

Abstract
Promoting brown adipose tissue (BAT) formation and function may reduce obesity. Recent data link retinoids to energy balance, but a specific role for retinoid metabolism in white versus brown fat is unknown. Retinaldehyde dehydrogenases (Aldhs), also known as aldehyde dehydrogenases, are rate-limiting enzymes that convert retinaldehyde (Rald) to retinoic acid. Here we show that Aldh1a1 is expressed predominately in white adipose tissue (WAT), including visceral depots in mice and humans. Deficiency of the Aldh1a1 gene induced a BAT-like transcriptional program in WAT that drove uncoupled respiration and adaptive thermogenesis. WAT-selective Aldh1a1 knockdown conferred this BAT program in obese mice, limiting weight gain and improving glucose homeostasis. Rald induced uncoupling protein-1 (Ucp1) mRNA and protein levels in white adipocytes by selectively activating the retinoic acid receptor (RAR), recruiting the coactivator PGC-1α and inducing Ucp1 promoter activity. These data establish Aldh1a1 and its substrate Rald as previously unrecognized determinants of adipocyte plasticity and adaptive thermogenesis, which may have potential therapeutic implications.
Obesity is closely associated with many disorders, including atherosclerosis and type 2 diabetes1,2. Although excess caloric intake and decreased energy expenditure promote overall weight gain, visceral adiposity is particularly associated with cardiometabolic risk3. As such, unique factors may control the development and function of specific fat depots. Whereas WAT stores energy in the form of triglycerides, BAT oxidizes fatty acids and dissipates energy through uncoupled respiration and heat production4–6. WAT is the main adipose depot in humans; however, recent work has established both the presence of BAT in humans and its association with leanness6,7. These distinct aspects of adipocyte biology require further understanding and may represent a therapeutic strategy to combat obesity and its complications. Although the presence of brown adipocytes in WAT and factors determining white versus brown adipogenesis have received considerable attention8–12, the exact origin of white and brown adipocytes and the potential for adipocyte plasticity in terms of white versus brown characteristics remains under debate13,14.
Retinoids–vitamin A metabolites with diverse, essential biological functions–have recently been linked to the control of adipogenesis and energy homeostasis, with effects on obesity, diabetes and cardiovascular disease15–19. Retinoids exert these actions largely by activating RAR and the retinoid X receptor (RXR), nuclear hormone receptors that regulate gene expression20,21. Retinoid formation relies on a network of enzymes in which dietary vitamin A, or retinol, is first oxidized to Rald by alcohol and retinol dehydrogenases. Subsequently, Aldhs irreversibly convert Rald to retinoic acid—the rate-limiting step of retinoic acid formation20–22.
We recently identified Rald as an active signaling metabolite in WAT and a modulator of adipogenesis in 3T3-L1 cells16. Deficiency in Aldh1a1 (also known as Raldh1), the main postnatal Aldh isoform, results in higher Rald concentrations16,23 and protects mice against diet-induced obesity and diabetes by increasing energy expenditure16. However, the mechanisms for this hypermetabolic phenotype, its dependency on high-fat feeding and its relevance to human adiposity has remained unclear.
Here we show that Aldh1a1 is a key determinant of WAT plasticity and the regulation of white versus brown adipocyte characteristics. In both mice and humans, Aldh1a1 was expressed primarily in visceral WAT, and its expression level was highly associated with obesity. In mice fed standard chow, Aldh1a1 deficiency strongly induced both expression of classic BAT markers and uncoupled respiration in WAT, increasing adaptive thermogenesis in vivo. Rald potently regulated transcription of Ucp1 in white adipocytes through effects on RAR and not RXR, including peroxisome proliferator–activated receptor-γ coactivator (PGC-1α) recruitment; RAR antagonist treatment reversed the thermogenic phenotype in Aldh1a1 deficiency. Notably, treatment of high-fat diet–fed mice with antisense oligonucleotides to Aldh1a1 repressed Aldh1a1 expression selectively in visceral WAT, induced a thermogenic program in WAT and conferred protection against cold exposure and obesity. These findings establish Aldh1a1 and its substrate Rald as previously unrecognized regulators of white adipose plasticity and adaptive thermogenesis and identify Aldh1a1 in visceral fat as a new potential target for treating obesity.
RESULTS
Aldh1a1 is predominantly expressed in visceral fat
To consider the possible role of Aldh1a1 in specific adipose stores, we analyzed its mRNA expression and protein levels in different fat depots of standard chow–fed C57BL/6J mice and metabolically healthy human subjects of normal weight. In C57BL/6J mice, Aldh1a1 expression was highest in perigonadal white adipose tissue (GWAT), a mouse visceral fat depot, significantly lower in inguinal subcutaneous white adipose tissue (SWAT) and barely detectable in interscapular BAT; Aldh1a1 protein levels followed a similar pattern (Fig. 1a). In humans, both ALDH1A1 mRNA and protein were robustly present in visceral adipose tissue (viscAT), with significantly lower levels in abdominal subcutaneous adipose tissue (Fig. 1b).
Figure 1.
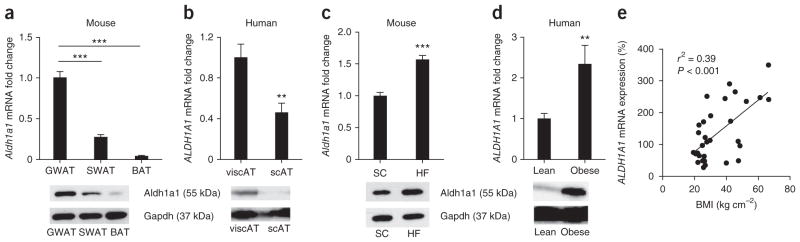
Aldh1a1 is present primarily in visceral fat, and its expression correlates with obesity. (a) Aldh1a1 mRNA and protein expression in different fat depots of female C57BL/6J mice (n = 10). (b) ALDH1A1 mRNA and protein expression in visceral adipose tissue (viscAT) and subcutaneous adipose tissue (scAT) from nonobese male and female subjects (n = 20). (c) Aldh1a1 mRNA and protein expression in GWAT from standard chow–fed (SC) lean and high-fat diet–fed (HF) obese C57BL/6J mice (n = 8 per group). (d) ALDH1A1 mRNA and protein expression in viscAT from nonobese (body-mass index (BMI) = 25.2 ± 0.15 kg m−2) and morbidly obese (BMI = 53.0 ± 0.55 kg m−2) subjects; n = 20 per group. Representative western blots are shown. (e) Linear regression analysis between BMI and ALDH1A1 mRNA expression in human viscAT (n = 40). **P < 0.01, ***P < 0.001. Data are given as mean ± s.e.m.
Given that visceral adiposity is strongly implicated in the pathogenesis of obesity-related metabolic disorders, we investigated whether adipose Aldh1a1 expression varied with obesity. Aldh1a1 expression was significantly higher in GWAT and viscAT of C57BL/6J mice fed a high-fat diet and in morbidly obese human subjects, respectively, compared to lean controls (Fig. 1c, d). Moreover, regression analysis revealed a positive association between ALDH1A1 mRNA expression in viscAT and body mass index (r2 = 0.39, P < 0.001) in 40 human subjects (Fig. 1e). These results implicate Aldh1a1 in obesity and, specifically, in visceral adiposity.
