

Abstract
The bone morphogenetic protein (BMP) type II receptor (BMPR2) has a long cytoplasmic tail domain whose function is incompletely elucidated. Mutations in the tail domain of BMPR2 are found in familial cases of pulmonary arterial hypertension. To investigate the role of the tail domain of BMPR2 in BMP signaling, we generated a mouse carrying a Bmpr2 allele encoding a non-sense mediated decay-resistant mutant receptor lacking the tail domain of Bmpr2. We found that homozygous mutant mice died during gastrulation, whereas heterozygous mice grew normally without developing pulmonary arterial hypertension. Using pulmonary artery smooth muscle cells (PaSMC) from heterozygous mice, we determined that the mutant receptor was expressed and retained its ability to transduce BMP signaling. Heterozygous PaSMCs exhibited a BMP7‑specific gain of function, which was transduced via the mutant receptor. Using siRNA knockdown and cells from conditional knockout mice to selectively deplete BMP receptors, we observed that the tail domain of Bmpr2 inhibits Alk2‑mediated BMP7 signaling. These findings suggest that the tail domain of Bmpr2 is essential for normal embryogenesis and inhibits Alk2‑mediated BMP7 signaling in PaSMCs.
Introduction
Bone morphogenetic proteins (BMPs) were initially identified as signaling factors involved in the formation of bone and cartilage. BMPs are now known to participate in a broad spectrum of biological activities during embryogenesis and organogenesis, as well as in the homeostasis of mature organs [1,2]. BMPs are members of the transforming growth factor beta family. BMPs bind to heterotetrameric receptor complexes formed by BMP type 2 and BMP type 1 serine–threonine kinases. Upon assembly of the BMP receptor complex by a BMP ligand, the constitutively active type 2 receptor phosphorylates the type 1 receptor, which in turn activates cytoplasmic BMP-responsive Smad signaling molecules—Smads 1, 5, and 8. Phosphorylated BMP-responsive Smads interact with Smad4 and translocate into the nucleus, where they modulate the transcription of BMP-responsive genes, such as Id1 and Smad6 [2-4].
BMP receptors include four type 1 (Alk1, Alk2, Alk3 and Alk6) and three type 2 kinases (Bmpr2, Acvr2a and Acvr2b) [2]. The expression of these receptors differs depending on the cell type or tissue. For example, mouse pulmonary artery smooth muscle cells (PaSMCs) express Bmpr2 and Acvr2a with lower amounts of Acvr2b; Alk2 and Alk3 are the predominant BMP type 1 receptors expressed in PaSMCs [5]. All BMP receptors have a similar structure including an extracellular ligand-binding domain, a transmembrane domain, and a cytoplasmic serine–threonine kinase domain. Unlike other BMP receptors, the predominantly expressed form of Bmpr2 (Bmpr2‑WT) contains a long cytoplasmic tail domain (Bmpr2‑TD) encoded by Bmpr2 exons 12 and 13 [6,7]. In a small fraction of Bmpr2 transcripts, exon 12 is alternatively spliced, resulting in a short-form variant of the receptor [7]. Although the Bmpr2‑TD has been reported to interact with several proteins that can modulate BMP signaling [8,9], the functional role of the Bmpr2‑TD remains to be fully defined.
BMPR2 is implicated in the development of pulmonary arterial hypertension (PAH) [10,11]. PAH is a disease of the pulmonary circulation characterized by neointimal formation, obstruction of vessels, plexiform lesions, and pruning of the small pulmonary arteries [12]. Heterozygous BMPR2 mutations have been reported in approximately 75% of patients with hereditary PAH and in 25% of idiopathic cases [13]. Seventy percent of BMPR2 mutations introduce a premature termination codon [14]. BMPR2 transcripts containing premature termination codons are subject to nonsense-mediated decay (NMD), an RNA surveillance mechanism that degrades aberrant mRNAs [15,16]. BMPR2 transcripts that undergo NMD lead to functional haploinsufficiency. However, some types of mutant transcripts can escape NMD, and the translated mutant receptor may exhibit a more deleterious phenotype by acting, for example, in a dominant-negative manner. It has been reported that PAH has an earlier onset and a worse prognosis in patients that carry NMD-resistant BMPR2 mutations than in patients who carry NMD-sensitive BMPR2 mutations [17].
In previous studies, we examined the impact of Bmpr2 haploinsufficiency on BMP signaling in PaSMCs isolated from genetically modified mice. We observed that PaSMCs isolated from heterozygous mice carrying a Bmpr2 mutant allele lacking exons 4 and 5 (Bmpr2 +/-) were less responsive to BMP4 and BMP7 than were PaSMCs isolated from WT mice [5,18]. We also investigated the impact of complete loss of Bmpr2 using PaSMCs from mice harboring mutant Bmpr2 alleles in which exon 4 and 5 were flanked with loxP sequences (Bmpr2 flox/flox) [5]. When both Bmpr2 alleles were disrupted (Bmpr2 del/del) in PaSMCs, BMP4 signaling was diminished, whereas BMP7 signaling was unexpectedly increased. We found that Acvr2a, but not Acvr2b, compensated for the absence of Bmpr2.
To investigate the role of the tail domain of Bmpr2 and model the impact of an NMD-resistant Bmpr2 mutation, we generated mice that carry a mutant Bmpr2 allele (Bmpr2 Δtd), which encodes a receptor lacking the tail domain of Bmpr2 (Bmpr2‑ΔTD). We isolated PaSMCs from mice carrying the Bmpr2 Δtd allele to characterize the role of Bmpr2-TD in BMP signaling.
Results
Generation and phenotype of mutant mice harboring the Bmpr2Δtd allele
The mutant Bmpr2 Δtd allele was generated by inserting a cassette encoding the enhanced green fluorescent protein (Egfp) and a stop codon in frame after exon 11. The strategy for generating mice carrying the mutant Bmpr2 Δtd allele is described in Methods and Figure S1. We observed that Bmpr2 Δtd/Δtd mice died early in embryogenesis (embryonic day (E) 7.5 to 8.5), revealing a previously unknown role for the Bmpr2‑TD in embryogenesis. In contrast, Bmpr2 Δtd/+ mice grow normally and have a lifespan similar to that of their WT littermates. Right ventricular systolic pressure and mean arterial pressure did not differ between Bmpr2 Δtd/+ and WT mice at 6 to 8 months of age (Figure S2). These findings suggest that Bmpr2 Δtd/+ mice do not spontaneously develop PAH.
Expression of Bmpr2‑ΔTD in PaSMCs
To begin to understand the impact of the Bmpr2 Δtd allele on BMP signaling, we sought to determine whether the mutant Bmpr2 gene is expressed. Bmpr2 + and Bmpr2 Δtd mRNA and protein levels were measured in PaSMCs using quantitative real-time PCR (qPCR) and immunoblot techniques, respectively. Total Bmpr2 mRNA levels did not differ between WT and Bmpr2 Δtd/+ PaSMCs, when determined using oligonucleotides spanning the Bmpr2 exon 6–7 junction (Figure 1A). Bmpr2 + mRNA levels in Bmpr2 Δtd/+ PaSMCs were half of those observed in WT cells when measured using oligonucleotides spanning the exon 12–13 junction. Immunoblot analysis showed that levels of Bmpr2‑WT were less in Bmpr2 Δtd/+ PaSMCs than in WT PaSMCs. The protein expressed by the mutant allele, Bmpr2‑ΔTD, in Bmpr2 Δtd/+ cells was smaller (~100 kDa) than Bmpr2‑WT (Figure 1B). These results show that the Bmpr2 Δtd allele is transcribed, Bmpr2 Δtd transcripts are resistant to NMD, and Bmpr2‑ΔTD protein is expressed in PaSMCs.
