

Abstract
During HIV-1 infection a population of latently infected cells is established. This population is the major obstacle preventing total eradication of the virus from AIDS patients. HIV-1 latency is thought to arise by various mechanisms including repressive chromatin modifications. Transcription factors such as YY1 have been shown to facilitate repressive chromatin modifications by the recruitment of histone deacetylases. In this study, we identified a novel binding site for YY1 on the HIV-1 LTR, 120 nucleotides upstream of the transcription start site. We show that YY1 can bind to this site in vitro and in vivo and that binding to the LTR is dissociated upon T cell activation. Overexpression of YY1 causes an increase in the proportion of cells that produce latent infections. These observations, in combination with previous results, demonstrate that YY1 plays a prominent role in controlling the establishment and maintenance of latent HIV-1 provirus in unstimulated cells.
Introduction
A population of cells containing transcriptionally repressed integrated provirus is established early in HIV-1 infection [1]. This latent HIV-1 reservoir is the major obstacle in developing a cure for HIV-1/AIDS. Although the latent viral reservoir tends to be small in patients on highly active antiretroviral therapy (HAART) (~1 x 106 infectious units per million CD4+ cells) [2], HIV-1 preferentially infects CD4+ memory T cells [3,4], which are maintained for many years [5]. There are also other cellular reservoirs that are sources for re-emergence of virus including monocytes, macrophages, astrocytes, dendritic cells, hematopoietic progenitor cells, natural killer cells, mast cells and neurons [6]. HAART can decrease the plasma viral load to undetectable levels [7], but upon interruption of treatment viremia rebounds as a result of replication from this minimal population of latent cells [8]. Consequently a major limitation of HAART is that it does not represent a cure for the disease since it does not target the latent population. Many strategies are currently being pursued to purge the latent viral reservoir, some of which focus on reversing the repressive effects of chromatin [9].
Repressive chromatin is associated with deacetylation of specific lysines within the N-terminal tails of histones by histone deacetylases (HDACs). HDACs are recruited to DNA by transcription factors involved in transcriptional repression. Many host cell transcription factors have been shown to negatively regulate transcription from the HIV-1 LTR, including NF-κB p50, SP1, CBF1 and Yin Yang 1 (YY1) [10]. Of these, YY1 was shown to be of particular importance [11]. YY1 was first identified with respect to HIV-1 transcription associated with a sequence within the -16 to +27 region on the HIV-1 LTR. It was shown to bind indirectly to DNA at this location in a complex with the late simian virus 40 (LSF) protein. This study showed that YY1, in association with LSF bound near the core promoter, is involved in repression of HIV-1 transcription by recruitment of histone deacetylase 1 (HDAC1) [12].
In previous studies, we have observed a YY1 complex formed in electrophoretic mobility shift assays (EMSA) using probes spanning the highly conserved binding site for USF1/2 and TFII-I (RBF-2), designated RBEIII [13,14]. The overall goal of this study was to characterize the role of YY1 binding to the RBEIII site. We show that YY1 directly binds sequences overlapping RBEIII in vitro and have identified mutations that prevent this interaction. YY1 also associated with the RBEIII-region of the LTR in cells using chromatin immunoprecipitation (ChIP) in unstimulated cells, but becomes dissociated from the HIV-1 LTR in stimulated cells. Furthermore, we show YY1 is associated with the LTR in cells that form latent provirus, whereas YY1 is absent from the LTR in the population of infected cells where transcription from the LTR is active. Additionally, overexpression of YY1 promotes silencing of HIV LTR expression after 48 hours and up to four weeks following infection, but a YY1 mutant defective for recruitment of HDAC1 has no effect. Taken together these results show that YY1 plays an important role in establishing and maintaining immediate latency and thereby this protein may be a potential therapeutic target for modulating the latent HIV reservoir in AIDS patients.
Materials and Methods
Recombinant DNA molecules
The YY1 ORF, produced by PCR using oligonucleotides WB005 and WB006 (Table S1) was cloned into the pFastbac plasmid (Invitrogen) at the EcoRI and XhoI restriction sites, to produce the YY1 baculovirus expression vector. The pTY-LAI HIV-1 LTR mini-virus reporter was derived from the lentiviral vector pTY-EFeGFP (NIH AIDS Reagent Program) as previously described [15] but with the luciferase reporter replaced with the dsRed ORF, inserted as an NsiI - SwaI fragment generated by amplification with oligonucleotides SAR17 and SAR18 (Table S1). The HIV-1 LTR YY1 binding site mutant (ACTGCTGA to ACTGCact) was produced using site directed mutagenesis with oligonucleotides TM237, TM238, WB140 and WB141 (Table S1). A Flag tag-encoding fragment was inserted into pEF4-his/myc using oligonucleotides WB089 and WB090 bearing Kpn1 and SpeI restriction sites to create pEFlag. The wild type YY1 ORF was amplified using oligonucleotides WB104 and WB137 (Table S1) and cloned into the SpeI and EcoRI restriction sites of pEFlag to produce pEFlag-YY1. The YY1 glycine/alanine/lysine mutant was created by deleting codons for amino acids 155-198 by generating PCR fragments with oligonucleotides WB104, WB196, and WB197, WB137. The reaction products were mixed together in a second reaction and amplified with oligonucleotides WB104 and WB137 and the resulting fragment was cloned into the SpeI and EcoRI sites of pEFlag to produce pEFlag-g/a/k.
