

Abstract
Birt–Hogg–Dubé (BHD) syndrome is an autosomal dominant disorder where patients are predisposed to kidney cancer, lung and kidney cysts and benign skin tumors. BHD is caused by heterozygous mutations affecting folliculin (FLCN), a conserved protein that is considered a tumor suppressor. Previous research has uncovered multiple roles for FLCN in cellular physiology, yet it remains unclear how these translate to BHD lesions. Since BHD manifests hallmark characteristics of ciliopathies, we speculated that FLCN might also have a ciliary role. Our data indicate that FLCN localizes to motile and non-motile cilia, centrosomes and the mitotic spindle. Alteration of FLCN levels can cause changes to the onset of ciliogenesis, without abrogating it. In three-dimensional culture, abnormal expression of FLCN disrupts polarized growth of kidney cells and deregulates canonical Wnt signalling. Our findings further suggest that BHD-causing FLCN mutants may retain partial functionality. Thus, several BHD symptoms may be due to abnormal levels of FLCN rather than its complete loss and accordingly, we show expression of mutant FLCN in a BHD-associated renal carcinoma. We propose that BHD is a novel ciliopathy, its symptoms at least partly due to abnormal ciliogenesis and canonical Wnt signalling.
INTRODUCTION
Birt–Hogg–Dubé (BHD) syndrome (MIM #135150) is a rare autosomal dominant disorder that was first described in 1975 by Hornstein and Knickenberg as a distinct disorder associated with intestinal polyps (1). Birt, Hogg and Dubé later reported the same disorder, but in association with medullary thyroid carcinoma (2). A clear association with kidney cancer, mostly of mixed clear cell/chromophobe histology (3), was recognized in 1999 (4) and has been extensively documented since. The prevalence of BHD is estimated at 1/200 000 and the majority of papers published to date put the lifetime risk of developing renal cell carcinoma (RCC) in BHD patients at ∼30% (5). Our own, more recent data suggest a range of 16–20% (3). A roughly similar risk exists for pneumothorax, possibly due to basal lung cysts that are present to a varying degree in almost all BHD patients. About 80% of BHD patients will develop benign skin lesions called fibrofolliculomas (5), generally after the age of 35. An emerging aspect of the BHD phenotype is cyst formation in kidney, liver and the pancreas [Fig. 1, and (6)]. BHD is caused by mostly truncating mutations in the gene coding for the protein FLCN (7), whose functions are largely unknown but which is considered a tumor suppressor (8,9). FLCN is an ancient and highly conserved protein, with multiple orthologs present in fungi and animals. Previous research suggests that FLCN is a downstream target of both AMP-dependent protein kinase (AMPK) and mammalian Target of Rapamycin complex 1 (mTORC1) signalling (10). FLCN might also modulate mTORC1, but conflicting data obtained in cells and tissues that lack FLCN show both up- and down-regulation of mTORC1 activity (9,11–13). We recently reported that the absence of FLCN causes aberrant hypoxia-inducible factor 1 transcriptional activity and the Warburg effect, where FLCN-deficient cells favoured aerobic glycolysis over oxidative phosphorylation (14). Deregulation of TGFβ signalling in FLCN-deficient cells has also been reported, although the reports are contradictory on the nature of FLCN's involvement (15,16). FLCN has recently been implicated in control of ribosomal RNA synthesis through an interaction with the protein RPT4 (17), a finding that might explain the aberrant transcriptional activity observed in a number of studies (14,15).
Figure 1.

BHD syndrome is associated with development of renal cysts. (A) CT scan of a BHD patient. Coronal plane. Arrows indicate cysts in liver and kidney. (B) Paraffin-embedded samples were obtained from a renal carcinoma from a BHD patient with a c.499C>T mutation (encoding pGln167X). Immunohistochemical staining with custom-made C terminal FLCN antibody revealed FLCN around kidney tubules and within the tumor. Highlighted area around the kidney cyst is shown in (C). Magnification ×50. Scale bar is 400 µm. (C) Magnification highlighted area B. Magnification ×400. (D) H&E stain of highlighted area B. Magnification 400×. (E) H&E stain (composite of three images) of a cyst from Nihon Rat kidney tissue. H&E, magnification ×50. Scale bar is 100 µm. (F) H&E stain (composite of four images) of a tumor from Nihon Rat kidney tissue. Magnification ×50. Scale bar is 100 µm. (G) H&E stain (composite of six images) of a cyst from Nihon Rat kidney tissue. There are clear cysts containing two distinct populations of cells. The first population with cuboidal morphology (arrow), and the second with an eosinophilic cytoplasm that protruded to into the cyst lumen (arrowhead). Magnification ×50. Scale bar is 100 µm.
The von Hippel-Lindau (VHL) and Tuberous Sclerosis Complex disease syndromes, predisposing to malignant and benign renal tumors, respectively, have previously been linked to impaired cilia function and cyst formation (18,19). Since BHD patients also develop cysts in multiple organs such as kidney, liver and lungs, we hypothesized that FLCN might similarly have a functional role in primary cilia and that ciliary dysfunction could contribute to the BHD phenotype. Cilia are microtubule-based structures that are enveloped by a highly specialized membrane and protrude from the apical cell membrane. Cilia formation is restricted to cells that have exited the cell cycle, allowing the centrosome to differentiate into a basal body that forms the base of the cilium (reviewed in 20). Different ciliary subtypes have been described, of which primary (non-motile) cilia are the most common and are considered to have primarily sensory functions. Motile cilia are predominantly involved with generating fluid flow (21). Dysfunction of (primary) cilia is a universal cause of the ciliopathies, which are characterized by pathophysiology associated with changes in the morphology and/or number of cilia. The large group of disorders classified as ciliopathies is particularly (but not exclusively) associated with cyst formation in several organs, including kidneys (22,23). In recent years, it has become apparent that cilia act as a master switch essential to maintain normal kidney morphology. The mechanisms involved are far from resolved, but many links have been made between ciliary signalling and planar cell polarity (PCP), the mechanism that defines the cellular orientation in tissue morphology (24). Defective PCP can cause renal tubule elongation defects, which are believed to underlie cystic growth initiation (25). Cysts may represent a potential pre-tumorigenic stage (26) and ciliary dysfunction is increasingly implicated in the pathogenesis of several cancer types, including renal carcinoma (27), melanoma (28), pancreatic ductal adenocarcinoma (29) and ovarian cancer (30,31).
We set out to test our hypothesis by examining FLCN's subcellular localization in various cellular models and its influence on ciliary number, morphology and function. Our data suggest that BHD syndrome can be considered a novel ciliopathy, and that FLCN has a role in regulating cellular morphology and PCP, possibly through interaction with Wnt signalling. We also provide evidence that BHD patient derived FLCN mutants retain functionality, which has important implications for understanding and treating BHD manifestations. If therapeutic or preventive approaches to BHD-associated pathology are to be developed, FLCN's functional role(s) must ultimately be elucidated.
RESULTS
BHD syndrome is associated with development of renal cysts
BHD patients develop cysts in multiple internal organs such as kidney and liver (Fig. 1A). Using a custom-made antibody directed against the C-terminus of FLCN, we stained kidney tumor material derived from a BHD patient with a c.499C>T mutation (that encodes for a truncated FLCN mutant, pGln167X) (Fig. 1B). Hence, this antibody will only detect the wild-type protein, not this mutant form. The kidney sample contains both unaffected kidney (Fig. 1B, upper section) and clearly demarcated renal tumor material with mixed chromophobe/clear cell histology (Fig. 1B, lower section). We observed strong expression within the normal kidney and clear staining for FLCN around the kidney tubules (Fig. 1B and C and Supplementary Material, Fig. S1). We also noted a cystic area devoid of cells (Fig. 1B–D), which represents a small kidney cyst. Previously, we determined that the remaining FLCN allele in this tumor contains a second hit of a 15 base pair deletion, predicted to result in the deletion of exon 6 (32). Within the tumor mass, we still observe staining for FLCN (Fig. 1B, lower section), suggesting the presence of a mutant form of the protein in this particular tumor. Next we examined kidney material obtained from the Nihon rat BHD animal model (33). The Nihon rat has a naturally occurring mutation in Flcn that is predicted to result in a severely truncated FLCN protein. Upon examination of the kidney, multiple cysts (Fig. 1E) as well as cancerous lesions were evident (Fig. 1F). Of note, the cysts contained two populations of cells—one population of clear cells with a cuboidal morphology that lined the cyst (Fig. 1G, see arrow) and another population with eosinophilic cytoplasm that grew into the cyst lumen (Fig. 1G, see arrowhead).
Thus, both humans and rats with BHD syndrome develop kidney cysts, highlighting that BHD might be a member of a larger group of disorders called ciliopathies, of which cysts in various organs, particularly the kidneys, are hallmark lesions. Ciliopathies are associated with cystic disease linked to changes in the number and/or shape of cilia (23). As ciliopathy proteins often localize to the cilium (although this is not always a prerequisite) (34), we hypothesized that FLCN might localize to cilia and proceeded to examine whether FLCN is a ciliary protein.