Figure 1. Bmpr2 expression in PaSMCs obtained from WT or Bmpr2 Δtd/+ mice.
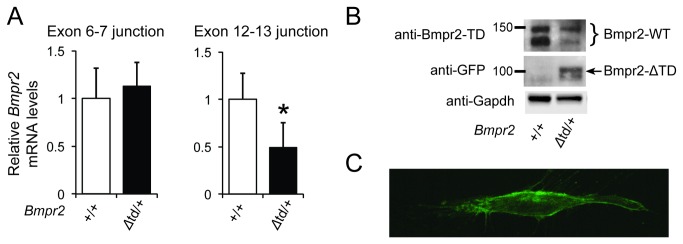
(A) Levels of Bmpr2 mRNA were measured in WT (Bmpr2+/+) or Bmpr2 Δtd/+ PaSMCs by qPCR using hydrolysis probes for Bmpr2 exon junctions 6–7 and 12–13. Bmpr2 mRNA levels were normalized to Gapdh and expressed as the fold-change relative to Bmpr2 +/+ PaSMCs. *P < 0.01 compared to Bmpr2 +/+ PaSMCs. (B) Immunoblots prepared from lysates of Bmpr2 +/+ and Bmpr2 Δtd/+ PaSMCs were incubated with an antibody directed against the tail domain of Bmpr2 to detect Bmpr2‑WT or with an anti-GFP antibody to detect Bmpr2‑ΔTD. Immunoblots were subsequently incubated with an antibody directed against Gapdh as a control for protein loading. (C) Confocal microscopy image of a PaSMC transiently transfected with a plasmid directing expression of Bmpr2 Δtd and reacted with an anti-GFP antibody showing localization of Bmpr2‑ΔTD at the cell membrane.
To determine whether Bmpr2‑ΔTD can localize to the cell membrane, PaSMCs were transfected with a plasmid directing the expression of Bmpr2 Δtd, following by immunostaining with an antibody directed against GFP. Confocal microscopy revealed that the mutant receptor localized to the cell membrane (Figure 1C). This finding suggests that the Bmpr2‑TD is not required for intracellular trafficking of Bmpr2‑WT to the cell membrane.
Bmpr2Δtd/+ PaSMCs exhibit a BMP ligand-specific gain of function
The observation that Bmpr2‑ΔTD localizes to the cell membrane suggested that the mutant receptor could participate in BMP signaling. To investigate the impact of the Bmpr2‑TD on BMP signaling, we compared PaSMCs from WT and Bmpr2 Δtd/+ mice. Incubation with BMP4 induced the phosphorylation of Smad1/5/8 and expression of the Id1 gene similarly in WT and Bmpr2 Δtd/+ PaSMCs (Figure 2A and B). In both WT and Bmpr2 Δtd/+ PaSMC, BMP4 induction of Id1 gene expression peaked at 4 hours (Figure 2A) and persisted for up to 24 hours (Figure S3). In contrast, incubation with BMP7 for 1.5 and 4 hours led to a greater induction of Smad1/5/8 phosphorylation and Id1 and Smad6 gene expression in Bmpr2 Δtd/+ PaSMCs than in WT cells (Figure 2B and C). After 8 hours of exposure to BMP7, levels of Id1 mRNA returned to baseline (Figure 2B). These findings suggest that loss of one copy of the Bmpr2‑TD leads to a BMP ligand-specific gain of function.
Figure 2. BMP7 signaling is enhanced in Bmpr2 Δtd/+ PaSMCs.

(A) Immunoblots of lysates of WT (Bmpr2+/+) or Bmpr2 Δtd/+ PaSMCs treated with BMP4 or BMP7 (10 ng/ml) for various times were reacted with antibodies directed against phosphorylated and total Smad1/5/8. Quantification of the ratio of phosphorylated Smad1/5/8 to total Smad1/5/8 (analysis of 3 independent experiments) demonstrated that BMP4 signaling is similar in Bmpr2 +/+ or Bmpr2 Δtd/+ PaSMCs, whereas BMP7 signaling is greater in Bmpr2 Δtd/+ PaSMCs. *P<0.05 compared to Bmpr2 +/+ PaSMC group treated with BMP7. Id1 (B) and Smad6 (C) mRNA levels were measured by qPCR in Bmpr2 +/+ or Bmpr2 Δtd/+ PaSMCs treated with BMP4 or BMP7 (10 ng/ml) for various times. Id1 and Smad6 gene expression was normalized to Gapdh and expressed as fold-change relative to control Bmpr2 +/+ PaSMC group. *P < 0.01 compared to Bmpr2 +/+ PaSMC group treated with BMP7.
Bmpr2‑ΔTD contributes to the increased responsiveness of Bmpr2Δtd/+ PaSMCs to BMP7
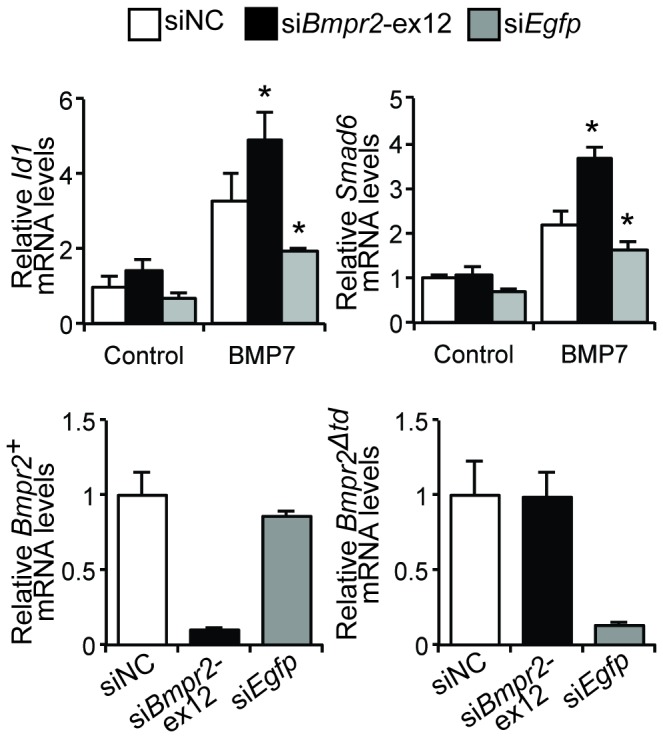
To investigate whether the enhanced responsiveness of Bmpr2 Δtd/+ PaSMCs to BMP7 depends on the presence of Bmpr2‑ΔTD, Bmpr2 Δtd/+ PaSMCs were treated with small interfering RNAs (siRNAs) to silence Bmpr2 + mRNA (targeting Bmpr2 exon 12; siBmpr2‑ex12) or Bmpr2 Δtd mRNA (targeting Egfp; siEgfp). The ability of BMP7 to induce Id1 and Smad6 gene expression was retained in Bmpr2 Δtd/+ PaSMCs treated with siBmpr2‑ex12, but decreased in cells treated siEgfp (Figure 3A). These data suggest that the enhanced BMP7 signaling seen in Bmpr2 Δtd/+ PaSMCs requires Bmpr2‑ΔTD.
Figure 3. Bmpr2‑ΔTD contributes to BMP7 signaling in Bmpr2 Δtd/+ PaSMCs.