Cell lines, transfections and cell culture
SF9 insect cells were cultured and infected for protein expression as previously described [13]. Jurkat-tat cells were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: Jurkat-tat from Drs. Antonella Caputo, William Haseltine, and Joseph Sodroski. Human embryonic kidney (HEK) 293T cells are from the American Type Culture Collection. Jurkat-tat cells were grown in RPMI 1640 (Sigma) with 10% fetal bovine serum (Sigma) supplemented with penicillin (100 U/mL), streptomycin (100 mg/mL) (Gibco), and Gentecin (0.8 mg/mL) (Invitrogen) and maintained in a humidified 37 °C, 5% CO2 atmosphere. HEK 293T were grown in DMEM (Sigma) with 10% fetal bovine serum (Sigma) supplemented with penicillin (100 U/mL) and streptomycin (100 U/mL). VSV pseudo typed pTY-LAI-dsRed virus was produced by co-transfection of Human embryonic kidney (HEK) 293T cells (ATCC) as previously described [16]. Viral stocks were purified through a 0.2 µM Whatman puradisc syringe filter and concentrated by centrifugation in Amicon Ultra-4 Centrifugal Filter Units (Millipore). One million Jurkat-tat cells were infected with 40 µL of concentrated virus (to yield ~ 10% infection rate as determined by expression of GFP) by spinoculation for 1 hour. Infected Jurkat-tat cells were monitored for GFP and dsRed expression by BD Bioscience Flow cytometer everyday for four days and once a week for a month. Cells were sorted using a BD Influx cell sorter. Jurkat-tat cells were transfected using an Amaxa Nucleofector (Lonza) according to the manufacture’s instructions. Stable cell lines were selected using 0.15 mg/mL zeocin (Invitrogen). Jurkat-tat cells were stimulated with 50 nM PMA (Sigma) for four hours. Data was analyzed using FlowJo analysis software (Tree Star).
EMSA assays
Jurkat nuclear extracts for EMSA reactions were prepared as previously described [17]. SF9 cells extracts for EMSA reactions were prepared as previously described [13]. EMSAs were performed as before [16]. Antibodies for super-shift assays are as follows: α-TFII-I (Santa Cruz), α-USF1 (Santa Cruz), α-USF2 (Santa Cruz), α-YY1 (AbCam). Double stranded oligonucleotides used for EMSA are listed below: RBEIII probe GATCCTTCAAGAACTGCTGACATCGAGCTTTCTC, RBEIII mutant probe GATCCTTCAAGAACTGCactCATCGAGCTTTCTC, and parovirus promoter YY1 binding sequence competitor oligonucleotide: GAAATGACGTAATTGTCCGCCATCTTGTACCGG.
ChIP assays
Jurkat-tat nuclei were extracted as follows: 3 x 107 cells were suspended in NP40 lysis buffer (0.5% NP40, 10 mM Tris HCl, pH 7.8, 3 mM MgCl2), 1X protease inhibitor cocktail (Roche) and incubated on ice for 5 minutes. The nuclei were pelleted for 5 minutes at 500 x g at 4°C and suspended in 1 mL of RPMI, 3 mM MgCl2, 1X protease inhibitor cocktail and 1% formaldehyde. The nuclei were cross-linked at room temperature for 10 minutes and the reaction was stopped by addition of 125 mM glycine. After 5 minutes the nuclei were pelleted as above, washed in cold PBS, suspended in sonication buffer (10 mM Tris, pH 7.8, 10 mM EDTA, 0.5% SDS) and sonicated to obtain DNA fragments < 200 base pairs. The ChIP assays were performed as previously described [15]. Statistical analysis using Student’s T-tests were performed using R 2.15.1. Antibodies used for ChIP assays are as follows: α-YY1 (Abcam), α-TFII-I (Santa Cruz) and α-Flag (Ablab, UBC). Oligonucleotides used for qPCR reactions are as follows: RBEI P1 AGTGGCGAGCCCTCAGAT, RBEI P2 AGAGCTCCCAGGCTCAAATC, ER P1 TTTCCGCTGGGGACTTTC, ER P2 CCAGTACAGGCAAAAAGCAG, RBEIII P1 TTTCCGCTGGGGACTTTC, RBEIII P2 CAGCGGAAAGTCCCTTGTAG.
Results
YY1 binds directly to the HIV-1 LTR near the RBEIII element in vitro
YY1 was previously shown to indirectly associate with the HIV-1 LTR between -16 and +27 position near the transcription start site through binding to LSF. This interaction was shown to cause repression of LTR-directed transcription by recruitment of HDAC1 [11]. Additionally, in previous studies using EMSA we observed a protein complex from Jurkat cells that was sensitive to YY1 antibody when using probes spanning the highly conserved RBF-2 (USF1/2-TFII-I) binding site called RBEIII (-120 to -140). It was shown that deletion of sequences 3’ of the core -ACTGCTGA- RBEIII element prevented formation of the putative YY1 complex [14]. In this report we examined the interaction of YY1 with this region of the HIV-1 LTR in more detail. Consistent with previous results, using a radiolabeled oligonucleotide spanning the RBEIII site in EMSA with Jurkat nuclear extracts, we observe a complex that is sensitive to the addition of YY1 antibody (Figure 1A, lane 5), but not to antibodies against TFII-I, USF1 or USF2 (Figure 1A, lanes 2-4). This indicates that YY1 is capable of forming a complex near the RBEIII element that is distinct from the previously described factor RBF-2, which we have shown to be comprised of USF1, USF2 and TFII-I [16]. To determine if YY1 is capable of binding directly to the HIV-1 LTR at this position, we expressed recombinant YY1 protein in insect cells using baculovirus (Figure 1B). We found that recombinant YY1 produced a complex in EMSA reactions with the radiolabeled RBEIII oligonucleotide probe that migrates identically with the YY1 antibody-sensitive protein complex produced in Jurkat nuclear extracts (Figure 1C, compare lanes 1 and 4). Furthermore, we found that the complex produced with recombinant YY1 from baculovirus could be super-shifted with YY1 antibody (Figure 1C, lane 2). Binding of recombinant YY1 to the RBEIII oligonucleotide was also strongly competed by unlabeled competitor oligonucleotide bearing a previously defined YY1 binding sequence from the B19 parovirus promoter [18](Figure 1D, lane 2). To identify nucleotides that are required for interaction of YY1 with the RBEIII oligonucleotide, we used competition experiments with unlabeled mutant oligonucleotides in EMSA with the labeled wild type RBEIII probe (Figure 1D, lanes 3-10). In these experiments we found four nucleotides that seem to be particularly important for binding of YY1 to this region, including the 3’ -TG- residues within the RBEIII core sequence (Figure 1D, -ACTGCTGA-, lanes 4-5) and the -CA- residues in the -CAT- sequence directly 3’ of the RBEIII site (Figure 1D, lanes 7-8). YY1 recognition sequences on other promoters contain a conserved -CAT- motif, which is the core consensus sequence for YY1 binding [19-22].