Aldh1a1 deficiency induces a BAT gene program in WAT
Given this association of Aldh1a1 with obesity (Fig. 1) and the enhanced energy expenditure in Aldh1a1-deficient (Aldh1a1−/−) mice fed a high-fat diet16, we studied mitochondrial uncoupling in WAT and BAT as a mechanism for energy dissipation in Aldh1a1 deficiency. Ucp1 mRNA expression and protein content were significantly induced in GWAT of standard chow–fed Aldh1a1−/− versus C57BL/6J wild-type (WT) control mice; however, this induction was significantly lessened in SWAT, and Ucp1 mRNA and protein remained unchanged in BAT (Fig. 2a, b). On immunohistochemistry images, lipid droplet size and adipocyte morphology appeared unaltered in Aldh1a1-deficient WAT, but Ucp1 was strongly induced as compared to WT adipose tissue (Fig. 2b).
Figure 2.
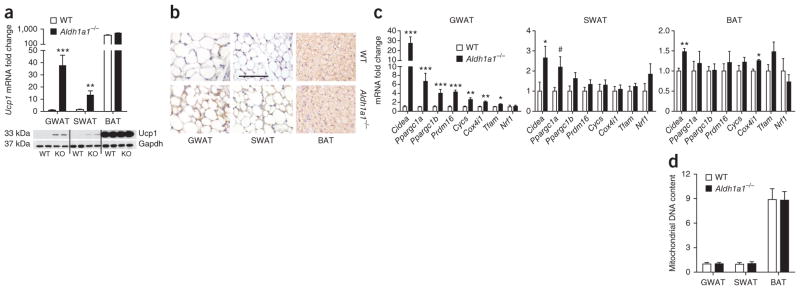
Aldh1a1 deficiency is characterized by increased transcription of brown fat markers in white fat. (a) Ucp1 mRNA expression and protein analysis in GWAT, SWAT and BAT of standard chow-fed WT and Aldh1a1−/− mice. (b) Immunohistochemical analysis of Ucp1 (brown staining) in GWAT, SWAT and BAT of WT and Aldh1a1−/− mice. Scale bar, 100 μm. (c) mRNA expression of classic brown fat markers in GWAT, SWAT and BAT of WT versus Aldh1a1−/− mice. (d) Mitochondrial DNA content (genomic ND1 expression) determined in GWAT, SWAT and BAT of WT versus Aldh1a1−/− mice. n = 6–8 per group. #P = 0.07, *P < 0.05, **P < 0.01, ***P < 0.001. Data are given as mean ± s.e.m.
To further characterize fat in the presence or absence of Aldh1a1, we measured mRNA expression of known BAT markers including cell-death activator (Cidea), PGC-1α and PGC-1β (encoded by Ppargc1a and Ppargc1b, respectively), PR domain–containing 16 (Prdm16), cytochrome c (Cycs), cytochrome c oxidase subunit 4i1 (Cox4i1), transcription factor A (Tfam) and nuclear respiratory factor 1 (Nrf1) in GWAT, SWAT and BAT from chow-fed WT and Aldh1a1−/− mice. Expression of all BAT marker genes was significantly increased in Aldh1a1-deficient GWAT, with Nrf1 being the only exception (Fig. 2c). In contrast, expression of this same gene panel was either unchanged or only modestly increased in SWAT and BAT of Aldh1a1−/− versus WT mice (Fig. 2c). We next tested whether altered mitochondrial biogenesis might explain this increase in BAT marker expression in white fat. In GWAT, SWAT and BAT, mitochondrial DNA content was unchanged between genotypes (Fig. 2d). Electron microscopy of GWAT revealed no differences in mitochondrial density or ultrastructure (Supplementary Fig. 1a). These data indicate that Aldh1a1 deficiency increases BAT marker expression in visceral WAT without altering mitochondrial biogenesis.
Aldh1a1 deficiency activates a thermogenic program in WAT
To test whether increased Ucp1 expression in GWAT from Aldh1a1−/− mice altered GWAT function, we assayed mitochondrial activity using citrate synthase activity, the rate-limiting step of the tricarboxylic acid cycle24,25. Citrate synthase activity was significantly increased in GWAT but not BAT from Aldh1a1−/− versus WT mice (Fig. 3a). Increased mitochondrial uncoupling drives oxidative phosphorylation, enhancing cellular respiration4. Oxygen consumption rates were significantly higher in Aldh1a1-deficient GWAT (1.7-fold) but not BAT (Fig. 3b) as compared to those in WT mice.
Figure 3.

Aldh1a1 deficiency activates a thermogenic program in white fat. (a, b) Citrate synthase activity (a) and oxygen consumption rate (b) in GWAT and BAT of WT versus Aldh1a1−/− mice as measured by enzymatic assays and the Seahorse Extracellular Flux Analyzer, respectively. (c) Core body temperature of WT versus Aldh1a1−/− mice. Results represent average body temperature over a 48-h period at 23 °C and 4 °C, respectively. (d, e) Representative Ucp1 western blot (d) and H&E and immunohistochemical Ucp1 staining (e) in GWAT, SWAT and BAT of WT versus Aldh1a1−/− (KO) mice after 48 h of cold stimulation. n = 6–8 per group, *P < 0.05. Scale bar, 100 μm. Data are given as mean ± s.e.m.
As uncoupled respiration dissipates energy through heat production, we next measured core body temperature in WT versus Aldh1a1−/− mice at room temperature (23 °C) and during 48 h of cold exposure (4 °C). At 23 °C, body temperature did not differ between genotypes. At 4 °C, body temperatures of WT mice dropped significantly; in contrast, mice lacking Aldh1a1 were completely protected against cold exposure (Fig. 3c). Consistent with these findings, cold exposure markedly increased Ucp1 expression in Aldh1a1-deficient GWAT and, to a minor extent, in SWAT, with no evidence for genotypic differences in BAT (Fig. 3d, e). Cold exposure induced the emergence of BAT-like Ucp1-positive multilocular adipocytes in Aldh1a1-deficient WAT (Fig. 3e).
We next asked whether altered adrenergic activation of BAT could have contributed to adaptive thermogenesis seen in Aldh1a1 deficiency. Expression of β-adrenergic receptors (Adrb1, Adrb2 and Adrb3) and the catecholamine-synthesizing enzyme tyrosine hydroxylase (Th) was unchanged in WT and Aldh1a1−/− BAT (Supplementary Fig. 1b, c). Likewise, baseline and isoproterenol-induced oxygen consumption in brown adipocytes and expression of BAT genes did not differ between genotypes (Supplementary Fig. 1d, e). These results argue for a role of Aldh1a1 in regulating uncoupled respiration and adaptive thermogenesis through effects in WAT but not BAT.
