

Abstract
Adenosine triphosphate-binding cassette subfamily G member 2 (ABCG2) plays a major role in cancer cell multidrug resistance, which contributes to low eifficacy of chemotherapy. Chalcones were recently found to be potent and specific inhibitors, but unfortunately display a significant cytotoxicity. A cellular screening against ABCG2-mediated mitoxantrone efflux was performed here by flow cytometry on 54 chalcone derivatives from three different series with a wide panel of substituents. The identified leads, with submicromolar IC50 (half maximal inhibitory concentration) values, showed that the previously identified 2′-OH-4′,6′-dimethoxyphenyl, as A-ring, could be efficiently replaced by a 2′-naphthyl group, or a 3′,4′-methylenedioxyphenyl with lower affinity. Such a structural variability indicates 3polyspecificity of the multidrug transporter for inhibitors. At least two methoxyl groups were necessary on B-ring for optimal inhibition, but substitution at positions 3, 4, and 5 induced cytotoxicity. The presence of a large O-benzyl substituent at position 4 and a 2′-naphthyl as A-ring markedly decreased the cytotoxicity, giving a high therapeutic ratio, which constitutes a critical requirement for future in-vivo assays in animal models.
Keywords: ABC transporters, BCRP/ABCG2, cancer chemotherapy, drug transport, inhibitory chalcone derivatives, multidrug resistance transporters
Introduction
Cancer cells display a strong ability to acquire resistance to antitumor drugs, termed multidrug resistance (MDR), which constitutes a critical hurdle to cancer therapy. While several mechanisms can induce cellular resistance to chemotherapeutics, the energy-dependent drug efflux, mediated by adenosine triphosphate-binding cassette (ABC) transporters, is recognized to play a major role. Overexpression of multidrug ABC transporters in tumors alters anticancer drug efficacy by significantly reducing their intracellular accumulation. ABC subfamily G member 2 (ABCG2), also called ABCP for its abundance in placenta,1 BCRP (breast cancer resistance protein),2 or MXR (mitoxantrone resistance protein),3 was the most recently discovered of the three main multidrug ABC transporters overexpressed in cancer cells, after ABC subfamily C member 1 (ABCC1)/MDR protein 1 (MRP1)4 and ABCB1/MDR1/P-glycoprotein.5 ABCG2 was shown to confer resistance to a wide variety of anticancer agents,3 revealing a characteristic polyspecificity toward transport drug substrates. Its clinical relevance was demonstrated in both adult and childhood acute myeloid leukemia,6,7 and it was clearly involved in drug bioavailability at various protection barriers.8,9 Overcoming MDR against anticancer agents, which is of importance for future clinical treatments, might be achieved through effective inhibitors of multidrug ABC transporters. ABCG2 inhibitors were therefore investigated in a number of studies, leading to identification of a few potent compounds.10,11 Different types of inhibitors were characterized: firstly, nonselective inhibitors already known as ABCB1 inhibitors and then considered as dual inhibitors, such as GF120918/elacridar12 and XR9576/tariquidar.13 A few inhibitors appeared to be selective: fumitremorgin C, from Aspergillus fumigatus,14 which was however highly neurotoxic and of which synthetic derivatives, such as Ko143, were prepared.15 Derivatives of XR9576/tariquidar16 and GF120918/elacridar17 were also ABCG2-specific, but displayed cytotoxicity and a limited in-vivo bioavailability.18 A flavonoid-binding site was identified for chrysin and other flavones,19–21 while methoxy-trans-stilbenes appeared less toxic.22 Lower-affinity rotenoids,23 acridone derivatives,17 and chromones24 appeared to bind to the same site, as for a number of flavones, and benzo-pyrane/furane derivatives, of which three-dimensional quantitative structure-activity relationship analyses allowed to establish a molecular model.25,26 Recent studies with chalcones and derivatives (Figure 1) reported a role of substituents, essentially OH, methoxyl (OMe), and Cl, on both A- and B-rings,27,28 but the results were not always easily interpretable since both rings were simultaneously substituted. In addition, we showed that OMe groups on phenyl B-ring, especially at position 4, were quite detrimental towards cytotoxicity.29 Our present aim was therefore to use unsubsti-tuted A-ring for studying the role of a large panel of B-ring substitutions, on both inhibition potency and cytotoxicity. We show here, with a number of chalcone derivatives, that large unsubstituted groups such as 2′-naphthyl or 3′,4′-methylenedioxy-phenyl efficiently replaced the phenyl A-ring, supporting polyspecificity of ABCG2 for inhibitors. At least two OMe were required on the phenyl B-ring for optimal inhibition, although inducing a significant cytotoxicity, whereas most other substituents were not, or only weakly, efficient toward inhibition. Using both a large O-benzyl substituent at critical B-ring position 4 and a 2′-naphthyl group as A-ring additively decreased cytotoxicity and gave a high therapeutic ratio (TR), making chalcone 45 a good candidate for future in-vivo trials in mice models.
Figure 1.