Figure 1. YY1 binds to the HIV-1 LTR near RBEIII.

Panel A: EMSA was performed with Jurkat nuclear extracts and radiolabeled RBEIII probe (core RBEIII site highlighted in red). Antibodies to TFII-I (lane 2), USF1 (lane 3), USF2 (lane 4), or YY1 (lane 5) were added to analyze the complex components. Free probe (FP) is shown in lane 6. Panel B: An extract from SF9 cells infected with a baculovirus expressing YY1 was separated on 12% SDS-PAGE and analyzed by immunoblotting with YY1 antibody (lane 2). Lane 1 contains a high molecular weight protein ladder. Panel C: EMSA was performed with recombinant YY1 produced by baculovirus (lanes 1 and 2) or Jurkat nuclear extracts (lane 4) with the radiolabeled RBEIII probe. The RBF2 complex in lane 4 is shown. YY1 antibody was added to the reaction in lane 2. Free probe is shown in lane 3. Panel D: (Top) EMSA was performed with recombinant YY1 and labeled RBEIII probe (lane 1), unlabeled competitor oligonucleotides were added to the reactions at 100-fold molar excess (lanes 2-10) (Bottom). Sequences of unlabeled competitor probes; 2, a known YY1 binding sequence oligonucleotide from the parvovirus promoter; 3, wild type RBEIII probe; 4-5, three nucleotide substitutions; 6-12, point substitutions of the RBEIII probe; 13 free probe (FP). Non-specific (NS) bands are also shown. Substitutions are indicated in lower case.
YY1 binds the HIV-1 LTR near RBEIII in vivo
To examine whether YY1 is capable of binding the LTR near RBEIII in vivo, we used a double-labeled mini-virus reporter, which allows detection of cells bearing integrated provirus independently of LTR activity. The virus contains eGFP expressed from an internal E1Fα promoter and dsRed expressed from the 5’ LTR as a fusion with p24Gag (Figure 2A). Since it was previously shown that YY1 is recruited to the LTR flanking the transcription start site through interaction with LSF [11], near an additional binding site for RBF-2 designated RBEI [23], we designed primers for ChIP to separately measure interaction of YY1 with the core promoter/RBEI region, the upstream RBEIII region, in addition to the enhancer region between these two elements (Figure 2B). We introduced a substitution of three nucleotides at the RBEIII site, shown to prevent binding of YY1 to the RBEIII oligonucleotide in vitro (Figure 2C). Virus was produced for both the wild type and RBEIII/YY1 mutant LTR reporter constructs and used to infect Jurkat-tat cells. Individual clones were isolated from the infected populations using fluorescence activated cell sorting (FACS) for eGFP expression. For ChIP assays, to enable detection of separate interactions with these regions we sheared DNA from cross-linked Jurkat-tat cells to less than ~200 nucleotides. In ChIP experiments with YY1 immunoprecipitated complexes, we observed a 2-fold lower signal with the primer set that amplifies the enhancer region of the wild type LTR relative to the primer sets that amplify the RBEI/ core promoter and RBEIII regions, indicating preferential interaction of YY1 with the two regions flanking the enhancer region (Figure 2C, closed bars). Furthermore, we also observed a 4-fold relative decrease in YY1 occupancy at the RBEIII region in Jurkat-tat cells bearing the RBEIII/YY1 LTR mutation (Figure 2C, RBEIII). Interestingly, the amount of YY1 binding near the transcription start site also decreased by approximately one half when the upstream RBEIII/YY1 site was mutated. This indicates that there may be cooperative binding of this factor between these two sites flanking the HIV enhancer.
Figure 2. Mutations at RBEIII prevent binding of YY1 in Jurkat-tat cells.

Panel A: Schematic representation of the pTY-LAI-dsRed reporter mini virus. The 5’ LTR controls expression of a p24 capsid-dsRed fusion, while the E1Fα promoter constitutively expresses eGFP. Panel B: Representation of primers used for ChIP analysis of the upstream RBEIII region, enhancer region (ER), and RBEI/ transcription start site region. Panel C: A representative pool of Jurkat-tat cells infected with the wild type and RBEIII/YY1 mutant reporter virus were used for ChIP analysis with YY1 antibody. Top, the core RBEIII sequence is shown in red and substitutions in the RBEIII/YY1 mutant indicated in lowercase. Bottom, Representative pools of Jurkat-tat cells infected with the wild type reporter virus or mutant RBEIII cell lines were used for ChIP analysis with YY1 antibody, using the primer sets indicated in Panel B. Error bars represent the standard deviation. ** Represents P values ≤ 0.01.
YY1 is dissociated from the HIV-1 LTR in stimulated T cells
YY1 was previously shown to interact with LSF near the transcription start site where it contributes to repression of transcription from the HIV-1 LTR [11]. Consequently, we examined whether binding of YY1 to the elements flanking the enhancer was affected in stimulated Jurkat-tat cells. Phorbol-12-myristate-13-acetate (PMA) causes activation of T cells by stimulating the RAS-MAPK and PKC-IKK pathways downstream of the T cell receptor, which causes activation of transcription from the HIV-1 LTR by the GABP/ Ets, AP1 and NF-kB transcription factors [24]. Accordingly, treatment of Jurkat-tat cells infected with the wild type LTR-dsRed reporter mini-virus with PMA causes induction of dsRed expression as detected by flow cytometry (Figure 3A). Using ChIP, we examined binding of YY1 to the LTR in both stimulated and unstimulated cells (Figure 3B). We found that association of YY1 with the LTR was decreased by approximately 6-fold at the RBEIII site in cells stimulated with PMA (Figure 3B, open bars). Binding of YY1 near the transcription start site was also decreased significantly in Jurkat-tat cells treated with PMA. This indicates that YY1 must be dissociated from the HIV-1 LTR to allow transcriptional activation and supports its role as a repressor in establishment of latent proviral populations. To determine if the loss of YY1 at the HIV-1 LTR was due to an alteration in YY1 stability or expression we performed Western blots on untreated and PMA treated cells, but did not observe a change in YY1 abundance between the two populations (Figure 3C and 3D). This indicates that dissociation of YY1 from its binding sites on the HIV-1 LTR is likely caused by a post-translational modification rather than targeted degradation.
