

Abstract
Juvenile xanthogranuloma is a type of non-Langerhans cell histiocytosis that occurs most frequently in infants and children. It is usually asymptomatic and can present as either a cutaneous or extracutaneous lesion. The present case is believed to be the first reported in the English literature of juvenile xanthogranuloma presenting as an ulcerated bleeding lesion on the dorsum of the nose.
Keywords: Bleeding, Juvenile, Nose, Xanthogranuloma
Abstract
Le xanthogranulome juvénile est un type d’histiocytose non langerhansienne qui affecte davantage les nourrissons et les enfants. Il est en général asymptomatique et peut se manifester par une lésion cutanée ou extracutanée. Le cas de xanthogranulome juvénile présenté ici serait le premier à avoir été signalé dans la littérature de langue anglaise sous la forme d’une lésion ulcérée sanguinolente au niveau de l’arête du nez.
Juvenile xanthogranuloma (JXG), previously known as nevoxanthoendothelioma, was first described by Adamson in 1905 (1). JXG is a type of non-Langerhans cell histiocytosis (non-LCH) that can either present as a single or multiple, cutaneous or extracutaneous lesion. It is a benign condition, usually asymptomatic, and frequently occurs in the head and neck area. A case of a patient with cutaneous JXG of the nose is presented, along with a discussion of the histological features and the different options of management.
CASE PRESENTATION
A 10-month-old male patient, previously healthy, was presented to the plastic surgery clinic at The Hospital for Sick Children, Toronto, Ontario with a two-month history of a gradually enlarging midline lesion on the dorsum of the nose. The lesion had a friable bleeding surface with no history of trauma. On examination, there was a 1.0 cm × 1.0 cm well-defined, red, papular, ulcerated lesion on the dorsum of the nose covered with a blood clot (Figure 1). The lesion was purely cutaneous and not attached to the underlying muscle or bone. The rest of the clinical examination revealed a large café-au-lait macule on the left thigh and Mongolian spots on the upper and lower back and left forehead. A resolving hemangioma was noticed on the right abdomen.
Figure 1).
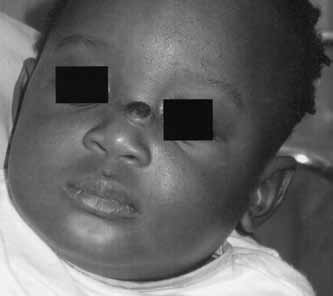
Preoperative photograph showing the ulcerated bleeding mass at the base of the nose
A magnetic resonance image showed a well-defined soft tissue lesion measuring 12 mm × 10 mm × 6.4 mm that was situated midline and to the left of the nose. The lesion was of intermediate signal intensity on T1 and T2 weighted imaging and enhanced moderately with contrast. It was of low signal on diffusion weighted imaging.
The decision was made to perform an excisional biopsy to achieve a definite diagnosis. A circular portion of skin, 1.1 cm in diameter and 0.9 cm in thickness, was received by the pathologist. The epidermal surface was markedly ulcerated, and on cut section, the dermis was replaced by a firm, yellow nodule that did not involve the deep margin. On histological examination, there was a highly cellular infiltrate immediately below the ulcerated epithelium. This dense infiltrate formed a solid mass occupying nearly the entire thickness of the dermis, but there was a sharp demarcation at the deep margin where the infiltrate had a rounded border and did not involve the subcutis. The lateral margins were involved.
This cellular lesion consisted of predominantly mononuclear cells with a bean-shaped or reniform nucleus and pale eosinophilic cytoplasm. There were focal areas where the mononuclear cells were intermixed with spindle cells, foam cells and multinucleated giant cells with Touton features (Figure 2). Mitoses were rare, and no atypical forms were seen. A few scattered lymphocytes, neutrophils and eosinophils were present in the background. Immunohistochemical studies confirmed that the majority of the cells forming the infiltrate were non-LCH: the cells were positive for vimentin, CD68 and factor XIIIa, and negative for CD1a, S100, CD45 and CD34. The morphology and immunohistochemical profile were diagnostic of a JXG. Consequently, there was no need to get free margins and the defect was left to heal by secondary intention (Figure 3). The patient was evaluated by the hematology oncology team and the neurofibromatosis team to role out associated neurofibromatosis type I or chronic myelogenous leukemia (CML).
Figure 2).
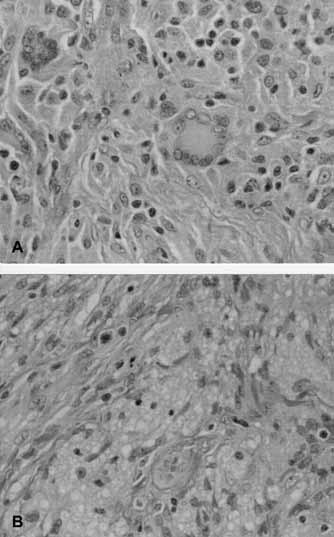
High magnification demonstrating cellular dermal infiltrate consisting of plump histiocytes, Touton multinucleated giant cells, scattered lymphocytes (A) and foamy macrophages (B) (hematoxylin and eosin stain, original magnification ×400)
Figure 3).