FLCN localizes to a number of microtubule-based structures
We determined the localization of endogenous FLCN in several different cell types that form primary cilia. To rule out the possibility of a non-specific staining, we utilized several different FLCN antibodies. In all cell lines and with all antibodies used, we observed a clear and specific staining of anti-FLCN antibodies to primary cilia (Fig. 2A and Supplementary Material, Fig. S2). To further rule out the possibility of a non-specific antibody stain, we stably expressed EGFP-tagged FLCN (hereafter referred to as EGFP-FLCN WT) in type II Madin-Darby Canine Kidney (MDCK) cells. In these cells, we observed EGFP-tagged FLCN localizing to cilia (Supplementary Material, Fig. S4A). In line with previous data, we observed FLCN localization throughout the cytoplasm and in the nucleus (10,35). Interestingly, we noted that anti-FLCN antibody also localized to 1 or 2 discrete foci within the cell. We subsequently stained with the centrosome marker gamma-tubulin, and found co-localization, indicating that FLCN is a centrosomal protein (Fig. 2B). We confirmed these results in the stable cell line, where EGFP-FLCN WT (in addition to cilia) localizes at 1 or 2 discrete foci, indicating that after centrosome duplication FLCN localizes to both the centrosomes (Fig. 2C). Next we considered the possibility that FLCN might be a universal constituent of both primary and motile cilia and their respective basal bodies. In primary human nasal epithelial (HNE) cells that contain beating motile cilia, we observed localization of FLCN to the cilia (Fig. 2D) and at the base plate, which is the basal body equivalent for motile cilia (Fig. 2E). In Drosophila melanogaster, BHD−/− males exhibit a fertility defect (36). We therefore asked whether FLCN is present in sperm flagellae or their associated structures. The sperm flagellum is structurally identical to motile cilia, although its beating pattern is different (37). Some ciliopathies are associated with sperm motility defects (38). As shown in Figure 2F, in human spermatozoa we observed clear staining of FLCN to the centriole (Fig. 2F, arrow) and annulus (Fig. 2F, arrowhead). The latter structure is equivalent to the ciliary diffusion barrier for non-motile cilia (37). Faint flagellar staining can also be observed, but due to the low level of staining it is not clear whether this is an artefact or genuine localization of FLCN in the flagellum.
Figure 2.

FLCN is a constitutive ciliary, basal body and centrosomal protein. In this figure, all endogenous FLCN stains were obtained using the FLCN AP antibody. (A) FLCN localizes to primary cilia. HK-2 cells serum starved for 48 h were fixed and stained for endogenous FLCN (green) and acetylated alpha tubulin (red). Nuclei were stained with DAPI (blue). Cilium in the white box is shown in orthogonal view below. Scale bar is 10 µm. (B) Endogenous FLCN localizes to the centrosome. IMCD3 cells were fixed and stained for gamma-tubulin (red) and endogenous FLCN (green). Nuclei are stained with DAPI (blue). Scale bar is 10 µm. (C) EGFP-FLCN WT localizes to the centrosome. MDCK cells stably expressing EGFP-tagged FLCN WT were fixed and counterstained with anti-gamma-tubulin (red) to mark the centrosomes (indicated by arrows). EGFP-FLCN WT localizes at centrosome before (B i, scale bar is 20 µm) and after centrosome duplication (B ii, scale bar is 10 µm). Nuclei are stained with DAPI (blue). (D) FLCN localizes to motile cilia. (i) Single stain. Primary HNE cells were fixed and stained for endogenous FLCN (green). Middle panel is DIC image for visualization of cilia (indicated by the arrow). Scale bar is 20 µm. (ii) Double stained. HNE cells were fixed and stained for endogenous FLCN (red) and acetylated alpha tubulin (green). Scale bar is 10 µm. (E) FLCN localizes to the basal body. HNE cells were fixed and stained for endogenous FLCN (red) and pericentrin (green) to mark the base plate. Scale bar is 20 µm. (F) In human sperm endogenous FLCN localizes to the centriole and annulus. Human sperm cells were fixed and stained for endogenous FLCN (red). Note the staining at the centriole (arrowhead) and annulus (arrow). Scale bar is 10 µm. (G) FLCN localizes to the mitotic spindle. HK-2 cells overexpressing EGFP-FLCN WT were fixed and counterstained with acetylated alpha tubulin (red) to mark the mitotic spindles (indicated by arrows) and DAPI (blue). Scale bar is 20 µm.
Finally, several known ciliary proteins such as IFT88, LKB1 and VHL (39–41) are known to localize to the mitotic spindle. To see if this is the case for FLCN, we examined mitotic cells expressing EGFP-FLCN WT and indeed, we observed the EGFP-tagged FLCN protein localizing to the mitotic spindle (Fig. 2G).
Disease-associated mutations of FLCN do not affect subcellular localization
Having observed full-length FLCN localizing to cilia, the basal body and the centrosome, we reasoned that disease-associated mutations might affect FLCN's subcellular localization. Therefore, we expressed EGFP-tagged versions of several disease-causing FLCN mutations in the immortalized proximal kidney tubule cell line HK-2 (42). The tagged versions of missense mutations p.His255Arg (H255R) and p.Lys508Arg (K508R) could be visualized at the centrosome (Supplementary Material, Fig. S3 and Supplementary Material, Table S1). Next, we examined EGFP-tagged patient mutations that result in C-terminal deletions, p.Tyr463Stop (Y463X), and the most common mutation, p.His429ProfsX27. Again, both of these truncating mutants localize to the centrosomes. These observations suggest that BHD-associated truncating FLCN mutations do not prevent centrosomal localization.
Unfortunately, due to consistently low expression levels and a deleterious effect on ciliation, localization of the H255R, Y463X and p.His429ProfsX27 mutant FLCN proteins to cilia and the mitotic spindle could not be determined. However, we successfully visualized EGFP-FLCN K508R in the cilia (Supplementary Material, Fig. S4A) and at the mitotic spindle (Supplementary Material, Fig. S4B). Thus, the localization of the K508R mutant seems comparable with that of the wild-type protein.
The BHD-derived renal cancer cell line UOK257 shows abnormal ciliation
As loss of cilia is a frequent occurrence in kidney cancer (27), we were interested whether BHD-derived renal cancer cells can form cilia. UOK257 is a BHD-derived kidney tumor cell line carrying the most common germline FLCN mutation, c.1285dupC (43), resulting in the FLCN truncation p.His429ProfsX27. Whereas BHD patients are heterozygotes, and still have one wild-type allele for the FLCN gene, the UOK257 cell line used in this study has been reported to have lost the wild-type allele and consequently only possesses the mutant copy (43). The presumed lack of functional FLCN might impair ciliogenesis. To test this idea, we compared the ability of UOK257 and UOK257-2 [UOK257 cells with re-expression of FLCN (10)] to ciliate. Neither cell line ciliated upon serum starvation, however, cilia were evident in both cell lines when left at full confluence for several days (Fig. 3A). Interestingly, we noted that after 6 days, confluent FLCN-rescued UOK257-2 cells exhibited fewer cilia than UOK257 cells. When we increased this time to 10 days, this difference between the cell lines was no longer evident (Fig. 3A), suggesting that overexpression of FLCN may impede formation of primary cilia. The length of the cilia observed in these UOK257 cells (6.3 ± 2.5 µm n = 74) was not significantly different to that observed in our control HK-2 cell line (7.0 µm ± 3.2 µm n = 147).
Figure 3.
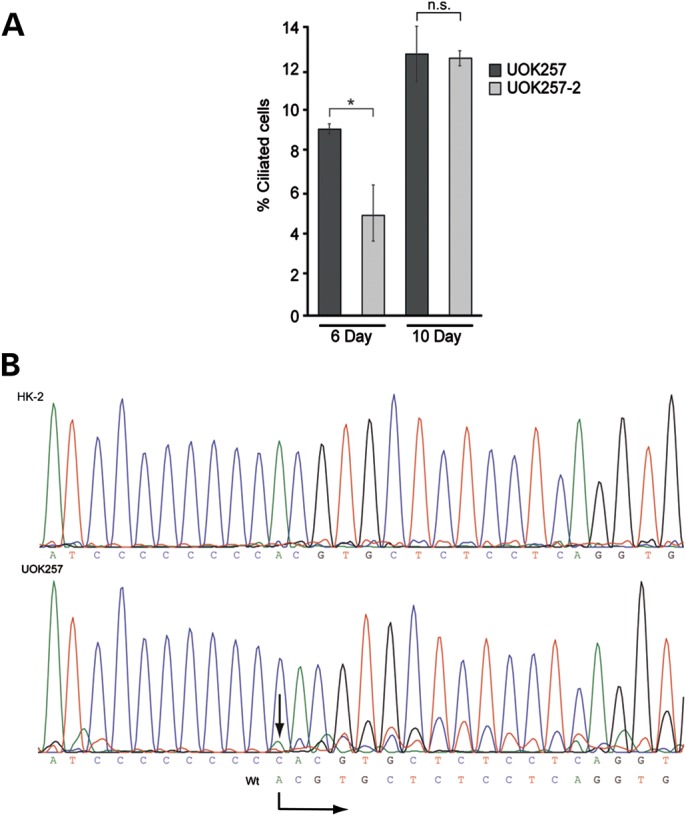
The BHD-derived renal cancer cell line UOK257 shows abnormal ciliation. (A) UOK257 and UOK257-2 cells were maintained at confluence for indicated amount of days, fixed and stained with acetylated alpha tubulin to mark the ciliary axoneme and pericentrin to mark the ciliary base. Nuclei were stained with DAPI and the percentage of ciliated cells was counted using ImageJ. After 6 days of confluence, UOK257 had a significantly reduced number of cilia compared with UOK257-2 (P = 0.023). After 10 days of confluence, both cell lines have a similar number of cilia (P = 0.422). Error bars represent standard error of the mean. A minimum of 365 cells were counted per cell line per time point. *P < 0.05. (B) Sequencing of genomic DNA isolated from HK-2 and UOK257 cells showing the presence of the wild-type allele (starting sequence indicated by the arrow).