(A) Bmpr2 Δtd/+ PaSMCs were transfected with negative control siRNA (siNC), siBmpr2‑ex12, or siEgfp (30 nM). After 48 h, the ability of BMP7 (10 ng/ml for 1.5 h) to induce Id1 and Smad6 mRNA expression was measured by qPCR, normalized to Gapdh and expressed as fold-change relative to Bmpr2 Δtd/+ PaSMCs transfected with siNC. *P < 0.01 compared to siNC group treated with BMP7, † P<0.01 compared to siBmpr2‑ex12 group treated with BMP7. Efficiency of silencing Bmpr2 + (siBmpr2‑ex12) and Bmpr2 Δtd (siEgfp) transcripts was measured by qPCR. (B) Bmpr2 Δtd/flox and Bmpr2 Δtd/del PaSMCs were treated with BMP4 or BMP7 (10 ng/ml) for 30 and 60 minutes, upon which the activation of Smad1/5/8 was evaluated by immunoblotting. Quantification of the Smad1/5/8 activation is plotted as the ratio of pSmad1/5/8 to total Smad1/5/8. (C) The ability of BMP4 or BMP7 to induce Id1 and Smad6 gene expression in Bmpr2 Δtd/flox and Bmpr2 Δtd/del PaSMCs was measured by qPCR, normalized to Gapdh and expressed as fold-change relative to untreated Bmpr2 Δtd/flox PaSMCs. *P < 0.01 compared to Bmpr2 Δtd/flox PaSMC group treated with BMP7.
To confirm that the increased BMP7 signaling seen in Bmpr2 Δtd/+ cells does not require expression of the wild-type allele, we infected PaSMCs from Bmpr2 Δtd/flox mice with an adenovirus specifying Cre recombinase (Ad-Cre) to delete the Bmpr2 flox allele (Bmpr2 Δtd/del PaSMCs). Bmpr2 Δtd/flox cells infected with an adenovirus specifying red fluorescent protein (Ad-RFP) were used as control. Bmpr2 Δtd/del PaSMCs did not express detectable Bmpr2‑WT protein (Figure S4). Incubation with BMP4 led to a similar induction of Smad1/5/8 phosphorylation (Figure 3B) and Id1 and Smad6 gene expression (Figure 3C) in Bmpr2 Δtd/flox and Bmpr2 Δtd/del PaSMCs. In contrast, incubation with BMP7 led to a greater increase in the phosphorylation of Smad1/5/8 and in Id1 and Smad6 gene expression in Bmpr2 Δtd/del than in Bmpr2 Δtd/flox PaSMCs. These results provide additional support for the concept that the presence of Bmpr2‑WT is not required for Bmpr2 Δtd/flox PaSMCs to signal in response to BMP7. Moreover, the presence of the Bmpr2 + allele appears to inhibit signaling via Bmpr2‑ΔTD.
Since Acvr2a, but not Acvr2b, can compensate for the absence of Bmpr2 in Bmpr2 del/del PaSMCs [5], we considered the possibility that Acvr2a was responsible for BMP signaling in Bmpr2 Δtd/+ or Bmpr2 Δtd/del PaSMCs. Silencing Acvr2a mRNA modestly increased the ability of BMP4 to induce Id1 gene expression in Bmpr2 Δtd/+ PaSMCs, as well as the ability of BMP4 to induce Smad6 gene expression in Bmpr2 Δtd/+ and Bmpr2 Δtd/del PaSMCs (Figure 4). These findings show that Acvr2a is not required for BMP4 signaling in Bmpr2 Δtd/+ or Bmpr2 Δtd/del PaSMCs. In contrast, silencing Acvr2a mRNA modestly decreased the ability of BMP7 to induce Id1 and Smad6 gene expressions in Bmpr2 Δtd/del PaSMCs (Figure 4), as well as decreasing the ability of BMP7 to induce Id1 gene expression in Bmpr2 Δtd/+ PaSMCs. These findings show that Bmpr2‑ΔTD and, to a lesser extent, Acvr2a can transduce BMP7 signaling in PaSMCs harboring the Bmpr2 Δtd allele.
Figure 4. BMP7 signaling in Bmpr2 Δtd/+ and Bmpr2 Δtd/del PaSMCs does not depend on the presence of Acvr2a.

(A) Bmpr2 Δtd/+ PaSMCs were treated with a siRNA specific for Acvr2a transcripts. The ability of BMP4 or BMP7 (10 ng/ml for 1.5 h) to induce Id1 and Smad6 gene expression was measured by qPCR, normalized to Gapdh and expressed as fold-change relative to Bmpr2 Δtd/+ PaSMCs treated with siNC. *P < 0.01 compared to siNC within BMP treatment. Silencing efficiency was quantified by measuring Acvr2a mRNA levels. (B) Bmpr2 Δtd/del PaSMCs were treated with siAcvr2a. The ability of BMP4 or BMP7 (10 ng/ml for 1.5 h) to induce Id1 and Smad6 gene expression was measured by qPCR, normalized to Gapdh and expressed as fold-change relative to Bmpr2 Δtd/del PaSMCs treated with siNC. *P < 0.01 compared to siNC within BMP treatment. Acvr2a silencing efficiency was measured by qPCR.
Alk2 is required for the response of Bmpr2Δtd/+ PaSMCs to BMP7
Based on our previous findings in Bmpr2 del/del PaSMCs [5] and the high affinity of BMP7 for Alk2 [19,20], we hypothesized that the response to BMP7 would be less in Bmpr2 Δtd/+ cells deficient in Alk2 than in Bmpr2 Δtd/+ cells that express Alk2. To test this hypothesis, we generated Bmpr2 Δtd/+ mice carrying Alk2, Alk3, or both alleles flanked by loxP sequences. PaSMCs from these mice were infected with Ad-Cre to delete alleles flanked by loxP sequences or Ad-RFP as a control. Incubation with BMP4 induced Id1 and Smad6 gene expression similarly in Bmpr2 Δtd/+ ; Alk2 flox/flox and Bmpr2 Δtd/+; Alk2 del/del PaSMCs (Figure 5A). In contrast, incubation with BMP4 induced Id1 and Smad6 gene expression less in Bmpr2 Δtd/+ ; Alk3 del/del than in Bmpr2 Δtd/+ ; Alk3 flox/flox PaSMCs (Figure 5B). The ability of BMP7 to induce Id1 and Smad6 gene expression was markedly less in Bmpr2 Δtd/+ ; Alk2 del/del than in Bmpr2 Δtd/+ ; Alk2 flox/flox PaSMCs and was similar in Bmpr2 Δtd/+ ; Alk3 flox/flox and Bmpr2 Δtd/+ ; Alk3 del/del PaSMCs (Figure 5). These results suggest that the enhanced responsiveness of Bmpr2 Δtd/+ PaSMCs to BMP7 requires the presence of Alk2. To corroborate these results, we examined the contribution of Bmpr2‑ΔTD or Bmpr2‑WT to mediate BMP7 signaling in PaSMCs predominantly expressing Alk2 (i.e. in Alk3‑deficient PaSMCs). Silencing of Bmpr2 + transcripts in Bmpr2 Δtd/+ ; Alk3 del/del PaSMCs augmented the ability of BMP7 to induce Id1 or Smad6 gene expression, whereas silencing of Bmpr2 Δtd transcripts reduced the responsiveness of Bmpr2 Δtd/+ ; Alk3 del/del PaSMCs to BMP7 (Figure 6). These results support the concept that BMP7 signaling is transduced by the mutant receptor Bmpr2‑ΔTD and Alk2, and suggest that the tail domain of Bmpr2 may inhibit BMP7 signaling via Alk2. Deletion of both Alk2 and Alk3 abrogated BMP4 and BMP7 signaling in Bmpr2 Δtd/+ PaSMCs, as well as in Bmpr2 +/+ PaSMCs (Figure S5). These findings suggested that the very low levels of Alk6 detected in PaSMCs are insufficient to transduce BMP signaling.
