. (A) 3D image of mushroom body neurons, color-coded for depth: posterior in blue, anterior in red. Kc, Kenyon cells; ca: calix, ped: pedunculus, α, β, γ: projections. (B–G) 3D surface visualization of PrP distribution in mushroom body dorsal projections. (B) PrP-WT colocalizes with GFP throughout the mushroom body projections. (C and D) Cross-sections through the dorsal lobes show co-distribution of PrP and GFP in α and α′ (arrow) branches. (E and E′) PrP-M205,212S does not form the cap (arrow), and PrP is not present in the α′ axons labeled with GFP (arrowhead). (F and G) Cross-sections show a lack of PrPs in the α′ branch (arrows). (H–M′) PrP-M205,212S disrupts the distribution of Syx and Nrg in cell bodies. (H and H′) PrP-WT accumulates in the Golgi and the membrane in Kc (arrow). (I and I′) PrP-M205,212S accumulates in small vesicles in the Kc (arrow). In flies expressing PrP-WT, Syx displays low levels of Syx in the Kc (J and J′, arrow) and Nrg accumulates in the membrane (L and L′, arrow). In contrast, flies expressing PrP-M205,212S, accumulates higher levels of Syx (K and K′, arrow) and dissociation of Nrg from the membrane (M and M′, arrow).
To further verify the disruption of the disulfide bond by the α3M>S substitutions, we used another conformational antibody, IPC2, which recognizes the two cysteines (C178 and C213) plus M212 (all mouse numbering) (11). Since an intact disulfide bond is critical for the IPC2 epitope, this is an ideal tool for evaluating the oxidative folding of PrP. As expected, IPC2 recognized two bands in PrP-WT corresponding to the most abundant un- and monoglycosylated glycoforms (Fig. 3C). Interestingly, IPC2 did not recognize PrP-M205S in western blot (Fig. 3C), suggesting that the M205S substitution prevented the formation of the disulfide bond. IPC2 did not recognize PrP-M205,212S either (Fig. 3C), but since the mutation in M212 altered the IPC2 epitope, we cannot interpret this negative result as a structural change in PrP. However, it is safe to assume that the double mutant undergoes even stronger conformational changes than M205S and, thus, also fails to form the disulfide bond. Overall, these results supported our hypothesis that the α3MS substitutions induce profound conformational changes on the globular domain of PrP, including preventing its oxidative folding.
To further characterize the conformational changes induced by the PrP-α3M>S substitutions, we stained brains with the amyloid fibril-labeling agent, thioflavin-S. As a positive control for the procedure, we used flies expressing the human amyloid-β1-42 peptide (23). As expected, flies expressing amyloid-β1-42 accumulated high levels of thioflavin-S signal specifically in the Kc, while control flies expressing LacZ did not (Supplementary Material, Fig. S2A and B). In contrast, flies expressing WT or mutant PrP produced no thioflavin-S signal in Kc (Supplementary Material, Fig. S2C–E), indicating that neither PrP-WT nor the α3M>S mutants promote the accumulation of amyloid fibrils.
PrP-α3M>S acquires partial resistance to proteinase K
To determine whether the aberrant behavior of the PrP-α3M>S mutants was associated with the production of proteinase K (PK)-resistant conformers, we subjected extracts from young (Day 1) and old (Day 30) flies to a gradient of PK at 25°C. As we have shown before, PrP-WT is highly sensitive to PK and is totally degraded with as little as 2.5 µg/ml at 25°C in both young and old flies (24) (Fig. 4A and B). In contrast, both PrP-α3M>S mutants displayed partial resistance to PK and produced a fragment of around 19 kDa, a pattern that increased with aging (Fig. 4A and B). Although both mutants produced the same PK-resistant fragment, PrP-M205,212S showed a higher PK resistance profile. This PK-resistance profile is not consistent with PrPSc, instead it evokes the accumulation of Ctm PrP, a rare transmembrane isoform associated with abnormal PrP processing in some genetic forms of prion diseases (4).
Figure 4.

PrP-α3M>S mutants acquire Ctm topology. (A and B) Tissue homogenates from flies expressing PrP-WT, -M205S and -M205,212S subjected to a PK gradient at Days 1 (A) and 30 (B). PrP-WT is degraded by the lowest concentration of PK regardless of age. PrP-M205S and -M205,212S are partially resistant to PK, which increases over time. (C) Protease protection in microsomes. In the absence of a detergent, microsomes protect full-length PrP-WT from PK in 1-day-old flies, but 30-day-old flies also accumulated Ntm and Ctm PrP. In young PrP-M205S and -M205,212S flies, microsomes reveal full-length and Ctm PrP, which increases in older flies. In the presence of detergent, PK degrades all PrP. (D) The SP antibody detects the two mutants (bottom), whereas 6D11 detects all PrPs in the same membrane (top) (E) Model of secreted and Ctm PrP indicating the TMD and the SP.
PrP-α3M>S induces Ctm PrP topology
To confirm that PrP-α3M>S mutants produce Ctm PrP topologies, we isolated microsomes and performed PK digestion in the presence or absence of a detergent. Microsomes should protect the complete protein when PrP is secreted; however, transmembrane isoforms would expose part of the protein to PK, resulting in specific degradation patterns (Fig. 4E). In young flies expressing PrP-WT, microsomes protected the full protein form PK digestion, consistent with its complete translocation into the ER lumen (Fig. 4C, top). Interestingly, PK digestion of microsomes from older flies produced secreted PrP plus two smaller bands of around 19 and 15 kDa (Fig. 4C, top). The use of detergent (0.5% triton) allowed PK to completely digest all PrP regardless of the age of the flies, confirming its PK sensitivity. Thus, the two smaller bands protected by microsomes were reminiscent of the two transmembrane PrP topologies, Ntm and Ctm (4). When we treated in the same way microsomes purified from flies expressing PrP-M205S, only secreted PrP was present in young flies, but was accompanied by a prominent 19 kDa band (Ctm) in older flies (Fig. 4C, center). Thus, the PrP-M205S mutant favored the formation of Ctm at the expense of Ntm topologies in aged flies. Finally, microsomes from young flies expressing PrP-M205,212S treated with PK produced both secreted and Ctm PrP, whereas older flies produced high levels of Ctm and lacked secreted PrP (Fig. 4C, bottom). Hence, PrP-M205,212S further favored the accumulation of Ctm, particularly in older flies, suggesting that the abnormal folding of α3 promotes the formation of Ctm PrP.
