

Abstract
Polycystic liver diseases (PLD) represent a group of genetic disorders in which cysts occur in the liver (autosomal dominant polycystic liver disease) or in combination with cysts in the kidneys (autosomal dominant polycystic kidney disease). Regardless of the genetic mutations, the natural history of these disorders is alike. The natural history of PLD is characterized by a continuous increase in the volume and the number of cysts. Both genders are affected; however, women have a higher prevalence. Most patients with PLD are asymptomatic and can be managed conservatively. Severe symptoms can affect 20% of patients who develop massive hepatomegaly with compression of the surrounding organs. Rrarely, patients with PLD suffer from acute complications caused by the torsion of hepatic cysts, intraluminal cystic hemorrhage and infections. The most common methods for the diagnosis of PLD are cross sectional imaging studies. Abdominal ultrasound and computerized tomography are the two most frequently used investigations. Magnetic resonance imaging is more sensitive and specific, and it is a valuable test for patients with intravenous contrast allergies or renal dysfunction. Different treatment modalities are available to physicians caring for these patients. Medical treatment has been ineffective. Percutaneous sclerotherapy, trans-arterial embolization, cyst fenestration, hepatic resection and liver transplantation are indicated to specific groups of patients and have to be tailored according to the extent of disease. This review outlines the current knowledge of the pathophysiology, clinical course, diagnosis and treatment strategies of PLD.
Keywords: Polycystic liver disease, Hepatic, Epidemiolgy, Classification, Therapy, Genetic
Core tip: The management of patients with symptomatic polycystic liver disease is challenging. Among several treatments options, the most common interventions are: percutaneous cyst aspiration, fenestration, hepatic resection and liver transplantation. There is no consensus on the best treatment options and the optimal timing for interventions in symptomatic patients. In vision of these limitations, we reviewed the most recent literature and present a comprehensive article on this topic.
INTRODUCTION
The association between polycystic liver disease (PLD) and autosomal dominant polycystic kidney disease (ADPKD) was described for the fist time by Bristowe in 1856[1,2]. Initially, it was thought that PLD could develop only in the context of ADPKD[3]. The notion that isolated PLD might be a separate condition was proposed in the 1950’s[4]. In 2003, a linkage analysis of eight Finnish families confirmed that PLD is genetically distinct from ADPKD[5]. Asymptomatic patients usually do not require any intervention[6]. In some patients, massive hepatomegaly can cause pain or compression of the adjacent gastrointestinal organs, vasculature, and diaphragm. This can have a significant effect on patients’ quality of life and performance status[6,7]. For these patients, the main aim is to reduce their symptoms by decreasing the liver volume[8-10]. Current surgical options include open or laparoscopic cyst fenestration with or without hepatic resection and orthotopic liver transplantation (OLT). Significant advances in surgical techniques have improved the outcomes of PLD patients. However, the selection of the appropriate approach remains a clinical challenge, and there is no consensus on the optimal timing and what represents the best therapeutic modality.
INCIDENCE AND GENETICS
ADPKD affects up to 0.2% of the general population[11]. On the other hand, isolated PLD has prevalence of less than 0.01%[12]. Both ADPKD and PLD are autosomal dominant and 75%-90% of patients with ADPKD have associated PLD[13]. In humans, PLD has been linked to mutations of four genes. Two genes (PKD1, locus 16p13.3, encoding polycystin-1 and PKD2, locus 4q21, and encoding polycystin-2) are predominantly associated with renal disease and less frequently with PLD. PKD1 mutations are more common and account for 85%-90% of the cases, whereas mutations in PKD2 affect approximately 10%-15% of patients[11]. The remaining two mutations (PRKCSH, locus 19p13.2, encoding the protein kinase C substrate 80K-H or hepatocystin and SEC63, locus 6q21, encoding the Sec63 protein) are linked only to the development of PLD[11]. However, these mutations explain just 25% to 40% of cases of PLD[14,15]. Comparative characteristics between ADPKD and PLD are summarized in Table 1.
Table 1.