Figure 5. BMP7 preferentially utilizes Alk2 in Bmpr2 Δtd/+ PaSMCs.
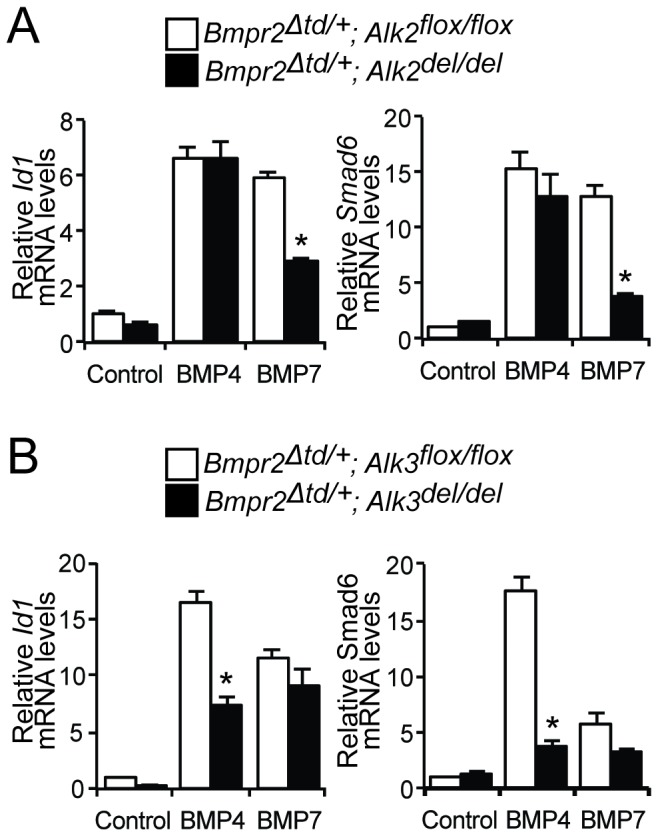
(A) The ability of BMP4 or BMP7 (10 ng/ml for 1.5 h) to induce Id1 and Smad6 gene expression, in Bmpr2 Δtd/+ PaSMCs deficient in Alk2 or expressing Alk2 was examined by qPCR. Id1 and Smad6 gene expression was normalized to Gapdh and expressed as fold-change relative to Bmpr2 Δtd/+ ; Alk2 flox/flox PaSMCs. *P < 0.01 compared to Bmpr2 Δtd/+ ; Alk2 flox/flox PaSMCs treated with BMP7. (B) The ability of BMP4 or BMP7 (10 ng/ml for 1.5 h) to induce Id1 and Smad6 gene expression, in Bmpr2 Δtd/+ PaSMCs deficient in Alk3 or expressing Alk3 was measured by qPCR. Id1 and Smad6 gene expression was normalized to Gapdh and expressed as fold-change relative to Bmpr2 Δtd/+ ; Alk3 flox/flox PaSMCs. *P < 0.01 compared to Bmpr2 Δtd/+ ; Alk3 flox/flox PaSMC treated with BMP4.
Figure 6. Bmpr2‑TD attenuates Alk2‑mediated BMP7 signaling in PaSMCs.
Alk3‑deficient Bmpr2 Δtd/+ PaSMCs were transfected with specific siRNA to silence Bmpr2 + (siBmpr2‑ex12) or Bmpr2 Δtd (siEgfp) transcripts. After 48 h, the ability of BMP7 to induce Id1 and Smad6 gene expression was measured by qPCR, normalized to Gapdh and expressed as fold-change relative to Bmpr2 Δtd/+; Alk3 del/del PaSMCs treated with siNC. *P < 0.01 compared to control cells (siNC) treated with BMP7. Silencing efficiency was quantified by qPCR.
Discussion
In this study, we report the generation of a genetically modified mouse that carries a Bmpr2 allele with an NMD-resistant mutation in the sequences encoding the Bmpr2‑TD. We found that Bmpr2 Δtd/Δtd mice die early in embryogenesis (E7.5‑8.5) and that Bmpr2 Δtd/+ mice appear to grow normally. RVSP is similar in Bmpr2 Δtd/+ mice and their WT littermates at 6 to 8 months of age. We observed that the receptor encoded by the mutant allele, Bmpr2‑ΔTD, is expressed and traffics to the membrane of PaSMCs. In PaSMCs from Bmpr2 Δtd/+ mice, we found a BMP7‑specific gain of signaling with preserved BMP4 signaling. Knockdown of Bmpr2 + transcripts in Bmpr2 Δtd/+ PaSMCs or deletion of the Bmpr2 flox allele in conditional Bmpr2 Δtdlflox PaSMCs showed that Bmpr2‑WT is not required for these cells to transduce signaling in response to BMP7. However, knockdown of Bmpr2 Δtd transcripts in Bmpr2 Δtd/+ PaSMCs inhibited BMP7 signaling. Finally, we determined that the increased responsiveness of Bmpr2 Δtd/+ PaSMCs to BMP7 relies on the presence of Alk2, thus revealing that the tail domain of Bmpr2 inhibits Alk2‑mediated signaling.
In the process of posttranscriptional regulation, the mRNA surveillance mechanism of NMD plays a critical role degrading aberrant transcripts prior to translation [16,17]. It was conceivable that transcripts generated by the Bmpr2 Δtd allele would undergo NMD. We observed, however, that Bmpr2 Δtd mRNA represented half of the Bmpr2 transcripts expressed in Bmpr2 Δtd/+ PaSMCs. Likewise, Bmpr2‑ΔTD protein expression was readily detected in lysates from PaSMCs expressing the Bmpr2 Δtd allele. These data show that transcripts from the Bmpr2 Δtd allele were resistant to NMD. Our mouse model differs from other genetically modified mice carrying mutations in the Bmpr2-TD. Mutant transcripts from heterozygous mice carrying a BMPR2 R899X knockin allele were found to be subject to NMD [21], rendering these knockin mice similar to haploinsufficient Bmpr2 +/- mice. In contrast, BMPR2 R899X protein was detected in the pulmonary vasculature of mice in which a transgene specifying the mutant protein was inducibly overexpressed in smooth muscle cells [22].
Mice with Bmpr2 mutations have been used to study how human BMPR2 mutations might predispose carriers to PAH. Bmpr2 +/- mice express about 50% of Bmpr2 + mRNA levels and manifest little [18] or no [23,24] pulmonary hypertension at baseline; however, pulmonary hypertension induced by an inflammatory stress [24] or an infusion of serotonin [23] is more marked in Bmpr2 +/- than in WT mice. Mice carrying one copy of a mutant Bmpr2 allele lacking exon 2 (Bmpr2 ΔE2) do not manifest pulmonary hypertension at baseline but develop more marked pulmonary hypertension after prolonged exposure to hypoxia [25]. Although the main objective of our work was to study the role of the tail domain of Bmpr2 using cells from Bmpr2 Δtd/+ mice, we did examine whether Bmpr2 Δtd/+ mice spontaneously develop pulmonary hypertension. At baseline, RVSP does not differ in 6- to 8-month-old Bmpr2 Δtd/+ and WT mice. The absence of pulmonary hypertension at baseline in mice carrying heterozygous Bmpr2 mutations (with mutant Bmpr2 alleles expressed at levels similar to those of the WT allele) is consistent with the observation that PAH occurs in only one-fifth of the individuals harboring BMPR2 mutations, suggesting that additional genetic or environmental factors (second hit) are involved in the clinical manifestation of the disease [14].