Core structure of generic chalcones (A) and substituted derivatives studied in our previous work (B–K).29
Abbreviations: OMe, methoxyl; Me, methyl.
Materials and methods
Synthesis, purification, and physicochemical analyses of chalcones
All reagents used were of analytical grade, and purchased from Merck KGaA, Darmstadt, Germany or Sigma-Aldrich (St Louis, MO, USA), except for 2,4,6-trimethoxy-acetophenone, xanthoxylin [2-hydroxy-4,6-dimethoxyaceto-phenone], 2-hydroxy-3-bromo-4,6-dimethoxyacetophenone, and 3-methoxy-4-(phenylmetho-xy)-benzaldehyde, which were prepared according to Figure 2, as previously described.30–34 The chalcones were prepared by magnetic stirring of the acetophenone derivative (1.2 mmol), methanol (20 mL), KOH 50% w/v (5 mL), and the corresponding aldehyde (1.2 mmol), at room temperature for 24 hours. Distilled water and hydrochloric acid (10%) were added to the reaction for total precipitation of the products. The compounds were then obtained by vacuum filtration and later recrystallized from dichloromethane and hexane. All chalcones were previously described in the literature.30,32,35–50 The purified compounds were obtained in yields ranging from 40% to 99%. Melting points were determined with a Microquímica MGAPF-301 apparatus (Monte Mor, São Paulo, Brazil). Infrared spectra were recorded with an Abb Bomen FTLA 2000 spectrometer (Zurich, Switzerland) on KBr disks. Nuclear magnetic resonance (1H and 13C) measurements were recorded on a Varian Inc. Oxford AS-400 (Palo Alto, CA, United States) (400 MHz) spectrometer, using tetramethylsilane as an internal standard. Elemental analysis was carried out with a CE Instruments CHNS EA 1110 (Hindley, United Kingdom). Percentages of C and H were in agreement with the product formula (within ± 0.4% of theoretical values to C).
Figure 2.

Synthesis of chalcones.
Notes: a = KOH 50% w/v, CH3OH, room temperature, 24 hours. In classical chalcones, both A and B are phenyl groups.
Compounds for biological assays
Mitoxantrone, calcein-AM, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich. All other reagents were commercial products with the highest available purity grade. All chalcone derivatives were dissolved in DMSO (dimethylsulfoxide) and then diluted in Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose medium. The stock solution was stored at −20°C and warmed to 25°C just before use.
Cell cultures
The human fibroblast HEK293 cell lines transfected with either ABCG2 (HEK293-ABCG2) or the empty vector (HEK293-pcDNA3.1 cells) were obtained as previously described.21 Flp-In-293, an isogenic HEK293 cell line, was co-transfected using Lipofectamine™ (Invitrogen, Carlsbad CA, USA) with either the empty vector pcDNAR/FRT or the pcDNA/FRT-ABCC1 vector, in combination with the Flp recombinase vector pOG44, resulting in targeted integration of the expression vector to the same locus in each cell. HEK293 cell lines transfected with ABCB1 (P-glycoprotein) were kindly provided by Dr SE Bates (National Cancer Institute [NCI] at the National Institutes of Health [NIH], Bethesda, MD, USA). All cells were maintained in DMEM high glucose, supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and supplemented in some cases with either 0.75 mg/mL G418 (for HEK293-pcDNA3.1 and HEK293-ABCG2), or 2 mg/mL G418 (for HEK293-ABCB1), or 200 μg/mL hygromycin B (for Flp-In-293-pcDNA5/FRT and Flp-In-293-ABCC1).
ABCG2- and ABCB1-mediated mitoxantrone transport
HEK293 cells were seeded at a density of 1 × 105 cells/well into 24-well culture plates. After 48 hours incubation, the cells were exposed to 5 μM mitoxantrone (HEK293-ABCG2 and HEK293-MDR1 cells) for 30 minutes at 37°C, in the presence or absence of compounds at various concentrations. After cell washing with phosphate buffer saline, the cells were trypsinized. The intracellular drug fluorescence was monitored by flow cytometry with a FACS Calibur cytometer (BD Biosciences, San Jose, CA, USA). At least 10,000 events were collected for which the maximal fluorescence (100%) was the difference between geometric mean fluorescence of cells incubated with 5 μM GF120918 and without inhibitor.23 For ABCB1-mediated mitoxantrone transport, the cells transfected with the empty vector were used as a control.
MRP1-mediated calcein transport
HEK293 cells transfected with either ABCC1 or the empty vector were exposed to 0.2 μM calcein-AM and analyzed by flow cytometry as described above. The maximal fluorescence (100%) was the difference between geometric mean fluorescence of control cells (HEK293-pcDNA3.1) and MRP1-transfected cells, incubated with substrate but without inhibitor.
Cytotoxicity assays
HEK293 cells transfected by either pcDNA3.1-ABCG2 (resistant cells) or the empty vector (control sensitive cells) were seeded into 96-well culture plates at a 1 × 104 cells/well density. After overnight incubation, the cells were treated with various concentrations of compounds for 72 hours at 37°C under 5% CO2. Cell viability was evaluated with an MTT colorimetric assay51 Control experiments were performed with DMEM high glucose containing 0.1% of DMSO (v/v). The results from at least three replicates were expressed as percentage of viable cells versus control cells, taken as 100%. The curves were fitted with the Sigma Plot™ (Systat Software Inc, San Jose, CA, USA) software.
Statistical analysis
Each experiment was performed at least in triplicate. The data are presented as mean ± standard deviation.
Results and discussion
New structure–activity relationships (SARS) among inhibitory chalcones
A total of 54 chalcone derivatives were investigated here, belonging to three different series listed in Tables 1–3: (1) the first series (Table 1), with 23 derivatives, containing a 3 ′,4′-methylenedioxy-phenyl unit as A-ring; (2) the second series (Table 2), with 22 derivatives, containing a 2′-naphthyl group as A-ring; and (3) the third series (Table 3), with nine derivatives, containing a 1-naphthyl group or other substituents as B-ring.
Table 1.
Inhibition of ABCG2-mediated mitoxantrone efflux by chalcones 1–23
| First series | ||
|---|---|---|