Given the apparent differences we observed in ciliation between the UOK cell lines, we then sought to confirm their identity by sequencing the FLCN locus. Upon genomic sequencing of all of the FLCN exons using intronic primers, the only FLCN mutation detected was His429ProfsX27. However, it was also evident that a low level of the wild-type allele was present in the cells (Fig. 3B). This finding was subsequently confirmed by both immunofluorescence and western blotting experiments (see Supplementary Material, Fig. S5). Thus, we conclude that UOK257 cells are not FLCN null, as originally reported, but rather still express full-length, wild-type FLCN.
Knockdown of FLCN in human kidney cells delays ciliogenesis
Considering that UOK257 cells express a lower level of full-length FLCN than seen in HK-2 cells (Supplementary Material, Fig. S5C), we wondered if the ciliary phenotype observed in these cells resulted from reduced levels of wild-type FLCN. Therefore, we generated stable knockdowns of FLCN in HK-2 cells (see Supplementary Material, Fig. S6). Subsequently, we selected FLCN knockdown HK-2 clone A5 (hereafter referred to as FLCN KD, estimated 50% knockdown) and the scrambled control HK-2 clone G10 (hereafter referred to as NT; Fig. 4A). Knockdown of FLCN did not alter cell morphology or growth rate (data not shown). Cells were plated at low density and subsequently serum-starved for 48, 72, 96 and 120 h to initiate ciliogenesis. As seen in Figure 4B, at 48 and 72 h, the FLCN KD cell line showed a significant reduction in number of ciliated cells compared with the NT control cells. Knockdown or knockout of other ciliary proteins can also cause changes in the length of cilia (20). Cilia in the HK-2 FLCN KD cells showed a slight but significant reduction in length versus the NT control cell line after 48 h of serum starvation. This was not evident after 72 h of serum starvation (Fig. 4C). At the later time points of 96 and 120 h, all cell lines exhibited more cilia, and no significant difference in the number of ciliated cells between FLCN KD and NT control was evident (Fig. 4C). Collectively, these data suggest that ciliogenesis is delayed rather than inhibited upon FLCN knockdown.
Figure 4.

Knockdown and exogenous expression of FLCN affects ciliogenesis in kidney cells. (A) Western blot of knockdown and control cells. Western blot on 30 µg of WCL showing stable knockdown of FLCN using shRNA in HK-2 clone FLCN KD compared with wild-type HK-2 and HK-2 with scrambled control shRNA clone NT (also see Supplementary Material, Fig. S6). Membrane was probed with FLCN AP antibody. Actin was used as a loading control. (B) Measurement of ciliogenesis in FLCN knockdown versus non target control. HK-2 FLCN KD and NT cells were serum starved for the indicated amount of hours, fixed and stained with acetylated alpha tubulin to mark the ciliary axoneme and pericentrin to mark the ciliary base. Nuclei were stained with DAPI and the percentage of ciliated cells and cilium length was measured using ImageJ. After 48 h (P < 0.001) and 72 h (P < 0.001) of serum starvation, HK-2 FLCN KD cells showed a statistically significant reduction in the percentage of ciliated cells compared with NT cells. After 96 h (P = 0.915) and 120 h (P = 0.067) of serum starvation, there was no difference in the percentage of ciliated cells between the two cell lines. Error bars represent standard error of the mean. Average of 350 cells counted per cell line per time point. *P < 0.05. (C) Measurement of cilia length in FLCN knockdown versus non-target control. Average cilium length of HK-2 FLCN KD cells (n = 40) was significantly decreased compared with cilia of NT cells (n = 124) after 48 h of serum starvation (P = 0.001). After 72 h of serum starvation, no difference in cilia length was observed between HK-2 FLCN KD (n = 24) and NT cells (n = 55) (P = 0.55). Error bars represent standard error of the mean. *P < 0.05. (D) Western blot of FLCN expressing cell lines. Western blot probed with EGFP antibody showing MDCK cells with stable expression of EGFP alone, EGFP-FLCN WT and the disease-causing FLCN missense variant EGFP-FLCN K508R. Actin was used as a loading control. Actin is not detected in the EGFP-only MDCK cells, as a result of 10-fold dilution of this sample due to very high EGFP expression in this cell line. (E) Growth curve in FLCN expressing cell lines. At a starting density of 105 cells per well, untransfected MDCK cells and MDCK cells with stable expression of EGFP, EGFP-FLCN WT or EGFP-FLCN K508R were plated. Cell number was determined at each time point. Each data point is the average of six independent counts. Error bars represent standard error of the mean. (F) Measurement of ciliogenesis in FLCN expressing cell lines. Untransfected MDCK (MDCK) and MDCK cells stably expression EGFP, EGFP-FLCN WT (WT) or EGFP-FLCN K508R (K508R) were maintained at confluence for 5 days, fixed and stained with acetylated alpha tubulin to mark the ciliary axoneme and with DAPI to stain nuclei. Cells expressing EGFP-FLCN WT showed a statistically significant reduction in the percentage of ciliated cells compared with cells expressing EGFP alone (P < 0.001) or EGFP-FLCN K508R (P < 0.001), as counted using ImageJ. Error bars represent standard error of the mean. *P < 0.05.
Exogenous expression of FLCN affects proliferation and ciliogenesis
Having noted that exogenous expression of FLCN in UOK257-2 cells seemed to reduce the number of cilia observed we then decided to examine the effect of exogenous FLCN expression on ciliation in more detail. To do this, we generated cell lines expressing EGFP alone or EGFP-FLCN WT from an expression construct. We also attempted to make stable cell lines expressing the different EGFP-tagged patient mutations described above. However, only the FLCN missense variant EGFP-FLCN K508R resulted in a usable stable cell line (Fig. 4D). We noted that the cell lines expressing exogenous EGFP-FLCN seem to grow more slowly than the cells expressing EGFP alone and we therefore measured the doubling time of these cells. As shown in Figure 4E and Supplementary Material, Table S2, expression of EGFP-FLCN WT and EGFP-FLCN K508R significantly increased the cellular doubling time, when compared with the control cell lines. This finding contrasts with previously published data, suggesting that a missense mutant (H255R) does not affect cell growth (8). Next, we tested the ability of these different cells to ciliate (Fig. 4F).
To initiate formation of cilia, all cells were cultured to 5 days post-confluence. Cells expressing EGFP-FLCN WT (52.5% ± 4.77 n = 705) exhibited a statistically significant reduction in the number of cilia relative to the EGFP alone control (EGFP 70% ± 0.12 n = 825). Interestingly, despite the apparently normal localization and similar growth defect to that observed for wild-type FLCN, EGFP-FLCN K508R (66.5 ± 2.03%, n = 670) did not change the percentage of ciliated cells when compared with untransfected or cells expressing EGFP alone (Fig. 4F).
Knockdown and exogenous expression of FLCN impairs kidney cell spheroid growth in three-dimensional culture
Having established that exogenous expression and knockdown of FLCN both adversely affect cilia number in monolayer culture, we then assessed the effect of modulating FLCN expression in a 3D culture system. Due to the complex signalling required for proper spatial orientation, a mild ciliopathy phenotype in monolayer culture can translate into a more severe one in 3D cultures (34). MDCK and IMCD3 cell lines cultured in 3D are both established models for renal morphogenesis, mimicking normal renal tubules by forming spheroids or tubules composed of polarized cells exhibiting a lumen lining a ciliated apical membrane. We considered that BHD cyst formation could be related to delayed ciliogenesis resulting from reduced levels of wild-type FLCN. Therefore, we assessed the effects of FLCN knockdown in detail in the 3D system. Spheroid formation in cells treated with siRNA against Flcn (SiFlcn) was severely impaired when compared with non-target siRNA (SiCON) (Fig. 5A i). The level of knockdown achieved was ∼60% (Fig. 5A ii) and in line with previous data, fewer cilia were observed in Flcn knockdowns compared with non-target controls (Fig. 5A iii). The number of cells composing the median section of a spheroid did not differ significantly in the SiFlcn-treated cells versus control knockdown (Fig. 5B i). We then determined the overall and lumen size of these spheroids. As shown in Figure 5B ii, while SiFlcn only resulted in a small decrease in the overall size of the spheroid, it did cause a marked and significant decrease in luminal size when compared with the control spheroids. In addition to the effects of Flcn knockdown on cilia frequency and lumen size, we observed an increase in improper orientation of cell divisions. In normal spheroids, cell division occurs in plane with the apical membrane, directed towards the lateral membranes (Fig. 5C i). By scoring the number of cells that are either actively dividing in the wrong orientation, or have ended up outside the normal plane, we found that Flcn knockdown causes a strong increase in abnormally orientated cell divisions (Fig. 5C ii). Having observed that knockdown of Flcn affects spheroid formation in 3D cultures, we then assessed the effect of exogenous expression of EGFP-FLCN WT in 3D spheroids. Consistent with the previously observed growth defect in cells expressing EGFP-FLCN WT, the number of cells composing an EGFP-FLCN WT spheroid was reduced when compared with the control cell line (Supplementary Material, Fig. S7A). We then compared the overall and lumen size of the EGFP-FLCN WT spheroids to that of the EGFP alone control cell line (Fig. 5E i). Interestingly, spheroid formation in cell types expressing EGFP-FLCN WT was severely impaired, with smaller spheroids exhibiting a reduction (or absence) in the lumen size. Thus, both knockdown and exogenous expression of FLCN perturb lumen formation, with the latter giving a more severe phenotype.
Figure 5.