Comparative epidemiological and genetic mutation characteristics of autosomal dominant polycystic kidney disease associated polycystic liver disease and isolated polycystic liver disease
| Characteristics | ADPKD associated PLD | Isolated PLD |
| Prevalence | 0.20% | < 0.01% |
| Type of inheritance | AD | AD |
| Gene mutated | PKD1; PKD2 | PRKCSH; SEC63 |
| Encoded product | Polycystin-1; Polycystin-2 | Hepatocystin; Sec63 protein |
| Chromosome locus | 216p13.3; 4q21 | 19p13.2; 6q21 |
AD: Autosomal dominant; ADPKD: Autosomal dominant polycystic kidney disease; PLD: Polycystic liver disease.
PATHOPHYSIOLOGY
Malformation of the hepatic ductal plate and cilia of cholangiocytes is the main characteristic linked to the pathophysiology of PLD (Figure 1).
Figure 1.
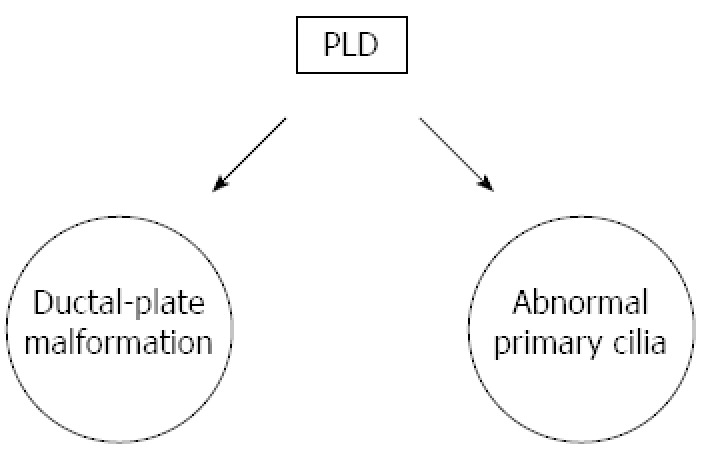
Pathophysiology of polycystic liver disease. PLD: Polycystic liver disease.
DUCTAL-PLATE MALFORMATION
The ductal plate is the anatomical template for the development of the intra-hepatic bile ducts[16]. Normal bile ducts arise from the ductal plate through a complex sequence of growth and apoptosis. Complexes of disconnected intralobular bile ductules (von Meyenburg complexes) are retained because they do not undergo apoptosis in PLD[10]. As a consequence, multiple cysts arise from progressive dilatation of these abnormal ductules[17-19] that display the same epithelium and structures of functioning cholangiocytes[20,21].
ABNORMAL PRIMARY CILIA
Cholangiocytes are the only ciliated cells in the liver. Cilia have mechanosensory capacity and modulate the intracellular levels of cAMP and Ca2+ when bent by the flow of bile. They also detect changes in osmolarity and composition of the bile[22-24]. Ciliary defects result in a decreased cytoplasmic level of Ca2+ and an increased cytoplasmic level of cAMP[25]. These changes are responsible for the hyperproliferation of cholangiocytes and for the cystogenesis that is a consequence of the altered balance between fluid secretion and absorption in the lumen of the biliary ducts[25].
NATURAL HISTORY AND RISK FACTORS FOR PLD
The natural history of PLD is characterized by a continuous increase in the volume and the number of cysts[11,26,27]. The annual growth of affected livers is in the range of 0.9%-3.2% of the initial hepatic volume[10,28-30]. Both genders are affected; however, women have a higher prevalence. Exposure to estrogen during pregnancies, the use of oral contraceptive pills or estrogen replacement therapy seems to accelerate the progression of the disease[1,27,31]. Other risk factors are the severity of renal dysfunction that is dependent on the volume of the cysts in the kidneys[1]. Table 2 summarizes the known factors that influence the progression of PLD.
Table 2.
Risk factors for liver-cyst growth in polycystic liver disease
| Risk factors for liver-cyst growth in polycystic liver disease |
| Advancing patient age |
| Female gender |
| Estrogen exposure: multiple pregnancies, OCPs, estrogen replacement therapy |
| Severity of renal dysfunction and renal cyst volume |
OCPs: Oral contraceptive pills.