The increased production of Ctm PrP by both α3M>S mutants explained the abnormal behavior of these proteins, including their increased PK resistance, hyperglycosylation and abnormal cellular distribution. However, these results could not explain the higher molecular weight of the α3M>S mutants after PNGase F treatment. We hypothesized that the increased electrophoretic mobility of the α3M>S mutants could be due to the retention of the SP, a phenomenon documented for the PrP-3AV and -L9R, 3AV mutants in mice (25). To evaluate the presence of the SP, we used a specific antibody generated by Harris and Stewart (25). We performed an immunoblot and found that neither LacZ nor PrP-WT flies produced immunoreactivity with the SP antibody, but both α3M>S mutants showed a band corresponding to the expected molecular weight of PrP (Fig. 4D). The identification of bands with the same size suggests that a subpopulation with exactly the same properties contain the SP in both mutants, which is in agreement with the coexistence of diverse PrP topologies described in Figure 4C. These results support the production of Ctm PrP topologies that retain the SP in vivo (Fig. 4E), although we cannot rule out the coexistence of Ctm isoforms with and without the SP.
PrP-α3M>S does not induce progressive neuronal dysfunction
We next characterized the effects of ubiquitously expressing PrP-M205S and PrP-M205,212S on the longevity of the flies. The control flies expressing LacZ lived for 35 days before they started dying, with 50% of the flies living for 47 days (Fig. 5A). In contrast, flies expressing PrP-WT exhibited significantly shortened lifespan, with 50% of the flies living <25 days (Fig. 5A). Surprisingly, PrP-M205S and PrP-M205,212S displayed survival curves similar to control flies (Fig. 5A), with 50% survival at 45 days.
Figure 5.
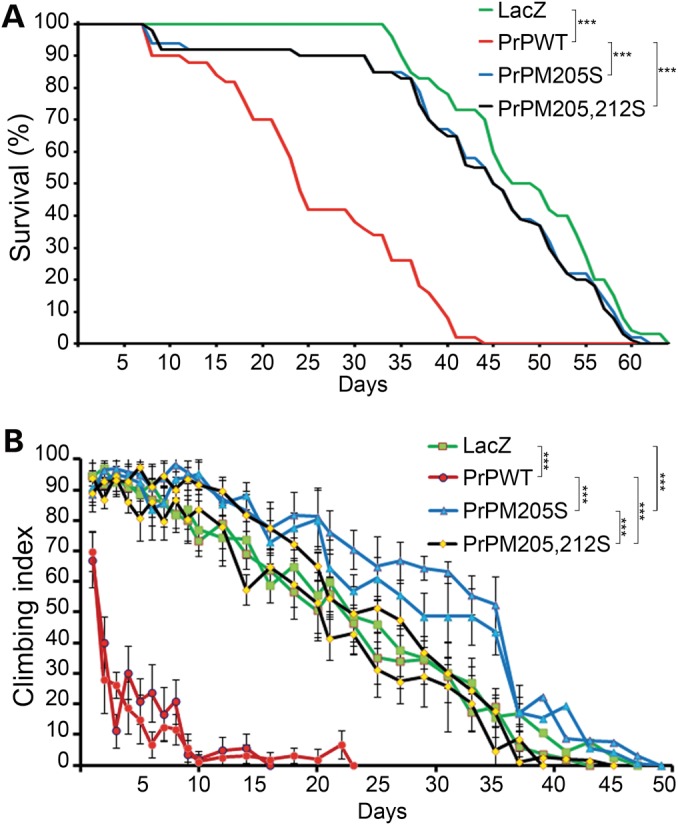
PrP-α3M>S mutants do not induce progressive neurodegeneration. (A) Adult females expressing PrP-WT (red) exhibit reduced survival compared with control flies expressing LacZ (green). Flies expressing either PrP-M205S (blue) or PrP-M205,212S (black) show similar longevity to control flies. (B) Adult females expressing PrP-WT (red) in motor neurons show 50% climbing by Day 2 and stop climbing by Day 10. Flies expressing PrP-M205S and PrP-M205,212S (black) show the same climbing ability as control flies expressing LacZ (green). ***P < 0.001
Then, we studied the effect of these mutations on the ability of PrP to cause locomotor dysfunction. For this, we expressed PrP-WT, -M205S and -M205,212S in motor neurons and measured locomotor activity over time. We had shown before that PrP-WT induces a rapid locomotor dysfunction, where >50% of the flies displayed the climbing deficit by Day 2 compared with the 35 days of control (LacZ) flies (Fig. 5B). Flies expressing either PrP-M205S or PrP-M205,212S performed better than PrP-WT and similar to controls (Fig. 5B). These two functional experiments indicated that the α3M>S substitutions did not show the progressive neuronal dysfunction induced by PrP-WT, confirming the profound effects of the polar substitutions in α3M on the biological activity of PrP.
PrP-M205,212S induces neurodevelopmental defects
The longevity and climbing assays indicated that the α3M>S mutations were less toxic than PrP-WT. However, previous work on Ctm PrP strongly suggested a role of this minor PrP topology in pathogenesis (2,7). To determine the neurotoxicity of α3M>S mutations in Drosophila brain neurons, we expressed PrP in the mushroom bodies. The mushroom body projections are relatively complex structures formed by 2500 neurons each (see Fig. 7A), which we have used previously to document the neurotoxicity of PrP from hamster, mouse and rabbit (19,24). As expected, expression of LacZ (control) did not affect the architecture of the mushroom body projections over time (Fig. 6A, E and I). Expression of PrP-WT had no effect in young flies, but induced mild degeneration in 40-day-old flies, as illustrated by thinner dorsal lobes with dystrophic axonal membranes (blebbing) (Fig. 6B, F and I). Young flies expressing PrP-M205S had normal mushroom bodies (Fig. 6C), while flies expressing PrP-M205,212S exhibited thinner projections that lacked the terminal caps (Fig. 6D). However, neither M205S nor M205,212S displayed significant changes during aging despite the obvious developmental phenotype of M205,212S (Fig. 6G–I). In summary, PrP-M205,212S induced aberrant development of mushroom body axonal projections, but the lack of progressive neurodegeneration was consistent with the normal longevity and locomotion (Fig. 5).
Figure 6.

PrP-α3M>S induces aberrant axonal development in the adult brain. (A–H) Expression of LacZ and PrP in the mushroom bodies labeled with GFP; micrographs show maximum projection of Z-stacks. (A and E) Control flies expressing LacZ display normal mushroom body projections that do not change over time. (B and F) Expression of PrP-WT results in normal development, followed by thinning and blebbing of the dorsal projections in older flies (F, arrowhead). (C and G) Expression of PrP-M205S results in normal development and no degenerative changes. (D and H) PrP-M205,212S induces abnormal development of the dorsal projections (D, arrow) that do not degenerate over time (H, arrow). **P < 0.01, ***P < 0.001.