Knockdown or expression of exogenous FLCN impairs kidney cell spheroid growth in three-dimensional culture. (A) Knockdown of FLCN impairs kidney cell spheroid growth in three-dimensional culture. (i) IMCD3 cells were treated with non-targeting RNAi (siCON) or FLCN RNAi (siFlcn) for 48 h. The cells were subsequently split into two equal parts, with the first half cultured for 3 days in matrigel and the remainder in 2D culture for quantification of knockdown shown in (ii). Cultures were fixed and stained. Tight-junction marker ZO-1 (orange) localizes to the apical side, β-catenin (green) is enriched at lateral cell–cell contacts and acetylated alpha tubulin stains cilia (red) that are present protruding into the lumen. Nuclei are stained with DAPI. Representative spheroids are shown. SiCON cells form a typical spheroid that is evenly rounded and has a clear lumen. Cells treated with SiFlcn form spheroids with impaired ciliogenesis, reduced overall and luminal sizes. Arrows indicate cilia containing β-catenin. Scale bar is 10 µm. (ii) Quantification of Flcn levels using RT-PCR in IMCD3 cells treated with siCON or siFlcn (normalized to Rpl19). The RNA was extracted from cells at the same time point as the 3D spheroids shown in (i) were fixed. (iii) Quantification of the number of cilia observed in non-target siCON or the siFlcn spheroids. Spheroids with reduced Flcn levels exhibit significantly decreased numbers of cilia compared with the siCON control (P > 0.0001). (B) Knockdown of FLCN in IMCD3 decreases spheroid and lumen size in three-dimensional culture. IMCD3 cells with siRNA-mediated knockdown of Flcn form spheroids that consist of the same number of cells (i) but are significantly smaller (ii) in size (P = 0.0058) and have a smaller lumen (ii, P = 0.0003) compared with control spheroids. (C) Knockdown of FLCN impairs spindle orientation in three-dimensional culture. (i) Representative image of misoriented, dividing cell and a cell that ended up outside the normal plane (white arrows) in siFLCN spheroid versus control. Cells are stained with DAPI. Scale bar 20 µm. (ii) Quantification of cell division orientation in IMCD3 spheroids treated with SiFlcn, which show a significant increase in the number of misorientated spindles per spheroid compared with control spheroids (P < 0.0001). (D) Expression of wild-type but not K508R FLCN impairs kidney cell spheroid growth in three-dimensional culture. MDCK cells stably expression EGFP, EGFP-FLCN WT or EGFP-FLCN K508R were grown in collagen for 11 days. Cultures were fixed and stained with DAPI (blue). Note the lack of lumen formation in the cells expression EGFP-FLCN WT in the 2D and 3D projection. Scale bar is 20 µm. (E) Quantification of spheroid parameters with exogenous FLCN expression in IMCD3. (i) IMCD3 cells with stable expression of EGFP-FLCN WT form spheroids that are significantly smaller in size (P < 0.0001) and have a smaller lumen (P < 0.0001) compared with control spheroids expressing EGFP only. (ii) Expression of EGFP-FLCN K508R results in normal-sized spheroids (P = 0.2381) with a slight reduction in lumen size (P < 0.0001) compared with control spheroids expressing EGFP only.
A BHD-associated FLCN missense mutation is partially dysfunctional with respect to spheroid formation
Next, we tested the ability of the K508R mutant to influence spheroid formation, as this mutation does not seem to affect ciliogenesis. In contrast to EGFP-FLCN WT, EGFP-FLCN K508R spheroids appear similar to normal spheroids (Fig. 5D), where a clear lumen can be observed. The size of the spheroid and lumen was determined and while the overall size was similar, the lumen size was found to slightly deviate from EGFP controls, suggesting that K508R may have a mild effect on lumenogenesis. In line with the previous growth rate data, the number of cells composing an EGFP-FLCN K508R spheroid was reduced when compared with the control cell line (Supplementary Material, Fig. S7B).
These observations further suggest that the spheroid formation defect observed upon FLCN knockdown or exogenous expression could be linked to ciliary dysfunction.
Flcn knockdown results in β-catenin delocalization and activates canonical Wnt signalling
In the spheroid assays, we used β-catenin as a standard marker for the baso-lateral membranes. When we performed these assays, we noted that β-catenin can often be observed in the cilium (Fig. 5Ai, indicated by arrow). Intriguingly, in the few cilia present in the spheroids treated with SiFlcn, we rarely observed this localization of β-catenin (Supplementary Material, Fig. S7C). The sequestration of β-catenin to cilia has been reported to dampen canonical Wingless signalling (44) and abnormal Wnt/β-catenin signalling has previously been implicated in renal cystogenesis (45). Therefore, we hypothesized that the observed defects in spheroid formation upon knockdown of Flcn might be a consequence of altered Wnt signalling. Thus, we examined canonical Wnt activity by looking at β-catenin phosphorylation and Axin2/CyclinD1 mRNA levels. Upon SiFlcn treatment, no reduction in the total amount of β-catenin was evident when compared with control RNAi (Fig. 6A). However, an antibody against the active, unphosphorylated form of β-catenin showed clear activation of β-catenin upon Flcn knockdown. Furthermore, quantitative RT-PCR of downstream target genes Axin2 and CyclinD1 shows up-regulation of these two genes upon Flcn knockdown, further supporting activation of the canonical Wnt pathway (Fig. 6B).
Figure 6.

FLCN knockdown causes activation of canonical Wnt signalling. (A) Knockdown of FLCN results in activation of β-catenin. Western blot on 30 µg of WCL from treated IMCD3 cells in 2D culture. Cells were harvested at 60 h post transfection. Cells were serum starved for 18 h prior to harvesting. IMCD3 express equal amounts of total β-catenin (middle panel) upon treatment with non-targeting SiCON or SiFlcn or treatment with 20 mm KCl or LiCl for 16 h. Using an antibody to detect the unphosphorylated, active β-catenin, cells treated with LiCl (an activator of β-catenin through inhibition of GSK3β) showed increased active β-catenin as did SiFlcn treated cells (upper panel). The β-tubulin (lower panel) was used as a loading control. (B) Activation of downstream targets of the Wnt pathway upon FLCN knockdown. The activation of the Wnt pathway was further confirmed using quantitative RT-PCR of downstream target genes Axin2 and CyclinD1. Upon Flcn knockdown, mRNA levels of both genes exhibited up-regulation.
DISCUSSION
BHD patients show cyst formation in lungs, kidney and liver (6). Cyst formation in multiple organ systems is a hallmark characteristic of ciliopathies (23), which led us to hypothesize that ciliary dysfunction could contribute to the BHD phenotype. Our results suggest that this is indeed the case and provide a possible explanation for key aspects of the BHD phenotype.
In a kidney sample taken from a BHD patient during surgery for renal cancer, we observed a cyst and a tumor in close proximity. Interestingly, we found similar arrangements in Nihon rat kidneys (data not shown). It is well established that in this model, cancer is preceded by the appearance of cuboidal cells with a clear cytoplasm lining dilated collecting ducts, followed by cells with acidophilic cytoplasm (33). The cancers that appear around 8 weeks consist of both clear and acidophilic cells. Large cysts such as the one shown in Figure 1E and G have not been reported as a microscopic finding, but macroscopic ones have (33). Of note, the cysts that we observed in the rat are lined with clear cuboidal cells; acidophilic cells grow into the cyst lumen (Fig. 1G). The tumors contain both cell types, with the clear cells consistently located towards the periphery. We note that human BHD RCC show the same arrangement of cells (3). We suggest from these observations that in humans with BHD, as in the Nihon rat, tumors develop from cysts. Hence, understanding cyst formation can help elucidate carcinogenesis in BHD.
FLCN is a dose-dependent regulator of cilia formation
Ciliogenesis is a process requiring complex regulation that is not yet fully understood. Entrance into the G0 phase of the cell cycle and establishment of cell polarity are considered to be the first prerequisites for ciliation (46). We observe that levels of FLCN affect the timing of ciliation. We also show that endogenously expressed FLCN, and tagged versions of FLCN, very specifically localize to tubulin-based structures such as motile and non-motile cilia, centrosomes and the mitotic spindle in a variety of cell types. HK-2 cells with reduced endogenous FLCN expression show reduced ciliation and a subtle but significant decrease in ciliary length at early starvation time points, which are subsequently overcome. These findings indicate that cells with less FLCN have a delay in ciliogenesis, compared with normal cells. IMCD3 spheroids recapitulate this reduction in ciliation upon knockdown of Flcn. Furthermore, the expression of exogenous wild-type FLCN also resulted in reduced ciliation, suggesting that tight regulation of FLCN levels is required for normal ciliogenesis.
We have not observed any obvious defects in the size or shape of matured cilia, indicating that once ciliogenesis has been initiated, FLCN's role is permissive. In line with other renal cell types examined (MDCK and HK-2), under conditions of confluence, UOK257 cells do have the ability to form cilia. However, despite the apparently normal appearance of these cilia in UOK257 cells, the actual percentage of cells exhibiting cilia is low relative to other cell lines examined. Similarly, ciliation is delayed in these UOK257 cells. The low level of and delay in ciliation observed may be a consequence of a reduced level of full-length FLCN expression in these cells. The abnormal ciliation in UOK257-2 is consistent with our observations in other cell types and suggests that FLCN expression levels are critically important for correct timing of ciliogenesis.
FLCN regulates canonical Wnt signalling and spindle orientation in spheroids
We previously described the IMCD3 spheroid culture system as a suitable method to assess ciliopathy genes and their effects on renal morphology (34). The siRNA-mediated knockdown of FLCN in IMCD3 cells demonstrates a role for the protein in establishing cellular orientation, as the spheroids formed by these knockdown cells exhibit an increased number of cells outside of the normal plane of division. This mitotic spindle misalignment may reflect disturbed PCP (controlled by non-canonical Wnt signalling), abnormal cortical microtubule function or mis-localized polarity proteins. Cilia have a role in the process of polarization by dampening the activity of the canonical Wnt pathway via sequestration of β-catenin (44). Accordingly, FLCN knockdown seems to reduce ciliary β-catenin retention and is associated with an increase in the level of unphosphorylated β-catenin. The lack of phosphorylation of β-catenin results in the stabilization of the protein and an increase in canonical Wnt activity (47). As a consequence, non-canonical PCP might be inhibited (48), resulting in spindle misalignment. In view of the increased Wnt activity, it is of considerable interest that recent observations clearly show an increased incidence of kidney cancer in people who chronically use lithium chloride for psychiatric conditions (49). Lithium activates canonical Wnt signalling through repression of GSK3β activity (50). The spectrum of renal tumors associated with lithium use is remarkably similar to that seen in BHD. Thus, we propose that carcinogenesis in BHD may be partly due to deregulated canonical Wnt signalling.