Rald promotes Ucp1 transcription in white adipocytes
To investigate mechanisms for increased uncoupling in GWAT, we used chemical and targeted genetic approaches in mouse and human adipocyte models. The C3H10T1/2 (10T1/2) cell line is an established model of adipogenesis26. In undifferentiated 10T1/2 cells, Aldh1a1 is not present but is robustly induced during adipogenesis (Supplementary Fig. 2a). Stimulation of 10T1/2 cells with the known Aldh inhibitor diethyl aminobenzaldehyde (DEAB, 1 μM)27 during adipogenic differentiation significantly induced Ucp1 gene and protein expression as compared to that in vehicle-treated cells (Fig. 4a). Similarly, stable repression of Aldh1a1 mRNA in 10T1/2 cells by shRNA-expressing lentivirus (shAldh1a1, 80% knockdown without a compensatory increase in Aldh1a2 or Aldh1a3; Supplementary Fig. 2b) induced Ucp1 mRNA and protein fivefold as compared to lentiviral-treated control cells (shCtrl, Fig. 4b). Given that Aldh1a1 deficiency increases endogenous Rald concentrations16,23, we tested Rald as a Ucp1 transcriptional regulator. Stimulation of 10T1/2 cells with all-trans-retinal (Rald, 1 μM) during adipogenic differentiation induced expression of Ucp1 (100-fold) and other BAT markers including Ppargc1b, Prdm16 and Cidea, whereas expression of the adipogenic genes peroxisome proliferator-activated receptor-γ (Pparg), fatty acid–binding protein 4 (Fabp4) and adiponectin (Adipoq) and lipid accumulation remained unaltered (Fig. 4c and Supplementary Fig. 2c, d). Similarly, Rald stimulation significantly increased Ucp1 expression in differentiating human stromal-vascular cells from subcutaneous fat biopsies (Fig. 4d).
Figure 4.
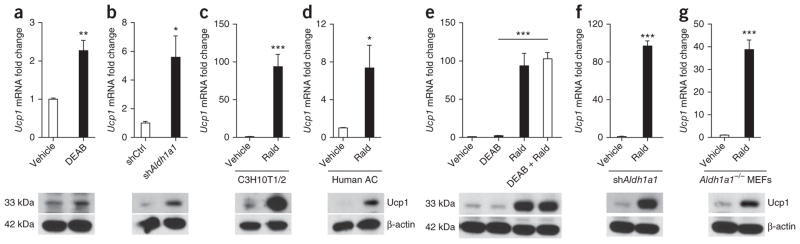
Rald induces Ucp1 expression in white adipocytes. (a) Ucp1 mRNA expression and protein levels in 10T1/2 cells after adipocyte differentiation in the presence or absence of the Aldh inhibitor DEAB (1 μM). (b) Ucp1 mRNA and protein analysis in differentiated 10T1/2 cells stably transfected with scrambled (shCtrl) or Aldh1a1-targeting (shAldh1a1) shRNA. (c, d) Ucp1 mRNA and protein analysis in 10T1/2 cells (c) and human stromal-vascular cells (d) from subcutaneous fat biopsies, differentiated into adipocytes in the presence or absence of Rald (1 μM). (e) Ucp1 gene expression and protein content in differentiated 10T1/2 cells stimulated with DEAB (1 μM), Rald (1 μM) or both. (f, g) shAldh1a1-transfected 10T1/2 cells (f) and MEFs (g) isolated from Aldh1a1-deficient embryos were stimulated with Rald (1 μM) during adipogenic differentiation followed by Ucp1 mRNA and protein analysis. n = 5 or 6 per condition, *P < 0.05, **P < 0.01, ***P < 0.001. Data are given as mean ± s.e.m.
To test whether Rald conversion to retinoic acid accounted for the increased Ucp1 expression seen, we studied Rald effects in three distinct in vitro models in which Aldh1a1 expression or function was repressed or absent. Co-stimulation with Rald and the Aldh inhibitor DEAB increased Ucp1 expression to the same extent as did Rald alone in differentiating 10T1/2 cells (Fig. 4e). Rald stimulation in shAldh1a1-treated 10T1/2 cells increased Ucp1 mRNA and protein expression 100-fold (Fig. 4f), an effect similar to Rald stimulation of untransfected 10T1/2 cells (Fig. 4c). Finally, Rald stimulation (24 h) in Aldh1a1−/− mouse embryonic fibroblast (MEF)-derived adipocytes increased Ucp1 gene expression 40-fold as compared to that in vehicle-treated cells (Fig. 4g). Together, these findings support Rald induction of Ucp1 expression in white adipocytes independent of its conversion to retinoic acid.
Rald regulates Ucp1 expression through RAR and PGC-1α
As retinoids regulate gene expression through retinoid receptor modulation, we next investigated retinoid receptor involvement in Rald effects on Ucp1 expression. We stimulated differentiating 10T1/2 cells with Rald in the presence or absence of known antagonists to either RAR (AGN193109) or RXR (HX531)28,29. The RAR antagonist AGN193109 completely inhibited Rald-mediated expression of Ucp1 and other BAT marker genes (Ppargc1b, Prdm16 and Cidea), whereas HX531 did not alter Ucp1 expression (Fig. 5a and Supplementary Fig. 3a, b). Notably, HX531 did significantly inhibit 9-cis-retinoic acid (9cisRA)-induced expression of the retinoic acid target gene Cyp26a1, verifying HX5131 as a functional RXR antagonist in this experimental model (Supplementary Fig. 3c). To further investigate RAR dependency through an alternative approach, we repeated 10T1/2 experiments in the presence of a validated siRNA to Rara (RARα), the main isoform expressed in these cells (data not shown) or control siRNA (siCtrl). Compared to siCtrl treatment, repression of Rara mRNA in 10T1/2 cells (60%, Supplementary Fig. 3d) significantly blunted Rald-mediated induction of Ucp1 and additional BAT markers (Ppargc1b, Prdm16 and Cidea) (Fig. 5b and Supplementary Fig. 3e, f). Rxra (RXRα) siRNA inhibited adipogenesis, resulting in extremely low Ucp1 expression in all treatment groups (data not shown). Similar experiments in shAldh1a1-transfected 10T1/2 cells also showed that increased Ucp1 expression was dependent on the presence of RAR but not RXR (Fig. 5c, d and Supplementary Fig. 3g).
Figure 5.

Rald-mediated Ucp1 expression is RAR-dependent and involves PGC-1α recruitment. (a) Ucp1 mRNA expression in 10T1/2 adipocytes stimulated with either the RAR antagonist AGN193109 (AGN), the RXR antagonist HX531, Rald (1 μM) or the combination of Rald + AGN or Rald + HX531 (each 1 μM). (b) Ucp1 mRNA quantification in 10T1/2 adipocytes transiently transfected with scrambled (siCtrl) or Rara siRNA (siRARα) and stimulated with Rald (1 μM). (c) Ucp1 mRNA quantification in shControl and shAldh1a1-transfected 10T1/2 adipocytes stimulated with AGN193109 or HX531 (both 1 μM). (d) Ucp1 mRNA analysis in Aldh1a1 knockdown 10T1/2 cells (shAldh1a1) transfected with siCtrl or siRARα. (e) Core body temperature in Aldh1a1−/− mice injected with the RAR antagonist AGN193109 or vehicle for 2 weeks. Results represent average body temperature over a 48-h period at 23 °C and 4 °C, respectively. (f) Western blot for Ucp1 in vehicle- and AGN-treated Aldh1a1−/− mice after cold exposure. (g) Ligand-dependent recruitment of PGC-1α to either the RARα-LBD or RXRα-LBD, respectively, as determined by cell-free TR-FRET assays. Dose-response curves for Rald, ATRA and 9-cis–retinoic acid (9-cis-RA) are shown. (h) Undifferentiated 10T1/2 cells, which lack Aldhs, were transfected with an RARE-luciferase construct (RARE-Luc) followed by stimulation (24 h) with increasing concentrations of Rald or ATRA and luciferase activity assays. (i) Undifferentiated 10T1/2 cells were transfected with a mouse 3.1-kb Ucp1 promoter luciferase construct (Ucp1-Luc) and subsequently stimulated (24 h) with cAMP (250 μM), Rald, ATRA and retinol (each 1 μM). Normalized luciferase activities are shown as fold change. (j) RARα and PGC-1α recruitment to the Ucp1 promoter region in Rald-stimulated (1 μM) 10T1/2 adipocytes analyzed by ChIP. Fold enrichment and a representative DNA gel pictures are given. n = 5 or 6 per condition, *P < 0.05,**P < 0.01, ***P < 0.001; Veh, vehicle. Data are given as mean ± s.e.m.