PrP-M205,212S perturbs vesicular trafficking to the membrane
To investigate the mechanism by which PrP-M205,212S disrupts axonal development, we studied the distribution of WT and mutant PrP in the large cluster of mushroom body neurons in young flies at Days 1–2 (Fig. 7A). The distribution of PrP-WT overlapped with membrane-anchored CD8-GFP in the mushroom body projections, reaching the distal domain of all the lobes (Fig. 7B). Cross-sections of the dorsal lobes show co-distribution of GFP and PrP in the α and α′ lobes (Fig. 7C and D). In contrast, PrP-M205,212S not only caused the development of thinner and shorter projections, but also failed to reach the α′ lobes labeled with CD8-GFP (Fig. 7E and E′). Cross-section of the dorsal projections demonstrated the presence of PrP-M205,212S in the α lobe, but not in the α′ (Fig. 7F and G), highlighting the abnormal distribution of mutant PrP. Together with the observation that PrP-M205,212S accumulates intracellularly (Fig. 2D), these phenotypes could be due to the induction of defective vesicular trafficking by PrP-M205,212S.
To test this idea, we analyzed the subcellular distribution of PrP and two neuronal markers in the Kc, the cell bodies of the mushroom body neurons also in young flies. As shown before, PrP-WT accumulated mostly in the Golgi and the membrane in the Kc (Fig. 7J and Supplementary Material, Fig. S1E). In contrast, PrP-M205,212S exhibited a punctate pattern that filled the whole cell body (Fig. 7P). As indicated above, PrP-M205S showed a mixed distribution between WT and M205,212S (Fig. 7M). We also analyzed the expression of Neuroglian (Nrg), an integral membrane glycoprotein with a role in cell adhesion and axonal growth (26). Cells expressing PrP-WT displayed membrane distribution of Nrg (Fig. 7H and H′), but expression of PrP-M205,212S altered the distribution of Nrg, which seemed to be retained in the cytoplasm (Fig. 7N and N′). In PrP-M205S flies, some cells showed a strong membrane distribution, while others displayed a more diffuse signal (Fig. 7K and K′). To confirm the aberrant distribution of key neuronal proteins by mutant PrP, we studied the distribution of Syntaxin (Syx), a synaptic protein with a key role in vesicle fusion and neurotransmission (27). Cells expressing PrP-WT contained small Syx-positive vesicles en route to the axonal terminals (Fig. 7I). However, cells expressing PrP-M205,212S showed the accumulation of Syx-positive vesicles in the cell bodies, suggesting that Syx is not properly transported to the distal axonal terminals (Fig. 7O). Expression of M205S induced subtle changes in the distribution of Syx consistent with a slight increase in cellular retention (Fig. 7L). These results suggested that the abnormal retention of PrP-M205,212S in secretory vesicles affected the distribution of other proteins with key function for axonal growth and function.
To confirm the abnormal distribution of PrP-M205,212S, we isolated lipid rafts from Drosophila heads at Days 1–2. PrP is known to populate the lipid raft, a domain critical for PrP biology and conversion (28). As we have shown before with hamster PrP, PrP-WT accumulated in lipid rafts, a floating domain mainly found in Fraction 3 of an Optiprep gradient characterized by the presence of receptors and synaptic proteins such as Syx (Fig. 8A) (24,29). Syx and Nrg also accumulated in lipid rafts in flies expressing PrP-WT, while the membrane ion pump α-ATPase did not accumulate in lipid rafts (Fig. 8A), confirming the correct purification of lipid rafts. However, PrP-M205,212S did not accumulate in the lipid raft domain, confirming its retention in secretory vesicles observed by immunofluorescence (Fig. 8B). Interestingly, neither Syx nor Nrg accumulated in lipid rafts in flies expressing PrP-M205,212S (Fig. 8B), further confirming that the abnormal distribution of PrP perturbs vesicular trafficking and alters the distribution of key membrane proteins. Overall, these observations indicate that the aberrant processing and membrane insertion of PrP result in dramatic consequences for the development, maintenance and/or function of neurons.
Figure 8.

PrP-M205,212S prevents the accumulation of Syx and Nrg in lipid rafts. Detection of proteins in lipid rafts. (A) PrP-WT, Syx and Nrg accumulate in lipid raft fractions (Fraction 3). (B) In flies expressing PrP-M205,212S, neither PrP and Syx, nor Nrg reach the lipid raft. As a control, we detected α-ATPase, a transmembrane protein that never accumulates in the lipid raft.
Ctm PrP enhances the neurotoxicity of PrP-WT
One possible role for Ctm PrP in disease is as a seed for the conversion of native PrP into neurotoxic conformations. This idea is supported by the strong neurodegeneration shown in mice expressing Ctm PrP conformers in a background expressing PrP-WT as opposed to the lack of neurodegeneration in mice that were Prnp−/− (7). To test the idea that Ctm PrP induces progressive degeneration through the conversion of PrP-WT, we co-expressed PrP-WT with the α3M mutants in the mushroom bodies. As controls, we generated flies expressing double amount of PrP-WT (2xWT) that showed normal development of mushroom body projections (Fig. 9A), but induced similar thinning of projections in older flies as the single copy, albeit with more intense membrane blebbing (Fig. 9A and B). Co-expression of PrP-WT and -M205S induced abnormal development of the dorsal projections (Fig. 9A and B), a new phenotype not observed in flies expressing only PrP-WT or -M205S (Fig. 6). Moreover, aging for 40 days induced further thinning of dorsal and medial projections (Fig. 9A and B). Finally, co-expression of PrP-WT and –M205,212S induced stronger developmental defects in the mushroom bodies, with shorter and thinner dorsal and medial projections (Fig. 9A and B). Aging of these flies resulted in strong neurodegenerative changes, including loss of axonal density and widespread membrane blebbing (Fig. 9A and B). Since the degenerative phenotypes observed in the flies co-expressing PrP-WT and –M205,212S were stronger than that observed in the flies expressing high levels of PrP-WT, we propose that Ctm PrP enhances the toxicity of the secreted PrP isoform, thus providing a mechanistic role for Ctm PrP in disease.
Figure 9.