| ||
| Compound | Ring B | % inhibition at 5 μM |
| 1 |

|
15.3 ± 6.7 |
| 2 |

|
26.5 ± 7.1 |
| 3 |

|
40.9 ± 1.0 |
| 4 |

|
12.8 ± 7.5 |
| 5 |

|
13.2 ± 5.2 |
| 6 |

|
28.8 ± 11.2 |
| 7 |

|
13.7 ± 7.5 |
| 8 |

|
24.7 ± 3.9 |
| 9 |
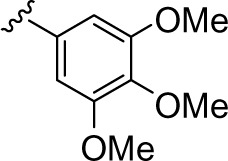
|
73.0 ± 10.1 |
| 10 |

|
15.1 ± 4.6 |
| 11 |

|
31.8 ± 3.8 |
| 12 |

|
8.3 ± 8.1 |
| 13 |

|
57.2 ± 8.3 |
| 14 |

|
33.3 ± 4.0 |
| 15 |

|
18.7 ± 2.7 |
| 16 |

|
25.4 ± 2.8 |
| 17 |

|
59.1 ± 11.2 |
| 18 |

|
29.8 ± 3.0 |
| 19 |

|
30.2 ± 6.4 |
| 20 |

|
18.2 ± 5.2 |
| 21 |

|
85.7 ± 6.7 |
| 22 |

|
79.5 ± 5.7 |
| 23 |

|
73.1 ± 4.5 |
Notes: The preparation of all compounds is described in the Materials and methods section. Each chalcone was assayed at 5 μM for its ability to inhibit mitoxantrone efflux from ABCG2-transfected HEK293 cells by flow cytometry as described. The relative inhibition was calculated by reference to 5 μM GF120918 producing 100% inhibition.
Abbreviations: OBut, buthoxyl; Me, methyl; OMe, methoxyl.
Table 3.
Inhibition by chalcones 46–54
| Third series | |||
|---|---|---|---|

| |||
| Compound | Ring A | Ring B | % inhibition at 5 μM |
| 46 |

|

|
88.2 ± 5.6 |
| 47 |

|

|
73.0 ± 2.6 |
| 48 |

|

|
49.2 ± 7.2 |
| 49 |

|

|
57.0 ± 6.7 |
| 50 |

|

|
82.3 ± 11.1 |
| 51 |

|

|
21.3 ± 9.0 |
| 52 |

|

|
60.9 ± 6.7 |
| 53 |

|

|
63.7 ± 12.7 |
| 54 |

|

|
55.3 ± 8.9 |
Note: The conditions were the same as in Table 1.
Abbreviation: OMe, methoxyl.
Table 2.
Inhibition by chalcones 24–45
| Second series | ||
|---|---|---|