To test in vivo whether the thermogenic phenotype observed in Aldh1a1 deficiency is mediated through RAR, we injected Aldh1a1-deficient mice with the RAR antagonist AGN193109 (0.5 mg per kg of body weight per day, intraperitoneally (i.p.)) or vehicle for 2 weeks before cold exposure studies and Ucp1 analysis. Core body temperature did not differ between groups at 23 °C ambient temperature. However, during cold exposure (4 °C), treatment of mice with RAR antagonist resulted in significantly lower body temperatures as compared to controls (Fig. 5e). Consistent with these temperature changes, AGN193109 treatment markedly reduced Ucp1 expression in WAT but not in BAT (Fig. 5f), suggesting that the thermogenic program in Aldh1a1 deficiency is RAR dependent.
Given these results, we next examined Rald interaction with RAR as a mechanism for increased Ucp1 expression. PGC-1α is a transcriptional coactivator that induces Ucp1 expression30. Using cell-free time-resolved fluorescence resonance energy transfer (TR-FRET) assays, we tested Rald-mediated recruitment of PGC-1α to either the RARα or RXRα ligand-binding domain (LBD). As expected, in response to their known natural ligands, all-trans–retinoic acid (ATRA) and 9cisRA potently recruited PGC-1α to RARα and RXRα, respectively. However, Rald alone also effectively recruited PGC-1α to RARα (half-maximal effective concentration (EC50) 82 nM) but had virtually no effect on PGC-1α recruitment to RXRα in these cell-free assays, which lack the ability to convert Rald to retinoic acid (Fig. 5g). Stimulation with either ATRA or Rald significantly increased activity of a canonical retinoic acid response element (RARE) luciferase reporter in a concentration-dependent manner (Fig. 5h).
As the Ucp1 promoter contains several RARE consensus sites31, we examined whether Rald can directly activate a RARE-containing Ucp1 promoter transfected into undifferentiated 10T1/2 cells, which lack retinoid-converting enzymes. Stimulation with cAMP (250 μM), a known Ucp1 transcriptional activator31, significantly increased Ucp1 reporter activity (Fig. 5i). Notably, Rald and ATRA, but not their precursor retinol, increased luciferase activity in a concentration-dependent manner (Fig. 5i and Supplementary Fig. 3h). Next, we used chromatin immunoprecipitation (ChIP) to study Rald-dependent transcription factor recruitment to the Ucp1 promoter region in 10T1/2 cells. A robust RARα signal was present at the Ucp1 promoter under basal conditions that did not increase further after Rald stimulation (Fig. 5j). The TR-FRET data above suggested that Rald may recruit PGC-1α to RARα already present on the Ucp1 promoter. Indeed, similar ChIP studies showed that Rald stimulation enriched PGC-1α occupancy at the Ucp1 promoter region five-fold (Fig. 5j). Taken together, these data suggest that Rald recruits PGC-1α to RARα at the Ucp1 promoter, thus inducing Ucp1 transcription.
Aldh1a1 antisense treatment induces a thermogenic prorgam
To test whether acute, tissue-selective loss of Aldh1a1 in vivo alters Ucp1 expression, thermogenesis and body weight, we injected a validated Aldh1a1 antisense oligonucleotide (ASO) or a control ASO twice a week for 6 weeks (35 mg per kg of body weight per dose, i.p.) into C57BL/6J mice fed standard chow. The Aldh1a1 ASO treatment significantly decreased Aldh1a1 mRNA and protein levels selectively in liver and GWAT; Aldh1a1 expression remained unchanged in SWAT, BAT, spleen, intestine and skeletal muscle as compared to treatment with control ASO (Fig. 6a, b and Supplementary Fig. 4a). Aldh1a1 ASO treatment significantly increased Ucp1 expression in GWAT but not SWAT or BAT (Fig. 6c, d). To test the functional impact of these effects, we measured core body temperature under ambient and cold-induced conditions. Core body temperature did not differ between mice treated with Aldh1a1 ASO or control ASO at ambient temperature (23 °C). Exposure to 4 °C (48 h) decreased body temperature in mice treated with control ASO; in contrast, Aldh1a1 ASO–treated mice were protected significantly against cold exposure (Fig. 6e).
Figure 6.

ASO-mediated Aldh1a1 knockdown in GWAT promotes white-fat thermogenesis and limits diet-induced obesity. C57BL/6J mice on standard chow diet were injected with Aldh1a1 ASO and control ASO (Ctrl ASO) for 6 weeks (two doses of 35 mg per kg of body weight per week, i.p., n = 8 per group). (a, b) Aldh1a1 mRNA (a) and protein (b) expression in the indicated tissues of mice treated with Aldh1a1 ASO or control ASO (Ctrl, Ctrl ASO; ASO, Aldh1a1 ASO). (c, d) Ucp1 mRNA (c) and protein (d) levels in GWAT, SWAT and BAT of mice treated with Aldh1a1 ASO versus control ASO. (e) Core body temperature of mice treated with Aldh1a1 ASO versus control ASO at 23 °C and at 4 °C over 48 h (n = 6 per group). (f–i) A cohort of C57BL/6J mice (n = 16) was fed a high-fat diet (HFD) for 8 weeks before initiation of Aldh1a1 ASO or control ASO treatment (n = 8 per group), continued HFD, and measurement of body weight gain (f, g), fat depot mass (h) and insulin tolerance testing (at 17 weeks) (i). *P < 0.05, **P < 0.01, ***P < 0.001. Data are given as mean ± s.e.m. NS, not significant.
To test whether GWAT-selective Aldh1a1 repression could modulate established obesity, we fed C57BL/6J mice a high-fat diet for 8 weeks before treating them with Aldh1a1 ASO and continuing the high-fat diet for an additional 9 weeks. Aldh1a1 ASO treatment significantly limited weight gain in obese mice and decreased GWAT mass (Fig. 6f–h), resulting in improved insulin and glucose tolerance (Fig. 6i and Supplementary Fig. 4b), all as compared to a treatment with control ASO. Thus, selective Aldh1a1 repression in visceral fat confers induction of a thermogenic program that inhibits body weight gain and improves glucose homeostasis in obesity.