CLINICAL PRESENTATION
PLD is asymptomatic in 80% of patients[8,9] and is usually diagnosed incidentally. Women present with massive and symptomatic cystic liver more frequently than men[32]. For 20% of patient, symptoms are typically caused by the compression of organs surrounding the liver, bleeding or infectious complications of the cysts. Compressive symptoms include abdominal distention, early satiety that can lead to decreased oral intake and severe malnutrition, gastro-esophageal reflux, dyspnea, hepatic venous-outflow obstruction (Budd-Chiari syndrome), inferior vena cava syndrome, portal-vein and bile-duct compression. Complications of liver cysts include infections, torsions, rupture and hemorrhage[1,18,33,34] (Table 3). In asymptomatic patients, serum laboratory studies are usually normal. In the presence of symptoms, 47% of patients have elevated serum alkaline phosphatase, 70% have elevated serum levels of gamma glutamil transferase[35-38], 27% have elevated serum levels of aspartate amino transferase and 15% have elevated serum levels of total bilirubin[35,36]. Liver synthetic function is typically preserved despite the presence of innumerable cysts[32] while 45% of patients might have elevated serum tumor marker CA19-9 without proof of malignancy[39]. Other tumor markers such as CA-125, carcinoembryonic antigen, and alpha-fetoprotein may also be elevated but less frequently than CA19-9[40-42].
Table 3.
Summary of the most frequent symptoms caused by polycystic liver disease
| Symptoms due to mass effect | Symptoms due to complications of the cysts |
| Abdominal distention | Infection |
| Early satiety | Torsion |
| Postprandial fullness | Rupture |
| Gastro-oesophageal reflux | Haemorrhage |
| Malnutrition | |
| Dyspnoea | |
| Hepatic venous-outflow obstruction (Budd-Chiari syndrome) | |
| Inferior vena cava syndrome | |
| Portal-vein compression | |
| Bile-duct compression |
ASSOCIATED EXTRA-HEPATIC DISEASES
Intracranial arterial aneurysms can affect 6% of patients without a family history of ADPKD and up to 16% of patients with family history of ADPKD. Other common conditions are mitral-valve prolapse and colonic diverticulosis that can be detected in 25% of patients with PLD[1,11,43-45]. Screening for intracranial aneurysm by magnetic resonance angiography (MRA) is recommended only for patients with ADPKD, older than 30 years or for those patients with family history of hemorrhagic strokes or intracranial arterial aneurysms[46]. Screening for intracranial arterial aneurysms is also warranted in cases of a sudden severe headache, or for candidates to liver or kidney transplantation. Screening for mitral-valve prolapse is not recommended unless a cardiac murmur is ascultated during routine clinical examinations[11,47]. Finally, patients with ADPKD may have asymptomatic cysts within other organs, such as the pancreas, spleen, ovaries, and lungs[48]. Pancreatic cysts are the most common with a reported incidence of 9% among ADPKD patients older than 30 years[49-51].
DIAGNOSIS
The most common methods for the diagnosis of PLD are cross sectional imaging studies. Abdominal ultrasound (US) and computerized tomography (CT) are the two most frequent investigations[52,53]. For hepatic cysts, MRI is more sensitive and specific, and it is a valuable test for patients with intravenous contrast allergies or renal dysfunction or when other studies are unable to satisfy the diagnostic needs[54]. Hepatic cysts have radiological characteristics identical to benign developmental cysts. On US, they appear anechoic and well-circumscribed[55]. On CT and MRI, they have non-enhancing, well-circumscribed round walls with hypodense content[55]. On T2-weighted MRI and CT scans, they appear homogenously enhanced spherical lesions[55] (Figure 2B and C). The distinction between isolated PLD and ADPKD relies on the number of renal cysts, age at presentation and family history (Table 4). In adults, younger than 30 years with a positive family history, the diagnosis of ADPKD is established by radiologic evidence of at least two unilateral or bilateral cysts. At least two cysts in each kidney are necessary for the diagnosis of patients between the age of 30 to 59 years, and at least four cysts in each kidney for patients 60 years or older[56]. It is worth noting that at least one third of patients with isolated PLD may also have a few kidney cysts[15,56,57]. It has been proposed that sporadic cases of PLD should be diagnosed when a patient has more than 15 to 20 cysts and no previous family history[1,18] while four cysts suffice in the presence of a positive familial history[1,18].
Figure 2.
