

Abstract
Parkinson’s disease (PD) is the second-most common age-dependent neurodegenerative disorder and is caused by severe degeneration of dopaminergic neurons in the substantia nigra pars compacta. Unfortunately, current treatment only targets symptoms and involves dopamine replacement therapy, which does not counteract progressive degeneration. MicroRNAs (miRNAs) are a class of small RNA molecules implicated in post-transcriptional regulation of gene expression during development. Recent studies show that miRNAs are playing an important role in the pathophysiology of PD. miRNA-based therapy is a powerful tool with which to study gene function, investigate the mechanism of the disease, and validate drug targets. In this review, we focus on the recent advances of the use of miRNAs in the pathogenesis of PD.
Keywords: miRNA, α-synuclein, LRRK2, miRNA-based therapy, pathophysiology
Introduction
Parkinson’s disease (PD) is the second-most common age-dependent neurodegenerative disorder and it affects roughly 1% of the population over 50.1 The disease is clinically characterized by bradykinesia, resting tremor, cogwheel rigidity, and eventually postural instability.2 PD was first described in 1817 by James Parkinson,3 and it was reported in 1893 that these symptoms were attributable to severe degeneration of dopaminergic neurons in the substantia nigra pars compacta.4 However, as the disease advances, the pathology spreads to involve other brain regions, including cingulate gyrus, the amygdala, and higher cortical regions, resulting in dementia and psychiatric manifestations in affected patients.
Pathologically, abnormal accumulation of the presynaptic protein α-synuclein in the brain, forming Lewy bodies, is a hallmark of disease.5 While PD is primarily a sporadic disease in nature, to date, at least five genes have been linked with this multigenic disease including α-synuclein, leucine-rich repeat kinase 2 (LRRK2), parkin, phosphatase and tensin homolog-induced kinase 1 (PINK1), and DJ-1.6–9 In addition, cumulative evidence indicates that dysfunction of these pathways plays a direct role in the etiology of Parkinson’s disease. Recent success in using microRNA (miRNA) as therapeutic targets holds substantial promise for treatment of the complex Parkinson’s disease.
miRNAs are a class of endogenous and small non-coding RNA molecules, approximately 21–24 nucleotides in length, which play an important function in post-transcriptional regulation of gene expression during development. The first miRNA, lin-4, was discovered in Caenorhabditis elegans in 1993.10 This class of small non-coding RNAs is highly evolutionarily conserved and induces translational repression or degradation of a target messenger (mRNA), according to the biogenesis and mechanism outlined below.11,12
miRNA genes are mainly transcribed by RNA polymerase II into a primary transcript forming a self-folded hairpin structure,13,14 which is recognized and processed by the RNAse III enzyme Drosha in the nucleus, to yield a 70 base pair (bp) precursor miRNA (pre-miRNA).15 Pre-miRNAs are subsequently transported from the nucleus to the cytoplasm by an exportin-5 dependent mechanism. Once in the cytosol, pre-miRNAs are further processed by a second RNAse III enzyme, Dicer, into a 23 bp double stranded miRNA/miRNA*.16 One strand is loaded into the RNA-induced silencing complex (RISC), which requires the presence of proteins belonging to the Argonaute family. Complementarily, mature miRNAs guide RISC to their target mRNAs via base-pairing interactions between the 5′ end of the miRNA and the 3′ untranslated region (3′UTR) of the target mRNA,11,12 which results in mRNA translational repression or degradation.17 Figure 1 shows the miRNA biogenesis pathway.18–20
Figure 1.
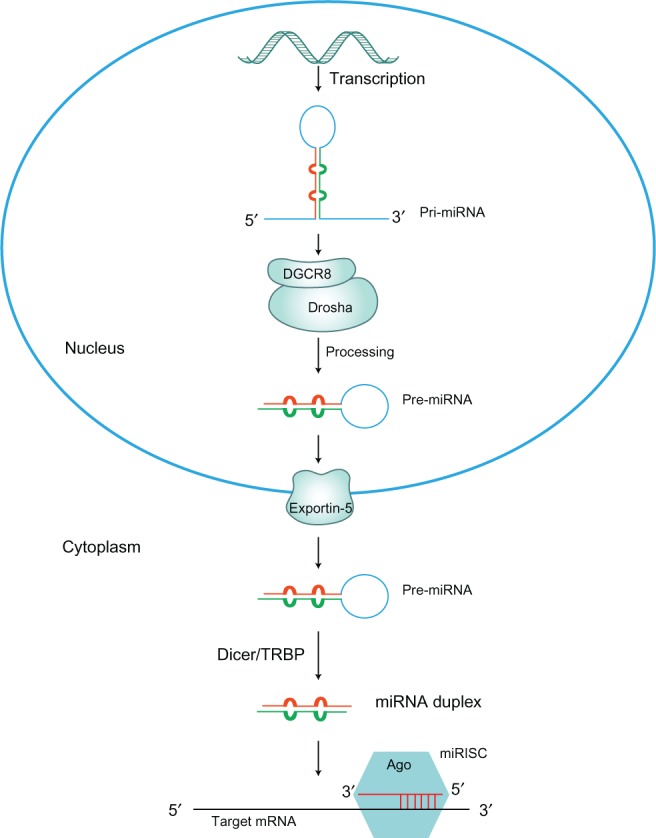
The miRNA biogenesis pathway.
Notes: The first step of microRNA biogenesis is the transcription of a long pri-miRNA that contains loop–stem structures. These loop–stem structures will be further cleaved-off, mediated by the Drosha-DGCR8 complex, a processing step that also takes place in the nucleus. The cleavage product is called pre-microRNA and has a typical two nucleotide 3′ overhang, essential for nuclear export by exportin-5. Pre-miRNA constitutes a transport complex together with exportin-5 and its cofactor Ran (the GTP-bound form). Following export, the pre-miRNA is cleaved by Dicer in combination with the TRBP. This yields an approximately 20 base pair miRNA duplex, which is separated and usually one strand is selected as the mature miRNA, whereas the other strand is degraded. After incorporation into miRISC and strand selection, the mature miRNA strand induces translational repression and/or mRNA cleavage, leading to reduction of the protein.18–20
Abbreviations: Ago, argonaute; GTP, guanosine triphosphate; miRISC, miRNA-induced silencing complex; miRNA, microRNA; pri-miRNA, primary miRNA; pre-miRNA, precursor miRNA; TRBP, trans-activation response element RNA binding protein.