BMPs are involved in numerous processes during early embryonic development including organogenesis and morphogenesis [1]. We previously demonstrated that Bmpr2 -/- embryos are arrested during gastrulation [26]. In contrast, mice homozygous for a hypomorphic Bmpr2 (Bmpr2 ΔE2/ΔE2), which appears to retain some BMP signaling capabilities, are able to complete gastrulation, but die during midgestation due to defects in the organogenesis of the cardiovascular and skeletal systems [27]. In the present study, we observed that homozygous Bmpr2 Δtd/Δtd mice die in gastrulation even though the Bmpr2‑ΔTD mutant retains the ability to activate Smads in response to BMP ligands. These observations show that embryogenesis not only requires Bmpr2 kinase activity, but also the presence of the tail domain of Bmpr2.
To begin to understand how BMP signaling is modulated by the absence of the tail domain of Bmpr2, we tested the responsiveness of Bmpr2 Δtd/+ PaSMCs to BMP4 and BMP7 and found an unexpected BMP7‑specific gain of function. We considered several possible mechanisms by which this gain of function might occur. First, we considered the possibility that the absence of the Bmpr2‑TD would alter the ability of the receptor to traffic and localize to the cell membrane. In Xenopus embryos, the neuroectodermal protein Jiraiya interacts with a motif in the Bmpr2‑TD to inhibit bmpr2 trafficking to the cell membrane [28], suggesting that loss of the tail domain may facilitate the trafficking of the receptor to the cellular membrane. Moreover, it has been reported that BMPR2 proteins with mutations in the tail domain can traffic to the cellular membrane and can transduce BMP signaling [29,30]. Similarly, we observed that Bmpr2‑ΔTD localized to the cell membrane of PaSMCs. Taken together, these findings demonstrate that the Bmpr2‑TD is not required for trafficking of the receptor to the cell surface.
Previous reports have identified several proteins that can interact with the tail domain of BMPR2 and regulate BMP signaling. Tribbles homolog 3 (Trib3) interacts with the BMPR2-TD and dissociates from the receptor upon BMP4 binding and activation of the receptor complex [8]. Once unbound, Trib3 promotes the ubiquitination of SMURF1 (SMAD-specific E3 ubiquitin-protein ligase 1), thereby enhancing BMP signaling by reducing the degradation of activated SMADs. Another protein interacting with the BMPR2-TD, cGMP-dependent protein kinase type I (PKG), phosphorylates the receptor leading to enhanced BMP signaling [9]. Following BMP2 binding to the receptor complex, PKG dissociates from the BMPR2-TD and binds to activated SMADs and enhances their transcriptional activity. However, loss of Trib3 or PKG binding to the tail domain of BMPR2 is unlikely to explain the BMP7‑specific increased responsiveness of cells expressing Bmpr2‑ΔTD.
We previously reported that Acvr2a transduced BMP4 signaling and was required for the enhanced BMP7 signaling found in PaSMCs lacking Bmpr2 (Bmpr2 del/del) [5]. We considered the possibility that the BMP7‑specific gain of function seen in PaSMCs carrying the Bmpr2 Δtd allele was exclusively transduced by Acvr2a. We observed, however, that silencing of the Bmpr2 Δtd allele in Bmpr2 Δtd/+ PaSMCs markedly reduced BMP7 signaling. Moreover, silencing Acvr2a transcripts only modestly affected the ability of Bmpr2 Δtd/+ or Bmpr2 Δtd/del PaSMCs to transduce BMP7 signaling. These results demonstrate that the enhanced BMP7 signaling seen in PaSMCs carrying the Bmpr2 Δtd allele is predominantly mediated by Bmpr2‑ΔTD rather than by Acvr2a. We previously reported that knockdown of Acvr2a expression reduced BMP signaling in Bmpr2 del/del PaSMC but not in Bmpr2 flox/flox cells. In our current studies, we observed that BMP7 signaling was greater in Bmpr2 Δtd/del PaSMCs than in Bmpr2 Δtd/flox PaSMCs. Taken together, these observations suggest that the tail domain of Bmpr2 can suppress BMP7 signaling transduced by either Acvr2a or Bmpr2‑ΔTD.
Different BMPs have distinct affinities for each of the BMP receptors. For example, BMP7 has a higher affinity for Alk2 than for other BMP type 1 receptors [19,20]. We therefore tested the hypothesis that the enhanced BMP7 signaling seen in cells expressing Bmpr2‑ΔTD is mediated by Alk2. In Bmpr2 Δtd/+ PaSMCs, deletion of Alk3 markedly reduced BMP4 signaling but not BMP7 signaling. In contrast, we observed that deletion of Alk2 markedly impaired the ability of Bmpr2‑ΔTD to transduce BMP7 signals. These results demonstrate that Bmpr2‑ΔTD and Alk2 mediate BMP7 signaling in cells harboring the Bmpr2 Δtd allele. Taken together with our observations in Bmpr2 del/del PaSMC, these findings suggest that the tail domain of Bmpr2 suppresses Alk2‑dependent BMP7 signaling by either Bmpr2‑ΔTD or Acvr2a and raise the possibility that the the tail domain of Bmpr2 directly inhibits Alk2 function. Unfortunately, currently available commercial antibodies detect BMP type I receptors only when they are overexpressed, hampering the detection of interactions of endogenously expressed BMP receptors.
In conclusion, we report the generation of a mouse harboring an NMD-resistant mutation in the sequences encoding for the Bmpr2‑TD. Mice homozygous for the mutant allele died early in embryogenesis, possibly because of a critical role for Bmpr2‑TD in gastrulation. Heterozygous mice grow normally and, as observed in genetically modified mice carrying other mutant Bmpr2 alleles, they did not spontaneously develop PAH. The BMP7‑specific gain of function observed in PaSMCs from heterozygous Bmpr2 Δtd/+ mice was mediated by the mutant receptor and the BMP type 1 receptor, Alk2. Our data suggest that BMP7 signaling is inhibited in WT PaSMCs by a restriction exerted by the Bmpr2‑TD over Alk2. These data also raise the possibility that some disease-causing BMPR2 mutations may alter BMP signaling in a BMP ligand-specific manner.
Material and Methods
Generation of mice carrying mutant BMP receptors
The strategy to create the Bmpr2 Δtd allele is shown in Figure S1A. The Bmpr2 Δtd-targeting vector carries sequences for Egfp (in frame after exon 11), the SV40 polyadenylation signal (SV40pA), and a phosphoglycerol kinase promoter-controlled neomycin-resistance gene (PGK-neo) cassette after exon 11. Mouse embryonic stem (ES) cells were transfected with the targeting vector, and Southern blot analysis identified an ES cell clone with homologous recombination (Figure S1B). The recombinant ES cells were injected into blastocysts and germline transmission was achieved. Removal of the PGK-neo cassette, flanked by loxP sequences, was achieved by mating the heterozygous Bmpr2 Δtd/+ mice with Ella-Cre transgenic mice. Mice heterozygous for the Bmpr2 Δtd allele were derived from crossing chimeric mice with C57BL/6 female mice. PCR analysis for genotyping purposes using DNA isolated from E7.5 embryos is shown in Figure S1C. Genotyping primers for the mutant Bmpr2 Δtd allele are 5’-GTGCTACAGGCAGTGAGGTCACTC-3’ and 5’-TAGGTCAGGGTGGTCACGAGGGTG-3’ (400-bp product). Genotyping primers for Bmpr2 + allele are 5’-GACTTCACACAGGCTGCAAATGGG-3’ and 5’-CATACTGGGTTGTGGCAGCATGGG-3’ (300-bp product).