Figure 3. YY1 dissociates from the HIV-1 LTR in stimulated Jurkat-tat cells.

Panel A: Flow analysis of the untreated control (left) and PMA (right) induced Jurkat-tat cells bearing integrated pTY-LAI-dsRed reporter mini-virus. Expression of eGFP is indicated on the x-axis and dsRed expression on the y-axis. Panel B: Representative clones of Jurkat-tat cells infected with the wild type reporter virus induced with PMA or untreated (control) were used for ChIP analysis with YY1 antibody, using the primers indicated in Figure 2B. Panel C: Immunoblots of control and PMA induced Jurkat-tat cell lysates with YY1 antibody. Panel D: Immunoblot quantification of control and PMA induced cells using the Odyssey Infrared Imaging System. The y-axis represents the amount of YY1 divided by the amount of GAPDH normalized to one. The error bars show the standard deviation. * Represents P values ≤ 0.05, ** represents P values ≤ 0.01, *** represents P values ≤ 0.001.
Twenty-four to seventy-two hours post infection with the double reporter virus, we find that the majority of the Jurkat-tat cells expressing eGFP (indicating viral integration), do not express dsRed from the LTR promoter, indicating early transcriptional latency. Considering that YY1 is dissociated from the LTR in PMA stimulated cells, we wondered whether YY1 would be associated with the LTR in unstimulated cells containing virus that had remained transcriptionally active after infection. For this experiment, we infected Jurkat-tat cells with the double reporter virus and used FACS to sort out infected cells with active LTR transcription (eGFP and dsRed positive cells) and infected cells without LTR expression (eGFP only). Since we used a HIV-1 mini-virus, which does not express viral gene products that cause cytotoxicity [25,26] we were able to expand these two cell populations for analysis to compare YY1 binding at the LTR using ChIP. We found that the amount of YY1 present on the HIV-1 LTR at both the RBEIII site and the transcriptional start site in Jurkat-tat cells expressing only eGFP, with an inactive LTR, was approximately 5-fold higher than the amount of YY1 present on the LTR that was transcriptionally active in the double positive (eGFP and dsRed positive) infected population (Figure 4B). This indicates that YY1 is not present on the HIV-1 LTR in Jurkat-tat cells that have established active HIV-1 infections immediately upon infection and supports the view that association of YY1 with the LTR has an important role in negatively regulating the HIV-1 LTR and establishing proviral latency when bound at both positions. Similar to the results of Figure 3C, we do not observe any differences in expression levels of YY1 in the two sorted cell populations (data not shown).
Figure 4. YY1 is not bound to actively transcribed HIV-1 LTR in unstimulated Jurkat-tat cells.

Panel A: A representative population of Jurkat-tat cells infected with the wild type reporter virus was sorted using by FACS and populations of cells expressing only eGFP (green) or both eGFP and dsRed (red) were collected and expanded in culture. Panel B: ChIP analysis was performed on the expanded populations of Jurkat-tat cells expressing eGFP (green) or both eGFP and dsRed (red) using YY1 antibodies and the primer sets as described in Figure 2B. * Represents P values ≤ 0.05, ** represents P values ≤ 0.01, *** represents P values ≤ 0.001.
TFII-I is bound constitutively to the HIV-1 LTR
TFII-I was previously shown to bind to the RBEIII and RBEI sites in association with USF1/2 [13,23]. We used ChIP to examine the effect of the triple mutation at RBEIII (Figure 2B), for binding of TFII-I to the LTR. Interestingly we found that the mutation caused a 10-fold decrease of TFII-I binding to the mutant RBEIII LTR, at both the upstream RBEIII element and the at the RBEI site near the transcriptional start (Figure 5A). This is similar to the effect that was observed with binding of YY1 to the LTR and suggests that there may be cooperative binding of these factors at these sites. We also compared association of TFII-I with the LTR in unstimulated cells and cells stimulated with PMA (Figure 5B). We found that unlike YY1, TFII-I was bound to the HIV-1 LTR in both conditions, at both the RBEI and RBEIII sites, consistent with previous observations [16]. Consequently, because TFII-I is constitutively present on the HIV-1 LTR, this factor may play a role in both repression and activation of transcription, unlike YY1 whose association with the LTR is primarily consistent with the function of a transcriptional repressor.
Figure 5. TFII-I binds to RBEIII constitutively.

Panel A: A representative pool of Jurkat-tat cells infected with the wild type (solid bars) and RBEIII/YY1 mutant (open bars) reporter virus were used for ChIP analysis with TFII-I antibody. Panel B: A representative clone of Jurkat-tat cells infected with the wild type reporter virus was induced with PMA or left untreated (control) and used for ChIP analysis with α-TFII-I antibodies, and the primers specific for RBEIII, the enhancer region (ER) and the RBEI site. * Represents P values ≤ 0.05, ** represents P values ≤ 0.01.