PrP-WT enhances the neurotoxicity of α3M>S mutants. (A) Micrographs show maximum projection of Z-stacks of mushroom body lobes. Two copies of PrP-WT (2×) result in normal mushroom bodies followed by generalized blebbing in older flies, arrowhead) with thinning of both dorsal and medial projections. Co-expression of PrP-WT and –M205S induces abnormal development of the dorsal lobe, arrow) and progressive degeneration of all lobes co-expression of PrP-WT and -M205,212S results in thin and short dorsal and medial projections, arrows) followed by potent degeneration of all lobes after 40 days, arrows) and overall blebbing, arrowhead). (B) Statistical analysis of the mushroom body surface. N = 12 for each group. ***P < 0.001. (C) Locomotor activity of flies co-expressing WT and mutant PrP. Control flies expressing LacZ under the control of BG380-Gal4 show strong climbing ability (black dashed line). Flies expressing 2×WT stop climbing at Day 6 (black lines). Flies expressing PrP-WT + M205S (grey line) stop climbing at Day 4. Flies co-expressing PrP-WT + M205,212S (black dotted line) start with lower climbing ability and stop climbing at Day 5. ***P < 0.001. (D and E) Immunoprecipitation with 15B3. (D) Using 6H4, PrP-WT and -M205S accumulate 15B3-positive conformations at Day 30, but 6H4 does not recognize PrP-M205,212S. (E) 6D11 does not recognize 15B3-positive conformations in PrP-WT flies, but detects them in PrP-M205S at Day 30. PrP-M205,212S immunoreacts with 15B3 at Day 1 and accumulates at higher levels at Day 30. On the left, controls with no 15B3 or no PrP demonstrate the specificity of the assay. (F) 15B3 immunoprecipitation in flies co-expressing WT and mutant PrPs. PrP-2×WT flies accumulate 15B3-positive isoforms at Day 30. PrP-WT + M205S flies show accumulation of 15B3 conformations at Day 1 corresponding to PrP-M205S. In Day 30 flies, both PrP-WT and -M205S are recognized by 15B3. In flies expressing PrP-WT + M205,212S, both PrP-WT and -M205,212S accumulate 15B3-positive isoforms from Day 1.
A prediction from these observations is that flies co-expressing WT and mutant PrP would exhibit accelerated locomotor dysfunction. To test this, we co-expressed PrP-WT with either -M205S or -M205,212S and compared the locomotor activity with flies expressing PrP-2×WT under the control of BG380-Gal4. As expected PrP-2×WT flies exhibited fast locomotor dysfunction due to the high levels of PrP (Fig. 9C). Flies co-expressing PrP-WT and -M205S started with a similar activity, but exhibited a steeper decline in their locomotor activity (Fig. 9C). In contrast, flies co-expressing PrP-WT and -M205,212S started with lower climbing ability and showed a rapid locomotor decline (Fig. 9C). These results agree with the observations in the mushroom bodies and suggest a role for CtmPrP in accelerating the misfolding of PrP-WT into pathogenic conformations.
Ctm PrP accelerates the conformational change of PrP-WT
To test directly the idea that Ctm PrP acts as a seed that promotes the misfolding of PrP-WT, we took advantage of a conformational antibody that specifically recognizes pathogenic, PrPSc-like isoforms (30). We have shown before that 15B3 immunoprecipitates aberrant conformations in older flies expressing PrP-WT from hamster and mouse, but not from rabbit, a rare mammal resistant to prion diseases (19). We first tested WT and mutant PrP using the 6H4 antibody, which showed that both PrP-WT and -M205S accumulated PrPSc-like conformations in older flies (Fig. 9D). However, PrP-M205,212S was not detected, which was consistent with the loss of the 6H4 epitope described in Figure 3. To detect all PrP immunoprecipitated by 15B3, we immunoblotted the membrane with 6D11. Under these conditions, PrP-WT was not detected in either young or old flies (Fig. 9E). However, PrP-M205S accumulated small amounts of 15B3-positive conformations in young flies, which increased in older flies (Fig. 9E). PrP-M205,212S produced a strong signal in young flies, which doubled in older flies (Fig. 9E). These results suggested that, although the 6D11 antibody is less sensitive against the 15B3 conformers, both α3M mutants promote the formation of these conformations compared with PrP-WT.
Finally, to determine the effect of the α3M mutants on the dynamics of PrP-WT, we examined 15B3 immunoreactivity in flies co-expressing WT and mutant PrP. As controls, we analyzed 15B3 reactivity in PrP-2×WT flies. As in the low dose, we found no signal in the younger flies (Fig. 9F), but older flies were positive for 15B3, indicating that expression levels are critical for promoting PrP misfolding. Young flies co-expressing PrP-WT and -M205S accumulated high levels of 15B3 conformers that run higher than PrP-WT, suggesting that this was only PrP-M205S. Compared with the flies only expressing PrP-M205S, this result suggested that PrP-WT promoted the accumulation of 15B3 conformers. In older flies of the same genotype, both PrP-WT and -M205S acquired 15B3 immunoreactivity, suggesting a cooperative effect of both proteins. Interestingly, flies co-expressing PrP-WT and -M205,212S accumulated high levels of 15B3 conformers at Day 1 and both proteins were recognized due to their different molecular weight (Fig. 9F). The accumulation of high amounts of PrP-WT with 15B3-positive conformations at Day 1 is a new phenotype that supports the seeding effect of the α3M mutants on the misfolding of the normal, secreted form of PrP. Thus, the accelerated accumulation of these pathogenic conformations agrees with the stronger neurodegenerative phenotypes observed in flies co-expressing WT and mutant PrP.
DISCUSSION
We present here the first in vivo study of the consequences of introducing polar substitutions in conserved methionines in α3 of PrP. In silico simulations found that PrP-M205S, -M212S, and -M205,212S disturbed the native folding and increased surface hydrophobicity, while full-length recPrP bearing α3M>S mutations demonstrated increased aggregation dynamics (15,16). These studies suggested that the α3M>S mutations promoted PrPC instability and favored its conversion to misfolded conformations, including PrPSc. In addition, cultured cells expressing these PrP mutants showed failure of oxidative folding and partial PK resistance (17). Several other studies had reported that α3M were sulfoxidized in PrPSc in human and animal brains, suggesting a connection between α3 hydrophobicity and PrP conversion (6,11,13). Together, these observations revealed a key role of α3M in regulating PrP folding, thus providing a pathogenic mechanism for prion diseases. However, no in vivo evidence was available for the effects of α3M>S mutations on PrP structure and neurotoxicity.
To our surprise, flies expressing α3M>S mutations accumulated Ctm PrP, a rare transmembrane topology favored by mutations in the SP and TMD (4,5). So far, only mutations in these two domains had been reported to induce Ctm PrP in cell-free translation systems, whereas several pathogenic mutations in α2 and α3, including D178N, V180I, H187R, F198S, E200K, D202N and V210I, did not (31,32). But, why do polar substitutions in distal α3M result in transmembrane translocation of the proximal TMD? Conformational antibodies provided a key clue to this question: α3M>S mutations prevented the oxidative folding of the globular domain. Since α3M are deeply buried in the hydrophobic core, they may stabilize the globular domain by anchoring interactions with distant residues in α2, loop 1 and loop 2. Thus, α3M>S mutations relax the interactions within the hydrophobic core and increase the distance between α2 and α3 to accommodate the new polar side chains. Since M212S is next to C213, changes in the position or orientation of M212S must affect the distance between the C213 and C178, thus preventing the formation of the disulfide bond. Although M205S is far from C178 and C213, computer simulations predict that even small alterations in the hydrophobic domain induce long distance effects (8). Moreover, the side chain of M212 is very close to M134 in PrPC, suggesting that M212 maintains β1 and loop 1 in close proximity to the hydrophobic core. Disrupting these interactions may release the distal portion of the TMD (111–135) from the globular domain and expose it to the membrane translocation machinery, resulting in Ctm topology. Interestingly, recent studies with recPrP have pointed to the formation of the disulfide bond as a key determinant of membrane insertion (33,34), supporting our observations that perturbations in the oxidative folding of PrP result in Ctm topologies. These observations reveal new, interesting clues about the role of the hydrophobic core in controlling PrP folding and preventing its membrane topology.