An alternative explanation for the spindle orientation defect could be that FLCN does not exert a direct effect on PCP signalling, but rather is actively involved in spindle orientation. The observation that EGFP-FLCN WT localizes to the centrosome and mitotic spindle is compatible with this idea. Other ciliary proteins have been shown to affect spindle positioning. For instance, the classic ciliary transport protein IFT88 localizes to the centrosome in mitosis, where it plays a fundamental role in microtubule transport (39). FLCN might exert a similar function in controlling essential processes at the spindle pole, defects of which result in misorientation of the mitotic spindle. Finally, a recent report describes that LKB1 regulates spindle orientation through AMPK, possibly through microtubule destabilization (40), and we have previously demonstrated an increase in AMPK phosphorylation in UOK257 cells (14).
Very recently, Nookala et al. (51) provided evidence that the C-terminal half of FLCN may act in vitro as a guanine nucleotide exchange factor for Rab35, a poorly characterized GTPase involved in endocytotic membrane recycling and in the final steps of cytokinesis (52). How this function relates to the pervasive cellular abnormalities caused by FLCN's absence and the BHD phenotype is not yet clear. However, both ciliogenesis and mitotic spindle positioning require vesicular transport, which exhibit significant overlap (53) and we show that these processes are affected in BHD. Thus, FLCN may have a function in intracellular transport events involved in ciliogenesis.
FLCN might exhibit a partial genotype–phenotype correlation
To date, no clear correlation has been found between mutations in FLCN and the severity of BHD symptoms observed in patients. However, our findings with FLCN K508R may indicate that there might be a milder form of BHD associated with specific missense mutations. The growth defect caused by K508R and its cellular localization are indistinguishable from the wild-type protein. However, in contrast to wild-type FLCN, expression of K508R does not seem to adversely affect ciliogenesis and only mildly affects the spheroid forming abilities of the cell. These results are suggestive of at least some (partial) functionality of K508R mutant. This is in line with the published clinical information (54). The patient, in whom the mutation was found, presented with bilateral multifocal benign renal oncocytomas and was only diagnosed with BHD upon sequencing of the oncocytomas. At the time of publication, they did not manifest any of the other known BHD symptoms (W.M. Linehan, personal communication, 2013). It is tempting to speculate that, if kidney cancer in BHD arises from cysts, mutations that do not affect ciliogenesis will not be associated with malignancy. Another missense mutation (p.Arg239Cys) has been reported in a patient presenting with kidney cancer (55). If our reasoning is correct, this particular mutant should affect ciliogenesis.
Mutant FLCN alleles might contribute to the phenotype
From our observations, we think that the clinical manifestations of BHD may not result from simple loss of one FLCN allele. Rather, the mutant versions of FLCN could also contribute to the BHD phenotype, perhaps in a dominant negative manner. Relative to the UOK257 cell lines, the restoration and exogenous expression of FLCN in UOK257-2 cells does not increase the low percentage of ciliated cells upon confluence. Mutant versions of FLCN that were tested localized to the centrosome and (at least for K508R) negatively impact cellular proliferation. Expression of truncated or wild-type FLCN seems to impede ciliogenesis (with the notable exception of K508R).
Complete loss of FLCN has been shown to be lethal (9,13) and we would therefore not expect to encounter it in patients or in any tissue. Accordingly, the majority of BHD-associated kidney tumors have somatic second hits affecting FLCN, rather than loss of the wild-type allele (56). We also show that truncated FLCN is still expressed in a renal cancer from a BHD patient (Fig. 1 and (32)). Finally, people with the Smith-Magenis syndrome (MIM 182290), a congenital disorder caused by heterozygous deletion of chromosome 17p11.2 (including the region containing the FLCN locus), do not develop symptoms consistent with BHD (57). In particular, patients do not exhibit an elevated risk of developing kidney cancer.
Based on these observations, we propose that a ‘just right’ hypothesis, as for the APC tumor suppressor (58), also holds true for FLCN.
In conclusion, we provide evidence that BHD syndrome is a novel ciliopathy. The causative protein, FLCN, localizes to motile and non-motile cilia, centrosomes and the mitotic spindle. It is involved in the initiation of ciliogenesis—once ciliation is underway, FLCN's role seems to be permissive. Knockdown of FLCN affects PCP, which might be due to the ciliation defect. Additional roles for FLCN in establishing cellular polarity seem likely and we have evidence for abnormal canonical Wnt signalling in the context of FLCN knockdown. Wnt signalling is potentially oncogenic and might thus present a new mechanism for carcinogenesis in BHD. Our data further suggest that the correct level of FLCN expression is critically important for its optimal function. Delayed ciliogenesis is likely to affect the ability of cells to orientate themselves in newly generated polarized tissues. The observations in Nihon rat kidney suggest that cyst formation may be a prerequisite to cancer development. Hence, FLCN mutations that do not affect ciliogenesis, such as K508R, might not cause cancer and it would be of considerable interest to test this hypothesis using knock-in mouse models.
The classification of BHD as a ciliopathy should help better understand its pathogenesis, as the study of BHD can now benefit from the intense research effort on ciliopathies.
MATERIALS AND METHODS
DNA constructs and antibodies
Human FLCN was cloned into pEGFP C1 (Clontech, Mountain View, CA, USA) using EcoRI. EGFP-tagged FLCN mutant constructs were made by site-directed mutagenesis using the Quikchange II Site Directed Mutagenesis kit according to the manufacturer's instructions (Agilent, Santa Clara, CA, USA) with the following primers (Supplementary Material, Table S3).
EGFP-tagged FLCN WT and mutants were cloned into the pQCXIP vector (Clontech) using AgeI and EcoRI.
Human-specific FLCN shRNA plasmid pLKO.1-puro-shRNA5968 and the Non Target shRNA plasmid pLKO.1-puro-NonTaget (Sigma) were a kind gift from Prof. A. Pause (McGill University, Canada). Murine-specific Flcn siRNA oligos were purchased from Dharmacon (Lafayette, CO, USA).
Primary antibodies used for immunofluorescence microscopy are anti-FLCN-AP (rabbit polyclonal, a kind gift from Prof. A. Pause, 1:200), anti-FLCN (rabbit monoclonal, 3697 Cell Signalling, Danvers, MA, USA 1:200), anti-FLCN [rabbit polyclonal, (12) a kind gift from Dr T. Cash 1:100], anti-pericentrin (rabbit polyclonal, ab4448 Abcam, Cambridge, UK, 1:1000), anti-gamma-tubulin (mouse monoclonal, T3559 Sigma 1:1000), anti-acetylated-alpha-tubulin (mouse monoclonal, ab24610 Abcam 1:200), anti-acetylated-alpha-tubulin (mouse, T7451 Sigma, 1:20 000), anti-β-catenin (rabbit, AHO0462 Invitrogen, Paisley, UK, 1:500) and anti-ZO-1 (rat, R40.76 Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:500). Secondary antibodies used were goat-anti-mouse Texas red (1010-07 Southern Biotech, Birmingham, AL, USA, 1:80), goat-anti-rabbit FITC (4050-02 Southern Biotech, 1:80), goat-anti-mouse Cy5 (1034-15, Southern Biotech, 1:80) and anti-Rabbit-488, anti-mouse-568 and anti-Rat-647 (all from Molecular Probes, Invitrogen).
Primary antibodies used for western blot were raised against EGFP (mouse, 11814460001 Roche, Basel, Switzerland 1:2000), actin (mouse monoclonal, A5441 Sigma 1:5000), beta-tubulin (ab6046, Abcam, 1:500), gamma-tubulin (mouse monoclonal, T3559 Sigma 1:1000), total β-catenin (Ab6302, Abcam, 1:400) and unphosphorylated β-catenin (ABC; clone 8E7, Millipore, 1:400).
Secondary antibodies used include HRP-conjugated goat-anti-mouse and goat-anti-rabbit (Jackson ImmunoResearch Laboratories, West Grove, PA, USA, 1:10 000).
Custom-made rabbit antibodies against the N-terminus and C-terminus of FLCN were generated and affinity purified by Eurogentec (Seraing, Belgium). The N-terminal antibody was raised against a peptide consisting of amino acids 29–41 (LPQGDGNEDSPGQCE) and the C-terminal antibody was raised against a peptide consisting of amino acids 522–536 (TKVDSRPKEDTQKLL) of human FLCN (NP_659434).