| ||
| Compound | Ring B | % inhibition at 5 μM |
| 24 |

|
39.7 ± 6.4 |
| 25 |

|
16.7 ± 6.8 |
| 26 |

|
9.2 ± 1.7 |
| 27 |

|
2.0 ± 1.4 |
| 28 |

|
42.9 ± 1.2 |
| 29 |

|
22.5 ± 8.8 |
| 30 |

|
23.8 ± 13.1 |
| 31 |

|
11.0 ± 2.8 |
| 32 |

|
49.7 ± 1.5 |
| 33 |

|
96.9 ± 7.4 |
| 34 |

|
17.4 ± 8.1 |
| 35 |

|
33.7 ± 8.7 |
| 36 |

|
14.6 ± 6.9 |
| 37 |

|
29.0 ± 11.0 |
| 38 |

|
6.7 ± 1.3 |
| 39 |

|
24.1 ± 6.9 |
| 40 |

|
1.2 ± 2.9 |
| 41 |

|
31.8 ± 5.3 |
| 42 |

|
7.5 ± 1.9 |
| 43 |

|
23.2 ± 7.7 |
| 44 |

|
42.1 ± 8.8 |
| 45 |

|
81.5 ± 4.4 |
Note: The conditions were the same as in Table 1.
Abbreviations: OBut, buthoxyl; Me, methyl; OMe, methoxyl.
New structure–activity relationships were identified by comparing the inhibitory activity of each compound at 5 μM against ABCG2-mediated mitoxantrone efflux, measured by flow cytometry and using 5 μM GF120918 as a reference for complete inhibition (Tables 1–3). The following substitutions were identified to positively contribute to inhibition.
A 3′,4′-methylenedioxy-phenyl unit, replacing A-ring in compounds of the first series (Table 1), was as efficient in 9 as the active 2′-OH-4′,6′-diOMe-phenyl (A-ring) substituent previously identified in chalcone i (Figure 1).29
A 2′-naphthyl group replacing A-ring in compounds of the second series (Table 2) was also quite efficient in 33, as indolyl in chalcones j and k (Figure 1).29 Such a polyspecificity toward A-ring chalcones is consistent with that observed for different classes of flavonoidic and flavonoid-like inhibitors, assumed to bind to the same site or to overlapping sites, such as hydrophobic flavones,19–21 rotenoids,23 acridones,17 tariquidar derivatives,16 methoxy trans-stilbenes,22 and other chalcones.27–29 Polyspecificity of inhibitory sites is also consistent with the well known polyspecificity of ABCG252 and other multidrug ABC transporters toward large panels of transport substrates. It is worthwhile mentioning that the gain-of-function R482T/G mutation in ABCG2, which was found to extend the transport substrate panel to rhodamine 123 and anthracyclins,53 also allowed inhibitors such as GF120918 and imatinib to be transported.52
In both series, at least two OMe (in 9, 22, 23, and 33) were required for optimal inhibition when comparing with compounds with only one OMe (8, 19, 32) or without any OMe (1–7, 10–18, 20, 24–31, 34–43). The only exception was 44, which suggested a negative contribution of OMe at position 6 versus position 5 in 33. One of the two OMe could be efficiently replaced by O-benzyl at position 4 (in 21 and 45). Interestingly, an O-benzyl substituent was also shown to positively contribute to ABCG2 inhibition in chromones.24
In contrast, all other tested substituents on B-ring were poorly efficient, such as Cl (in 1–4, 17, and 26–29), Br (in 7 and 38), F (in 15, 16, 36, and 37), NO2 (in 5, 6, 24, and 25), methyl (Me) (in 10, 18, 31, and 43), N(Me)2 (in 11 and 34) and buthoxyl (in 14 and 35), similarly as using a naphthyl group to replace B-ring (in 12, 13, 40, and 41).
Comparable positive contributions were observed from the third series of chalcones (Table 3): the requirement for at least two OMe (in 46, 50, 52, 53, and 54), or one OMe and one O-benzyl (in 47), while the low efficiency of 51 was most likely related to the charged carboxyl preventing diffusion through the plasma membrane and therefore the compound uptake.
Selectivity for ABCG2 and cytotoxicity
The 54 chalcones described in this work were then investigated for their ability to inhibit mitoxantrone efflux by ABCB1/MDR1/P-glycoprotein, and calcein efflux by ABCC1/MRP1. As shown in Figure 3, all chalcones were essentially specific for ABCG2, since no significant inhibition was produced in most cases. A low inhibition of ABCB1 activity was however observed at 10 μM with two strong ABCG2 inhibitors, 9 and 33, both containing three OMe groups on the B-ring. Three other compounds slightly inhibited ABCC1-mediated drug efflux, by reference to MK571: 34 and 11 containing a 4-N(Me)2 group on B-ring, and 48 containing a 2′-OH on A-ring. The N(Me)2 substituent present in 11 and 34 might allow these chalcones to be transported, as observed for ABCB1 transport substrates,54 which could explain lower specificity. In contrast, the three potent ABCG2 inhibitors containing a large 4-O-benzyl (ie, 21, 45, and 47) were highly selective and did not inhibit ABCB1 nor ABCC1.
Figure 3.