Bmpr2 Δtd/+ mice were backcrossed more than 9 times onto a C57BL/6 background. Bmpr2 flox/flox mice [31] bred onto C57BL/6 background were bred to Bmpr2 Δtd/+ mice to generate Bmpr2 Δtd/flox mice. Alk2 flox/flox mice on a mixed C57BL/6; SV129 background [32] or Alk3 flox/flox mice on a C57BL/6 background [33] were bred to Bmpr2 Δtd/+ mice to generate Bmpr2 Δtd/+ ; Alk2 flox/flox, or Bmpr2 Δtd/+ ; Alk3 flox/flox mice, respectively.
All animal experiments were conducted under protocols reviewed and approved by the Subcommittee on Research and Animal Care of the Massachusetts General Hospital.
Hemodynamic measurements in Bmpr2Δtd/+ and WT mice
Mice were anesthetized with ketamine (100 mg/kg) and fentanyl (250 µg/kg) intraperitoneally, intubated, and mechanically ventilated (10 µl/g, 100 breaths per minute; FiO2 = 1). Pancuronium (2 mg/kg) was administered intraperitoneally, and a PE‑10 polyethylene catheter was placed in the left carotid artery for continuous measurement of heart rate and systemic arterial pressure. A 1.2F high-fidelity pressure catheter (FTS‑1211B‑0018; Scisense, London, ON, Canada) was advanced into the right ventricle via the jugular vein to measure right ventricular systolic pressure (RVSP), as an estimate of pulmonary arterial systolic pressure. All signals were recorded and analyzed using a data acquisition system (A D Instruments, Colorado Springs, CO). At the end of the study mice were euthanized with an intraperitoneal injection of pentobarbital (200 mg/kg).
PaSMC isolation and culture
Mice were euthanized with an intraperitoneal injection of pentobarbital (200 mg/kg). Pulmonary arteries (PA) were isolated and incubated individually in trypsin‑EDTA for 10 min at 37°C. PAs were cut into ~1 mm3 pieces and enzymatically digested using a solution of collagenase, papain, elastase, and soybean trypsin inhibitor for 30 min at 37°C. After dissociation, cells were washed twice in DMEM containing 20% fetal bovine serum. After the final wash, cells were resuspended in DMEM with 20% FBS and antibiotics (penicillin – streptomycin) and were cultured at 37°C in 10% CO2. After the first passage, cells were grown in DMEM containing 10% FBS. Cells were used for experiments between passages 3 and 10.
Adenovirus infection
To disrupt Bmpr2, Alk2, or Alk3 genes in PaSMCs isolated from mice harboring alleles carrying loxP sequences, cells were infected with Ad-Cre or Ad-RFP, as a control, at a multiplicity of infection of 150. After cells recovered from infection, efficiency of recombination of the Bmpr2 allele flanked by loxP sequences was determined by PCR and immunoblot techniques, as reported previously [31]. Efficiency of recombination of Alk2 or Alk3 alleles flanked by loxP sequences was determined by qPCR using hydrolysis probes.
Small interfering RNA inhibition of BMP receptors
Silencer® Select siRNA (Applied Biosystems, Life Technologies) specific for Bmpr2, Egfp, and Acvr2a or negative control siRNA (30-50 nM) were transfected into PaSMCs using Pepmute siRNA transfection reagent (SignaGen Laboratories), as described by the manufacturer. After 48 hours, transfected cells were starved in DMEM with 0.1% FBS for 12 to 16 hours and then treated with BMP4 or BMP7.
Gene expression
Total RNA was extracted by guanidine isothiocyanate/phenol method. cDNA was synthesized using M-MLV reverse transcriptase and random primers (Promega). Id1, Smad6, Bmpr2, Egfp, Acvr2a, Alk2, Alk3 and Gapdh transcript levels were measured by qPCR in a Mastercycler ep realplex 2 (Eppendorf) using hydrolysis probes (TaqMan® Gene Expression Assays, Applied Biosystems, Life Technologies) and Probe Fast Master Mix (Kapa Biosystems). qPCR reactions were prepared using the specific FAM-labeled hydrolysis probes and the Gapdh VIC-labeled primer-limited hydrolysis probe, as internal reference gene. Changes in relative gene expression normalized to Gapdh mRNA levels were determined using the relative CT method.
Immunoblot techniques
Confluent PaSMCs were incubated with DMEM with 0.1% FBS for 12 to 16 hours and then treated for various times with BMP4 or BMP7. Cells were lysed with RIPA buffer containing proteinase and phosphatase inhibitor cocktails (Sigma). Lysates were mixed with NuPAGE® LDS sample buffer (Invitrogen, Life Technologies) containing 1mM DTT. Proteins were separated by NuPAGE® Bis‑Tris gels (Invitrogen, Life Technologies), transferred to polyvinylidene difluoride membranes (Immobilon‑FL, Millipore), and blocked in TBS containing 5% skim milk and 0.1% Tween 20. Membranes were reacted with antibodies directed against phosphorylated Smad1/5/8, Gapdh (Cell Signaling); as well as total Smad1/5/8 (Santa Cruz Biotechnology), the tail domain of Bmpr2 (BD Transduction Laboratories), or GFP (Roche). After incubation with HRP-conjugated IgG secondary antibodies (Epitomics and Cell Signaling) and ECL Plus reagent (GE Healthcare Life Sciences), chemifluorescence signals were detected with a Versadoc® Imaging Systems. Captured images were analyzed with ImageJ software (NIH).
Immunofluorescence labeling
PaSMCs were transiently transfected with a plasmid carrying the sequences of Bmpr2‑ΔTD. After 16 hours, cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.1% Triton X-100 in PBS. Immunohistochemical staining was performed with a mouse anti-GFP antibody (Invitrogen, Life Technologies), followed by incubation with fluorescein isothiocyanate-labeled goat anti-mouse IgG. Subcellular localization of Bmpr2‑ΔTD was determined by confocal laser scanning microscopy.
Statistics
Differences between groups were determined using two-way ANOVA for experiments using cultured PaSMCs. Figures show results representative of three or more PaSMC isolates for each genotype. For qPCR experiments, each experimental condition was performed in quadruplicate. All data are expressed as means ± standard deviation. The Student t‑test was used to analyze mouse hemodynamic measurements (HR, MAP, RVSP). A value of p < 0.05 indicated a significant difference.
Supporting Information
Bmpr2Δtd gene-targeting strategy. (A) Schematic diagrams (from top to bottom) of the wild-type Bmpr2 gene, the targeting vector, and the mutant Bmpr2 Δtd allele after homologous recombination. The entire tail domain of Bmpr2 is encoded by exon 12 and 13. A genomic fragment containing intron 11 and exon 12 was replaced by the sequence of Egfp (in frame after exon 11) followed by SV40 polyA signal and a PGK-neo cassette. (B) Southern blot analysis of DNA isolated from ES clones. (C) PCR genotyping analysis of E7.5 embryos generated by intercrosses of F1 heterozygotes.
(TIF)
Hemodynamic measurements in Bmpr2+/+ and Bmpr2Δtd/+ mice. Heart rate (HR), mean systemic arterial pressure (MAP), and right ventricular systolic pressure (RVSP) were measured in 6- to 8-month-old mice (littermates).