YY1 is involved in immediate silencing of the HIV-1 LTR upon infection
To further characterize the role of YY1 in causing the immediate repression of HIV-1 expression in infected cells, we examined the effect of overexpressing YY1. For these experiments we transfected Jurkat-tat cells with expression plasmids producing wild type YY1 (pEFlag-YY1) and a YY1 mutant containing a deletion of the glycine/alanine/lysine rich region of YY1 (Figure 6A), previously shown to interact with HDAC1 [12] (pEFlag-g/a/k) and a vector control (pEFlag). Stably transfected lines were isolated by selection with zeocin. Over-expression of wild type YY1 and the YY1 g/a/k mutant was verified by immunoblotting with α-Flag antibody (Figure 6B). When analyzed by ChIP, we observed that both wild type YY1 and the YY1 g/a/k mutant bound to both the RBEI and RBEIII sites in Jurkat-tat cells containing the integrated virus (Figure 6C). The stable Jurkat-tat cell lines were infected with the HIV-1 double reporter virus; eGFP and dsRed expression was analyzed by flow cytometry every 24 hours for the first four days and once a week thereafter for a month. The percent of infected cells with actively transcribed virus was calculated as a ratio of dsRed expression relative to the total eGFP-expressing population. Approximately 5% of infected cells bearing the vector control express dsRed one day post infection. This proportion of cells with actively transcribing virus rises to ~20-25% within a week and remains stable at this proportion for at least a month (Figure 6D, pEFlag). Jurkat-tat cells over-expressing YY1 contain ~5% of infected cells expressing dsRed one day post infection, which nearly identical as the control cells, this proportion does not increase throughout the course of culturing these cells for a month (Figure 6D, YY1). This supports the notion that YY1 plays an important role in repression of the LTR to produce and maintain latently infected cells. In Jurkat-tat cells overexpressing the YY1 g/a/k deletion mutant, we observe a result nearly identical to cells bearing the vector control, where ~5% of infected cells express dsRed one day post infection, a proportion which increases to 20-25% after a week (Figure 6D, g/a/k). These results suggest that YY1’s role in repressing transcription from the HIV-1 LTR and establishment of latency early after infection may involve recruiting HDAC1.
Figure 6. YY1 overexpression increases the proportion of virus that maintains latent infection.

Panel A: Schematic representation of expression constructs: wild type YY1 (YY1), a YY1 deletion mutant lacking the HDAC1 interaction region (g/a/k) and vector control (pEFlag) used to produce stable cell lines. Indicated are a histidine rich region (His), the glycine/alanine (GA) and glycine/lysine (GK) rich regions, and the zinc finger domains (ZF) DNA binding domain. Panel B: Immunoblot of cell extracts from stable lines transfected with the YY1 (lane 1), g/a/k YY1 mutant (lane 2) and vector control (lane 3) with α-Flag antibody. Panel C: ChIP was performed on pools of stably transfected Jurkat-tat cells overexpressing YY1, the g/a/k YY1 mutant or the vector control, infected with the wild type reporter virus using α-Flag antibody, and the primer sets as described in Figure 2B. Panel D: Stably transfected Jurkat-tat cells overexpressing YY1, the g/a/k YY1 mutant or the vector control were infected with the wild type pTY-LAI-dsRed reporter virus construct. Flow analysis was performed every 24 hours for 4 days and every week for a month post infection. The % active infection was calculated by dividing the number of cells with active HIV-1 LTR (eGFP and dsRed expressing cells) by the number of infected cells (total eGFP expressing cells).
Discussion
A significant proportion of cells infected with HIV-1 establish a latent population within days of infection [1,27]. Furthermore, in cells where viral transcription is initially active upon infection after integration into the chromosome, expression of viral RNAs invariably becomes repressed over the next several weeks [28,29]. The mechanisms contributing to repression of viral transcription and establishment of latency involve, but are not limited to, the local chromatin environment, integration site and the available host cell transcription factors [30]. Expression from the HIV-1 LTR is regulated by many host cell transcription factors, which can function as either transcriptional repressors or activators [10].
YY1 was one of the first host cell transcription factors demonstrated to be involved in repression of the HIV-1 LTR. It was shown that YY1 interacts with the transcription factor LSF, which binds near the transcription start site on the HIV-1 LTR promoter [11]. More recently YY1 was shown to function in transcriptional repression at this location by recruitment of HDAC1 [12]. We have previously observed YY1 forming a complex from Jurkat nuclear extracts in EMSA reactions with probes spanning the highly conserved upstream RBEIII element [13,14]. We extended this observation in the present study by the identification of a novel YY1 binding site that overlaps RBEIII at approximately 120 base pairs upstream from the transcriptional start site. Using EMSA we have identified specific nucleotides for direct binding of YY1 to the LTR and have shown using ChIP that mutation of these residues inhibit interaction of YY1 with the HIV-1 LTR promoter in cells. Interestingly, this upstream binding site for YY1 overlaps the previously described highly conserved RBEIII element, which binds a complex of factors that include USF1, USF2 and TFII-I (designated RBF-2) [13]. Nucleotides required for interaction of YY1 are located at the 3’ end of the core RBEIII sequence and include a -CAT- sequence, which was previously shown to be present as a core sequence for binding of YY1 to elements on other promoters [19,20,22,31]. In fact, the binding site for YY1 we have identified in vitro seems to completely overlap that for the RBF-2 co-factor TFII-I [16]. However, binding of YY1 is dissociated in cells stimulated with PMA, whereas TFII-I is constitutively bound.
The cis-element designated as RBEIII was initially identified for its high conservation on the HIV-1 LTR from patient samples [32] and its requirement for stimulation of LTR transcription by Ras-MAPK signaling [33]. Including the results shown here, this region of the HIV-1 LTR, immediately upstream of the enhancer, seems abundantly crowded with multiple factors that bind overlapping elements. USF1/2 binds the -ACTGCTGA- core RBEIII sequence [13], but only in association with TFII-I, which likely contacts the immediately adjacent CATC residues [16]. This 3’ flanking sequence overlaps residues important for binding of YY1 in vitro as described in this study (Figure 7). However, we note that overlapping interactions of multiple transcription factors is not without precedent for the HIV-1 LTR. For example, NF-kB and NFAT bind to identical sequences within the enhancer region and these elements overlap binding sites for GABP/ Ets as well as CBF-1 [24]. At present we have not established whether USF1/2 in combination with TFII-I and YY1 can interact at RBEIII simultaneously, or whether binding is mutually exclusive. If the latter is true, then it is possible that populations of latent HIV-1 may bind different combinations of factors at this location. As further complication, it was recently shown that HIV subtypes A and C have sequence polymorphisms 3’ flanking the RBEIII site (Figure 7, TGACAca) that forms an overlapping AP1 binding site [34]. Based on our study YY1 should also bind to these two subtypes, which indicates that YY1 and AP1 may have cooperative or redundant roles in repressing the HIV-1 LTR. AP1 was shown to modulate latency upon initial infection and deletion of this region in HIV-1 subtype E causes a decrease in the frequency of latent infection [34], which is consistent with the role of YY1 bound to this region in establishment of latency. These observations might indicate that establishment of latency by different HIV subtypes might involve different combinations of mechanisms.