Why is membrane translocation the solution to the structural perturbations induced by α3M>S? Another clue to the aberrant biogenesis of α3M>S was the partial preservation of the SP, which suggested that during Ctm PrP biosynthesis the N-terminus of the nascent chains never translocated into the ER. Thus, mutations that disrupt the oxidative folding of the distal globular domain interfere with the translocation of the nascent chain to the ER. This result is consistent with observations by Stewart et al. using the L9R and 3AV mutants in vitro and in mice (5). The work by the Hegde laboratory supports a model of membrane translocation of PrP coupled to the translation of the nascent chains (co-translational translocation) (35). In this model, the translocation machinery sequentially recognizes the SP and TMD; thus, mutations that reduce the hydrophobicity of the SP or increase the helicity of the TMD result in retention of the SP and membrane translocation of the TMD with Ctm topology. Our results mostly agree with this model, yet incorporate an additional role for oxidative folding in controlling the exposure of the TMD to the translocon. Our results suggest that most of the protein has to be translated for the globular domain to contribute to the interaction of the TMD with the translocation machinery, thus arguing for a post-translational translocation model (35). The use of different mutations (SP, TMD or α3) could explain the disagreement in the timing and mechanism of ER translocation. In the case of the SP mutants, aberrant processing of the SP leads to cytosolic localization and recognition of the TMD as a transmembrane signal (5). The TMD mutants optimize PrP targeting to the translocon complex, which displaces the SP and prevents the translocation of the N-terminus into the ER lumen (4,5,35). Lastly in the α3M>S mutants the SP can initiate its interaction with the translocon, but the lack of oxidative folding leaves the TMD exposed in the cytosolic side, leading to its recognition by the translocation machinery and membrane insertion.
In addition to these intrinsic factors (SP, TMD and α3M), our results support the contribution of extrinsic factors (age) in PrP translocation. Whereas young flies expressing PrP-WT produced mostly secreted PrP, older flies accumulated significant amounts of Ctm and Ntm topologies. Thus, normal aging contributes to the aberrant biogenesis of PrP. α3M>S mutants also increased the production of Ctm over time, suggesting that cellular aging is an important factor in PrP processing. Although we have no mechanistic explanation for this phenomenon, it is likely linked to the challenging nature of the PrP structure, including the weak SP (36) and a buried TMD that is not supposed to be inserted in the membrane. Thus, age-related inefficient recognition of nascent PrP chains by cytoplasmic chaperones and the translocation machinery may lead to exposed TMD and transmembrane translocation.
The relevance of this minor PrP topology is based on its proposed role as a neurotoxic agent in at least some inherited forms of prion diseases (2,4). Mice expressing mutant PrP that produced high amounts of Ctm PrP (KH>II, 3AV, and A117V) showed neurodegeneration with astrocytic gliosis in the absence of PrPSc. On the other hand, mutations that prevented the formation of Ctm PrP (STE-TM1 and G123P) showed no neurodegeneration (2,4). These data strongly suggested the role of Ctm PrP in neurodegeneration. To assess the physiological relevance of these artificial mutations favoring Ctm, the authors confirmed that human brains from patients harboring the GSS-linked A117V mutation also accumulated Ctm PrP, arguing that Ctm topologies can mediate the human disease (4). Moreover, mice expressing PrP-A117V inoculated with prions developed disease much faster than PrP-ΔSTE mice, suggesting that Ctm PrP increases susceptibility to PrPSc (2). These results led to the proposal that Ctm PrP is the neurotoxic agent in all prion diseases, including infectious, sporadic and genetic forms. However, none of the pathogenic mutations tested outside of the SP and the TMD produced significant amounts of Ctm topology in vitro, arguing against a generalized role for Ctm PrP in prion diseases (5). Here, we identified a toxic activity of Ctm PrP associated with its retention in secretory vesicles that prevents the normal transport of key membrane proteins to the lipid raft resulting in aberrant axonal growth. In contrast, the same mutants showed no effect in longevity and locomotor assays. These apparently contradictory results are explained by the use of different Gal4 strains for each experiment. The mushroom body driver line (OK107-Gal4) is restricted to those neurons, but is expressed at high levels, leading to the aberrant development of the axonal projections. In contrast, the ubiquitous driver used for measuring longevity (da-Gal4) is very weak, allowing for normal development of the central nervous system. The driver used for motor neurons (BG380-Gal4) is also fairly strong, but the lack of phenotypes with the α3M mutants suggests different expression dynamics that avoids the sensitive growth of the axons. Alternatively, motor neurons could be less sensitive to the specific perturbations induced by mutant PrP than the cholinergic neurons of the mushroom bodies. Although we cannot explain all these differences, the importance of these experiments is that they showed significant differences between PrP-WT and the α3M mutants. Fortunately, the biochemical analysis of PrP supported the different behavior of mutant PrP and its potential relevance to disease. Thus, the α3M mutants induced axonal transport phenotypes that can contribute to explain sporadic prion diseases. In addition to these neurodevelopmental phenotypes, Ctm PrP can sensitize neurons to the neurotoxic activity of smaller amounts of pathogenic PrP conformations.
Interestingly, the generation of mice carrying mutations that favor Ctm PrP (L9R, 3AV) showed that neurodegeneration is highly dependent on the co-expression of PrP-WT (7). This result suggested that Ctm PrP might not be toxic on its own, but it could induce the conversion of secreted PrP. We confirmed this requirement of PrP-WT in flies and described strong conformational alterations associated with Ctm PrP. Recent structural studies found that the folding of Ctm PrP was perturbed by the membrane insertion of the TMD, which reduces the helix content without an increase in β-sheet (33). It is possible, then, that this abnormal conformation/topology may serve as a seed for the conversion of secreted PrP, thus potentially explaining a role for Ctm PrP in disease (see Fig. 10). In fact, our data with the 15B3 antibody support this seeding effect of Ctm PrP. Both PrP-α3M are recognized by 15B3, which is proposed to interact only with PrPSc-like conformations. So, in addition to the aberrant membrane translocation, the globular domain of these mutants acquires a pathogenic conformation (Fig. 10C). Consistent with the in vitro findings, the lack of the disulfide bond and the structural constraints of the membrane translocation result in a conformation consistent with pathogenic conformations (PrP* or PrPL). Based on these findings, it is not surprising that the interaction of PrP-WT with the α3M mutants promotes the formation of pathogenic conformations that are secreted and induce potent degeneration (Fig. 10B). Although these results are fairly significant, they do not demonstrate a direct interaction between PrP-WT and Ctm PrP. This type of evidence may be better collected in in vitro conversion assays where the constituents in the reaction can be carefully controlled. Overall, the role of Ctm PrP in disease deserves serious consideration because it induces perturbations in vesicular transport (this work) and contributes to the formation of pathogenic PrP conformations when it interacts with PrP-WT (this work and refs 2,7). Since Ctm PrP can accumulate in older flies spontaneously, Ctm topologies can play a significant role in both inherited and sporadic prion diseases, and increase the susceptibility to transmitted forms of the disease.