Cell lines and cell culture
Normal Human Dermal Fibroblasts (NHDF) were purchased from Lonza (Blackley, UK). MDCK cells type II were a gift from P. Zimmermann (Department of Human Genetics, University of Leuven, Belgium). HK-2 cells were provided by Prof. Dr M. van Engeland (Department of Pathology, Maastricht University Medical Center). UOK257 and UOK257-2 cell lines were kindly supplied by Dr L. Schmidt (NIH, USA). All cells were cultured in tissue culture grade plastics, in Dulbecco's modified Eagle medium (DMEM) containing 4.5 g/l d-glucose and l-glutamine (Lonza) supplemented with 10% FCS (Lonza) and 50 μg/ml gentamycin (Lonza) at 37°C in 5% CO2 (humidified atmosphere). The culture medium was changed every 2 to 3 days. At confluence, cells were split using trypsin-EDTA (Lonza). Primary ciliated HNE cells were obtained from consenting donors with no known history of BHD under local ethical approval from East of Scotland Research Ethics Service (EoSRES) REC1 (12/ES/0081). A 2 mm diameter cytology brush was inserted into each nostril and cellular material was recovered by abrasion of the nasal turbinate. This was washed with several changes of serum-free Medium 199 containing 1 mM Dithiothreitol (Sigma) and Primocin antibiotic/antimycotic (Invivogen, Toulouse, France). Cells were either processed immediately for immunofluorescence or maintained in culture for up to 1 week by placing into Bronchial Epithelial Growth Medium (Lonza), supplemented with SingleQuot Kit Supplements and appropriate growth factors. Human sperm was obtained from volunteer donors (healthy men with no known fertility problems randomly selected from the general public) who were recruited in accordance with the HFEA Code of Practice (version 8) under ethical approval (08/S1402/6) from the EoSRES REC1. Capacitated sperm cells were prepared using a swim-up assay (59) and were then immediately processed for immunofluorescence by spreading 10 µl sperm solution onto Polysine slides (VWR, Leicestershire, UK). Slides were air-dried prior to fixation in Methanol:Acetone (1vol:1vol) for 20 min at −20°C and staining was performed as described for 2D cultures.
Immunocytochemistry of 2D cultures
For fluorescent microscopy, cells were cultured on glass cover slips. Cilium formation was induced by culturing the cells for at least 4 days after reaching confluence or by serum starving the cells for at least 48 h in DMEM without FCS, depending upon the cell type. When ready for staining, cells were fixed using either ice-cold 100% methanol or 4% formaldehyde at room temperature and permeabilized using 0.2% Triton X-100, depending upon the primary antibody. Cells were incubated overnight at 4°C in primary antibody appropriately diluted in 3% BSA in TBS block buffer followed by 1 h incubation at room temperature in secondary antibody appropriately diluted in block buffer. DNA was stained with DAPI. Fluorescent samples were visualized using a Leica TCS SPE confocal laser scanning fluorescence microscope (Leica DMRBE, Mannheim, Germany). Images were further processed using ImageJ (NIH) software to determine the percentage of ciliated cells (n = 3, average 200 cells per group per experiment) and cilia length (n = 3, average 40 cilia per group per experiment).
Immunohistochemistry
Sections of 4 μm were processed using standard methodology; antigen retrieval was performed after blocking in H2O2, using 10 mm citrate buffer pH 6.0. For FLCN stain of BHD kidney sections, the custom-made C-terminal, N-terminal antibodies, or FLCN AP antibody was diluted in 3% BSA in PBS. To visualize we used the Dako REAL EnVision Detection System Peroxidase/DAB + Rabbit (Dako Heverlee, Belgium). The slides were counterstained with haematoxylin and embedded in Entellan.
Transfection and generation of stable cell lines
Transient transfections were performed using FuGENE HD Transfection Reagent (Roche) according to the manufacturer's instructions. To generate EGFP expressing stable cell lines, cells were transfected with appropriate plasmid DNA using FuGENE HD. Twenty-four hours post transfection cells were selected using 3 µg/ml puromycin (Invivogen). Clonal cell populations were picked using cloning cylinders and analysed for the expression of the protein of interest by western blot. To generate FLCN knockdown and non-target cell lines, HK-2 cells were transfected with appropriate plasmid DNA using FuGENE HD. Twenty-four hours after transfection selection of 1 µg/ml puromycin (Invivogen) was added. Clonal cell populations were picked using cloning cylinders and subsequently analysed by western blot for FLCN expression.
Growth curve
MDCK cells were seeded into six-well plates at a starting density of 105 cells/well. The cell number was determined at 48 h intervals using a haemocytometer. After counting cells were reseeded into a fresh six-well plate at an appropriate sub-confluent density and allowed to grow for another 48 h.
3D spheroid growth
MDCK cells were resuspended in a collagen matrix and seeded on a glass cover slip. The mixture was allowed to polymerize for 30 min at 37°C, and fresh medium was put on top to cover the matrix. After 11 days spheroids had formed. IMCD3 cells were cultured and reverse transfected with 50 nm On-TargetPlus siRNA murine-specific oligos (Dharmacon) using Lipofectamine RNAimax (Invitrogen) according to the manufacturer's recommendations. Twenty-four hours later, cells were collected and resuspended, mixed 1:1 with growth factor-depleted Matrigel (BD Bioscience, San Jose, CA, USA) and seeded in a glass-bottom Lab-tek II chamber (Nunc Thermo Scientific). The mixture was allowed to polymerize for 30 min at 37°C, and fresh medium was put on top to cover the matrix. After 3 days spheroids had formed.
Immunocytochemistry of 3D cultures
After spheroid formation, the medium was removed and the gels containing the spheroids were washed with warm PBS supplemented with calcium and magnesium and fixed in fresh 4% PFA for 30 min at room temperature. After washing with PBS, the cells were permeabilized for 20 min in gelatin dissolved in warm PBS (7 mg/ml) with 0.5% Triton X-100 added. Primary antibodies were diluted in the permeabilization gelatin buffer and the gels were incubated at 4°C overnight. After washing the spheroids three times for 30 min in permeabilization buffer, secondary antibodies were diluted in permeabilization buffer and the gels were incubated overnight at 4°C. The next day, the gels were washed three times in permeabilization buffer immersed in Fluoromount-G (Cell Lab, Beckman Coulter, Brea, CA, USA). Images were taken with a Zeiss LSM510 inverted confocal microscope. Using confocal laser scanning microscopy, several parameters of these spheroids can be determined, including the number of cells forming the median section of a spheroid. Additionally, the total spheroid size (basal membrane) and the luminal size (apical membrane) can be determined by measuring the distance of three lines spanning the maximal distance at a roughly 120° angle of one another. Also the number of cilia present parallel to the transverse section (over a 6–8 μm Z-section depending on spheroid size) can be counted.
Statistical analyses
The percentage of ciliated cells (n = 3, average 200 cells per group per experiment) was analysed using a two-tailed Pearson's Chi-square test for categorical data (significant P < 0.05). Cilium length (number of experiments = 3, average 40 cilia per group per experiment) and all spheroid parameters were analysed using non-parametric two-tailed Mann–Whitney tests (significant P < 0.05). For SiCON, SiFlcn and EGFP-FLCN K508R cells 30 spheroids were analysed, for EGFP cells 36 spheroids were analysed and for EGFP-FLCN WT 43 spheroids were analysed. Distances are averages of three measurements at a 120° angle of each other in μm.
RNA isolation and quantitative RT-PCR
RNA was isolated using the RNA isolation kit from Qiagen (Hilden, Germany), according to the manufacturer's recommendations. Probes Flcn mm00840973_m1 and Rpl19 Mm01606037_g1 for mRNA analysis were purchased from Applied Biosystems. Real-time PCR was performed in triplicate using the one-step RT-PCR kit and the 7500 system from Applied Biosystems. Normalized Ct values were used to calculate relative expression levels per condition and to determine the standard deviation (P = 0.05; n = 3). Real-time PCR for Axin2 and CyclinD1 was performed as previously described (60) using the primers listed in Supplementary Material, Table S4.
Sequencing
1 × 106 UOK257 and HK-2 cells were harvested using trypsin-EDTA. Genomic DNA was isolated using the Genomic DNA purification kit from Gentra Systems (Minneapolis, MN, USA) according to the manufacturer's recommendations. Subsequently, the BHD locus was sequenced using primers directed to intronic sequences adjacent to the FLCN exons.
Western blotting
Cells were harvested using typsin-EDTA and whole cell lysate (WCL) was obtained using NP-40 lysis buffer (150 mm NaCl, 1% NP-40, 250 mm Tris pH 7.3) supplemented with protease and phosphatase inhibitors (Roche). Protein concentration was determined using the Bradford protein assay (Sigma). An appropriate volume of Laemmli sample buffer was added to equal amounts of total protein followed by boiling for 5 min. Samples were subjected to SDS–PAGE electrophoresis before transfer to an Amersham Hybond™-PVDF membrane (GE Healthcare Life Sciences, Buckinghamshire, UK). After overnight incubation at 4°C in primary antibody appropriately diluted in 0.5% BSA in TBS block buffer, membranes were incubated with secondary antibody appropriately diluted in block buffer. Signal was detected using enhanced chemiluminescence (ECL) system (Thermo Scientific, Waltham, MA, USA).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by Dutch Cancer Society (grant KWF U2009-4352), Association for International Cancer Research (grant AICR 11-0687), Annadal Foundation and GROW (M.V.S. ,B.J.C, M.L., T.C. and M.V.), the European Community's Seventh Framework Programme FP7/2009 agreement no: 241955, SYSCILIA and the Netherlands Organization for Scientific grant NWO Vidi-917.66.354 (R.H.G.), Myrovlytis Trust (R.P.H.), the Wellcome Trust (grant 088032/z/08/z) (S.L.), Myrovlytis Trust (R.N.); Association of International Cancer Research Career Development Fellowship (06-914/915), AICR (grant 11-0687) and Myrovlytis Trust (A.T.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Prof. P.J. van Diest for pathological analysis of immunohistochemistry and F. Meeuwsen for technical support. The authors are grateful to Dr L. Schmidt (NIH) for kindly providing the UOK257 and UOK257-2 cell lines. We thank Prof. A. Pause for FLCN AP antibody, Dr T. Cash for FLCN antibody. MDCK type II cells were a kind gift from P. Zimmermann. HK-2 cells were a kind gift from Prof. M. van Engeland.