Two common mechanisms for miRNA mediated gene regulation have been described above: translational repression and mRNA degradation.21 In fact, a recent study showed that decreasing miRNA levels by destabilization of mammalian target mRNAs is a major mechanism in miRNA gene repression.22 An individual miRNA is able to target up to a few hundred different mRNAs23 and therefore, is able to regulate the expression of multiple and diverse proteins involved in a biological process.24 Recent evidence suggests that translation regulation is an important process in the pathophysiology of PD. It is hardly surprising that the number of roles assigned to miRNAs in the pathogenesis of PD is rapidly expanding.
In this review we focus on the recent advances in the use of miRNAs in pathological processes of PD.
miRNAs and SNCA
The α-synuclein gene (SNCA), encoding the protein SNCA, is one of the key genes playing a central role in the pathogenesis of PD.25 In PD, α-synuclein is the key protein that aggregates and accumulates in fibrillar form in the pathological hallmark lesions known as Lewy bodies and Lewy neuritis in vulnerable neurons.26,27 It is almost certain that the presence of α-synuclein-rich Lewy bodies is caused by inefficient clearance of α-synuclein. In in vitro studies, α-synuclein protein was shown to undergo concentration-dependent self-aggregation by a nucleation-dependent mechanism.28 Factors that reduce protein clearance efficiency will tip the balance in favor of protein aggregation and will promote the development of inclusions (Figure 2).29 Additionally, it was shown in a series of works that excess α-synuclein is deleterious to neurons and in particular dopamine neurons, the death of which is responsible for the characteristic motor features of PD.30–33
Figure 2.

Putative mechanisms of α-synuclein neurodegeneration in familial PD.
Notes: The presence of Lewy bodies in PD is caused by a failure to properly dispose of α-synuclein. Point mutations in α-synuclein and the recently described Iowan functional duplication lead to excessive intracellular accumulation of α-synuclein, resulting in spontaneous oligomerization of this protein. Mutant α-synuclein can inhibit proteasomal activity, potentially leading to enhanced accumulation; soluble α-synuclein protofibrils and intracellular protein aggregates have been shown to impair the proteasome. Dopamine may also form stable adducts with protofibrils, potentially enhancing this toxicity. Genetic mutations associated with the UPP pathway, including parkin and UCH-L1, are associated with familiar PD. Combined with other cellular stress, this leads to neuronal loss.
Copyright © 2003. Elsevier. Reproduced with permission from Eriksen JL, Dawson TM, Dickson DW, Petrucelli L. Caught in the act: α-synuclein is the culprit in Parkinson’s disease. Neuron. 2003;40(3):453–456.29
Abbreviations: PD, Parkinson’s disease; UCH-L1, ubiquitin carboxy-terminal hydrolase L1; UPP, ubiquitin proteasome pathway.
To date, two specific miRNAs were recently shown to regulate α-synuclein levels post-transcriptionally: miR-734 and miR-153,35 which are abundantly expressed in the brain. The human α-synuclein mRNA has a 3′-UTR, which has potential functional significance in regulating the stability of the mRNA and the rate of protein translation. Further, it was shown that the brain-enriched miRNAs, miR-7 and miR-153, preferentially bind to the SNCA mRNA 3′-UTR and significantly downregulate α-synuclein expression in PD mouse models (Figure 3).34,35 Recently, miR-7 has been reported to be specifically expressed during neuroblastoma differentiation, cortical development, embryonic stem cell differentiation, and control neurite outgrowth in vitro.36 Therefore, it is conceivable that depletion of certain miRNA species resulting in upregulated expression of α-synuclein levels in a PD brain would subsequently indicate a functional role of these miRNAs in the disease process.
Figure 3.
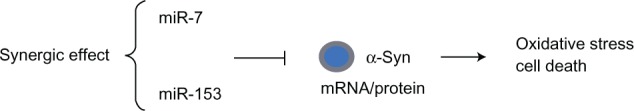
MicroRNA (miR-7 and miR-153) regulation of SNCA in PD.
Notes: The brain-enriched miRNAs, miR-7 and miR-153, are predicted to bind to the α-syn mRNA 3′-UTR as their targets. There is a significant synergy effect between miR-7 and miR-153 in down regulating α-syn mRNA levels and protein expressions. On the contrary, depletion of these miRNAs results in a concomitant increase in α-syn levels in a PD brain. miR-7 has a protective role by preventing oxidative stress and miR-7 inhibition causes cell death.
Abbreviations: 3′-UTR, 3′ untranslated region; α-syn, α-synuclein; mRNA, messenger RNA; miRNA, microRNA; PD, Parkinson’s disease.
miR-7, which is expressed mainly in neurons, is predicted to bind to the α-synuclein 3′-UTR with its seed sequence complementary to nucleotides 119–127, which is conserved in humans, rats, mice, dogs, and chickens.34 Transfection of α-synuclein-expressed cells with miR-7 showed that α-synuclein protein expressions and mRNA levels were both significantly reduced, indicating that miR-7 promotes the degradation of its target mRNA. Thus, the main mechanism of the downregulation of α-synuclein expression by miR-7 is decreased mRNA stability rather than alteration of translation rate. Furthermore, this effect would be lost with mutation of the miR-7 target sequence and in the absence of the 3′-UTR, which confirmed that repression of α-synuclein expression by miR-7 requires the 3′-UTR of the α-synuclein mRNA. It has been confirmed that miR-7 actively represses α-synuclein protein expression; on the contrary, endogenous levels of α-synuclein protein are significantly increased by using specific anti-miRNA inhibitors against miR-7.31 Further, the expression of miR-7 is decreased in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) intoxicated mouse model of PD, which may contribute to the increased α-synuclein expression in this model. The role of miR-7 in downregulating α-synuclein expression post-transcriptionally has been confirmed in several cellular systems.35 Importantly, miR-7 mediated repression of α-synuclein expression protects against the impaired proteasomal function37,38 and cytotoxicity39,40 associated with α-synuclein overexpression. Antisense inhibition of miR-7 in HeLa cells has also been found to downregulate cell growth and increase apoptosis,41 suggesting that miR-7 has a protective role by preventing oxidative stress.34,42
miR-153 is another miRNA species shown to down-regulate α-synuclein expression by targeting the α-synuclein 3′-UTR where the seed match is located between bases 462 and 468, a sequence that is conserved across vertebrate species.35 In particular, it was shown that miR-153 is also predominantly expressed in the brain,43 specifically binding to the α-synuclein mRNA 3′-UTR and downregulating its mRNA and protein levels.35 Overexpression of miR-153 significantly reduces endogenous α-synuclein levels, whereas miR-153 inhibition enhances translation of a luciferase construct bearing the α-synuclein 3′-UTR in primary neurons.