Inhibition by chalcones of P-glycoprotein (ABCB1)- and MRP1 (ABCC1)-drug efflux activity. All chalcones from the three series were checked individually at 10 μM by flow cytometry for their ability to inhibit either ABCB1-mediated mitoxantrone efflux (grey bars) or ABCC1-mediated calcein efflux (black bars). The control of complete inhibition was performed with either 5 μM GF120919 or 30 μM MK571, respectively.
Abbreviations: ABC, adenosine triphosphate-binding cassette; CTRL, control.
The most potent compounds, inhibiting by more than 70% at 5 μM, were further analyzed for both their IC50 (half maximal inhibitory concentration) values by flow cytometry (Table 4), and cytotoxicity by an MTT cell survival test (Figure 4A and B). The different compounds greatly varied in cytotoxicity, with 9, containing a 3′,4′-methylenedioxy-phenyl as A-ring and a 3,4,5-triOMe substituted B-ring, being the most cytotoxic with an IG50 (concentration producing 50% growth inhibition) value of 4.4 μM, giving a low TR (TR = IG50/IC50) of 7, similarly to 22 with a 2,5-diOMe substitution (TR = 8). A rather high cytotoxicity was also observed for 23, with 3,4-diOMe substitution (IG50 = 12.9 μM, TR = 24), and 46, with a 3,4-methylenedioxy-phenyl instead of B-ring, which confirmed the cytotoxic role of OMe at positions 3, 4, and 5 of phenyl B-ring previously observed when A-ring was an OH- and OMe-substituted phenyl.29 Therefore, the 3′,4′-methylenedioxy-phenyl as A-ring behaved similarly to the above substituted phenyl, but with a 3–4-fold lower affinity. By contrast, the addition of O-benzyl at position 4 was positive in 21, versus 23, by increasing threefold the TR (70 versus 24), as due to both increased affinity for inhibition and decreased cytotoxicity (Figure 4A). The effect was even more marked in 47 with a 4′-Br-phenyl as A-ring (TR = 99). Quite interestingly, the presence of a naphthyl group not only increased inhibition affinity but also lowered the cytotoxic effect of OMe substituents, especially when present as A-ring in 33 (IG50 = 34.9 μM and TR = 152) versus B-ring in 50 (IG50 = 20.6 μM and TR = 94) (Figure 4B and Table 4). This naphthyl positive effect on cytotoxicity appeared additive to that induced by 4-O-benzyl in 45 (IG50 > 100 μM, TR > 435). Such decreases in cytotoxicity might be possibly due to the prevention of interactions with unknown cellular target(s) inducing cell death.
Table 4.
TR values of potent chalcone inhibitors
| Compound | A-ring substituents | B-ring substituents | IG50 (μM) | IC50 (μM) | TR |
|---|---|---|---|---|---|
| 9 | methylenedioxy-phenyl | 3,4.5-triOMe-phenyl | 4.4 ± 0.5 | 0.64 ± 0.16 | 7 |
| 22 | methylenedioxy-phenyl | 2,5-diOMe-phenyl | 8.1 ± 1.3 | 1.07 ± 0.20 | 8 |
| 46 | 2′,4′-diOMe-phenyl | methylenedioxy-phenyl | 11.7 ± 1.8 | 0.64 ± 0.12 | 18 |
| 23 | methylenedioxy-phenyl | 3,4-diOMe-phenyl | 12.9 ± 1.6 | 0.54 ± 0.06 | 24 |
| 21 | methylenedioxy-phenyl | 3-OMe,4-OBz-phenyl | 17.5 ± 1.2 | 0.25 ± 0.12 | 70 |
| 47 | 4′-Br-phenyl | 3-OMe,4-OBz-phenyl | 28.7 ± 5.4 | 0.29 ± 0.04 | 99 |
| 50 | 2′-Oh, 4′,6′-diOMe-phenyl | 1-naphthyl | 20.6 ± 0.8 | 0.22 ± 0.07 | 94 |
| 33 | 2-naphthyl | 2,4,5-triOMe-phenyl | 34.9 ± 2.5 | 0.23 ± 0.06 | 152 |
| 45 | 2-naphthyl | 3-OMe,4-OBz-phenyl | >100 | 0.23 ± 0.07 | >435 |
Notes: The cytotoxicity IG50 values were evaluated from Figure 4A and B. The IC50 values were determined with increasing inhibitor concentrations up to 10 μM by flow cytometry as in Tables 1–3.
Abbreviations: IC50, half maximal inhibitory concentration; IG50, concentration producing 50% growth inhibition; OBz, O-benzyl; OMe, methoxyl; TR, therapeutic ratio.
Figure 4.