(TIF)
Id1 gene expression in Bmpr2+/+ and Bmpr2Δtd/+ PaSMCs after 24 hours treatment with BMP4 or BMP7 (10 ng/ml; *p < 0.01 versus without BMP ligand).
(TIF)
Immunoblotting of Bmpr2Δtd/flox and Bmpr2Δtd/del PaSMCs with anti-Bmpr2‑TD to detect Bmpr2‑WT or anti-GFP to detect Bmpr2‑ΔTD. Gapdh was used as loading control.
(TIF)
Concomitant loss of Alk2 and Alk3 prevents BMP signaling in PaSMCs. (A) Bmpr2 ∆td/+ ; Alk2 flox/flox ; Alk3 flox/flox [+] or Bmpr2 ∆td/+ ; Alk2 del/del ; Alk3 del/del [-] PaSMCs were stimulated with BMP4 or BMP7 (10 ng/ml) for 30 min or 1 h. Immunoblotting for pSmad1/5/8 and Smad1/5/8 show that Bmpr2 ∆td/+ PaSMCs lacking the expression of Alk2 and Alk3 have lost the ability to phosphorylate BMP-responsive Smad1/5/8. (B) Bmpr2 +/+ ; Alk2 flox/flox ; Alk3 flox/flox or Bmpr2 +/+ ; Alk2 del/del ; Alk3 del/del PaSMCs were stimulated with BMP4 or BMP7 (10 ng/ml) for 2 h, and the ability to induce Id1 and Smad6 gene expression was measured by qPCR. Bmpr2 +/+ PaSMCs lacking expression of Alk2 and Alk3 have lost the ability to induce Id1 and Smad6 gene expression in response to BMP ligands. (C) Bmpr2 ∆td/+ ; Alk2 flox/flox ; Alk3 flox/flox or Bmpr2 ∆td/+ ; Alk2 del/del ; Alk3 del/del PaSMCs were stimulated with BMP4 or BMP7 (10 ng/ml) for 2 h, and the ability to induce Id1 and Smad6 gene expression was measured by qPCR. Bmpr2 ∆td/+ PaSMCs lacking expression of Alk2 and Alk3 have lost the ability to induce Id1 and Smad6 gene expression in response to BMP ligands.
(TIF)
Acknowledgments
The authors thank Yushi Mishina and Vesa Kaartinen for providing Alk2+/flox and Alk3+/flox mice, respectively; Noriko Kawai, Sharon M. Cawley and Andrea U. Steinbicker for valuable technical advice; and Francesco Nordio and Andrea Bellavia for statistical analyses.
Funding Statement
Funding came from National Institutes of Health Heart, Lung and Blood Institute HL074352, http://www.nhlbi.nih.gov, DE-1685-1/1, Deutsche Forschungsgemeinschaft, http://www.dfg.de/en/. The funders had no role in study design, data collection and analysis, decisio to publish, or preparation of the manuscript.
References
- 1. Kishigami S, Mishina Y (2005) BMP signaling and early embryonic patterning. Cytokine Growth Factor Rev 16: 265-278. doi: 10.1016/j.cytogfr.2005.04.002. PubMed: 15871922. [DOI] [PubMed] [Google Scholar]
- 2. Miyazono K, Kamiya Y, Morikawa M (2010) Bone morphogenetic protein receptors and signal transduction. J Biochem 147: 35-51. doi: 10.1093/jb/mvp148. PubMed: 19762341. [DOI] [PubMed] [Google Scholar]
- 3. Schmierer B, Hill CS (2007) TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 8: 970-982. doi: 10.1038/nrm2297. PubMed: 18000526. [DOI] [PubMed] [Google Scholar]
- 4. Sieber C, Kopf J, Hiepen C, Knaus P (2009) Recent advances in BMP receptor signaling. Cytokine Growth Factor Rev 20: 343-355. doi: 10.1016/j.cytogfr.2009.10.007. PubMed: 19897402. [DOI] [PubMed] [Google Scholar]
- 5. Yu PB, Beppu H, Kawai N, Li E, Bloch KD (2005) Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J Biol Chem 280: 24443-24450. doi: 10.1074/jbc.M502825200. PubMed: 15883158. [DOI] [PubMed] [Google Scholar]
- 6. Kawabata M, Chytil A, Moses HL (1995) Cloning of a novel type II serine/threonine kinase receptor through interaction with the type I transforming growth factor-beta receptor. J Biol Chem 270: 5625-5630. doi: 10.1074/jbc.270.10.5625. PubMed: 7890683. [DOI] [PubMed] [Google Scholar]
- 7. Beppu H, Minowa O, Miyazono K, Kawabata M (1997) cDNA cloning and genomic organization of the mouse BMP type II receptor. Biochem Biophys Res Commun 235: 499-504. doi: 10.1006/bbrc.1997.6816. PubMed: 9207184. [DOI] [PubMed] [Google Scholar]
- 8. Chan MC, Nguyen PH, Davis BN, Ohoka N, Hayashi H et al. (2007) A novel regulatory mechanism of the bone morphogenetic protein (BMP) signaling pathway involving the carboxyl-terminal tail domain of BMP type II receptor. Mol Cell Biol 27: 5776-5789. doi: 10.1128/MCB.00218-07. PubMed: 17576816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schwappacher R, Weiske J, Heining E, Ezerski V, Marom B et al. (2009) Novel crosstalk to BMP signalling: cGMP-dependent kinase I modulates BMP receptor and Smad activity. EMBO J 28: 1537-1550. doi: 10.1038/emboj.2009.103. PubMed: 19424179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC et al. (1997) Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31-q32. Circulation 95: 2603-2606. doi: 10.1161/01.CIR.95.12.2603. PubMed: 9193425. [DOI] [PubMed] [Google Scholar]
- 11. Nichols WC, Koller DL, Slovis B, Foroud T, Terry VH et al. (1997) Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-32. Nat Genet 15: 277-280. doi: 10.1038/ng0397-277. PubMed: 9054941. [DOI] [PubMed] [Google Scholar]
- 12. McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW et al. (2009) ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol 53: 1573-1619. doi: 10.1016/j.jacc.2009.01.004. PubMed: 19389575. [DOI] [PubMed] [Google Scholar]
- 13. Fessel JP, Loyd JE, Austin ED (2011) The genetics of pulmonary arterial hypertension in the post-BMPR2 era. Pulm Circ 1: 305-319. doi: 10.4103/2045-8932.87293. PubMed: 22140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Machado RD, Aldred MA, James V, Harrison RE, Patel B et al. (2006) Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat 27: 121-132. doi: 10.1002/humu.20285. PubMed: 16429395. [DOI] [PubMed] [Google Scholar]
- 15. Cogan JD, Pauciulo MW, Batchman AP, Prince MA, Robbins IM et al. (2006) High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med 174: 590-598. doi: 10.1164/rccm.200602-165OC. PubMed: 16728714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hamid R, Hedges LK, Austin E, Phillips JA 3rd, Loyd JE et al. (2010) Transcripts from a novel BMPR2 termination mutation escape nonsense mediated decay by downstream translation re-initiation: implications for treating pulmonary hypertension. Clin Genet 77: 280-286. doi: 10.1111/j.1399-0004.2009.01311.x. PubMed: 20095988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Austin ED, Phillips JA, Cogan JD, Hamid R, Yu C et al. (2009) Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir Res 10: 87. doi: 10.1186/1465-9921-10-87. PubMed: 19785764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beppu H, Ichinose F, Kawai N, Jones RC, Yu PB et al. (2004) BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol 287: L1241-L1247. doi: 10.1152/ajplung.00239.2004. PubMed: 15286002. [DOI] [PubMed] [Google Scholar]