Figure 10.

Model of proposed interactions of Ctm and secPrP. (A–C) Biogenesis, distribution and age-dependent changes in secreted (WT) PrP (A), Ctm PrP (C) and the combination of Ctm and secPrP (B). Sec PrP accumulates in the extracellular space and slowly misfolds into pathogenic conformations. However, in the presence of Ctm PrP, sec PrP misfolds more efficiently and produces larger amounts of pathogenic PrP. The circle represents the signal peptide in the Ctm PrP.
MATERIALS AND METHODS
Generation of transgenic flies and genetics
MoPrP-M205S and -M205,212S complementary DNAs (cDNAs) were synthesized at GenScript and cloned between EcoRI and NotI sites onto the pUAST Drosophila expression vector (18). The pUAST-based constructs were injected into yw embryos at Rainbow Transgenics following standard procedures (37) to generate multiple independent transgenic lines for each plasmid. Flies carrying UAS-MoPrP-WT were obtained from Supattapone et al. (38). The driver strains OK107-Gal4 (mushroom bodies), BG380-Gal4 (motor neurons), and da-Gal4 (ubiquitous), and the reporters UAS-LacZ and UAS-CD8-GFP were obtained from the Bloomington Drosophila Stock Center. Fly stocks were maintained on standard Drosophila medium at 25°C. For experiments, homozygous females for the Gal4 strains were crossed with UAS males to generate progeny expressing PrP in the desired tissue. Crosses were placed at 25°C for 2 days, moved to 28°C until the progeny had completed development, and adults were aged at 28°C, unless otherwise indicated. All assays were performed using females.
Locomotor and longevity assays
Flies carrying LacZ (control) or PrP transgenes were crossed with BG380-Gal4 at 25°C and the progeny was tested for their ability to climb (climbing assay) at 25°C (39). Briefly, 25 newborn adult females were placed in empty vials in duplicate and forced to the bottom by firmly tapping against the surface. After 10 s, the number of flies that climb >5 cm was recorded. This was repeated 10 times to obtain the average climbing index each day. At the end of the assay, the climbing index (flies above line/total flies × 100) was plotted as a function of age in Excel. For the longevity assay, flies carrying PrP transgenes or LacZ were crossed with the da-Gal4 driver. Fifty females of each genotype were collected and kept in groups of 10 at 28°C. Flies were counted every day and transferred to a new tube until all the flies died. Then, longevity was calculated as percentage of flies alive at each time point and plotted in Excel. Finally, data series for both locomotor activity and longevity were analyzed for statistical significance in GraphPad Prism using a two-way ANOVA with the Bonferroni multiple comparisons test.
Drosophila homogenates and western blot
One fly per genotype and time point was used for analysis. Single flies were homogenized in 30 µl of RIPA buffer containing complete protease inhibitors (Roche) and centrifuged for 1 min at 1000 rpm. Then, 25 µl of the supernatant was mixed with loading buffer and resolved by SDS–PAGE, transferred to nitrocellulose membrane, immunoblotted, and the signal was revealed by chemiluminescence (Pierce). The primary antibodies were: anti-PrP 6D11 (1:10 000, Covance), anti-PrP 6H4 (1:10 000, Prionics), anti-PrP IPC2 (1:500, (11)), anti-PrP SP (1:500, (25)), anti-αTub (1:250 000, Sigma), anti-Syx (1:500) and anti-NrgII (1:500) from the Developmental Studies Hybridoma Bank. The secondary antibody was anti-Mouse-HRP (1:2000) (Sigma).
PrP processing
For de-glycosylation assays, 9 µl of cleared homogenates were incubated in the presence or absence of PNGase F or Endo H (NEB) according to the manufacturer's instructions and analyzed by immunoblotting. For protease digestions, cleared homogenates (∼3% w/v) were incubated with 0–20 µg/ml PK for 30 min at 25°C, and the digestions were stopped by adding LDS sample buffer (Invitrogen) and analyzed by immunoblotting.
Protease protection assays
Five flies of each genotype were homogenized in 70 µl of BIB buffer (24) and centrifuged for 30 min at 5000 rpm at 4°C. Supernatants were centrifuged for 90 min at 14 800 rpm at 4°C, and the resulting pellets (microsomes) were resuspended in 75 µl of BIB buffer. Resuspended microsomes were split in three equal parts and incubated in the absence or presence of PK (5 µg/ml), with and without 0.5% Triton X-100, for 30 min at 25°C. Reactions were stopped by adding LDS sample buffer and analyzed by immunoblotting.
Lipid rafts purification
Lipid rafts were isolated following the protocol previously published (24). Briefly, 40 heads of flies expressing PrP-WT and PrP-M205, 212S under the control of OK107-Gal4 were homogenized in 500 µl of TNET buffer. Then, 200 µl of homogenate were mixed with 400 of Optiprep at 60% (Sigma), followed by 1.8 ml of Optiprep at 30% and 600 µl of Optiprep at 5%. The gradient was centrifuged at 139 000g for 5 h at 4°C. Nine 290 µl fractions were collected from the top (1 through 9), precipitated with cold methanol, resuspended and analyzed by western blotting.