Intrinsic cytotoxicity on HEK293 cells and sensitization of BCRP (ABCG2)-transfected cells to mitoxantrone. The survival upon 72-hour culture of control sensitive cells in the presence of increasing concentrations of the indicated chalcones was quantified by MTT assays, as described in the Materials and methods section. Compared intrinsic cytotoxicities are shown in (A) for 21 (black triangles), 22 (black circles), 23 (white circles), and 47 (white triangles), and in (B) for 9 (black circles), 33 (white triangles), 45 (black squares), 46 (white circles), and 50 (black triangles). The sensitization to mitoxantrone, in (C) of the growth of ABCG2-transfected resistant cells (black circles) by 1 μM 50 (black triangles) reached a similar level as control sensitive cells (white circles).
Abbreviations: ABC, adenosine triphosphate-binding cassette; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide; HEK293, human embryonic kidney HEK293.
The potent inhibition of lead compounds against ABCG2-mediated mitoxantrone efflux in flow cytometry experiments was confirmed by their ability to chemosensitize cell growth of resistant cells (IG50 = 82.8 ± 21.7 nM) to the cytotoxic drug, as illustrated in Figure 4C for 50: a 1 μM noncytotoxic concentration of the chalcone efficiently chemosensitized cell growth to mitoxantrone, giving an IG50 value of 17.5 ± 2.2 nM, similar to that of sensitive control cells (14.5 ± 3.6 nM). A comparable efficiency was observed with 45 (IG50 = 16.4 ± 3.1 nM), whereas 22 only produced a partial sensitization (26.7 ± 11.4 nM) (data not shown) in agreement to its 4–5-fold lower affinity for inhibition (see Table 4). The fact that the same low concentrations of 50 and 45 chemosensitized cell growth in MTT assays as efficiently as they inhibited drug efflux in flow cytometry suggests that these chalcones were not mainly metabolized nor transported during the 72-hour timescale of the experiments. Chalcone 45, which is a potent and selective inhibitor with low toxicity, therefore constitutes a good candidate for future in-vivo assays in animal models.
Conclusion
Original data about ABCG2 inhibitory sites and molecular mechanism have been obtained in the present work, thanks to a wide variety of chalcones and derivatives. Firstly, the flavonoid-specific inhibitory site appeared to be polyspecific since it could accommodate diverse large substituents instead of the original phenyl A-ring, with 2′-naphthyl and 3′,4′-methylenedioxy-phenyl behaving quite efficiently toward inhibition. Secondly, substitution at position 4 of the phenyl B-ring was quite critical toward both inhibition and cytotoxicity, with a major role played by an O-benzyl group. The present data of cytotoxicity obtained from in-vitro screening on culture cells constitute an interesting first step that requires to be further developed for the identified lead compounds, such as chalcone 45, toward in-vivo pharmacokinetics and pharmacodynamics in mice.
Acknowledgments
Drs RW Robey and SE Bates, NCI-NIH, Bethesda, MD, USA, are acknowledged for providing HEK-293 cells transfected by ABCB1 (MDR-19), and GlaxoSmithKline for GF120918. LPR and EW were recipient of mobility fellowships from the Brazilian Federal Agency for the Support and Evaluation of Graduate Education (CAPES) (Process numbers 1842/08-0 and 8792127, respectively), and CG was recipient of a doctoral fellowship from the Ligue Nationale Contre le Cancer. Financial support was provided by the CNRS and Université Lyon 1 (UMR 5086), and grants from the Région Rhône-Alpes (CIBLE 2010), the Ligue Nationale Contre le Cancer (Equipe labellisée Ligue 2013) and International ANR-NIH (2010-INTB-1101) to ADP.
Footnotes
Disclosure
The authors declare no conflicts of interest in this work.
References
- 1.Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58(23):5337–5339. [PubMed] [Google Scholar]
- 2.Doyle LA, Yang W, Abruzzo LV, et al. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95(26):15665–15670. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyake K, Mickley L, Litman T, et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59(1):8–13. [PubMed] [Google Scholar]
- 4.Endicott JA, Ling V. The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem. 1989;58:137–171. doi: 10.1146/annurev.bi.58.070189.001033. [DOI] [PubMed] [Google Scholar]
- 5.Cole SPC, Bhardwaj G, Gerlach JH, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258(5088):1650–1654. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- 6.Steinbach D, Sell W, Voigt A, Hermann J, Zintl F, Sauerbray A. BCRP gene expression is associated with a poor response to remission induction therapy in childhood acute myeloid leukemia. Leukemia. 2002;16(8):1443–1447. doi: 10.1038/sj.leu.2402541. [DOI] [PubMed] [Google Scholar]
- 7.Ho MM, Hogge DE, Ling V. MDR1 and BCRP1 expression in leukemic progenitors correlates with chemotherapy response in acute myeloid leukemia. Exp Hematol. 2008;36(4):433–442. doi: 10.1016/j.exphem.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 8.Seamon JA, Rugg CA, Emanuel S, et al. Role of the ABCG2 drug transporter in the resistance and oral bioavailability of a potent cyclin-dependent kinase/Aurora kinase inhibitor. Mol Cancer Ther. 2006;5(10):2459–2467. doi: 10.1158/1535-7163.MCT-06-0339. [DOI] [PubMed] [Google Scholar]