In a recent work, quantitative analysis of gene transcripts and SNCA protein showed that miR-7 and miR-153 have an additive effect in downregulating α-synuclein expression.35 miR-7 and miR-153 alone produced a 33% and a 12% decrease, respectively, in reporter gene expression, while the combination of the two microRNAs, miR-153/7, resulted in 47% repression, indicating that there is significant synergy between miR-7 and miR-153 to repress α-synuclein mRNA levels and protein expression. These findings reveal that first, miR-7 and miR-153 are synergistic in their effect; second, the 3′-UTR of SNCA mRNA is necessary to regulate SNCA protein expression; third, miRNAs do not interact with the coding region of SNCA to regulate SNCA protein expression; and fourth, miRNAs act at the pre-translational level.35 Regulation of this mRNA level is likely due to miR-7- and miR-153-induced elevation of mRNA deadenylation and uncapping, as they completely complement SNCA mRNA.44,45 Specifically, both miR-7 and miR-153, as well as α-synuclein mRNA and protein, are found in highest levels in nervous tissue, particularly in the midbrain, hippocampus, and cortex, and their expression is restricted to neurons rather than astrocytes.34,35 However, in other organs, such as the lungs and heart, the level of these RNA transcripts was considerably lower. Interestingly, miR-7, miR-153, and SNCA mRNA show highest expression in the midbrain, indicating that deregulation of their expression levels may be important in the pathogenesis of PD. This correspondence in expression pattern is maintained during prenatal and postnatal development, suggesting that miR-7 and miR-153, which are co-expressed with SNCA through a transcription feed-forward loop,35 play a significant role in regulation of α-synuclein levels in patients with familial or sporadic PD.
miRNAs and LRRK2
LRRK2, the protein known as dardarin, was identified in 2004 as being responsible for the most common known genetic form of PD, with autosomal dominant inheritance and low penetrance.46,47 In addition to the growing body of knowledge showing that specific miRNAs target and regulate the expression of disease-associated genes, LRRK2 in a recent study that while specific miRNAs regulate the expression of disease-associated genes, a disease-linked protein can also conversely regulate the miRNA pathway.48
LRRK2 is supposed to exhibit GTPase activity, which is typical of ROCO family protein members. In the Drosophila model,49 analysis of transgenic Drosophilia expressing the pathogenic mutant forms of human LRRK2 gene, I1915T or G2019S, demonstrated that defective LRRK2 does not possess GTPase activity. The LRRK2 gene therefore derepressed miRNA-mediated inhibition of the transcription factors E2F1 and DP,48 which are involved in the control of cell cycle and survival,49,50 and thereby increased dopaminergic neuronal death.
miRNAs of the let-7 and miR-184* families were found to repress the translation of the two factors E2F1 and DP, respectively.48 Pathogenic LRRK2 inhibited let-7 and miR-184* function and consequently E2F1 and DP were upregulated; such effects were dependent on LRRK2 kinase activity. Deletion of let-7, antagomir-mediated inhibition of let-7 and miR-184* actions, and blockage of binding sites for let-7 and miR-184* in 3′-UTRs of their target mRNAs, led to increased expression of E2F1/DP and development of toxic effects similar to that associated with pathogenic LRRK2, which resulted in reduced locomotor activity and dopamine neuron count (Figure 4). On the other hand, increasing the levels of these two miRNAs partially attenuated the unfavorable effect of mutant LRRK2.48 Recently, it has been shown that let-7b, a member of the let-7 miRNA family, regulates neural stem cell proliferation and differentiation by targeting the stem cell regulator TLX and the cell cycle regulator cyclin D1.51 Overexpression of let-7b led to reduced neural stem cell proliferation and increased neural differentiation, whereas antisense knockdown of let-7b resulted in enhanced proliferation of neural stem cells.51
Figure 4.
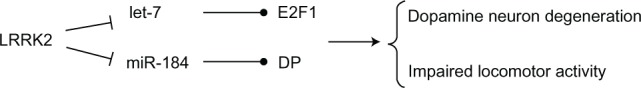
LRRK2 regulates the microRNA (let-7 and miR-184*) network.
Notes: Pathogenic LRRK2 inhibited let-7 and miR-184* function and consequently transcription factors E2F1 and DP were upregulated, and such effects were dependent on LRRK2 kinase activity. Overexpression of E2F1 and DP may lead to dopamine neuron degeneration and impaired locomotor activity.
Abbreviation: LRRK2, leucine-rich repeat kinase 2.
The effect of action of pathogenic LRRK2 mutants on miRNA regulation might depend on their co-immunoprecipitation with components of the RISC. In addition, the RISC core protein argonaute-1 levels are negatively regulated by LRRK2 in aged, but not young, flies, and promoted the assocation of argonaute-1 with the increased phosphorylation of 4E-BP in relation to LRRK2.48 These findings indicate that LRRK2 is playing an important role in the pathogenesis of PD, which is mediated through the miRNA pathway, thus paving the way for new therapeutic strategies for PD.
Although these studies demonstrate that translational regulation of E2F1 and DP by miRNAs let-7 and miR-184* is involved in LRRK2-mediated neurodegeneration, the complex mechanism by which mutant LRRK2 inhibits the miRNA pathway and causes neuronal death in the Drosophila still needs to be delineated in a mammalian system.