- 19. Aoki H, Fujii M, Imamura T, Yagi K, Takehara K et al. (2001) Synergistic effects of different bone morphogenetic protein type I receptors on alkaline phosphatase induction. J Cell Sci 114: 1483-1489. PubMed: 11282024. [DOI] [PubMed] [Google Scholar]
- 20. ten Dijke P, Yamashita H, Sampath TK, Reddi AH, Estevez M et al. (1994) Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J Biol Chem 269: 16985-16988. PubMed: 8006002. [PubMed] [Google Scholar]
- 21. Nasim MT, Ogo T, Chowdhury HM, Zhao L, Chen CN et al. (2012) BMPR-II deficiency elicits pro-proliferative and anti-apoptotic responses through the activation of TGFbeta-TAK1-MAPK pathways in PAH. Hum Mol Genet 21: 2548-2558. doi: 10.1093/hmg/dds073. PubMed: 22388934. [DOI] [PubMed] [Google Scholar]
- 22. West J, Harral J, Lane K, Deng Y, Ickes B et al. (2008) Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol 295: L744-L755. doi: 10.1152/ajplung.90255.2008. PubMed: 18723761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Long L, MacLean MR, Jeffery TK, Morecroft I, Yang X et al. (2006) Serotonin increases susceptibility to pulmonary hypertension in BMPR2-deficient mice. Circ Res 98: 818-827. doi: 10.1161/01.RES.0000215809.47923.fd. PubMed: 16497988. [DOI] [PubMed] [Google Scholar]
- 24. Song Y, Jones JE, Beppu H, Keaney JF Jr., Loscalzo J et al. (2005) Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation 112: 553-562. doi: 10.1161/CIRCULATIONAHA.104.492488. PubMed: 16027259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frank DB, Lowery J, Anderson L, Brink M, Reese J et al. (2008) Increased susceptibility to hypoxic pulmonary hypertension in Bmpr2 mutant mice is associated with endothelial dysfunction in the pulmonary vasculature. Am J Physiol Lung Cell Mol Physiol 294: L98-109. PubMed: 18024717. [DOI] [PubMed] [Google Scholar]
- 26. Beppu H, Kawabata M, Hamamoto T, Chytil A, Minowa O et al. (2000) BMP type II receptor is required for gastrulation and early development of mouse embryos. Dev Biol 221: 249-258. doi: 10.1006/dbio.2000.9670. PubMed: 10772805. [DOI] [PubMed] [Google Scholar]
- 27. Délot EC, Bahamonde ME, Zhao M, Lyons KM (2003) BMP signaling is required for septation of the outflow tract of the mammalian heart. Development 130: 209-220. doi: 10.1242/dev.00181. PubMed: 12441304. [DOI] [PubMed] [Google Scholar]
- 28. Aramaki T, Sasai N, Yakura R, Sasai Y (2010) Jiraiya attenuates BMP signaling by interfering with type II BMP receptors in neuroectodermal patterning. Dev Cell 19: 547-561. doi: 10.1016/j.devcel.2010.09.001. PubMed: 20951346. [DOI] [PubMed] [Google Scholar]
- 29. Nishihara A, Watabe T, Imamura T, Miyazono K (2002) Functional heterogeneity of bone morphogenetic protein receptor-II mutants found in patients with primary pulmonary hypertension. Mol Cell Biol 13: 3055-3063. doi: 10.1091/mbc.E02-02-0063. PubMed: 12221115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R et al. (2002) Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet 11: 1517-1525. doi: 10.1093/hmg/11.13.1517. PubMed: 12045205. [DOI] [PubMed] [Google Scholar]
- 31. Beppu H, Lei H, Bloch KD, Li E (2005) Generation of a floxed allele of the mouse BMP type II receptor gene. Genesis 41: 133-137. doi: 10.1002/gene.20099. PubMed: 15736264. [DOI] [PubMed] [Google Scholar]
- 32. Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V (2004) Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech Dev 121: 173-182. doi: 10.1016/j.mod.2003.12.003. PubMed: 15037318. [DOI] [PubMed] [Google Scholar]
- 33. Mishina Y, Hanks MC, Miura S, Tallquist MD, Behringer RR (2002) Generation of Bmpr/Alk3 conditional knockout mice. Genesis 32: 69-72. doi: 10.1002/gene.10038. PubMed: 11857780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bmpr2Δtd gene-targeting strategy. (A) Schematic diagrams (from top to bottom) of the wild-type Bmpr2 gene, the targeting vector, and the mutant Bmpr2 Δtd allele after homologous recombination. The entire tail domain of Bmpr2 is encoded by exon 12 and 13. A genomic fragment containing intron 11 and exon 12 was replaced by the sequence of Egfp (in frame after exon 11) followed by SV40 polyA signal and a PGK-neo cassette. (B) Southern blot analysis of DNA isolated from ES clones. (C) PCR genotyping analysis of E7.5 embryos generated by intercrosses of F1 heterozygotes.
(TIF)
Hemodynamic measurements in Bmpr2+/+ and Bmpr2Δtd/+ mice. Heart rate (HR), mean systemic arterial pressure (MAP), and right ventricular systolic pressure (RVSP) were measured in 6- to 8-month-old mice (littermates).
(TIF)
Id1 gene expression in Bmpr2+/+ and Bmpr2Δtd/+ PaSMCs after 24 hours treatment with BMP4 or BMP7 (10 ng/ml; *p < 0.01 versus without BMP ligand).
(TIF)
Immunoblotting of Bmpr2Δtd/flox and Bmpr2Δtd/del PaSMCs with anti-Bmpr2‑TD to detect Bmpr2‑WT or anti-GFP to detect Bmpr2‑ΔTD. Gapdh was used as loading control.
(TIF)
Concomitant loss of Alk2 and Alk3 prevents BMP signaling in PaSMCs. (A) Bmpr2 ∆td/+ ; Alk2 flox/flox ; Alk3 flox/flox [+] or Bmpr2 ∆td/+ ; Alk2 del/del ; Alk3 del/del [-] PaSMCs were stimulated with BMP4 or BMP7 (10 ng/ml) for 30 min or 1 h. Immunoblotting for pSmad1/5/8 and Smad1/5/8 show that Bmpr2 ∆td/+ PaSMCs lacking the expression of Alk2 and Alk3 have lost the ability to phosphorylate BMP-responsive Smad1/5/8. (B) Bmpr2 +/+ ; Alk2 flox/flox ; Alk3 flox/flox or Bmpr2 +/+ ; Alk2 del/del ; Alk3 del/del PaSMCs were stimulated with BMP4 or BMP7 (10 ng/ml) for 2 h, and the ability to induce Id1 and Smad6 gene expression was measured by qPCR. Bmpr2 +/+ PaSMCs lacking expression of Alk2 and Alk3 have lost the ability to induce Id1 and Smad6 gene expression in response to BMP ligands. (C) Bmpr2 ∆td/+ ; Alk2 flox/flox ; Alk3 flox/flox or Bmpr2 ∆td/+ ; Alk2 del/del ; Alk3 del/del PaSMCs were stimulated with BMP4 or BMP7 (10 ng/ml) for 2 h, and the ability to induce Id1 and Smad6 gene expression was measured by qPCR. Bmpr2 ∆td/+ PaSMCs lacking expression of Alk2 and Alk3 have lost the ability to induce Id1 and Smad6 gene expression in response to BMP ligands.
(TIF)