Figure 7. Multiple transcription factors, including YY1, directly bind near the conserved RBEIII element.
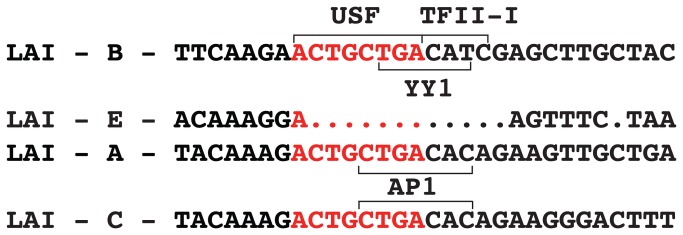
The RBEIII sequence (red) and flanking sequences are shown, indicating the positions bound by USF1/2, TFII-I and YY1. Sequence variations observed in HIV-1 subtypes LAI-B (pTY-LAI-dsRed), LAI-A, LAI-C and LAI-E are indicated. A binding site for AP1 identified in subtypes A and C is indicated [34].
We find that a mutation that prevents binding of YY1 in vitro at RBEIII causes a decrease in occupancy of YY1 at the RBEIII site in cells, in addition to a decrease in YY1 binding near the transcription start site. A similar effect was observed with TFII-I, which shows a loss of binding at RBEI when RBEIII is mutated. We expect that this represents cooperative DNA binding between these factors and consequently this might produce interaction between proteins bound to the two sites resulting in a higher order “loop” of DNA structure. In support of this possibility, YY1 has been shown to bend DNA in vitro [35,36]. Therefore, it is possible that looping between the core promoter and RBEIII site may be representative of repressive state on the LTR [24]. We have tried to examine this directly using chromatin conformation capture (3C) but this proved to be technically challenging because of the relatively close spacing between these sites.
The possibility of higher order structure proposed from this research may not be exclusive to the HIV-1 LTR. There are many viral promoters that contain multiple YY1 binding sites. Also, the role of YY1 in controlling latency of HIV-1 seems to extend to other latent viral promoters. There are a number of viruses that form latent infections where YY1 is involved in repression, including but not limited to adenovirus [37], Epstein barr virus [38], cytomegalovirus [39] and human papilloma virus [40]. Most of these contain multiple YY1 binding sites where one of the sites is at the transcription start site and another is upstream. All of these viral promoters contain the core –CAT- YY1 binding sequence, however the upstream YY1 binding sites are typically more similar to the site seen at RBEIII (-TGACAT-) [41,42].
YY1 is a ubiquitously expressed transcription factor that can cause activation or repression of transcription depending on promoter context. Our results extend upon earlier observations indicating that YY1 has a repressive role in HIV-1 transcription [11,12,43]. We show that this factor is a critical determinant for establishment of latency early after infection. We show that this factor is a critical determinant for establishment of latency early after infection. YY1 is bound to the LTR, at both the RBEIII and core promoter (RBEI) in unstimulated cells and is dissociated upon stimulation with PMA. Because the amount of YY1 was not reduced in activated cells, dissociation from the LTR may be caused by a modification such as acetylation of the C-terminal zinc fingers, which was previously shown to inhibit DNA binding [44].
Upon infection of unstimulated cells with the dual reporter virus, we find that the active infection of Jurkat-tat cells increases from 5% on day one to ~ 30% after one week post infection. The proportion of actively infected cells does not change for at least a month in culture. Because we have used a mini-virus reporter, which does not express the viral accessory factors, whose expression causes cellular toxicity [25,26]we were able to separately expand the populations of cells that actively transcribed the LTR and those that harbor latent provirus for ChIP analysis to analyze naturally active populations. We found that YY1 is associated with the LTR in the population of cells where the LTR is repressed, but not in the cell population where expression from the LTR has remained active. These results are consistent with previous studies showing a role for YY1 occupancy on the HIV-1 LTR as a contributing mechanism for repression [12,45] and may suggest that the post-translational state of YY1 at the time of infection may influence establishment of latency. The role of YY1 in controlling HIV-1 latency likely involves recruitment of HDAC1 because overexpression of a YY1 mutant lacking the HDAC1 interaction domain (glycine/alanine/lysine rich region) did not affect the proportion of infected cells that remain latent over the course of one month (Figure 6D).
Use of a mini-virus lacking expression of most HIV accessory factors, including tat, for these experiments allowed expansion of cells that were productively infected for analysis by ChIP (Figure 4). However, this necessitated use of Jurkat-tat cells to allow transcription elongation from the 5’ LTR. We note that constitutive tat expression in these cells bypasses the normal positive feedback loop for expression, and therefore could potentially decrease the amount of latently infected cells. Previous studies comparing HIV-1 infection in Jurkat and Jurkat-tat cell lines found that there is no difference between the two cell lines with respect to infection and integration but the latter cells produced a smaller latent population [46]. Consequently, the effect of YY1 on establishment of latency may be underrepresented in cells constitutively expressing tat, and it will be important to determine the role of YY1 in latency in primary cells. Additionally, multiple studies have shown that loss of tat function is essential to establish complete shutdown of provirus expression to form latency using systems involving a single reporter expressed from the LTR [30,46,47]. With our dual labeled virus, where we can detect infection independently from LTR activity we observe the majority of the newly infected population has produced latent infections within 24-72 hours in Jurkat-tat cells where tat is produced constitutively. We observe this effect with the mini-virus described here (Figure 4), and with a full-length dual reporter HIV construct [27], (Hashemi and Sadowski, unpublished). This indicates that the early latent population established shortly after infection may involve mechanisms that somehow can bypass the function of tat protein. We are currently examining details of these potential mechanisms.
In conclusion, in this study we report a novel YY1 binding site on the HIV-1 LTR 120 bps upstream of the transcriptional start site that is involved in formation of latent HIV provirus. Our results suggest that binding of YY1 and other factors near the RBEIII site may cause the formation of higher order structures on the LTR between the upstream RBEIII and transcriptional start site that may be involved in LTR repression. We are currently seeking ways to address this possibility directly, as well as define the mechanism(s) by which association of YY1 with the LTR is controlled for establishment of latency and the role of YY1 in HIV-1 in primary cells.