- 9.Breedveld P, Beijnen JH, Schellens JHM. Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol Sci. 2006;27(1):17–24. doi: 10.1016/j.tips.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed-Belkacem A, Pozza A, Macalou S, Perez-Victoria JM, Boumendjel A, Di Pietro A. Inhibitors of cancer cell multidrug resistance mediated by breast cancer resistance protein (BCRP/ABCG2) Anticancer Drugs. 2006;17(3):239–244. doi: 10.1097/00001813-200603000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Boumendjel A, Macalou S, Valdameri G, et al. Targeting the multidrug ABCG2 transporter with flavonoidic inhibitors: in vitro optimization and in vivo validation. Curr Med Chem. 2011;18(22):3387–3401. doi: 10.2174/092986711796504736. [DOI] [PubMed] [Google Scholar]
- 12.de Bruin M, Miyake K, Litman T, Robey RW, Bates SE. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. 1999;146(2):117–126. doi: 10.1016/s0304-3835(99)00182-2. [DOI] [PubMed] [Google Scholar]
- 13.Robey RW, Steadman K, Polgar O, et al. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004;64(4):1242–1246. doi: 10.1158/0008-5472.can-03-3298. [DOI] [PubMed] [Google Scholar]
- 14.Rabindran SK, He H, Singh M, et al. Reversal of a novel multidrug resistance mechanism in human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998;58(24):5850–5858. [PubMed] [Google Scholar]
- 15.Allen JD, van Loevezijn A, Lakhai JM, et al. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol Cancer Ther. 2002;1(6):417–425. [PubMed] [Google Scholar]
- 16.Kühnle M, Egger M, Muller C, et al. Potent and selective inhibitors of breast cancer resistance protein (ABCG2) derived from the P-glycoprotein (ABCB1) modulator tariquidar. J Med Chem. 2009;52(4):1190–1197. doi: 10.1021/jm8013822. [DOI] [PubMed] [Google Scholar]
- 17.Boumendjel A, Macalou S, Ahmed-Belkacem A, Blanc M, Di Pietro A. Acridone derivatives: design, synthesis, and inhibition of breast cancer resistance protein ABCG2. Bioorg Med Chem. 2007;15(8):2892–2897. doi: 10.1016/j.bmc.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 18.Arnaud O, Boumendjel A, Gèze A, et al. The acridone derivative MBLI-87 sensitizes breast cancer resistance protein-expressing xenografts to irinotecan. Eur J Cancer. 2011;47(4):640–648. doi: 10.1016/j.ejca.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang S, Yang X, Morris ME. Flavonoids are inhibitors of breast cancer resistance protein (ABCG2)-mediated transport. Mol Pharmacol. 2004;65(5):1208–1216. doi: 10.1124/mol.65.5.1208. [DOI] [PubMed] [Google Scholar]
- 20.Zhang S, Yang X, Coburn RA, Morris ME. Structure activity relationships and quantitative structure activity relationships for the flavonoid-mediated inhibition of breast cancer resistance protein. Biochem Pharmacol. 2005;70(4):627–639. doi: 10.1016/j.bcp.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 21.Ahmed-Belkacem A, Pozza A, Munoz-Martinez F, et al. Flavonoid structure-activity studies identify 6-prenylchrysin and tectochrysin as potent and specific inhibitors of breast cancer resistance protein ABCG2. Cancer Res. 2005;65(11):4852–4860. doi: 10.1158/0008-5472.CAN-04-1817. [DOI] [PubMed] [Google Scholar]
- 22.Valdameri G, Pereira Rangel L, Sapatafora C, et al. Methoxy stilbenes as potent, specific, untransported and noncytotoxic inhibitors of breast cancer resistance protein. ACS Chem Biol. 2012;7(2):322–330. doi: 10.1021/cb200435y. [DOI] [PubMed] [Google Scholar]
- 23.Macalou S, Ahmed-Belkacem A, Borelli F, et al. Non-prenylated rotenoids, a new class of potent breast cancer resistance protein inhibitors. J Med Chem. 2007;50(8):1933–1938. doi: 10.1021/jm061450q. [DOI] [PubMed] [Google Scholar]
- 24.Valdameri G, Genoux-Bastide E, Peres B, et al. Substituted chromones as highly-potent, nontoxic inhibitors, specific for the breast cancer resistance protein ABCG2. J Med Chem. 2012;55(2):966–970. doi: 10.1021/jm201404w. [DOI] [PubMed] [Google Scholar]
- 25.Nicolle E, Boccard J, Guilet D, et al. Breast cancer resistance protein (BCRP/ABCG2): New inhibitors and QSAR studies by a 3D linear solvation-energy approach. Eur J Pharm Sci. 2009;38(1):39–46. doi: 10.1016/j.ejps.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 26.Nicolle E, Boumendjel A, Macalou S, et al. QSAR analysis and molecular modeling of ABCG2-specific inhibitors. Adv Drug Deliver Rev. 2009;61(1):34–46. doi: 10.1016/j.addr.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Han Y, Riwanto M, Go ML, Ee PLR. Modulation of breast cancer resistance protein (BCRP/ABCG2) by non-basic chalcone analogues. Eur J Pharm Sci. 2008;35(1–2):30–41. doi: 10.1016/j.ejps.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Juvale K, Pape VFS, Wiese M. Investigation of chalcones and benzochalcones as inhibitors of breast cancer resistance protein. Bioorg Med Chem. 2012;20(1):346–355. doi: 10.1016/j.bmc.2011.10.074. [DOI] [PubMed] [Google Scholar]
- 29.Valdameri G, Gauthier C, Terreux R, et al. Investigation of chalcones as selective inhibitors of the breast cancer resistance protein ABCG2: critical role of methoxy substituents in both inhibition and cytotoxicity. J Med Chem. 2012;55(7):3193–3200. doi: 10.1021/jm2016528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiaradia LD, dos Santos R, Vítor CE, et al. Synthesis and pharmacological activity of chalcones derived from 2,4,6-trimethoxyacetophenone in RAW 264.7 cells stimulated by LPS: quantitative structure-activity relationships. Bioorg Med Chem. 2008;16(2):658–667. doi: 10.1016/j.bmc.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 31.Gulati KC, Seth SR, Venkataraman K. Phloracetophenone. Organic Synth. 1943;2:522. [Google Scholar]