Parkin, DJ-1, and miRNAs
In accordance with numerous studies, the two genes PAPK2 and PAPK7 are also associated with the pathogenesis of recessive parkinsonism.52–56 Parkin protein is encoded by the PAPK2 gene, whose mutations on chromosome 6q25.2-27 will cause autosomal recessive juvenile parkinsonism (AR-JP), one of the most common familial forms of PD.53 It has been reported that parkin participates in the proteasome-mediated degradation as a ubiquitin-protein ligase collaborating with the ubiquitin-conjugating enzyme UbcH7.53 Furthermore, a study of more than 100 families with AR-JP described mutations in parkin, which showed loss of the ubiquitin-protein ligase activity, indicating that mutations in the PARK2 gene are the cause of the development of PD.54
DJ-1, whose protein is encoded by the PARK7 gene, is a second protein involved in autosomal recessive primary parkinsonism with its homozygous mutations.55,56 DJ-1 plays a causal role in the cellular oxidative stress response, in that its deficiency displays proteasomal inhibition and an apparently normal initial burst of reactive oxygen species (ROS),57 leading to oxidative stress and apoptosis.58 Proteasomal inhibition and ROS have previously been correlated with PD pathology,59 thus the hypothesis that DJ-1 mutations lead to PD because of an increased sensitivity to such stressors.60 Some recent studies demonstrated that DJ-1 deficiency caused severe mitochondria fragmentation,61–63 which suggested that DJ-1 is also involved in the regulation of mitochondrial dynamics.64 Additionally, some data indicates that DJ-1 can bind to parkin during oxidative stress and protect mitochondria from damage, and showing the common neuroprotective role of these two proteins.65
In PD patients, miR-34b and miR-34c are downregulated compared with healthy controls at early (premotor) stages in brain samples,66 accompanied by a significant decrease in the concentrations of parkin and DJ-1 proteins.67,68 The levels of miR-34b and miR-34c are decreased by 40%–65% in affected brain areas such as the amygdala, substantia nigra, frontal cortex, and cerebellum.66 In addition, depletion of miR-34b/miR-34c in differentiated SH-SY5Y neuroblastoma cells resulted in cell death associated with impaired mitochondrial function and oxidative stress, which are consistently recognized biochemical defects in PD brains (Figure 5).66 Nevertheless, it remains to be proven whether the decreased expression of these miRNAs results from the death of dopaminergic neurons or from their specific downregulation in surviving neurons.66 Characterizing the physiological targets of miR-34b and miR-34c in neurons also needs to be better understood.
Figure 5.
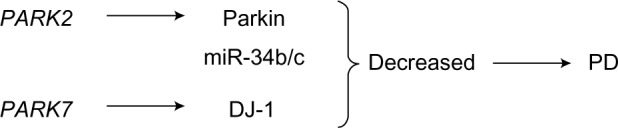
The role of PARK2 and PARK7 in the pathogenesis of PD.
Notes: In accordance with numerous studies, the genes PARK2 and PARK7 are also involved in the pathogenesis of PD. PARK2 encodes the parkin protein, and the DJ-1 protein is encoded by PARK7. In the brains of PD patients, the levels of miR-34b and miR-34c are significantly decreased, decreased levels of miR-34b and miR-34c are also accompanied by decreased concentrations of parkin and DJ-1 proteins. Nevertheless, there is no experimental evidence for miR-34b/miR-34c targeting to genes encoding parkin and DJ-1.
Abbreviation: PD, Parkinson’s disease.
miRNA and other candidate genes
Study of the relative abundance of miRNAs in affected brain regions in patients with PD and their comparison to non-affected controls has been one strategy to understand the role of specific miRNAs in the disease. The work of Kim et al was the first study to focus on the expression profiles of 224 different miRNAs precursors obtained from brain regions of patients with PD and control subjects.69–70 This finding has been confirmed by quantitative reverse transcription polymerase chain reaction, ribonuclease protection assay, and Northern blotting. Within this miRNA panel, eight miRNAs appeared to be specifically enriched in the midbrain compared to the cerebral cortex and cerebellum. The expression of one of these miRNAs, miR-133b, has been found to be significantly deficient in the midbrain of PD patient samples compared to controls.70 In addition, miR-133b appears to function as a negative regulator of dopaminergic neuron development, suggesting a direct link to dopamine homeostasis. The elevated level of miR-133b in primary embryonic rat midbrain cultures results in the decrease in dopamine release from neurons, while its lowered level has the opposite effect in number of dopamine neurons.69
miR-133b is also downregulated in mouse dopamine neuron deficient models (Aphakia strain,69 a naturally occurring mouse mutant, which can be used as a model of neuronal loss in human PD).71 Aphakia mice have a mutation in pituitary homeobox 3 (Pitx3), which is considered to be a PD candidate gene and a marker of DA-neurons. Pitx3, a homeobox transcription factor that is required for dopaminergic neuron differentiation in the midbrain substantia nigra,72–74 was recently identified as an miR-133b target.69 Additionally, two polymorphisms in Pitx3 have been associated with sporadic PD in humans.75,76
A murine model of PD suggested that miR-133b and Pitx3 regulate expression of each other via a negative feedback loop:69 the Pitx3 gene induces miR-133b transcription, which in turn represses Pitx3 expression.69 Thus, loss of Pitx3 also results in the loss of miR-133b in the PD model. Nevertheless, an important question remains unanswered concerning whether the loss of the miRNA contributes to the development of PD. However, in the in vitro dopamine neuron (DN) differentiation assay, overexpression of miR-133b targeted Pitx3 and resulted in the loss of DN differentiation.77 Conversely, inhibition of miR-133b led to increased expression of DN markers, indicating enhanced DN differentiation. This negative feedback mechanism regulates midbrain dopaminergic neurons activity and terminal differentiation (Figure 6). Although a miR-133b knockout mouse model has not yet been developed,78 PD probably results from the loss of Pitx3-dependent gene expression rather than the lack of miR-133b. Therefore, whether miR-133b might have protective potential against dopaminergic neuronal neurodegeneration, remains to be verified.
Figure 6.
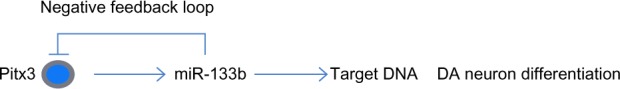
miR-133b and Pitx3 form a negative feedback loop.
Notes: Summary of the regulation of DA neuronal events in the brain within a negative feedback loop. The transcription factor Pitx3 induces transcription of miR-133b during neural differentiation, which in turn represses Pitx3.
Abbreviations: DA, dopaminergic; Pitx3, pituitary homeobox 3.