DISCUSSION
Understanding determinants of energy storage and utilization is crucial for addressing the epidemic of obesity and its complications. Energy dissipation through uncoupled respiration and increased thermogenesis are key components of energy balance. Promoting BAT or BAT-like characteristics may represent a therapeutic strategy for treating excess adiposity32. Despite this, activation of BAT differentiation and function remains poorly understood. Similarly, mechanisms that manipulate WAT to acquire BAT-like characteristics remain an alternative but even less well-characterized approach to enhancing energy expenditure and decreasing obesity33.
Retinoids, and the enzymes that control retinoid formation, represent a complex, highly regulated system that controls fundamental biological processes, including fuel metabolism15,17,21. We previously reported that mice lacking Aldh1a1, the rate-limiting enzyme in Rald conversion to retinoic acid, show enhanced energy expenditure when fed a high-fat diet16, suggesting that they have altered BAT activity. Here we report that Aldh1a1 shows an adipose depot-specific expression pattern in mice fed a chow diet, with expression lowest in BAT and highest in visceral WAT. Moreover, genetic Aldh1a1 deficiency increased expression of Ucp1 and other classical BAT markers in mouse GWAT, with minimal changes in SWAT and BAT. Aldh1a1 deficiency increased mitochondrial enzyme activity and oxygen consumption in GWAT, but not BAT, consistent with increased thermogenic capacity in white fat. Indeed, Aldh1a1−/− mice were completely resistant to cold exposure, establishing Aldh1a1 as a central regulator of WAT thermogenesis independent of BAT changes.
Other pathways that can induce brown-like transformation of white fat have been reported, although these mechanisms predominately altered subcutaneous fat, with the visceral depot considered less susceptible to acquiring BAT characteristics34,35. In GWAT of Aldh1a1−/− mice, the abundance of Ucp1-expressing unilocular white adipocytes under basal conditions and the increased presence of multilocular brown adipocytes after cold exposure suggest that Aldh1a1 deficiency fosters thermogenic activation in visceral fat. This latent thermogenic potential of Aldh1a1-deficient visceral fat is unmasked in response to cold or high-fat feeding. Although the exact origins of BAT-like white adipocytes remains under debate, the lack of bona fide BAT cells, despite high Ucp1 expression in Aldh1a1-deficient WAT at baseline, suggests ‘browning’ of white adipocytes rather than recruitment of BAT cells in this model. Our findings show that altered retinoid metabolism can promote functional plasticity of adipocytes, and identify Aldh1a1 as a previously unrecognized determinant of white versus brown fat transformation. This selective action of Aldh1a1 in GWAT may derive from the distinct expression pattern of this enzyme in different fat depots.
The effect of Aldh1a1 deficiency on white versus brown characteristics in adipocyte seems to involve Rald’s action as a transcriptional mediator that modulates retinoid receptor activity. Although other Aldh1a1 effects are difficult to exclude in contributing to the in vivo effects seen here, Aldh1a1 deficiency is an established model of elevated endogenous Rald concentrations, especially in tissues with high Aldh1a1 expression, such as WAT16,23. Rald can impair adipogenesis in 3T3-L1 cells and in vivo in response to high-fat feeding16. However, as studied here, in distinct pluripotent cell models and in the absence of the high-fat feeding, adipogenesis did not seem altered. We identify Rald as a positive transcriptional regulator of a classic BAT gene program including Ucp1 in mouse and human white adipocytes through RAR- but not RXR-dependent mechanisms. Rald recruited the coactivator PGC-1α to the Ucp1 promoter in white adipocytes, a seminal event in BAT activation30. Those in vitro observations suggested that RAR activation might mediate the thermogenic phenotype in Aldh1a1 deficiency. Indeed, Aldh1a1−/− mice treated with RAR antagonist had reduced Ucp1 expression in WAT and no longer manifested increased thermogenesis, consistent with RAR-dependent adaptive thermogenesis in this in vivo model. Previously, the RAR ligand retinoic acid has been shown to activate Ucp1 transcription31,36.
Our observation that Rald’s effects on Ucp1 transcription were similar in the presence or absence of the Rald-converting enzyme Aldh1a1 suggests Rald as a mediator in this model. In contrast to Rald, retinol failed to induce Ucp1 promoter activity. The fact that both retinol and Rald can bind RAR in ligand-displacement assays37, but only Rald activates Ucp1 expression in these experimental models, argues for Rald-specific functional effects in WAT. Whether other Aldh1a1 effects or Ucp1-independent pathways might contribute to thermogenesis in Aldh1a1 deficiency cannot be completely excluded. Similarly, the precise mechanisms through which Rald-RAR interaction potently induce a BAT-like thermogenic program in WAT warrants further investigation, but could include synergistic effects between Rald and retinoic acid, differential RXR and RAR modulation, or altered intracellular retinoid concentrations. In addition, intracellular or extracellular binding proteins, including retinol-binding protein-4 or cellular retinoic acid–binding protein II, which influence partitioning of retinoid ligands to specific nuclear receptors38, may influence Rald effects. Indeed, we have previously observed reduced retinol- binding protein-4 plasma concentrations in Aldh1a1−/− mice16, a pattern consistent with improved insulin sensitivity39.
Dissipating energy stores by promoting thermogenic capacity in WAT has been proposed as an approach to treating obesity and its complications33. Although gene repression through antisense therapies has possible limitations40, the impact of an Aldh1a1 ASO on body-weight control and glucose metabolism in obese mice identifies Aldh1a1 as a therapeutic target. Although ASO-mediated reduction of hepatic Adlh1a1 expression makes other effects, including altered glucose metabolism, difficult to exclude, selective Aldh1a1 repression in GWAT through ASO treatment supports the prospect of specifically targeting visceral fat depots, including induction of BAT-like characteristics and thermogenesis.
Here we establish Aldh1a1 and its substrate Rald as previously unidentified determinants of functional plasticity in WAT that can activate a thermogenic program, promote energy dissipation and limit obesity. Disruption of Aldh1a1 expression or function in visceral fat could offer new opportunities in targeting and treating specific adipose depots and obesity-related complications.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Animals, Aldh1a1 antisense oligonucleotide, RAR antagonist
We purchased C57BL/6J WT mice from the Jackson Laboratory. Aldh1a1-deficient (Aldh1a1−/−) mice, provided by G. Duester, were backcrossed to a C57BL/6J background (>20 generations). Mice were kept on a standard chow diet with a vitamin A content of 15 IU g−1 or on a high-fat diet (60% of kilocalories as fat, Research Diets). Mice had free access to food and water. Experiments were conducted using female mice at 12–14 weeks old, unless otherwise indicated. All animal studies were approved by the Harvard Medical School Institutional Animal Care and Use Committee.
An ASO targeting mouse Aldh1a1 and a control ASO not hybridizing to any known mouse RNA sequences were synthesized and purified by Isis Pharmaceuticals as described41. In vitro characterization including knockdown validation of the Aldh1a1 ASO was done in mouse primary hepatocytes. ASOs (70 mg per kg of body weight per week) were administered via i.p. injection twice per week.
RAR antagonist AGN193109 (0.5 mg per kg of body weight, Santa Cruz Biotechnology) was injected i.p. daily for 14 d.