Conflict of Interest statement. None declared.
References
- 1.Hornstein O.P., Knickenberg M. Perifollicular fibromatosis cutis with polyps of the colon—a cutaneo-intestinal syndrome sui generis. Arch. Dermatol. Res. 1975;253:161–175. doi: 10.1007/BF00582068. doi:10.1007/BF00582068. [DOI] [PubMed] [Google Scholar]
- 2.Birt A.R., Hogg G.R., Dube W.J. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch. Dermatol. 1977;113:1674–1677. doi:10.1001/archderm.1977.01640120042005. [PubMed] [Google Scholar]
- 3.Houweling A.C., Gijezen L.M., Jonker M.A., van Doorn M.B., Oldenburg R.A., van Spaendonck-Zwarts K.Y., Leter E.M., van Os T.A., van Grieken N.C., Jaspars E.H., et al. Renal cancer and pneumothorax risk in Birt-Hogg-Dube syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br. J. Cancer. 2011;105:1912–1919. doi: 10.1038/bjc.2011.463. doi:10.1038/bjc.2011.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toro J.R., Glenn G., Duray P., Darling T., Weirich G., Zbar B., Linehan M., Turner M.L. Birt-Hogg-Dube syndrome: a novel marker of kidney neoplasia. Arch. Dermatol. 1999;135:1195–1202. doi: 10.1001/archderm.135.10.1195. doi:10.1001/archderm.135.10.1195. [DOI] [PubMed] [Google Scholar]
- 5.Menko F.H., van Steensel M.A., Giraud S., Friis-Hansen L., Richard S., Ungari S., Nordenskjold M., Hansen T.V., Solly J., Maher E.R. Birt-Hogg-Dube syndrome: diagnosis and management. Lancet Oncol. 2009;10:1199–1206. doi: 10.1016/S1470-2045(09)70188-3. doi:10.1016/S1470-2045(09)70188-3. [DOI] [PubMed] [Google Scholar]
- 6.Kluger N., Giraud S., Coupier I., Avril M.F., Dereure O., Guillot B., Richard S., Bessis D. Birt-Hogg-Dube syndrome: clinical and genetic studies of 10 French families. Br. J. Dermatol. 2010;162:527–537. doi: 10.1111/j.1365-2133.2009.09517.x. doi:10.1111/j.1365-2133.2009.09517.x. [DOI] [PubMed] [Google Scholar]
- 7.Nickerson M.L., Warren M.B., Toro J.R., Matrosova V., Glenn G., Turner M.L., Duray P., Merino M., Choyke P., Pavlovich C.P., et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2:157–164. doi: 10.1016/s1535-6108(02)00104-6. doi:10.1016/S1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 8.Hong S.B., Oh H., Valera V.A., Stull J., Ngo D.T., Baba M., Merino M.J., Linehan W.M., Schmidt L.S. Tumor suppressor FLCN inhibits tumorigenesis of a FLCN-null renal cancer cell line and regulates expression of key molecules in TGF-beta signaling. Mol. Cancer. 2010;9:160. doi: 10.1186/1476-4598-9-160. doi:10.1186/1476-4598-9-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hudon V., Sabourin S., Dydensborg A.B., Kottis V., Ghazi A., Paquet M., Crosby K., Pomerleau V., Uetani N., Pause A. Renal tumor suppressor function of the Birt-Hogg-Dube syndrome gene product folliculin. J. Med. Genet. 2010;47:182–189. doi: 10.1136/jmg.2009.072009. doi:10.1136/jmg.2009.072009. [DOI] [PubMed] [Google Scholar]
- 10.Baba M., Hong S.B., Sharma N., Warren M.B., Nickerson M.L., Iwamatsu A., Esposito D., Gillette W.K., Hopkins R.F., 3rd, Hartley J.L., et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl Acad. Sci. USA. 2006;103:15552–15557. doi: 10.1073/pnas.0603781103. doi:10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baba M., Furihata M., Hong S.B., Tessarollo L., Haines D.C., Southon E., Patel V., Igarashi P., Alvord W.G., Leighty R., et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Nat. Cancer Inst. 2008;100:140–154. doi: 10.1093/jnci/djm288. doi:10.1093/jnci/djm288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartman T.R., Nicolas E., Klein-Szanto A., Al-Saleem T., Cash T.P., Simon M.C., Henske E.P. The role of the Birt-Hogg-Dube protein in mTOR activation and renal tumorigenesis. Oncogene. 2009;28:1594–1604. doi: 10.1038/onc.2009.14. doi:10.1038/onc.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasumi Y., Baba M., Ajima R., Hasumi H., Valera V.A., Klein M.E., Haines D.C., Merino M.J., Hong S.B., Yamaguchi T.P., et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc. Natl Acad. Sci. USA. 2009;106:18722–18727. doi: 10.1073/pnas.0908853106. doi:10.1073/pnas.0908853106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Preston R.S., Philp A., Claessens T., Gijezen L., Dydensborg A.B., Dunlop E.A., Harper K.T., Brinkhuizen T., Menko F.H., Davies D.M., et al. Absence of the Birt-Hogg-Dube gene product is associated with increased hypoxia-inducible factor transcriptional activity and a loss of metabolic flexibility. Oncogene. 2011;30:1159–1173. doi: 10.1038/onc.2010.497. doi:10.1038/onc.2010.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong S.B., Oh H., Valera V.A., Baba M., Schmidt L.S., Linehan W.M. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS ONE. 2010;5:e15793. doi: 10.1371/journal.pone.0015793. doi:10.1371/journal.pone.0015793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cash T.P., Gruber J.J., Hartman T.R., Henske E.P., Simon M.C. Loss of the Birt-Hogg-Dube tumor suppressor results in apoptotic resistance due to aberrant TGFbeta-mediated transcription. Oncogene. 2011;30:2534–2546. doi: 10.1038/onc.2010.628. doi:10.1038/onc.2010.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaur K., Li J., Wang D., Dutta P., Yan S.J., Tsurumi A., Land H., Wu G., Li W.X. The Birt-Hogg-Dube tumor suppressor Folliculin negatively regulates ribosomal RNA synthesis. Hum. Mol. Genet. 2013;22:284–299. doi: 10.1093/hmg/dds428. doi:10.1093/hmg/dds428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esteban M.A., Harten S.K., Tran M.G., Maxwell P.H. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J. Am. Soc. Nephrol. JASN. 2006;17:1801–1806. doi: 10.1681/ASN.2006020181. doi:10.1681/ASN.2006020181. [DOI] [PubMed] [Google Scholar]
- 19.Hartman T.R., Liu D., Zilfou J.T., Robb V., Morrison T., Watnick T., Henske E.P. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 2009;18:151–163. doi: 10.1093/hmg/ddn325. doi:10.1093/hmg/ddn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishikawa H., Marshall W.F. Ciliogenesis: building the cell's antenna. Nat. Rev. Mol. Cell Biol. 2011;12:222–234. doi: 10.1038/nrm3085. doi:10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- 21.Roy S. The motile cilium in development and disease: emerging new insights. BioEssays: News Rev. Mol. Cell. Dev. Biol. 2009;31:694–699. doi: 10.1002/bies.200900031. doi:10.1002/bies.200900031. [DOI] [PubMed] [Google Scholar]
- 22.Patel V., Li L., Cobo-Stark P., Shao X., Somlo S., Lin F., Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum. Mol. Genet. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. doi:10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hildebrandt F., Benzing T., Katsanis N. Ciliopathies. N. Engl. J. Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. doi:10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischer E., Pontoglio M. Planar cell polarity and cilia. Semin. Cell Dev. Biol. 2009;20:998–1005. doi: 10.1016/j.semcdb.2009.09.016. doi:10.1016/j.semcdb.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 25.McNeill H. Planar cell polarity and the kidney. J. Am. Soc. Nephrol. JASN. 2009;20:2104–2111. doi: 10.1681/ASN.2008111173. doi:10.1681/ASN.2008111173. [DOI] [PubMed] [Google Scholar]
- 26.Montani M., Heinimann K., von Teichman A., Rudolph T., Perren A., Moch H. VHL-gene deletion in single renal tubular epithelial cells and renal tubular cysts: further evidence for a cyst-dependent progression pathway of clear cell renal carcinoma in von Hippel-Lindau disease. Am. J. Surg. Pathol. 2010;34:806–815. doi: 10.1097/PAS.0b013e3181ddf54d. doi:10.1097/PAS.0b013e3181ddf54d. [DOI] [PubMed] [Google Scholar]
- 27.Basten S.G., Willekers S., Vermaat J.S., Slaats G.G., Voest E.E., van Diest P.J., Giles R.H. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia. 2013;2:2. doi: 10.1186/2046-2530-2-2. doi:10.1186/2046-2530-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J., Dabiri S., Seeley E.S. Primary cilium depletion typifies cutaneous melanoma in situ and malignant melanoma. PLoS ONE. 2011;6:e27410. doi: 10.1371/journal.pone.0027410. doi:10.1371/journal.pone.0027410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Asselt S.J., de Vries E.G., van Dullemen H.M., Brouwers A.H., Walenkamp A.M., Giles R.H., Links T.P. Pancreatic cyst development: insights from von Hippel-Lindau disease. Cilia. 2013;2:3. doi: 10.1186/2046-2530-2-3. doi:10.1186/2046-2530-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bezginov A., Clark G.W., Charlebois R.L., Dar V.U., Tillier E.R. Coevolution reveals a network of human proteins originating with multicellularity. Mol. Biol. Evol. 2013;30:332–346. doi: 10.1093/molbev/mss218. doi:10.1093/molbev/mss218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Egeberg D.L., Lethan M., Manguso R., Schneider L., Awan A., Jorgensen T.S., Byskov A.G., Pedersen L.B., Christensen S.T. Primary cilia and aberrant cell signaling in epithelial ovarian cancer. Cilia. 2012;1:15. doi: 10.1186/2046-2530-1-15. doi:10.1186/2046-2530-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Menko F.H., Johannesma P.C., van Moorselaar R.J., Reinhard R., van Waesberghe J.H., Thunnissen E., Houweling A.C., Leter E.M., Waisfisz Q., van Doorn M.B., et al. A de novo FLCN mutation in a patient with spontaneous pneumothorax and renal cancer; a clinical and molecular evaluation. Familial Cancer. 2012 doi: 10.1007/s10689-012-9593-8. doi:10.1007/s10689-012-9593-8. [DOI] [PubMed] [Google Scholar]