Supporting Information
Oligonucleotides used in this study.
(DOCX)
Acknowledgments
We thank Andy Johnson and Justin Wong of the UBC Flow Cytometry Facility for performing FACS, as well as assistance with flow cytometry. We thank Farhad Hashemi for his critical review of this manuscript.
Funding Statement
This research was supported by Canadian Institutes for Health Research Operating Grant HOP-120237 (to IS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Chun TW, Engel D, Berrey MM, Shea T, Corey L et al. (1998) Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci U S A 95: 8869-8873. doi: 10.1073/pnas.95.15.8869. PubMed: 9671771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K et al. (2003) Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9: 727-728. doi: 10.1038/nm880. PubMed: 12754504. [DOI] [PubMed] [Google Scholar]
- 3. Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A et al. (1990) HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61: 213-222. doi: 10.1016/0092-8674(90)90802-L. PubMed: 2331748. [DOI] [PubMed] [Google Scholar]
- 4. Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA (1990) HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J 9: 1551-1560. PubMed: 2184033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K et al. (1999) Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5: 512-517. doi: 10.1038/8394. PubMed: 10229227. [DOI] [PubMed] [Google Scholar]
- 6. Alexaki A, Liu Y, Wigdahl B (2008) Cellular reservoirs of HIV-1 and their role in viral persistence. Curr HIV Res 6: 388-400. doi: 10.2174/157016208785861195. PubMed: 18855649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fernández Guerrero ML, Rivas P, Molina M, Garcia R, De Górgolas M (2005) Long-term follow-up of asymptomatic HIV-infected patients who discontinued antiretroviral therapy. Clin Infect Dis 41: 390-394. doi: 10.1086/431487. PubMed: 16007538. [DOI] [PubMed] [Google Scholar]
- 8. Joos B, Fischer M, Kuster H, Pillai SK, Wong JK et al. (2008) HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc Natl Acad Sci U S A 105: 16725-16730. doi: 10.1073/pnas.0804192105. PubMed: 18936487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matalon S, Rasmussen TA, Dinarello CA (2011) Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol Med 17: 466-472. PubMed: 21424110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pereira LA, Bentley K, Peeters A, Churchill MJ, Deacon NJ (2000) A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res 28: 663-668. doi: 10.1093/nar/28.3.663. PubMed: 10637316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Margolis DM, Somasundaran M, Green MR (1994) Human transcription factor YY1 represses human immunodeficiency virus type 1 transcription and virion production. J Virol 68: 905-910. PubMed: 8289393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM et al. (2000) The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol 74: 6790-6799. doi: 10.1128/JVI.74.15.6790-6799.2000. PubMed: 10888618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J, Malcolm T, Estable MC, Roeder RG, Sadowski I (2005) TFII-I regulates induction of chromosomally integrated human immunodeficiency virus type 1 long terminal repeat in cooperation with USF. J Virol 79: 4396-4406. doi: 10.1128/JVI.79.7.4396-4406.2005. PubMed: 15767439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malcolm T, Chen J, Chang C, Sadowski I (2007) Induction of chromosomally integrated HIV-1 LTR requires RBF-2 (USF/TFII-I) and Ras/MAPK signaling. Virus Genes 35: 215-223. doi: 10.1007/s11262-007-0109-9. PubMed: 17546494. [DOI] [PubMed] [Google Scholar]
- 15. Bernhard W, Barreto K, Saunders A, Dahabieh MS, Johnson P et al. (2011) The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett 585: 3549-3554. doi: 10.1016/j.febslet.2011.10.018. PubMed: 22020221. [DOI] [PubMed] [Google Scholar]
- 16. Malcolm T, Kam J, Pour PS, Sadowski I (2008) Specific interaction of TFII-I with an upstream element on the HIV-1 LTR regulates induction of latent provirus. FEBS Lett 582: 3903-3908. doi: 10.1016/j.febslet.2008.10.032. PubMed: 18976654. [DOI] [PubMed] [Google Scholar]
- 17. Li YC, Ross J, Scheppler JA, Franza BR (1991) An in vitro transcription analysis of early responses of the human immunodeficiency virus type 1 long terminal repeat to different transcriptional activators. Mol Cell Biol 11: 1883-1893. PubMed: 2005886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Momoeda M, Kawase M, Jane SM, Miyamura K, Young NS et al. (1994) The transcriptional regulator YY1 binds to the 5'-terminal region of B19 parvovirus and regulates P6 promoter activity. J Virol 68: 7159-7168. PubMed: 7933098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hahn S (1992) The Yin and the Yang of mammalian transcription. Curr Biol 2: 152-154. doi: 10.1016/0960-9822(92)90268-F. PubMed: 15335993. [DOI] [PubMed] [Google Scholar]
- 20. Hyde-DeRuyscher RP, Jennings E, Shenk T (1995) DNA binding sites for the transcriptional activator/repressor YY1. Nucleic Acids Res 23: 4457-4465. doi: 10.1093/nar/23.21.4457. PubMed: 7501470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shrivastava A, Calame K (1994) An analysis of genes regulated by the multi-functional transcriptional regulator Yin Yang-1. Nucleic Acids Res 22: 5151-5155. doi: 10.1093/nar/22.24.5151. PubMed: 7816599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yant SR, Zhu W, Millinoff D, Slightom JL, Goodman M et al. (1995) High affinity YY1 binding motifs: identification of two core types (ACAT and CCAT) and distribution of potential binding sites within the human beta globin cluster. Nucleic Acids Res 23: 4353-4362. doi: 10.1093/nar/23.21.4353. PubMed: 7501456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dahabieh MS, Ooms M, Malcolm T, Simon V, Sadowski I (2011) Identification and functional analysis of a second RBF-2 binding site within the HIV-1 promoter. Virology 418: 57-66. doi: 10.1016/j.virol.2011.07.002. PubMed: 21813151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sadowski I, Lourenco P, Malcolm T (2008) Factors controlling chromatin organization and nucleosome positioning for establishment and maintenance of HIV latency. Curr HIV Res 6: 286-295. doi: 10.2174/157016208785132563. PubMed: 18691027. [DOI] [PubMed] [Google Scholar]