Human adipose tissue samples
Paired samples of visceral (omental) and subcutaneous adipose tissue were obtained from men (n = 8) and women (n = 32) of European descent. Morbidly obese subjects (BMI = 53.0 ± 0.55 kg m−2, n = 20) undergoing laparoscopic gastric banding were matched by age and sex to lean control subjects (BMI = 25.2 ± 0.15 kg m−2, n = 20) undergoing laparoscopic cholecystectomy or fundoplication. Adipose tissue samples were taken from similar locations in all patients. After excision, tissue specimens were washed in saline buffer, visible blood vessels were excised and the tissue was immediately snap frozen in liquid nitrogen. Subjects with any infectious, inflammatory, neoplastic or systemic disease, diabetes or other uncontrolled endocrine disease, or those receiving antibiotics, anti-inflammatory or antiobesity drugs were excluded. The study was approved by the Medical University of Vienna Ethics Committee. All subjects provided written informed consent.
Body temperature measurements
Core body temperature was assessed in mice as described42,43. WT and Aldh1a1−/− mice (12 weeks old, n = 6 per group) were killed (ketamine/xylazine/acepromazine, 100/10/3 mg per kgof body weight i.p.) and a telemetric temperature probe (E-mitter, MiniMitter) was implanted intra-abdominally. One week after surgery, mice were single housed in open-circuit Oxymax chambers as part of the Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments)44. Oxymax chambers were kept in a temperature enclosure with 12-h light and dark cycles at 23 °C, and mice had ad libitum access to food and water. After a 48-h acclimatization phase, core body temperature was recorded every 10 min for 48 h at ambient temperature (23 °C), followed by 48 h of cold stimulation (4 °C). After killing, fat depots were dissected and analyzed.
Glucose and insulin tolerance tests
Glucose tolerance tests and insulin tolerance tests were performed on mice after fasting (12 h and 6 h, respectively). Mice were injected i.p. either with d-glucose (Sigma, 0.75 g per kg of body weight) or recombinant human regular insulin (0.5 U per kg of body weight, Humulin R, Eli Lilly); blood glucose concentrations were measured periodically up to 120 min by a glucometer (Abbott Laboratories).
Cell culture reagents
Except as noted, media were purchased from Invitrogen, and retinoids and other chemicals from Sigma. RAR antagonist AGN193109 was obtained from Santa Cruz Biotechnology. RXR antagonist HX531 was provided by H. Kagechika.
Isolation of human and mouse adipose tissue stromal-vascular fraction and mouse embryonic fibroblasts
Abdominal subcutaneous fat biopsies were obtained from a 22-year-old woman undergoing gastric bypass surgery who gave written informed consent. Human subcutaneous fat biopsies and mouse BAT were immediately washed with PBS, minced and digested (1 mg ml−1 collagenase, 45 min, 37 °C) in DMEM containing 1% (wt/vol) BSA. Digested tissues were filtered through sterile 150-μm nylon mesh and centrifuged (250g, 5 min). The floating fractions containing adipocytes were discarded, and the stromal-vascular fraction pellet was resuspended in erythrocyte lysis buffer (154 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA for 10 min) to remove red blood cells. The cells were further centrifuged at 500g for 5 min, plated on a 24-well culture dish, and grown and differentiated to mature adipocytes as described below.
MEFs were isolated from Aldh1a1−/− embryos at embryonic day 14.5 using standard techniques45.
In vitro adipocyte differentiation
C3H10T1/2, isolated human stromal-vascular cells and Aldh1a1−/− MEFs were cultured and differentiated as described8. Briefly, cells were grown to confluence in DMEM, 10% (vol/vol) FBS and 1% (vol/vol) penicillin-streptomycin followed by standard adipogenic induction (20 nM insulin, 1 nM T3, 0.5 mM isobutylmethylxanthine, 1 μM dexamethasone, 0.125 mM indomethacin) and stimulation with retinoids, retinoid antagonists or vehicle (DMSO) as indicated. After a 48-h induction phase, growth medium was supplemented with insulin, T3 and the indicated retinoids, which were changed every other day up to day 6.
Cell transfection, reporter assays, siRNA and lentivirus infection
Undifferentiated C3H10T1/2 cells (which do not express Aldhs) were transfected with pGL3-RARE (Addgene) or Ucp1 promoter luciferase reporter constructs (from R. Koza) using FuGene HD (Roche) as described46. At 24 h after transfection, cells were stimulated with retinoids or cyclic AMP as indicated in Figure 5 and Supplementary Figure 3. Cell lysates were harvested 48 h after transfection, and luciferase activity was quantified by standard luminometer assay and normalized to β-galactosidase.
Mission shRNA constructs targeting Aldh1a1 and a nontargeting control shRNA were purchased from Sigma. To generate lentivirus particles, shRNA constructs were transfected into HEK293TN cells (SA Biosciences) along with the packaging vectors psPAX and pM2DG. After 72 h, virus-containing supernatant was precipitated using polyethylene glycol (SA Biosciences) and concentrated in PBS (30×). For stable cell line generation, C3H10T1/2 cells were infected with lentivirus and selected with puromycin (5 μg ml−1) over a 2-week period. For transient knockdown, C3H10T1/2 and shAldh1a1 cells were transfected with siRNA constructs against RARα and RXRα (Ambion) using siDeliverX reagent (Panomics) following manufacturer’s protocol. Knockdown was confirmed by mRNA and protein expression.
Chromatin immunoprecipitation
ChIP was performed in Rald-stimulated versus vehicle-stimulated C3H10T1/2 adipocytes using the EZ-Magna ChIP kit following the manufacturer’s instructions (Millipore) and antibodies to RARα (C-20), PGC-1α (H-300) or rabbit IgG as a negative control (all Santa Cruz). We used 4 μg antibody per confluent 10-cm cell-culture dish. Immunoprecipitated DNA was amplified by real-time PCR using primers specific for the mouse Ucp1 promoter region. Relative PGC-1α and RARα occupancy at the Ucp1 promoter was determined and normalized to input DNA.
Reverse transcription and gene expression
Total RNA was extracted using Trizol (Invitrogen), treated with DNase (Qiagen) and reverse transcribed to cDNA according to manufacturer’s instructions. Gene expression, normalized to 36B4, was analyzed by quantitative real-time RT-PCR (Sybr Green, 96-well plates) using a MyiQ cycler (Bio-Rad). Primer sequences are available upon request.
Mitochondrial DNA content
Genomic DNA was isolated from GWAT, SWAT and BAT of WT and Aldh1a1−/− mice (n = 8 per group) using the DNeasy Blood & Tissue Kit (Qiagen) following manufacturer’s instructions. Quantitative real-time PCR assessed genomic expression of mitochondrial NADH dehydrogenase subunit 1 (ND1) normalized to β-globin.
Immunoblotting
Homogenized tissue was lysed in RIPA buffer (Boston Bioproducts) containing protease and phosphatase inhibitors. Standard western blotting was performed using rabbit polyclonal antibodies to Ucp1 (ab23841, Abcam) and GAPDH (FL-335, Santa Cruz Biotechnology) rabbit monoclonal antibody to Aldh1a1 (EP1933Y, Abcam), and mouse monoclonal antibody to β-actin (C-4, both Santa Cruz Biotechnology). All antibodies were used at a dilution of 1:1,000. Proteins were detected by chemiluminescence (GE HealthCare).