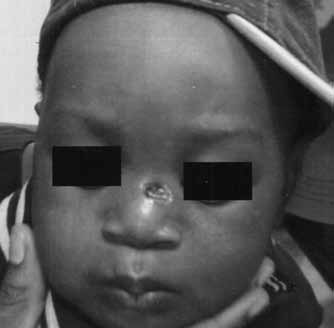
Two weeks postoperative photograph of the defect left to heal by secondary intention
DISCUSSION
JXG, although an uncommon condition, is considered to be the most common non-LCH lesion, and usually occurs in the head and neck region. It can either present as a solitary lesion (in up to 80% of the cases) or multiple well-demarcated round or oval lesions (2). They first appear as red-brown in colour but they rapidly acquire a yellow hue (3). The lesions most commonly occur in the head and neck area (4), followed by the upper torso, the upper extremities (5) and the lower extremities (6). In 5% of patients, JXG involves extracutaneous areas, with a high tendency to affect the ocular region. Other sites reported were the pericardium, liver, lung, kidneys, ovary and testes (7). In the head and neck region, areas reported were the oral cavity, nasal cavity (7,8), external auditory canal (9), temporal bone, nasopharynx and larynx (7,10).
The age of onset follows a bimodal distribution, with a peak in infancy and childhood and another between the third and fourth decades (8). Although the infant type may regress spontaneously, adult types rarely do. Male sex is dominant in the infant type (8). The triple association between JXG and both neurofibromatosis type I and juvenile CML is an accepted principle by most authors (11–14). Children with JXG and café-au-lait spots have a 20- to 32-fold higher risk of having CML (12).
JXG can be divided into three categories depending on the size of the lesion (15): papular (less than 10 mm); nodular (10 mm to 20 mm); and macronodular (greater than 20 mm)
Benign cephalic histiocytosis, generalized eruptive histiocytoma, papular xanthoma, xanthoma disseminatum and tuberous xanthoma are lesions to be considered in the differential diagnoses. In addition, malignant tumours such as rhabdomyosarcoma, fibrosarcoma and malignant fibrohistiocytoma should be especially considered when JXG is located in the deep tissues (16).
Histologically, JXG is a cellular histiocytic lesion consisting of one or more of the three basic cellular types: mononuclear cells with a reniform nucleus, multinucleated cells with or without Touton features and spindle cells. It is important to realize that JXG may have a varied microscopic appearance that likely reflects the life cycle of the lesion and its capacity for self-healing or regression (17). This presents a diagnostic challenge to the pathologist, especially in lesions without the giant cells or foam cells (18). Early lesions may show large accumulations of histiocytes without any lipid infiltration intermingled with only a few lymphocytes, plasma cells and eosinophils. In mature lesions, a granulomatous infiltrate is usually present, containing foam cells, foreign-body and/or Touton giant cells, histiocytes, lymphocytes and eosinophils. In the cutaneous lesions, there is often global replacement of the dermis by this cellular infiltrate, and a pushing cellular border is seen between the dermal and subcutaneous tissues.
Although the histological pattern may be inconsistent among different JXG lesions, the immunophenotype is relatively constant with the characteristic profile for non-LCH: positive reactivity for vimentin, CD68 and factor XIIIa and negative for CD1a. It is important to differentiate between LCH and JXG, because the latter has a greater potential for spontaneous regression than does LCH (15,17,19). The pathogenesis is not clear. Some reports have associated JXG with infection but most evidence showed that it is a reactive granulomatous response (5,7).
Options in treating this condition depend on the size and the clinical presentation, and can be summarized as follows:
Conservative: especially in the asymptomatic infantile type lesions that are self-limiting in a period of three to six years (18);
Steroids: early treatment with subconjunctival injection of steroids and topical steroid drops can result in regression of lesions involving the iris (20). The use of systemic steroids in these cases has been also reported (21); and
Surgical excision: the most common form of treatment is an excisional biopsy, and it is recommended for either symptomatic lesions or for cosmetic purposes.
CONCLUSION
The pathological nature of JXG was discussed and an unusual case of an ulcerated nasal JXG was presented. JXG is an uncommon benign lesion, commonly occurring in the head and neck area. Surgical excision without achieving a completely free margin is an acceptable method of treatment. The wounds can be left to heal by secondary intention with a satisfactory aesthetic result.
Acknowledgments
We are grateful to Dr Glenn Taylor, MD for his help in preparing the slides and reviewing the pathology section.