Another important candidate gene associated with the development of PD is the fibroblast growth factor 20 gene (FGF20), a neurotrophic factor preferentially expressed in the substantia nigra, stimulating the maturation of dopaminergic neurons.79,80 Single nucleotide polymorphisms (SNPs) are small genetic variations in chromosomal DNA sequences where a single nucleotide is replaced by one of the other three nucleotides. SNPs in miRNA target sites will affect transcription and processing of miRNAs, as well as the interactions between miRNAs and their targets, potentially leading to various human diseases ranging from cancer to PD.81 van der Walt et al demonstrated that the polymorphic variations in the FGF20 gene influence the risk of PD development, including two SNPs, rs1721100 and ss20399075, located within highly conserved sequences of the 3′-UTR.82
More recently, another SNP, rs12720208, which is also located in the 3′-UTR of the FGF20 mRNA, was identified to have the strongest association with risk for PD.83 This SNP lies within a predicted binding site for miR-433, which is highly expressed in the brain, leading to reduced miR-433 binding efficiency.84 In an experimental model, the risk allele rs12720208 decreased the binding of the mRNA with miR-433 and resulted in the overexpression of FGF20. Furthermore, increased levels of FGF20 led to a concomitant increase in α-synuclein levels, which has previously been shown to cause PD through both overexpression and point mutations (Figure 7).85 Human brain samples with the miR-433 SNP demonstrated upregulation of both FGF20 and α-synuclein.84 Thus, it is predicted that elevated FGF20 might predispose to PD through long-term upregulation of α-synuclein and further enhance neurogenesis early in life, but also create chronic elevation leading to degeneration during later life stages.83 Conversely, subsequent reports found no association between FGF20 polymorphism and PD risk, or a relationship between the rs12720208 genotype, FGF20, and α-synuclein protein levels.86,87 Thus, FGF20 probably plays an important role in subpopulations of PD and should be a focus for future study.
Figure 7.
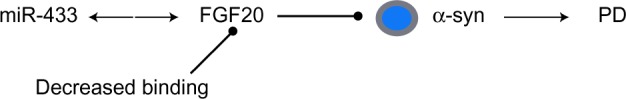
MicroRNA (miR-433) regulation of FGF20 in PD.
Notes: FGF20 disruptions are associated with a higher risk of PD, and one of its SNPs is within a miR-433 binding site. Decreased miR-433 binding efficiency results in FGF20 overexpression and a concomitant increase in α-syn protein levels.
Abbreviations: α-syn, α-synuclein; FGF20, fibroblast growth factor 20; PD, Parkinson’s disease; SNP, single-nucleotide polymorphism.
miRNA and PD therapy
The design of effective miRNA-based PD therapeutics faces an immediate challenge because of the complexity of neurodegenerative diseases. RNA interference has previously been exploited with great success in knocking down the expression of selected target genes and reducing protein levels involved in PD.88,89 As an example, targeting α-synuclein by infusion of chemically modified short interfering RNA90 or injection of small-hairpin-RNA-specific lentiviral vector systems91 notably decreased protein levels. The expectation has been proposed that RNA-interference-based therapies will be one of the major classes of drugs in the future,92 which strongly supports the notion that miRNAs may be better candidates for regulating endogenous gene expression in RNA-based therapies.93 As miRNA drugs do not appear to have the same issues with toxicity as short interfering RNA or small hairpin RNA based therapies,94,95 they are gaining more attention for their special potential in PD therapy.
In order to manipulate the expression levels of disease-related miRNAs in the hope of curing or prolonging disease onset, two miRNA-based therapeutic approaches are being investigated in vitro and in vivo. On the one hand, introduction of mature miRNA mimics, which are small RNA molecules that resemble miRNA precursors, can be used to effectively downregulate the expression of specific target proteins. The target protein can be any gene involved in disease pathogenesis, or the disease gene itself having a gain-of-function pathogenic mutation.96 However, there are also two main challenges that must be conquered in the use of this miRNA-based approach before assessing their efficacy in humans. For one thing, delivery of any drug to the brain by peripheral administration faces the particular technical hurdle posed by delivery across the blood–brain barrier. For another, the off-target physiological effect is another downfall in manipulating the levels or function of a specific miRNA. Although synthetic miRNA mimics are frequently used in culture research, the efficacy of miRNA mimics in in vivo conditions have not been studied extensively.97–99 Presumably, adequate dose optimization of the miRNA mimic must be achieved to create persistent effects before clinical application.96
On the other hand, creating a loss-of-function in the miRNA of interest can be achieved by exploiting synthetic anti-miRNA molecules.100 These are single-stranded antisense oligonucleotides with complementary sequence that bind to and inactivate the miRNA target, thus blocking these overexpressed miRNAs. In a previous study, Stoffel’s group designed chemically modified, cholesterol-conjugated single-stranded RNA analogs complementary to miRNAs, which were termed as “antagomirs,” to help the RNA enter cells.101 Another example is of modified locked nucleic acids anti-miRs that were designed specifically against miR-122, an abundant liver-specific miRNA.102,103 This approach has also been successfully used to reduce the amount of miR-122 without showing any toxicity in nonhuman primates and mice.102,103 Intravenous administration of chemically engineered oligonucleotides against several certain miR-NAs led to a marked reduction of corresponding miRNA levels in many organs, and the silencing of these miRNAs is specific, efficient, and long-lasting.101 An elegant study further demonstrated the therapeutic feasibility and safety of miRNA inhibition of this approach in a primate disease model.104 Therefore, the modified oligonucleotides, termed antagomirs, are efficient and specific silencers of endogenous miRNAs through intravenous injections or cerebrospinal fluid infusion, providing a potential role in miRNA-based therapy. The locked-nucleic-acids-modified oligonucleotides targeting miR-122 (SPC3649) method is currently being used in Phase I clinical trials for hepatitis C virus infection, probably becoming the first miRNA therapeutic target in humans.
Although miRNA-based therapies pose many unsolved challenges, including the specificity of miRNA function and the specific noninvasive delivery to the central nervous system, they still present exciting opportunities to combat humankind’s debilitating diseases.
Conclusion
In the past few years, the field of miRNA study has revealed highly significant molecular mechanisms relevant to PD pathogenesis. Emerging evidence strongly indicates the important role of miRNAs in the pathogenesis of this disorder and the disturbance of processes associated with regulation of the expression of genes implicated in its development. However, complex challenges also remain and miRNA therapy or diagnosis may be many years away from entering the clinic (Figure 8).105 This review represents the tip of the iceberg in the identification of miRNAs and their targets involved in PD, which has been summarized in Table 1. We strongly believe that miRNA-based techniques will be helpful in addressing unresolved questions concerning PD via in vitro and in vivo approaches. Given the pace of new findings and the discovery of applications, miRNA is likely to remain a major new therapy for the foreseeable future.
Figure 8.