Immunohistochemistry and Oil Red O
Paraffin sections were prepared from mouse GWAT, SWAT and BAT after fixation (10% (vol/vol) neutral-buffered formalin). Sections were stained with rabbit polyclonal antibody to Ucp1 (ab23841, Abcam, 1:1,000) or tyrosine hydroxylase (Millipore) and biotinylated secondary goat antibody to rabbit IgG (Vector Laboratories Inc., 1:1,000). Control staining was performed on selected sections with isotype control. Differentiated C3H10T1/2 cells were stained with Oil Red O (Thermo Fisher Scientific Inc.) as per the manufacturer’s protocol. Micrographs were taken with an Olympus Q-color 3 digital camera attached to an Olympus BX50 microscope.
Electron microscopy
GWAT from WT and Aldh1a1−/− mice (n = 4 per group) was fixed in 4% (vol/vol) formaldehyde, 5% (vol/vol) glutaraldehyde in 100 mM phosphate buffer, pH 7.2, for 12 h at 4 °C. Ultra-thin sections were cut, and micrographs were generated with a transmission electron microscope (JEOL).
Adipose tissue citrate synthase activity
GWAT and BAT from WT and Aldh1a1−/− mice (n = 6 per group) were homogenized in protein extraction buffer (50 mM Tris-HCl, 250 mM mannitol, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 10% (vol/vol) glycerol, 1% (vol/vol) Triton and protease inhibitors) to obtain protein lysates. After centrifugation and removal of the lipid layer, protein concentration was determined using the BCA Protein Assay (Thermo Fisher Scientific Inc.). Protein lysate (8 μg) was used for the citrate synthase activity assay following the manufacturer’s protocol (Sigma).
Adipose tissue and adipocyte oxygen consumption
Oxygen consumption rate (OCR) was determined in mouse adipose tissue and stromal-vascular cells derived from brown adipocytes using a modified protocol47. Briefly, freshly isolated GWAT and BAT from WT and Aldh1a1−/− mice (n = 5 per genotype) were rinsed with unbuffered KHB medium containing 111 mM NaCl, 4.7 mM KCl, 2 mM MgSO4, 1.2 mM Na2HPO4, 0.5 mM carnitin and 2.5 mM glucose. Adipose tissue was cut into small pieces and rinsed with KHB medium, and 10 mg of tissue was placed in each well of a XF24-well Islet Flux plate (Seahorse Bioscience). Then, 450 μl of KHB medium was added to each well and samples were analyzed in an XF24 Extracellular Flux Analyzer (Seahorse Bioscience) at 37 °C (ref. 48). The XF24 Analyzer mixed the media in each well three times for 2 min before measurements to allow oxygen partial pressure to equilibrate. Basal OCR was measured simultaneously in all wells three times. Five tissue replicates from five mice per genotype were analyzed in independent experiments and results were normalized to tissue weight. For brown adipocytes OCR, stromal-vascular cells from WT and Aldh1a1−/− mice were plated in a 24-well XF24 V28 cell culture microplate followed by adipogenic differentiation. Basal and isoproterenol-stimulated (1 μM) OCR were determined in an XF24 Extracellular Flux Analyzer. Five tissue replicates from four mice per genotype were analyzed in independent experiments.
Time-resolved fluorescence energy transfer
Lanthascreen TR-FRET RARα and RXRβ coactivator assays (Invitrogen) were performed as per the manufacturer’s protocol using a PerkinElmer EnVision fluorescence plate reader. Briefly, a glutathione-S-transferase (GST)-tagged recombinant RARα LBD or RXRα LBD was incubated with a terbium-labeled anti-GST antibody and a fluorescein-labeled coactivator peptide (PGC-1α) as well as increasing concentrations of the given retinoids. Binding of exogenous agonist (retinoids) to the LBD causes a conformational change that increases the affinity of the nuclear receptor (RARα or RXRα) for the coactivator peptide (PGC-1α). The close proximity of the fluorescently labeled coactivator peptide to the terbium-labeled antibody enhances the TR-FRET signal measured by the emission ratio of 520 nm/495 nm.
Statistical analyses
All data are given as mean ± s.e.m. Comparisons between two groups were assessed by the unpaired two-tail Student’s t test after normal distribution was confirmed. Linear regression analysis was performed to evaluate association between ALDH1A1 mRNA expression in human fat and BMI. A P value of 0.05 or less was considered statistically significant.
Supplementary Material
Acknowledgments
We thank H. Wang, G. Sukhova, E. Shvartz, T.A. Dang and V. Demchev for excellent technical support, R. Koza (Pennington Biomedical Research Center) for providing the Ucp1 promoter luciferase construct, H. Kagechika (University of Tokyo) for providing HX531 and G. Duester (Burnham Medical Research Institute) for the Aldh1a1−/− mice and helpful discussions. This work was supported by the US National Institutes of Health grants HL048743, AR054604-03S1, 5P30DK057521-12 (J.P.); Mary K. Iacocca Professorship DK082659 and the National Institute of Diabetes and Digestive and Kidney Diseases DK056626 (C.R.K.); DK048873 and DK048873-14S2 (D.E.C.); the Austrian Science Fund (FWF); J3107-B19 (F.W.K.).
Footnotes
Note: Supplementary information is available in the online version of the paper.