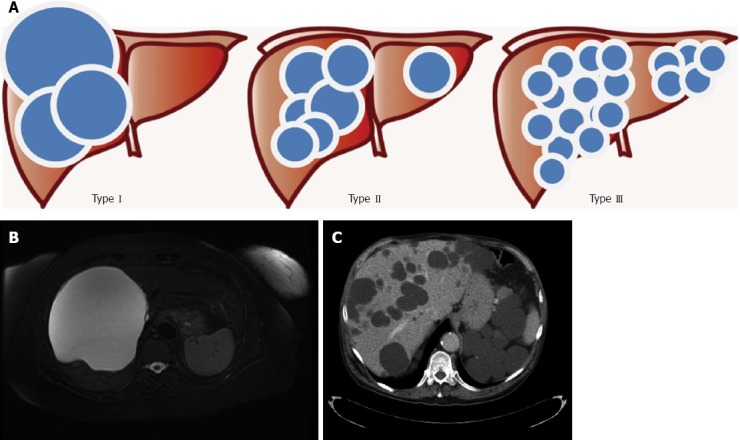
Gigot’s classification for polycystic liver diseases. A: Graphical representation; B: Abdominal magnetic resonance imaging of a patient affected by Gigot I cystic liver disease; C: Abdominal computerized tomography of a patient affected by Gigot II cystic liver disease.
Table 4.
The ravine diagnostic criteria for autosomal dominant polycystic kidney disease
| Patient’s age (yr) |
Number of cysts |
|
| Positive family history | Negative family history | |
| ≤ 30 | At least 2 cysts affecting 1 or both kidneys | At least 5 cysts |
| 31-59 | At least 2 cysts in each kidney | At least 5 cysts |
| ≥ 60 | At least 4 cysts in each kidney | At least 8 cysts |
INFECTED CYSTS
Hepatic cysts may become infected, and cause life-threatening sepsis[58,59]. Often, infected hepatic cysts are responsible for recurrent episodes of fever without any other signs or symptoms. In these circumstances, the diagnosis can be quite difficult as the accuracy of imaging tests remain low due to the altered anatomy of the liver parenchyma[60]. A promising investigation technique for suspected infected hepatic cysts is In-111 WBC scan[61]. Several other tracers such as 99mTc-diphosphonates, 67Gacitrate, and 111In- or 99mTc-labeled leukocytes have also been used[62]. Although labeled leukocyte imaging is theoretically the test of choice for detecting most infections, it is labor intensive, not always available and involves direct handling of potentially infected blood products. Therefore, considerable effort has been devoted to search for alternatives to this procedure such as the use of 67Gallium scintigraphy and 18F-FDG-positron emission tomography (PET). In recent years, PET has become the most commonly used diagnostic test for the detection of infected renal and hepatic cysts[60,62,63]. However, the accuracy of this technique is still under investigation. The literature on the treatment of infected cysts in PLD patients is very scarce and based only on a few case reports. Most of patients will need parenteral broad spectrum antibiotic therapy with percutaneous drainage of the content of the cyst if their symptoms persists.
CLASSIFICATION
Several clinical classifications have been proposed to grade the severity of PLD.
GIGOT’S CLASSIFICATION
Gigot’s classification relies on imaging findings and was designed to identify the best candidates for fenestration of symptomatic cysts[38] (Figure 3): Type I: presence of less than 10 large hepatic cysts measuring more than 10 cm in maximum diameter. Type II: diffuse involvement of liver parenchyma by multiple cysts with remaining large areas of non-cystic liver parenchyma. Type III: presence of diffuse involvement of liver parenchyma by small and medium-sized liver cysts with only a few areas of normal liver parenchyma.
Figure 3.