- 33.Kouchi M., Okimoto K., Matsumoto I., Tanaka K., Yasuba M., Hino O. Natural history of the Nihon (Bhd gene mutant) rat, a novel model for human Birt-Hogg-Dube syndrome. Virchows Arch. 2006;448:463–471. doi: 10.1007/s00428-005-0142-9. doi:10.1007/s00428-005-0142-9. [DOI] [PubMed] [Google Scholar]
- 34.Sang L., Miller J.J., Corbit K.C., Giles R.H., Brauer M.J., Otto E.A., Baye L.M., Wen X., Scales S.J., Kwong M., et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. doi:10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takagi Y., Kobayashi T., Shiono M., Wang L., Piao X., Sun G., Zhang D., Abe M., Hagiwara Y., Takahashi K., et al. Interaction of folliculin (Birt-Hogg-Dube gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene. 2008;27:5339–5347. doi: 10.1038/onc.2008.261. doi:10.1038/onc.2008.261. [DOI] [PubMed] [Google Scholar]
- 36.Singh S.R., Zhen W., Zheng Z., Wang H., Oh S.W., Liu W., Zbar B., Schmidt L.S., Hou S.X. The Drosophila homolog of the human tumor suppressor gene BHD interacts with the JAK-STAT and Dpp signaling pathways in regulating male germline stem cell maintenance. Oncogene. 2006;25:5933–5941. doi: 10.1038/sj.onc.1209593. doi:10.1038/sj.onc.1209593. [DOI] [PubMed] [Google Scholar]
- 37.Fisch C., Dupuis-Williams P. Ultrastructure of cilia and flagella - back to the future! Biol. Cell. 2011;103:249–270. doi: 10.1042/BC20100139. doi:10.1042/BC20100139. [DOI] [PubMed] [Google Scholar]
- 38.Adams N.A., Awadein A., Toma H.S. The retinal ciliopathies. Ophthalmic Genet. 2007;28:113–125. doi: 10.1080/13816810701537424. doi:10.1080/13816810701537424. [DOI] [PubMed] [Google Scholar]
- 39.Delaval B., Bright A., Lawson N.D., Doxsey S. The cilia protein IFT88 is required for spindle orientation in mitosis. Nat. Cell Biol. 2011;13:461–468. doi: 10.1038/ncb2202. doi:10.1038/ncb2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei C., Bhattaram V.K., Igwe J.C., Fleming E., Tirnauer J.S. The LKB1 tumor suppressor controls spindle orientation and localization of activated AMPK in mitotic epithelial cells. PLoS ONE. 2012;7:e41118. doi: 10.1371/journal.pone.0041118. doi:10.1371/journal.pone.0041118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thoma C.R., Matov A., Gutbrodt K.L., Hoerner C.R., Smole Z., Krek W., Danuser G. Quantitative image analysis identifies pVHL as a key regulator of microtubule dynamic instability. J. Cell Biol. 2010;190:991–1003. doi: 10.1083/jcb.201006059. doi:10.1083/jcb.201006059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryan M.J., Johnson G., Kirk J., Fuerstenberg S.M., Zager R.A., Torok-Storb B. HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 1994;45:48–57. doi: 10.1038/ki.1994.6. doi:10.1038/ki.1994.6. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y., Padilla-Nash H.M., Vira M.A., Abu-Asab M.S., Val D., Worrell R., Tsokos M., Merino M.J., Pavlovich C.P., Ried T., et al. The UOK 257 cell line: a novel model for studies of the human Birt-Hogg-Dube gene pathway. Cancer Genet. Cytogenet. 2008;180:100–109. doi: 10.1016/j.cancergencyto.2007.10.010. doi:10.1016/j.cancergencyto.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lancaster M.A., Schroth J., Gleeson J.G. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat. Cell Biol. 2011;13:700–707. doi: 10.1038/ncb2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lancaster M.A., Gleeson J.G. Cystic kidney disease: the role of Wnt signaling. Trends Mol. Med. 2010;16:349–360. doi: 10.1016/j.molmed.2010.05.004. doi:10.1016/j.molmed.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plotnikova O.V., Golemis E.A., Pugacheva E.N. Cell cycle-dependent ciliogenesis and cancer. Cancer Res. 2008;68:2058–2061. doi: 10.1158/0008-5472.CAN-07-5838. doi:10.1158/0008-5472.CAN-07-5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacDonald B.T., Tamai K., He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. doi:10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wallingford J.B., Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011;25:201–213. doi: 10.1101/gad.2008011. doi:10.1101/gad.2008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rookmaaker M.B., van Gerven H.A.J.M., Goldschmeding R., Boer W.H. Solid renal tumors of collecting duct origin in patients on chronic lithium therapy. Clin. Kidney J. 2012;5:412–415. doi: 10.1093/ckj/sfs091. doi:10.1093/ckj/sfs091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hedgepeth C.M., Conrad L.J., Zhang J., Huang H.C., Lee V.M., Klein P.S. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev. Biol. 1997;185:82–91. doi: 10.1006/dbio.1997.8552. doi:10.1006/dbio.1997.8552. [DOI] [PubMed] [Google Scholar]
- 51.Nookala R.K., Langemeyer L., Pacitto A., Ochoa-Montano B., Donaldson J.C., Blaszczyk B.K., Chirgadze D.Y., Barr F.A., Bazan J.F., Blundell T.L. Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. Open Biol. 2012;2:120071. doi: 10.1098/rsob.120071. doi:10.1098/rsob.120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chua C.E., Lim Y.S., Tang B.L. Rab35—a vesicular traffic-regulating small GTPase with actin modulating roles. FEBS Lett. 2010;584:1–6. doi: 10.1016/j.febslet.2009.11.051. doi:10.1016/j.febslet.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 53.Smith K.R., Kieserman E.K., Wang P.I., Basten S.G., Giles R.H., Marcotte E.M., Wallingford J.B. A role for central spindle proteins in cilia structure and function. Cytoskeleton. 2011;68:112–124. doi: 10.1002/cm.20498. doi:10.1002/cm.20498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Toro J.R., Wei M.H., Glenn G.M., Weinreich M., Toure O., Vocke C., Turner M., Choyke P., Merino M.J., Pinto P.A., et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J. Med. Genet. 2008;45:321–331. doi: 10.1136/jmg.2007.054304. doi:10.1136/jmg.2007.054304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woodward E.R., Ricketts C., Killick P., Gad S., Morris M.R., Kavalier F., Hodgson S.V., Giraud S., Bressac-de Paillerets B., Chapman C., et al. Familial non-VHL clear cell (conventional) renal cell carcinoma: clinical features, segregation analysis, and mutation analysis of FLCN. Clin. Cancer Res. 2008;14:5925–5930. doi: 10.1158/1078-0432.CCR-08-0608. doi:10.1158/1078-0432.CCR-08-0608. [DOI] [PubMed] [Google Scholar]
- 56.Vocke C.D., Yang Y., Pavlovich C.P., Schmidt L.S., Nickerson M.L., Torres-Cabala C.A., Merino M.J., Walther M.M., Zbar B., Linehan W.M. High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dube-associated renal tumors. J. Natl Cancer Inst. 2005;97:931–935. doi: 10.1093/jnci/dji154. doi:10.1093/jnci/dji154. [DOI] [PubMed] [Google Scholar]
- 57.Edelman E.A., Girirajan S., Finucane B., Patel P.I., Lupski J.R., Smith A.C., Elsea S.H. Gender, genotype, and phenotype differences in Smith-Magenis syndrome: a meta-analysis of 105 cases. Clin. Genet. 2007;71:540–550. doi: 10.1111/j.1399-0004.2007.00815.x. doi:10.1111/j.1399-0004.2007.00815.x. [DOI] [PubMed] [Google Scholar]
- 58.Albuquerque C., Breukel C., van der Luijt R., Fidalgo P., Lage P., Slors F.J.M., Leitão C.N., Fodde R., Smits R. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the β-catenin signaling cascade. Hum. Mol. Genet. 2002;11:1549–1560. doi: 10.1093/hmg/11.13.1549. doi:10.1093/hmg/11.13.1549. [DOI] [PubMed] [Google Scholar]
- 59.Lefievre L., Nash K., Mansell S., Costello S., Punt E., Correia J., Morris J., Kirkman-Brown J., Wilson S.M., Barratt C.L., et al. 2-APB-potentiated channels amplify CatSper-induced Ca(2+) signals in human sperm. Biochem. J. 2012;448:189–200. doi: 10.1042/BJ20120339. doi:10.1042/BJ20120339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu J., Dong A., Fernandez-Ruiz V., Shan J., Kawa M., Martinez-Anso E., Prieto J., Qian C. Blockade of Wnt signaling inhibits angiogenesis and tumor growth in hepatocellular carcinoma. Cancer Res. 2009;69:6951–6959. doi: 10.1158/0008-5472.CAN-09-0541. doi:10.1158/0008-5472.CAN-09-0541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.