- 25. Kolesnitchenko V, King L, Riva A, Tani Y, Korsmeyer SJ et al. (1997) A major human immunodeficiency virus type 1-initiated killing pathway distinct from apoptosis. J Virol 71: 9753-9763. PubMed: 9371641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Casella CR, Rapaport EL, Finkel TH (1999) Vpu increases susceptibility of human immunodeficiency virus type 1-infected cells to fas killing. J Virol 73: 92-100. PubMed: 9847311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dahabieh MS, Ooms M, Simon V, Sadowski I (2013) A double-fluorescent HIV-1 reporter shows that the majority of integrated HIV-1 is latent shortly after infection. J Virol 87: 4716-4727. doi: 10.1128/JVI.03478-12. PubMed: 23408629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Song SK, Li H, Cloyd MW (1996) Rates of shutdown of HIV-1 into latency: roles of the LTR and tat/rev/vpu gene region. Virology 225: 377-386. doi: 10.1006/viro.1996.0612. PubMed: 8918924. [DOI] [PubMed] [Google Scholar]
- 29. Li XD, Moore B, Cloyd MW (1996) Gradual shutdown of virus production resulting in latency is the norm during the chronic phase of human immunodeficiency virus replication and differential rates and mechanisms of shutdown are determined by viral sequences. Virology 225: 196-212. doi: 10.1006/viro.1996.0588. PubMed: 8918547. [DOI] [PubMed] [Google Scholar]
- 30. Donahue DA, Wainberg MA (2013) Cellular and molecular mechanisms involved in the establishment of HIV-1 latency. Retrovirology 10: 11. doi: 10.1186/1742-4690-10-11. PubMed: 23375003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shrivastava A, Calame K (1994) An analysis of genes regulated by the multi-functional transcriptional regulator Yin Yang-1. Nucleic Acids Res 22: 5151-5155. doi: 10.1093/nar/22.24.5151. PubMed: 7816599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Estable MC, Bell B, Merzouki A, Montaner JS, O’Shaughnessy MV et al. (1996) Human immunodeficiency virus type 1 long terminal repeat variants from 42 patients representing all stages of infection display a wide range of sequence polymorphism and transcription activity. J Virol 70: 4053-4062. PubMed: 8648743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bell B, Sadowski I (1996) Ras-responsiveness of the HIV-1 LTR requires RBF-1 and RBF-2 binding sites. Oncogene 13: 2687-2697. PubMed: 9000143. [PubMed] [Google Scholar]
- 34. Duverger A, Wolschendorf F, Zhang M, Wagner F, Hatcher B et al. (2012) An AP-1 binding site in the enhancer/core element of the HIV-1 promoter controls the ability of HIV-1 to establish latent infection. J Virol 86: 9055-9069. doi: 10.1128/JVI.00793-12. PubMed: 22696646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Natesan S, Gilman Z (1993) DNA bending and orientation-dependent function of YY1 in the c-fos promoter. Genes Dev 7: 2497-2509. doi: 10.1101/gad.7.12b.2497. PubMed: 8276234. [DOI] [PubMed] [Google Scholar]
- 36. Kim J, Shapiro DJ (1996) In simple synthetic promoters YY1-induced DNA bending is important in transcription activation and repression. Nucleic Acids Res 24: 4341-4348. doi: 10.1093/nar/24.21.4341. PubMed: 8932392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi Y, Seto E, Chang LS, Shenk T (1991) Transcriptional repression by YY1, a human GLI-Krüppel-related protein, and relief of repression by adenovirus E1A protein. Cell 67: 377-388. doi: 10.1016/0092-8674(91)90189-6. PubMed: 1655281. [DOI] [PubMed] [Google Scholar]
- 38. Montalvo EA, Cottam M, Hill S, Wang YJ (1995) YY1 binds to and regulates cis-acting negative elements in the Epstein-Barr virus BZLF1 promoter. J Virol 69: 4158-4165. PubMed: 7769675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu R, Baillie J, Sissons JG, Sinclair JH (1994) The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic Acids Res 22: 2453-2459. doi: 10.1093/nar/22.13.2453. PubMed: 8041605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bauknecht T, Angel P, Royer HD, zur Hausen H (1992) Identification of a negative regulatory domain in the human papillomavirus type 18 promoter: interaction with the transcriptional repressor YY1. EMBO J 11: 4607-4617. PubMed: 1330541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zalani S, Coppage A, Holley-Guthrie E, Kenney S (1997) The cellular YY1 transcription factor binds a cis-acting, negatively regulating element in the Epstein-Barr virus BRLF1 promoter. J Virol 71: 3268-3274. PubMed: 9060693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. O’Connor MJ, Tan SH, Tan CH, Bernard HU (1996) YY1 represses human papillomavirus type 16 transcription by quenching AP-1 activity. J Virol 70: 6529-6539. PubMed: 8794287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Romerio F, Gabriel MN, Margolis DM (1997) Repression of human immunodeficiency virus type 1 through the novel cooperation of human factors YY1 and LSF. J Virol 71: 9375-9382. PubMed: 9371597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yao YL, Yang WM, Seto E (2001) Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol 21: 5979-5991. doi: 10.1128/MCB.21.17.5979-5991.2001. PubMed: 11486036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mahmoudi T, Parra M, Vries RG, Kauder SE, Verrijzer CP et al. (2006) The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J Biol Chem 281: 19960-19968. doi: 10.1074/jbc.M603336200. PubMed: 16687403. [DOI] [PubMed] [Google Scholar]
- 46. Donahue DA, Kuhl BD, Sloan RD, Wainberg MA (2012) The viral protein Tat can inhibit the establishment of HIV-1 latency. J Virol 86: 3253-3263. doi: 10.1128/JVI.06648-11. PubMed: 22238306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pearson R, Kim YK, Hokello J, Lassen K, Friedman J et al. (2008) Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol 82: 12291-12303. doi: 10.1128/JVI.01383-08. PubMed: 18829756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotides used in this study.
(DOCX)