REFERENCES
- 1.Adamson HG. Society intelligence: The Dermatological Society of London. Br J Dermatol. 1905;17:222. [Google Scholar]
- 2.Sangueza OP, Salmon JK, White CR, Jr, Beckstead JH. Juvenile xanthogranuloma: A clinical, histopathologic and immunohistochemical study. J Cutan Pathol. 1995;22:327–35. doi: 10.1111/j.1600-0560.1995.tb01415.x. [DOI] [PubMed] [Google Scholar]
- 3.Caputo R, Grimalt R, Gelmetti C, Cottoni F. Unusual aspects of juvenile xanthogranuloma. J Am Acad Dermatol. 1993;29:868–70. doi: 10.1016/0190-9622(93)70259-v. [DOI] [PubMed] [Google Scholar]
- 4.Cohen BA, Hood A. Xanthogranuloma: Report on clinical and histologic findings in 64 patients. Pediatr Dermatol. 1989;6:262–6. doi: 10.1111/j.1525-1470.1989.tb00909.x. [DOI] [PubMed] [Google Scholar]
- 5.Piraccini BM, Fanti PA, Iorizzo M, Tosti A. Juvenile xanthogranuloma of the proximal nail fold. Pediatr Dermatol. 2003;20:307–8. doi: 10.1046/j.1525-1470.2003.20405.x. [DOI] [PubMed] [Google Scholar]
- 6.Whittam LR, Higgins EH. Juvenile xanthogranuloma on the sole. Pediatr Dermatol. 2000;17:460–2. doi: 10.1046/j.1525-1470.2000.01822.x. [DOI] [PubMed] [Google Scholar]
- 7.Saravanappa N, Rashid AM, Thebe PR, Davis JP. Juvenile xanthogranuloma of the nasal cavity. J Laryngol Otol. 2000;114:460–1. doi: 10.1258/0022215001905841. [DOI] [PubMed] [Google Scholar]
- 8.Fang TJ, Lin CZ, Li HY. Juvenile xanthogranuloma of the nasal cavity. Int J Pediatr Otorhinolaryngol. 2003;67:297–9. doi: 10.1016/s0165-5876(02)00385-3. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida GY. Xanthogranuloma of the external auditory canal. Otolaryngol Head Neck Surg. 1995;112:626–7. doi: 10.1177/019459989511200424. [DOI] [PubMed] [Google Scholar]
- 10.Benjamin B, Motbey J, Ivers C, Kan A. Benign juvenile xanthogranuloma of the larynx. Int J Pediatr Otorhinolaryngol. 1995;32:77–81. doi: 10.1016/0165-5876(94)01107-9. [DOI] [PubMed] [Google Scholar]
- 11.Iyengar V, Golomb CA, Schachner L. Neurilemmomatosis, NF2, and juvenile xanthogranuloma. J Am Acad Dermatol. 1998;39:831–4. doi: 10.1016/s0190-9622(98)70359-7. [DOI] [PubMed] [Google Scholar]
- 12.Zvulunov A, Barak Y, Metzker A. Juvenile xanthogranuloma, neurofibromatosis, and juvenile chronic myelogenous leukemia. World statistical analysis. Arch Dermatol. 1995;131:904–8. [PubMed] [Google Scholar]
- 13.Thami GP, Kaur S, Kanwar AJ. Association of juvenile xanthogranuloma with cafe-au-lait macules. Int J Dermatol. 2001;40:283–5. doi: 10.1046/j.1365-4362.2001.01212-2.x. [DOI] [PubMed] [Google Scholar]
- 14.Cooper PH, Frierson HF, Kayne AL, Sabio H. Association of juvenile xanthogranuloma with juvenile myeloid leukemia. Arch Dermatol. 1984;120:371–5. [PubMed] [Google Scholar]
- 15.Shapiro NL, Malis DJ, Charon CC, Billman GF, Kearns DB. Giant juvenile xanthogranuloma of the tongue. Am J Otolaryngol. 1999;20:241–4. doi: 10.1016/s0196-0709(99)90007-8. [DOI] [PubMed] [Google Scholar]
- 16.Hernandez-Martin A, Baselga E, Drolet BA, Esterly NB. Juvenile xanthogranuloma. J Am Acad Dermatol. 1997;36:355–67. doi: 10.1016/s0190-9622(97)80207-1. [DOI] [PubMed] [Google Scholar]
- 17.Dehner LP. Juvenile xanthogranulomas in the first two decades of life: A clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol. 2003;27:579–93. doi: 10.1097/00000478-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Arceci RJ. The histiocytoses: The fall of the Tower of Babel. Eur J Cancer. 1999;35:747–67. doi: 10.1016/s0959-8049(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 19.Zelger B, Cerio R, Orchard G, Wilson-Jones E. Juvenile and adult xanthogranuloma. A histological and immunohistochemical comparison. Am J Surg Pathol. 1994;18:126–35. doi: 10.1097/00000478-199402000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Casteels I, Olver J, Malone M, Taylor D. Early treatment of juvenile xanthogranuloma of the iris with subconjunctival steroids. Br J Ophthalmol. 1993;77:57–60. doi: 10.1136/bjo.77.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karcioglu ZA, Mullaney PB. Diagnosis and management of iris juvenile xanthogranuloma. J Pediatr Ophthalmol Strabismus. 1997;34:44–51. doi: 10.3928/0191-3913-19970101-10. [DOI] [PubMed] [Google Scholar]