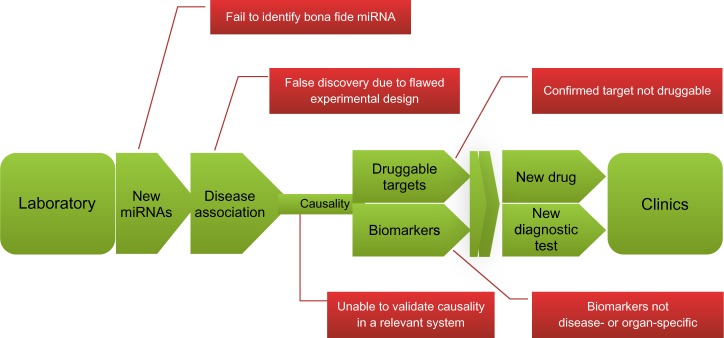
The road from laboratory to clinic: the promises and challenges of miRNA research.
Notes: The hopscotch course in green is a layout of an ideal path of miRNA research evolved from basic research to clinical practice. Red boxes indicate major challenges at different steps.
Copyright © 2003. Elsevier. Adapted with permission from Li Y, Kowdley KV. MicroRNAs in Common Human Diseases. Genomics Proteomics Bioinformatics. 2012;10(5):246–253.105
Abbreviation: miRNA, microRNA.
Table 1.
miRNAs potentially involved in PD pathophysiology
| miRNA | Target genes | Function | Reference |
|---|---|---|---|
| miR-7 | SNCA | Downregulate α-synuclein expression in nervous system, protect cells against oxidative stress | 34 |
| miR-153 | SNCA | Directly reduce SNCA protein expression. miR-7 and miR-153 are synergistic in their effect | 35 |
| let-7 | E2F1a, pdr-1 | Co-underexpress in the alpha-synuclein and pdr-1 strains; influence on pathogenicity of mutant LRRK2 via E2F1 and DP | 48,106 |
| miR-184* | DPa | Regulate DAb neurons survival and activity: influence on pathogenicity of mutant LRRK2 via E2F1 and DP | 48 |
| miR-34b | Not found | Regulate expression of DJ1 and Parkin, two proteins associated to familial forms of PD | 66 |
| miR-34c | Not found | Regulate expression of DJ1 and Parkin, two proteins associated to familial forms of PD | 66 |
| miR-133b | Pitx3c | Regulation of maturation and functioning of midbrain dopaminergic neurons within a negative feedback circuit | 69 |
| miR-433 | FGF20d | Increase translation of FGF20, which is correlated with increased α-synuclein expression | 83 |
Notes:
E2F1 and DP (transcription factors);
DA (dopaminergic);
Pitx3 (pituitary homeobox 3);
FGF20 (fibroblast growth factor 20).
Abbreviations: DA, dopaminergic; FGF20, fibroblast growth factor 20; LRRK2, leucine-rich repeat kinase 2; Pitx3, pituitary homeobox 3; PD, Parkinson’s disease.
Acknowledgments
The study was supported by the Projects of National Science Foundation of China (numbers 81071025, 81171203, 81171204, and 81200871), and Projects of the Shanghai Committee of Science and Technology, People’s Republic of China (numbers 11 nm0503300, 11410708900, and 12XD1403800).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Robinson PA. Protein stability and aggregation in Parkinson’s disease. Biochem J. 2008;413(1):1–13. doi: 10.1042/BJ20080295. [DOI] [PubMed] [Google Scholar]
- 2.Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116(7):1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parkinson J. An Essay on the Shaking Palsy. London: Sherwood, Neely and Jones; 1817. [Google Scholar]
- 4.D’Amelio M, Ragonese P, Sconzo G, Aridon P, Savettieri G. Parkinson’s disease and cancer: insights for pathogenesis from epidemiology. Ann N Y Acad Sci. 2009;1155:324–334. doi: 10.1111/j.1749-6632.2008.03681.x. [DOI] [PubMed] [Google Scholar]
- 5.Harraz MM, Dawson TM, Dawson VL. MicroRNAs in Parkinson’s disease. J Chem Neuroanat. 2011;42(2):127–130. doi: 10.1016/j.jchemneu.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42(9):781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302(5646):819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 8.Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med. 2009;11:e22. doi: 10.1017/S1462399409001148. [DOI] [PubMed] [Google Scholar]
- 9.Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41(12):1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 12.Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 13.Lee Y, Kim M, Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23(20):4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9(10):775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13(12):1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 16.Lee YS, Nakahara K, Pham JW, et al. Distinct roles for Drosophila Dicer-1 and Dicer-2 in the siRNA/miRNA silencing pathways. Cell. 2004;117(1):69–81. doi: 10.1016/s0092-8674(04)00261-2. [DOI] [PubMed] [Google Scholar]
- 17.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abe M, Bonini NM. MicroRNAs and neurodegeneration: role and impact. Trends Cell Biol. 2013;23(1):30–36. doi: 10.1016/j.tcb.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuss AW, Chen W. MicroRNAs in brain function and disease. Curr Neurol Neurosci Rep. 2008;8(3):190–197. doi: 10.1007/s11910-008-0031-0. [DOI] [PubMed] [Google Scholar]
- 20.Enciu AM, Popescu BO, Gheorghisan-Galateanu A. MicroRNAs in brain development and degeneration. Mol Biol Rep. 2012;39(3):2243–2252. doi: 10.1007/s11033-011-0973-1. [DOI] [PubMed] [Google Scholar]
- 21.Kosik KS. The neuronal microRNA system. Nat Rev Neurosci. 2006;7(12):911–920. doi: 10.1038/nrn2037. [DOI] [PubMed] [Google Scholar]
- 22.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microR-NAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 24.Liu NK, Xu XM. MicroRNA in central nervous system trauma and degenerative disorders. Physiol Genomics. 2011;43(10):571–580. doi: 10.1152/physiolgenomics.00168.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24(30):6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spillantini MG, Goedert M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann N Y Acad Sci. 2000;920:16–27. doi: 10.1111/j.1749-6632.2000.tb06900.x. [DOI] [PubMed] [Google Scholar]
- 27.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 28.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97(2):571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eriksen JL, Dawson TM, Dickson DW, Petrucelli L. Caught in the act: alpha-synuclein is the culprit in Parkinson’s Disease. Neuron. 2003;40(3):453–456. doi: 10.1016/s0896-6273(03)00684-6. [DOI] [PubMed] [Google Scholar]
- 30.Mutez E, et al. SNCA locus duplication carriers: from genetics to Parkinson disease phenotypes. Hum Mutat. 2011;32(4):E2079–E2090. doi: 10.1002/humu.21459. [DOI] [PubMed] [Google Scholar]
- 31.Sekine T, et al. Clinical course of the first Asian family with Parkinsonism related to SNCA triplication. Mov Disord. 2010;25(16):2871–2875. doi: 10.1002/mds.23313. [DOI] [PubMed] [Google Scholar]
- 32.Cullen V, et al. Acid beta-glucosidase mutants linked to gaucher disease, parkinson disease, and lewy body dementia alter alpha-synuclein processing. Ann Neurol. 2011;69(6):940–953. doi: 10.1002/ana.22400. [DOI] [PubMed] [Google Scholar]