AUTHOR CONTRIBUTIONS
F.W.K. and J.P. designed the study; F.W.K., C.V., P.O., S.S., J.D.B., S.N. and M.Z. researched data; F.W.K. wrote the manuscript; C.V., J.D.B., S.N., M.Z., T.M.S., D.E.C., C.R.K. and J.P. reviewed the manuscript; D.E.C. and C.R.K. contributed to discussion.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Mensah GA, et al. Obesity, metabolic syndrome, and type 2 diabetes: emerging epidemics and their cardiovascular implications. Cardiol Clin. 2004;22:485–504. doi: 10.1016/j.ccl.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Bray GA, Bellanger T. Epidemiology, trends, and morbidities of obesity and the metabolic syndrome. Endocrine. 2006;29:109–117. doi: 10.1385/ENDO:29:1:109. [DOI] [PubMed] [Google Scholar]
- 3.Klein S, et al. Waist circumference and cardiometabolic risk: a consensus statement from shaping America’s health: Association for Weight Management and Obesity Prevention; NAASO, the Obesity Society; the American Society for Nutrition; and the American Diabetes Association. Diabetes Care. 2007;30:1647–1652. doi: 10.2337/dc07-9921. [DOI] [PubMed] [Google Scholar]
- 4.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 5.Farmer SR. Molecular determinants of brown adipocyte formation and function. Genes Dev. 2008;22:1269–1275. doi: 10.1101/gad.1681308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seale P, Kajimura S, Spiegelman BM. Transcriptional control of brown adipocyte development and physiological function—of mice and men. Genes Dev. 2009;23:788–797. doi: 10.1101/gad.1779209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cypess AM, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tseng YH, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seale P, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vegiopoulos A, et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science. 2010;328:1158–1161. doi: 10.1126/science.1186034. [DOI] [PubMed] [Google Scholar]
- 11.Hansen JB, et al. Retinoblastoma protein functions as a molecular switch determining white versus brown adipocyte differentiation. Proc Natl Acad Sci USA. 2004;101:4112–4117. doi: 10.1073/pnas.0301964101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang S, et al. Corepressor SMRT promotes oxidative phosphorylation in adipose tissue and protects against diet-induced obesity and insulin resistance. Proc Natl Acad Sci USA. 2011;108:3412–3417. doi: 10.1073/pnas.1017707108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cinti S. Between brown and white: novel aspects of adipocyte differentiation. Ann Med. 2011;43:104–115. doi: 10.3109/07853890.2010.535557. [DOI] [PubMed] [Google Scholar]
- 14.Park KW, Halperin DS, Tontonoz P. Before they were fat: adipocyte progenitors. Cell Metab. 2008;8:454–457. doi: 10.1016/j.cmet.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Schwarz EJ, Reginato MJ, Shao D, Krakow SL, Lazar MA. Retinoic acid blocks adipogenesis by inhibiting C/EBPβ-mediated transcription. Mol Cell Biol. 1997;17:1552–1561. doi: 10.1128/mcb.17.3.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziouzenkova O, et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med. 2007;13:695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kane MA, et al. CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Mol Cell Biol. 2011;31:3277–3285. doi: 10.1128/MCB.05516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov. 2007;6:793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- 19.Villarroya F, Iglesias R, Giralt M. Retinoids and retinoid receptors in the control of energy balance: novel pharmacological strategies in obesity and diabetes. Curr Med Chem. 2004;11:795–805. doi: 10.2174/0929867043455747. [DOI] [PubMed] [Google Scholar]
- 20.Ross AC. Overview of retinoid metabolism. J Nutr. 1993;123:346–350. doi: 10.1093/jn/123.suppl_2.346. [DOI] [PubMed] [Google Scholar]
- 21.Ziouzenkova O, Plutzky J. Retinoid metabolism and nuclear receptor responses: New insights into coordinated regulation of the PPAR-RXR complex. FEBS Lett. 2008;582:32–38. doi: 10.1016/j.febslet.2007.11.081. [DOI] [PubMed] [Google Scholar]
- 22.Duester G, Mic FA, Molotkov A. Cytosolic retinoid dehydrogenases govern ubiquitous metabolism of retinol to retinaldehyde followed by tissue- specific metabolism to retinoic acid. Chem Biol Interact. 2003;143–144:201–210. doi: 10.1016/s0009-2797(02)00204-1. [DOI] [PubMed] [Google Scholar]
- 23.Molotkov A, Duester G. Genetic evidence that retinaldehyde dehydrogenase Raldh1 (Aldh1a1) functions downstream of alcohol dehydrogenase Adh1 in metabolism of retinol to retinoic acid. J Biol Chem. 2003;278:36085–36090. doi: 10.1074/jbc.M303709200. [DOI] [PubMed] [Google Scholar]
- 24.Wiegand G, Remington SJ. Citrate synthase: structure, control, and mechanism. Annu Rev Biophys Biophys Chem. 1986;15:97–117. doi: 10.1146/annurev.bb.15.060186.000525. [DOI] [PubMed] [Google Scholar]
- 25.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 26.Tang QQ, Otto TC, Lane MD. Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci USA. 2004;101:9607–9611. doi: 10.1073/pnas.0403100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chute JP, et al. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci USA. 2006;103:11707–11712. doi: 10.1073/pnas.0603806103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson AT, Wang L, Gillett SJ, Chandraratna RA. High affinity retinoic acid receptor antagonists: analogs of AGN 193109. Bioorg Med Chem Lett. 1999;9:573–576. doi: 10.1016/s0960-894x(99)00047-5. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi B, et al. Novel retinoid X receptor antagonists: specific inhibition of retinoid synergism in RXR-RAR heterodimer actions. J Med Chem. 2002;45:3327–3330. doi: 10.1021/jm0255320. [DOI] [PubMed] [Google Scholar]
- 30.Puigserver P, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 31.Alvarez R, et al. A novel regulatory pathway of brown fat thermogenesis. Retinoic acid is a transcriptional activator of the mitochondrial uncoupling protein gene. J Biol Chem. 1995;270:5666–5673. doi: 10.1074/jbc.270.10.5666. [DOI] [PubMed] [Google Scholar]
- 32.Cypess AM, Kahn CR. Brown fat as a therapy for obesity and diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:143–149. doi: 10.1097/MED.0b013e328337a81f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langin D. Recruitment of brown fat and conversion of white into brown adipocytes: strategies to fight the metabolic complications of obesity? Biochim Biophys Acta. 2010;1801:372–376. doi: 10.1016/j.bbalip.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J Clin Invest. 1998;102:412–420. doi: 10.1172/JCI3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seale P, et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest. 2011;121:96–105. doi: 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silva JE, Rabelo R. Regulation of the uncoupling protein gene expression. Eur J Endocrinol. 1997;136:251–264. doi: 10.1530/eje.0.1360251. [DOI] [PubMed] [Google Scholar]
- 37.Repa JJ, Hanson KK, Clagett-Dame M. All-trans-retinol is a ligand for the retinoic acid receptors. Proc Natl Acad Sci USA. 1993;90:7293–7297. doi: 10.1073/pnas.90.15.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007;129:723–733. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Q, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 40.Crooke ST. Progress in antisense technology. Annu Rev Med. 2004;55:61–95. doi: 10.1146/annurev.med.55.091902.104408. [DOI] [PubMed] [Google Scholar]
- 41.Baker BF, et al. 2′-O-(2-methoxy)ethyl-modified anti-intercellular adhesion molecule 1 (ICAM-1) oligonucleotides selectively increase the ICAM-1 mRNA level and inhibit formation of the ICAM-1 translation initiation complex in human umbilical vein endothelial cells. J Biol Chem. 1997;272:11994–12000. doi: 10.1074/jbc.272.18.11994. [DOI] [PubMed] [Google Scholar]
- 42.Wernstedt I, et al. Reduced stress- and cold-induced increase in energy expenditure in interleukin-6-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2006;291:R551–R557. doi: 10.1152/ajpregu.00514.2005. [DOI] [PubMed] [Google Scholar]
- 43.Hodges MR, et al. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008;28:2495–2505. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kennedy AR, et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am J Physiol Endocrinol Metab. 2007;292:E1724–E1739. doi: 10.1152/ajpendo.00717.2006. [DOI] [PubMed] [Google Scholar]
- 45.Xu J. Preparation, culture and immortalization of mouse embryonic fibroblasts. Curr Protoc Mol Biol. 2005;70:28.1.1–28.1.8. doi: 10.1002/0471142727.mb2801s70. [DOI] [PubMed] [Google Scholar]
- 46.Brown JD, Oligino E, Rader DJ, Saghatelian A, Plutzky J. VLDL hydrolysis by hepatic lipase regulates PPARδ transcriptional responses. PLoS ONE. 2011;6:e21209. doi: 10.1371/journal.pone.0021209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yehuda-Shnaidman E, Buehrer B, Pi J, Kumar N, Collins S. Acute stimulation of white adipocyte respiration by PKA-induced lipolysis. Diabetes. 2010;59:2474–2483. doi: 10.2337/db10-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu M, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.