Immunofluorescence
We characterized the subcellular distribution of WT and mutant PrP by co-expression with CD8-GFP in interneurons of the ventral ganglion of the larval brain under the control of OK107-Gal4. For analysis of the adult mushroom bodies, flies co-expressing CD8-GFP with MoPrP or LacZ under the control of OK107-Gal4 were collected at day one post eclosion. Whole-mount immunohistochemistry of adult brains and larval tissues was conducted as described previously (Fernandez-Funez et al., 2000). We used the following primary antibodies at the indicated dilutions: anti-PrP 6D11 (1:1000, Covance), anti-PrP 6H4 (1:1000, Prionics), anti-Rab11 (1:100, (40)), anti-Lamp1 (1:100, Abcam), anti-p120 (1:500, Calbiochem (20)), anti-Lva (1:250, (21)), anti-GM130 (1:1000, Abcam), anti-Syx (1:5) and anti-Neuroglian II (1:25) from the Developmental Studies Hybridoma Bank. Anti-mouse-Cy3 and anti-rabbit-Alexa fluor-468 (Molecular Probes) were used at 1:600 and 1:200, respectively. We collected fluorescent images with AxioVision (Zeiss) in an Axio-Observer Z1 microscope (Zeiss) by optical sectioning using ApoTome (structured light microscopy) with 40× NA: 1.3 and 63× NA: 1.4 oil objectives. Images were combined using Adobe Photoshop and processing included brightness/contrast adjustment to whole images. For surface quantification of mushroom bodies, we collected images from comparable sets using the same exposure and Z-step (0.5 µm). Then, we created maximum projections (orthoview) in AxioVision, drew the outline of the lobes manually, quantified surfaces in ImageJ and analyzed the statistical significance by two-way ANOVA with the Bonferroni multiple comparisons test. 3D images were created from Z-stacks using Zen (Fig. 7A) and AxioVision (Fig. 7B–G) software from Zeiss. For Figures 8B and C, we visualized the Z-stacks with the Surface option and created XZ clipping to observe protein distribution inside the mushroom body lobes.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health, grant DP2 OD002721-01, to P.F.-F., and start-up funds from the UF department of Neurology to P.F.-F. and D.R.-L. J.S.-G. was supported by a postdoctoral fellowship from the Basque Government and D.A. was supported by a University Scholars Program undergraduate award from UF.
Supplementary Material
ACKNOWLEDGEMENTS
We thank J. Herrera for his technical assistance, M. Gasset for helpful discussions and sharing of unpublished data, and D. Harris, R. Gavizon, M.G. Gaitan, J.C. Sisson, A. Raeber, the Bloomington Drosophila Stock Center and the Developmental Studies Hybridoma Bank for reagents.
Conflict of interest statement. None declared.
REFERENCES
- 1.Colby D.W., Prusiner S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. doi:10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hegde R.S., Tremblay P., Groth D., DeArmond S.J., Prusiner S.B., Lingappa V.R. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature. 1999;402:822–826. doi: 10.1038/45574. doi:10.1038/45574. [DOI] [PubMed] [Google Scholar]
- 3.Ma J., Wollmann R., Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–1785. doi: 10.1126/science.1073725. doi:10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- 4.Hegde R.S., Mastrianni J.A., Scott M.R., DeFea K.A., Tremblay P., Torchia M., DeArmond S.J., Prusiner S.B., Lingappa V.R. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. doi:10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- 5.Stewart R.S., Drisaldi B., Harris D.A. A transmembrane form of the prion protein contains an uncleaved signal peptide and is retained in the endoplasmic Reticulum. Mol. Biol. Cell. 2001;12:881–889. doi: 10.1091/mbc.12.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stahl N., Baldwin M.A., Teplow D.B., Hood L., Gibson B.W., Burlingame A.L., Prusiner S.B. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. doi:10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 7.Stewart R.S., Piccardo P., Ghetti B., Harris D.A. Neurodegenerative illness in transgenic mice expressing a transmembrane form of the prion protein. J. Neurosci. 2005;25:3469–3477. doi: 10.1523/JNEUROSCI.0105-05.2005. doi:10.1523/JNEUROSCI.0105-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Kamp M.W., Daggett V. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J. Mol. Biol. 2010;404:732–748. doi: 10.1016/j.jmb.2010.09.060. doi:10.1016/j.jmb.2010.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Requena J.R., Groth D., Legname G., Stadtman E.R., Prusiner S.B., Levine R.L. Copper-catalyzed oxidation of the recombinant SHa(29–231) prion protein. Proc. Natl. Acad. Sci. USA. 2001;98:7170–7175. doi: 10.1073/pnas.121190898. doi:10.1073/pnas.121190898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canello T., Engelstein R., Moshel O., Xanthopoulos K., Juanes M.E., Langeveld J., Sklaviadis T., Gasset M., Gabizon R. Methionine sulfoxides on PrPSc: a prion-specific covalent signature. Biochemistry. 2008;47:8866–8873. doi: 10.1021/bi800801f. doi:10.1021/bi800801f. [DOI] [PubMed] [Google Scholar]
- 11.Canello T., Frid K., Gabizon R., Lisa S., Friedler A., Moskovitz J., Gasset M. Oxidation of Helix-3 methionines precedes the formation of PK resistant PrP. PLoS Pathog. 2010;6:e1000977. doi: 10.1371/journal.ppat.1000977. doi:10.1371/journal.ppat.1000977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breydo L., Bocharova O.V., Makarava N., Salnikov V.V., Anderson M., Baskakov I.V. Methionine oxidation interferes with conversion of the prion protein into the fibrillar proteinase K-resistant conformation. Biochemistry. 2005;44:15534–15543. doi: 10.1021/bi051369+. doi:10.1021/bi051369+ [DOI] [PubMed] [Google Scholar]