- 32.Boeck P, Bandeira Falcão CAB, Leal PC, et al. Synthesis of chalcone analogues with increased antileishmanial activity. Bioorg Med Chem. 2006;14(5):1538–1545. doi: 10.1016/j.bmc.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Cechinel-Filho V, Vaz ZR, Zunino L, Calixto JB, Yunes RA. Synthesis of xanthoxyline derivatives with antinociceptive and antioedematogenic activities. Eur J Med Chem. 1996;31(10):833–839. [Google Scholar]
- 34.Tsai S, Klinman JP. De Novo design and utilization of photolabile caged substrates as probes of hydrogen tunneling with horse liver alcohol dehydrogenase at sub-zero temperatures: a cautionary note. Bioorg Chem. 2003;31(2):172–190. doi: 10.1016/S0045-2068(03)00018-X. [DOI] [PubMed] [Google Scholar]
- 35.Insuasty B, Orozco F, Quiroga J, Abonia R, Nogueras M, Cobo J. Microwave induced synthesis of novel 8,9-dihydro-7H-pyrimido[4,5-b] [1,4]diazepines as potential antitumor agents. Eur J Med Chem. 2008;43(9):1955–1962. doi: 10.1016/j.ejmech.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 36.Borchhardt DM, Mascarello A, Chiaradia LD, et al. Biochemical evaluation of a series of synthetic chalcone and hydrazide derivatives as novel inhibitors of cruzain from Trypanosoma cruzi. J Braz Chem Soc. 2010;21(1):142–150. [Google Scholar]
- 37.Damazio RG, Zanatta AP, Cazarolli LH, et al. Nitrochalcones: Potential in vivo insulin secretagogues. Biochimie. 2009;91(11–12):1493–1598. doi: 10.1016/j.biochi.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 38.Sadashivamurthy B, Basavaraju YB. New tetralone esters as intermediates for the synthesis of podophyllotoxin analogs. Indian J Heterocycl Chem. 2006;15(3):259–262. [Google Scholar]
- 39.Sathisha AD, Hemakumar KH, Basavaraju YB. Synthesis of new tetralone ester intermediates for podophyllotoxin analogues. Indian J Heterocycl Chem. 2007;17(1):15–18. [Google Scholar]
- 40.Parmar VS, Sharma S, Rathore JS, et al. Carbon-13 nuclear magnetic resonance studies on 1,3-diphenylprop-2-enones. Magn Reson Chem. 1990;28(5):470–474. [Google Scholar]
- 41.Wattanasin S, Murthy WS. An improved procedure for the preparation of chalcones and related enones. Synthesis. 1980;8:647–650. [Google Scholar]
- 42.Damazio RG, Zanatta AP, Cazarolli LH, et al. Antihyperglycemic activity of naphthylchalcones. Eur J Med Chem. 2010;45(4):1332–1337. doi: 10.1016/j.ejmech.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 43.Thirunarayanan G. Effects of substituent on 1H and 13C NMR chemical shifts in substituted styryl 2-naphthyl ketones. Acta Cienc Indica Chem. 2003;29(3):147–150. [Google Scholar]
- 44.Winter E, Chiaradia LD, de Cordova CAS, Nunes RJ, Yunes RA, Crezynski-Pasa TB. Naphthylchalcones induce apoptosis and caspase activation in a leukemia cell line: The relationship between mitochondrial damage, oxidative stress, and cell death. Bioorg Med Chem. 2010;18(22):8026–8034. doi: 10.1016/j.bmc.2010.09.025. [DOI] [PubMed] [Google Scholar]
- 45.Robinson TP, Hubbard RB, Ehlers TJ, Arbiser JL, Goldsmith DJ, Bowen JP. Synthesis and biological evaluation of aromatic enones related to curcumin. Bioorg Med Chem Lett. 2005;13(12):4007–4013. doi: 10.1016/j.bmc.2005.03.054. [DOI] [PubMed] [Google Scholar]
- 46.Patel HS, Patel VK, Dixit BC. Synthesis and antimicrobial activity of novel 2-amino-4-naphthyl-6-substituted phenylpyrimidine. Orient J Chem. 2001;17(3):142–150. [Google Scholar]
- 47.Chiaradia LD, Martins PGA, Cordeiro MNS, et al. Synthesis, biological evaluation, and molecular modeling of chalcone derivatives as potent inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatases (PtpA and PtpB) J Med Chem. 2012;55(1):390–402. doi: 10.1021/jm2012062. [DOI] [PubMed] [Google Scholar]
- 48.Formento Navarini AL, Chiaradia LD, Mascarello A, et al. Hydroxychalcones induce apoptosis in B16-F10 melanoma cells via GSH and ATP depletion. Eur J Med Chem. 2009;44(4):1630–1637. doi: 10.1016/j.ejmech.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 49.Chiaradia LD, Mascarello A, Purificaçao M, et al. Synthetic chalcones as efficient inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorg Med Chem Lett. 2008;18(23):6227–6230. doi: 10.1016/j.bmcl.2008.09.105. [DOI] [PubMed] [Google Scholar]
- 50.Campos-Buzzi F, Pereira de Campos J, Tonini PP, et al. Antinociceptive effects of synthetic chalcones obtained from xanthoxyline. Arch Pharm. 2006;339(7):361–365. doi: 10.1002/ardp.200600049. [DOI] [PubMed] [Google Scholar]
- 51.Mosmann T. Rapid colorimetric assay for cellular growth and survival. Application to proliferation and cyto-toxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 52.Telbisz A, Hegedus C, Ozvegy-Laczka C, et al. Antibody binding shift assay for rapid screening of drug interactions with the human ABCG2 multidrug transporter. Eur J Pharm Sci. 2012;45(1–2):101–109. doi: 10.1016/j.ejps.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 53.Sarkadi B, Ozvegy-Laczka C, Nemet K, Varadi A. ABCG2 – a transporter for all seasons. FEBS Lett. 2004;567(1):116–120. doi: 10.1016/j.febslet.2004.03.123. [DOI] [PubMed] [Google Scholar]
- 54.Ford JM, Prozialeck WC, Hait WN. Structural features determining activity of phenothiazines and related drugs for inhibition of cell growth and reversal of multidrug resistance. Mol Pharmacol. 1989;35(1):105–115. [PubMed] [Google Scholar]