- 33.Yavich L, Tanila H, Vepsalainen S, Jäkälä P. Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci. 2004;24(49):11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106(31):13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285(17):12726–12734. doi: 10.1074/jbc.M109.086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen H, Shalom-Feuerstein R, Riley J, et al. miR-7 and miR-214 are specifically expressed during neuroblastoma differentiation, cortical development and embryonic stem cells differentiation, and control neurite outgrowth in vitro. Biochem Biophys Res Commun. 2010;394(4):921–927. doi: 10.1016/j.bbrc.2010.03.076. [DOI] [PubMed] [Google Scholar]
- 37.Petrucelli L, O’Farrell C, Lockhart PJ, et al. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36(6):1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 38.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21(24):9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang H, Wu YC, Nakamura M, et al. Parkinson’s disease genetic mutations increase cell susceptibility to stress: mutant alpha-synuclein enhances H2O2- and Sin-1-induced cell death. Neurobiol Aging. 2007;28(11):1709–1717. doi: 10.1016/j.neurobiolaging.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 40.Junn E, Mouradian MM. Human alpha-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci Lett. 2002;320(3):146–150. doi: 10.1016/s0304-3940(02)00016-2. [DOI] [PubMed] [Google Scholar]
- 41.Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33(4):1290–1297. doi: 10.1093/nar/gki200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lau P, de Strooper B. Dysregulated microRNAs in neurodegenerative disorders. Semin Cell Dev Biol. 2010;21(7):768–773. doi: 10.1016/j.semcdb.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Farh KK, Grimson A, Jan C, et al. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310(5755):1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 44.Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 2006;20(14):1885–1898. doi: 10.1101/gad.1424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eulalio A, Huntzinger E, Nishihara T, Rehwinkel J, Fauser M, Izaurralde E. Deadenylation is a widespread effect of miRNA regulation. RNA. 2009;15(1):21–32. doi: 10.1261/rna.1399509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 47.Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466(7306):637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Girling R, Partridge JF, Bandara LR, et al. A new component of the transcription factor DRTF1/E2F. Nature. 1993;362(6415):83–87. doi: 10.1038/362083a0. [DOI] [PubMed] [Google Scholar]
- 50.Ingram L, Munro S, Coutts A, La Thangue N. E2F-1 regulation by an unusual DNA damage-responsive DP partner subunit. Cell Death Differ. 2011;18(1):122–132. doi: 10.1038/cdd.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao C, Sun G, Li S, et al. MicroRNA let-7b regulates neural stem cell proliferation and differentiation by targeting nuclear receptor TLX signaling. Proc Natl Acad Sci U S A. 2010;107(5):1876–1881. doi: 10.1073/pnas.0908750107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cookson MR. Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb Perspect Med. 2012;2(9):a009415. doi: 10.1101/cshperspect.a009415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimura H, Hattori N, Kubo S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25(3):302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 54.Illarioshkin SN, Periquet M, Rawal N, et al. Mutation analysis of the parkin gene in Russian families with autosomal recessive juvenile parkinsonism. Mov Disord. 2003;18(8):914–919. doi: 10.1002/mds.10467. [DOI] [PubMed] [Google Scholar]
- 55.Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299(5604):256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 56.Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SM. Neuroprotective Function of DJ-1 in Parkinson’s Disease. Oxid Med Cell Longev. 2013;2013:683920. doi: 10.1155/2013/683920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004;2(11):e362. doi: 10.1371/journal.pbio.0020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Demasi M, Davies KJ. Proteasome inhibitors induce intracellular protein aggregation and cell death by an oxygen-dependent mechanism. FEBS Lett. 2003;542(1–3):89–94. doi: 10.1016/s0014-5793(03)00353-3. [DOI] [PubMed] [Google Scholar]
- 59.Dauer W, Przedborski S. Parkinson’s disease: Mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 60.Billia F, Hauck L, Grothe D, et al. Parkinson-susceptibility gene DJ-1/PARK7 protects the murine heart from oxidative damage in vivo. Proc Natl Acad Sci U S A. 2013;110(15):6085–6090. doi: 10.1073/pnas.1303444110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Irrcher I, Aleyasin H, Seifert EL, et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet. 2010;19(19):3734–3746. doi: 10.1093/hmg/ddq288. [DOI] [PubMed] [Google Scholar]
- 62.Krebiehl G, Ruckerbauer S, Burbulla LF, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One. 2010;5(2):e9367. doi: 10.1371/journal.pone.0009367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thomas KJ, McCoy MK, Blackinton J, et al. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet. 2011;20(1):40–50. doi: 10.1093/hmg/ddq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem. 2012;121(5):830–839. doi: 10.1111/j.1471-4159.2012.07734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mandemakers W, Morais VA, De Strooper B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci. 2007;120(Pt 10):1707–1716. doi: 10.1242/jcs.03443. [DOI] [PubMed] [Google Scholar]
- 66.Miñones-Moyano E, Porta S, Escaramís G, et al. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet. 2011;20(15):3067–3078. doi: 10.1093/hmg/ddr210. [DOI] [PubMed] [Google Scholar]
- 67.Shapshak P. Molecule of the month: miRNA and Parkinson’s disease protein PARK2. Bioinformation. 2013;9(8):381–382. doi: 10.6026/97320630009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saito Y, Saito H. MicroRNAs in cancers and neurodegenerative disorders. Front Genet. 2012;3:194. doi: 10.3389/fgene.2012.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim J, Inoue K, Ishii J, et al. A microRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317(5842):1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hebert SS, De Strooper B. Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci. 2009;32(4):199–206. doi: 10.1016/j.tins.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 71.Van den Munckhof P, Luk KC, Ste-Marie L, et al. Pitx3 is required for motor activity and for survival of a subset of midbrain dopaminergic neurons. Development. 2003;130(11):2535–2542. doi: 10.1242/dev.00464. [DOI] [PubMed] [Google Scholar]
- 72.Hwang DY, Ardayfo P, Kang UJ, Semina EV, Kim KS. Selective loss of dopaminergic neurons in the substantia nigra of Pitx3-deficient aphakia mice. Brain Res Mol Brain Res. 2003;114(2):123–131. doi: 10.1016/s0169-328x(03)00162-1. [DOI] [PubMed] [Google Scholar]