- 13.Colombo G., Meli M., Morra G., Gabizon R., Gasset M. Methionine sulfoxides on prion protein Helix-3 switch on the alpha-fold destabilization required for conversion. PLoS One. 2009;4:e4296. doi: 10.1371/journal.pone.0004296. doi:10.1371/journal.pone.0004296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolschner C., Giese A., Kretzschmar H.A., Huber R., Moroder L., Budisa N. Design of anti- and pro-aggregation variants to assess the effects of methionine oxidation in human prion protein. Proc. Natl. Acad. Sci. USA. 2009;106:7756–7761. doi: 10.1073/pnas.0902688106. doi:10.1073/pnas.0902688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Younan N.D., Nadal R.C., Davies P., Brown D.R., Viles J.H. Methionine oxidation perturbs the structural core of the prion protein and suggests a generic misfolding pathway. J. Biol. Chem. 2012 doi: 10.1074/jbc.M112.354779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lisa S., Meli M., Cabello G., Gabizon R., Colombo G., Gasset M. The structural intolerance of the PrP alpha-fold for polar substitution of the helix-3 methionines. Cell Mol. Life Sci. 2010;67:2825–2838. doi: 10.1007/s00018-010-0363-1. doi:10.1007/s00018-010-0363-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lisa S., Domingo B., Martinez J., Gilch S., Llopis J.F., Schatzl H.M., Gasset M. Failure of prion protein oxidative folding guides the formation of toxic transmembrane forms. J. Biol. Chem. 2012;287:36693–36701. doi: 10.1074/jbc.M112.398776. doi:10.1074/jbc.M112.398776. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 19.Fernandez-Funez P., Zhang Y., Casas-Tinto S., Xiao X., Zou W.Q., Rincon-Limas D.E. Sequence-dependent prion protein misfolding and neurotoxicity. J. Biol. Chem. 2010;285:36897–36908. doi: 10.1074/jbc.M110.174391. doi:10.1074/jbc.M110.174391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stanley H., Botas J., Malhotra V. The mechanism of Golgi segregation during mitosis is cell type-specific. Proc. Natl. Acad. Sci USA. 1997;94:14467–14470. doi: 10.1073/pnas.94.26.14467. doi:10.1073/pnas.94.26.14467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sisson J.C., Field C., Ventura R., Royou A., Sullivan W. Lava lamp, a novel peripheral golgi protein, is required for Drosophila melanogaster cellularization. J. Cell. Biol. 2000;151:905–918. doi: 10.1083/jcb.151.4.905. doi:10.1083/jcb.151.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan J., Kinter M., McGeehan J., Perry G., Kneale G., Gambetti P., Zou W.Q. Concealment of epitope by reduction and alkylation in prion protein. Biochem. Biophys. Res. Commun. 2005;326:652–659. doi: 10.1016/j.bbrc.2004.11.088. doi:10.1016/j.bbrc.2004.11.088. [DOI] [PubMed] [Google Scholar]
- 23.Casas-Tinto S., Zhang Y., Sanchez-Garcia J., Gomez-Velazquez M., Rincon-Limas D.E., Fernandez-Funez P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 2011;20:2144–2160. doi: 10.1093/hmg/ddr100. doi:10.1093/hmg/ddr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez-Funez P., Casas-Tinto S., Zhang Y., Gomez-Velazquez M., Morales-Garza M.A., Cepeda-Nieto A.C., Castilla J., Soto C., Rincon-Limas D.E. In vivo generation of neurotoxic prion protein: role for hsp70 in accumulation of misfolded isoforms. PLoS Genet. 2009;5:e1000507. doi: 10.1371/journal.pgen.1000507. doi:10.1371/journal.pgen.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart R.S., Harris D.A. A transmembrane form of the prion protein is localized in the Golgi apparatus of neurons. J. Biol. Chem. 2005;280:15855–15864. doi: 10.1074/jbc.M412298200. doi:10.1074/jbc.M412298200. [DOI] [PubMed] [Google Scholar]
- 26.Bieber A.J., Snow P.M., Hortsch M., Patel N.H., Jacobs J.R., Traquina Z.R., Schilling J., Goodman C.S. Drosophila neuroglian: a member of the immunoglobulin superfamily with extensive homology to the vertebrate neural adhesion molecule L1. Cell. 1989;59:447–460. doi: 10.1016/0092-8674(89)90029-9. doi:10.1016/0092-8674(89)90029-9. [DOI] [PubMed] [Google Scholar]
- 27.Schulze K.L., Broadie K., Perin M.S., Bellen H.J. Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. doi:10.1016/0092-8674(95)90414-X. [DOI] [PubMed] [Google Scholar]
- 28.Baron G.S., Wehrly K., Dorward D.W., Chesebro B., Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. Embo J. 2002;21:1031–1040. doi: 10.1093/emboj/21.5.1031. doi:10.1093/emboj/21.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhai L., Chaturvedi D., Cumberledge S. Drosophila wnt-1 undergoes a hydrophobic modification and is targeted to lipid rafts, a process that requires porcupine. J. Biol. Chem. 2004;279:33220–33227. doi: 10.1074/jbc.M403407200. doi:10.1074/jbc.M403407200. [DOI] [PubMed] [Google Scholar]
- 30.Korth C., Stierli B., Streit P., Moser M., Schaller O., Fischer R., Schulz-Schaeffer W., Kretzschmar H., Raeber A., Braun U., et al. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature. 1997;390:74–77. doi: 10.1038/36337. doi:10.1038/36337. [DOI] [PubMed] [Google Scholar]
- 31.Stewart R.S., Harris D.A. Most pathogenic mutations do not alter the membrane topology of the prion protein. J. Biol. Chem. 2001;276:2212–2220. doi: 10.1074/jbc.M006763200. doi:10.1074/jbc.M006763200. [DOI] [PubMed] [Google Scholar]
- 32.Ashok A., Hegde R.S. Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog. 2009;5:e1000479. doi: 10.1371/journal.ppat.1000479. doi:10.1371/journal.ppat.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin J.Y., Shin J.I., Kim J.S., Yang Y.S., Shin Y.K., Kim K.K., Lee S., Kweon D.H. Disulfide bond as a structural determinant of prion protein membrane insertion. Mol. Cells. 2009;27:673–680. doi: 10.1007/s10059-009-0089-9. doi:10.1007/s10059-009-0089-9. [DOI] [PubMed] [Google Scholar]
- 34.Shin J.I., Shin J.Y., Kim J.S., Yang Y.S., Shin Y.K., Kweon D.H. Deep membrane insertion of prion protein upon reduction of disulfide bond. Biochem. Biophys. Res. Commun. 2008;377:995–1000. doi: 10.1016/j.bbrc.2008.10.095. doi:10.1016/j.bbrc.2008.10.095. [DOI] [PubMed] [Google Scholar]
- 35.Kim S.J., Hegde R.S. Cotranslational partitioning of nascent prion protein into multiple populations at the translocation channel. Mol. Biol. Cell. 2002;13:3775–3786. doi: 10.1091/mbc.E02-05-0293. doi:10.1091/mbc.E02-05-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rane N.S., Chakrabarti O., Feigenbaum L., Hegde R.S. Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J. Cell. Biol. 2010;188:515–526. doi: 10.1083/jcb.200911115. doi:10.1083/jcb.200911115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubin G.M., Spradling A.C. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–353. doi: 10.1126/science.6289436. doi:10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- 38.Gavin B.A., Dolph M.J., Deleault N.R., Geoghegan J.C., Khurana V., Feany M.B., Dolph P.J., Supattapone S. Accelerated accumulation of misfolded prion protein and spongiform degeneration in a Drosophila model of Gerstmann-Straussler-Scheinker syndrome. J. Neurosci. 2006;26:12408–12414. doi: 10.1523/JNEUROSCI.3372-06.2006. doi:10.1523/JNEUROSCI.3372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Bourg E., Lints F.A. Hypergravity and aging in Drosophila melanogaster. 4. Climbing activity. Gerontology. 1992;38:59–64. doi: 10.1159/000213307. [DOI] [PubMed] [Google Scholar]
- 40.Emery G., Hutterer A., Berdnik D., Mayer B., Wirtz-Peitz F., Gaitan M.G., Knoblich J.A. Asymmetric Rab 11 endosomes regulate delta recycling and specify cell fate in the Drosophila nervous system. Cell. 2005;122:763–773. doi: 10.1016/j.cell.2005.08.017. doi:10.1016/j.cell.2005.08.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.