- 73.Martinat C, Bacci JJ, Leete T, et al. Cooperative transcription activation by Nurr1 and Pitx3 induces embryonic stem cell maturation to the midbrain dopamine neuron phenotype. Proc Natl Acad Sci U S A. 2006;103(8):2874–2879. doi: 10.1073/pnas.0511153103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nunes I, Tovmasian LT, Silva RM, Burke RE, Goff SP. Pitx3 is required for development of substantia nigra dopaminergic neurons. Proc Natl Acad Sci U S A. 2003;100(7):4245–4250. doi: 10.1073/pnas.0230529100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fuchs J, Mueller JC, Lichtner P, et al. The transcription factor PITX3 is associated with sporadic Parkinson’s disease. Neurobiol Aging. 2009;30(5):731–738. doi: 10.1016/j.neurobiolaging.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 76.Bergman O, Håkansson A, Westberg L, et al. PITX3 polymorphism is associated with early onset Parkinson’s disease. Neurobiol Aging. 2010;31(1):114–117. doi: 10.1016/j.neurobiolaging.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 77.Li J, Dani JA, Le WD. The Role of Transcription Factor Pitx3 in Dopamine Neuron Development and Parkinson’s Disease. Curr Top Med Chem. 2009;9(10):855–859. [PMC free article] [PubMed] [Google Scholar]
- 78.Nelson PT, Wang WX, Rajeev BW. MicroRNAs (miRNAs) in Neurodegenerative Diseases. Brain Pathol. 2008;18(1):130–138. doi: 10.1111/j.1750-3639.2007.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ohmachi S, Watanabe Y, Mikami T, et al. FGF-20, a novel neurotrophic factor, preferentially expressed in the substantia nigra pars compacta of rat brain. Biochem Biophys Res Commun. 2000;277(2):355–360. doi: 10.1006/bbrc.2000.3675. [DOI] [PubMed] [Google Scholar]
- 80.Ohmachi S, Mikami T, Konishi M, Miyake A, Itoh N. Preferential neurotrophic activity of fibroblast growth factor-20 for dopaminergic neurons through fibroblast growth factor receptor-1c. J Neurosci Res. 2003;72(4):436–443. doi: 10.1002/jnr.10592. [DOI] [PubMed] [Google Scholar]
- 81.Sethupathy P, Collins FS. MicroRNA target site polymorphisms and human disease. Trends Genet. 2008;24(10):489–497. doi: 10.1016/j.tig.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 82.van der Walt JM, Noureddine MA, Kittappa R, et al. Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. Am J Hum Genet. 2004;74(6):1121–1127. doi: 10.1086/421052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang WX, Rajeev BW, Stromberg AJ, et al. The expression of microRNA miR-107 decreases early in Alzheimer‘s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28(5):1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davis TH, Cuellar TL, Koch SM, et al. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci. 2008;28(17):4322–4330. doi: 10.1523/JNEUROSCI.4815-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang G, van der Walt JM, Mayhew G, et al. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am J Hum Genet. 2008;82(2):283–289. doi: 10.1016/j.ajhg.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wider C, Dachsel JC, Soto AI, et al. FGF20 and Parkinson‘s disease: no evidence of association or pathogenicity via alpha-synuclein expression. Mov Disord. 2009;24(3):455–459. doi: 10.1002/mds.22442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Mena L, Cardo LF, Coto E, et al. FGF20 rs12720208 SNP and microRNA-433 variation: no association with Parkinson’s disease in Spanish patients. Neurosci Lett. 2010;479(1):22–25. doi: 10.1016/j.neulet.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 88.Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2(12):711–719. doi: 10.1038/nchembio839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de Fougerolles A, Vornlocher HP, Maraganore J, Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov. 2007;6(6):443–453. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lewis J, Melrose H, Bumcrot D, et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol Neurodegener. 2008;3:19. doi: 10.1186/1750-1326-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sapru MK, Yates JW, Hogan S, Jiang L, Halter J, Bohn MC. Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp Neurol. 2006;198(2):382–390. doi: 10.1016/j.expneurol.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 92.Blow N. Small RNAs: delivering the future. Nature. 2007;450(7172):1117–1120. doi: 10.1038/4501117a. [DOI] [PubMed] [Google Scholar]
- 93.Mack G. MicroRNA gets down to business. Nat Biotechnol. 2007;25(6):631–638. doi: 10.1038/nbt0607-631. [DOI] [PubMed] [Google Scholar]
- 94.McBride JL, Boudreau RL, Harper SQ, et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A. 2008;105(15):5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boudreau RL, McBride JL, Martins I, et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther. 2009;17(6):1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Junn E, Mouradian MM. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol Ther. 2012;133(2):142–150. doi: 10.1016/j.pharmthera.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boissonneault V, Plante I, Rivest S, Provost P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem. 2009;284(4):1971–1981. doi: 10.1074/jbc.M807530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hébert SS, Horré K, Nicolai L, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105(17):6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao X, He X, Han X, et al. MicroRNA-mediated control of oligodendrocyte differentiation. Neuron. 2010;65(5):612–626. doi: 10.1016/j.neuron.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10(3):544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Krützfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 102.Elmén J, Lindow M, Schütz S, et al. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452(7189):896–899. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 103.Elmén J, Lindow M, Silahtaroglu A, et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36(4):1153–1162. doi: 10.1093/nar/gkm1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327(5962):198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li Y, Kowdley KV. MicroRNAs in common human diseases. Genomics Proteomics Bioinformatics. 2012;10(5):246–253. doi: 10.1016/j.gpb.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Asikainen S, Rudgalvyte M, Heikkinen L, et al. Global microRNA expression profiling of Caenorhabditis elegans Parkinson’s disease models. J Mol Neurosci. 2010;41(1):210–218. doi: 10.1007/s12031-009-9325-1. [DOI] [PubMed] [Google Scholar]