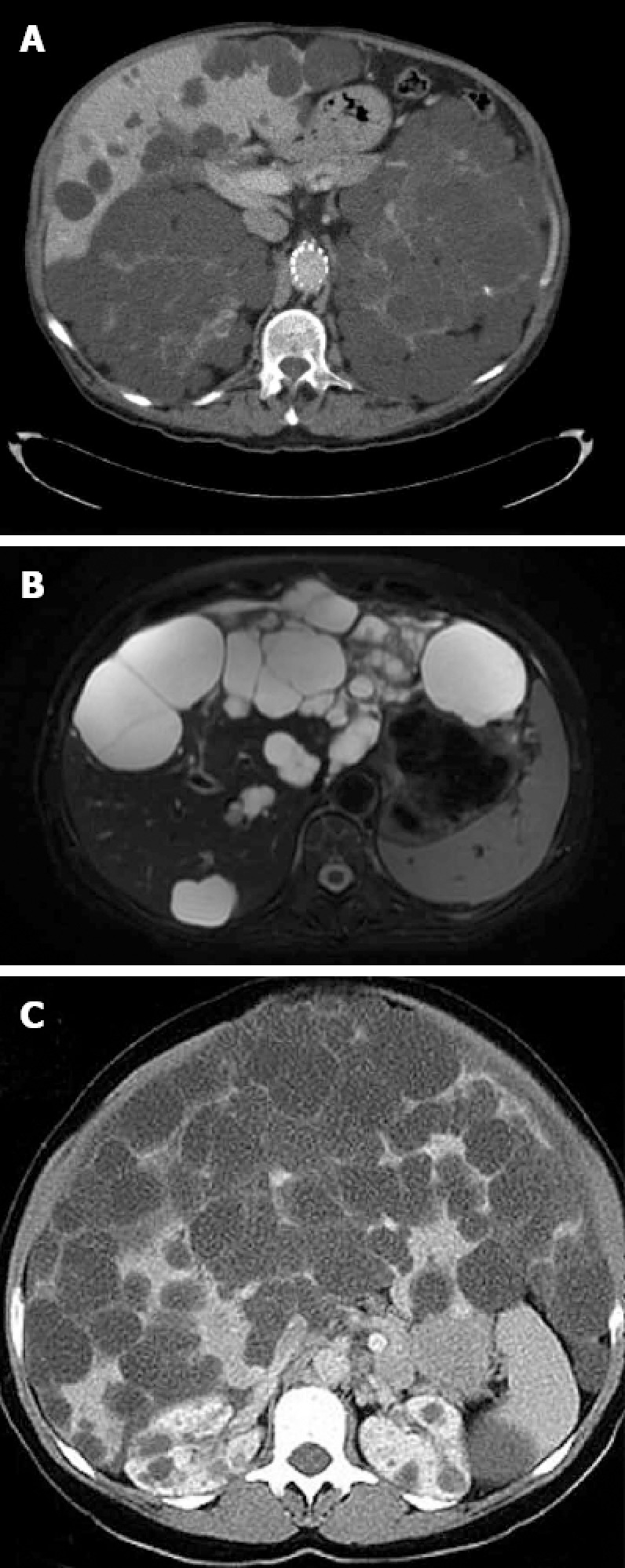
Gigot’s classification relies on imaging findings and was designed to identify the best candidates for fenestration of symptomatic cysts. A: Intravenous contrast enhanced computerized tomography (CT) of a patient affected by polycystic liver and renal disease. The cysts appears hypoattenuating with smooth and regular walls; B: T2 magnetic resonance imaging of a patient with multiple hepatic cysts. The cystic fluid appears bright on T2 images; C: Abdominal CT of a patient affected by Gigot III cystic liver disease.
QUIAN’S CLASSIFICATION
Qian’s classification has been used in the context of familial screening and relies on the number of cysts and the presence of symptomatic hepatomegaly[18]: (1) grade 0 - 0 cysts; (2) grade 1 - 1 to 10 cysts; (3) grade 2 - 11 to 20 cysts; (4) grade 3 - more than 20 cysts; and (5) grade 4 - more than 20 cysts and symptomatic hepatomegaly.
SCHNELLDORFER’S CLASSIFICATION
Schnelldorfer’s classification aims at differentiating patients who could benefit from resection or transplantation as summarized in Table 5[64].
Table 5.
Summary of Schnelldorfer’s classification that aims at differentiating patients who could benefit from resection or transplantation
| Type A | Type B | Type C | Type D | |
| Symptoms | Absent or mild | Moderate or severe | Severe (or moderate) | Severe (or moderate) |
| Cyst characteristics | Any | Limited No. large cysts | Any | Any |
| Areas of relative normal liver parenchyma | Any | ≥ 2 sectors | ≥ 1 sector | < 1 sector |
| Presence of portal vein or hepatic vein occlusion in the preserved hepatic sectors | Any | Absent | Absent | Present |
| Recommended therapy | Observation or medical therapy | Cyst fenestration | Partial hepatectomy with possible fenestration of remnant cysts | Liver Transplantation |
TREATMENT
Most patients with PLD are asymptomatic and do not require any intervention[6]. However, symptomatic PLD patients might require treatment when they experience severe dysfunction of organs around the liver due to the increased hepatic volume or when one or more cysts get torted, infected or develop intra-cystic hemorrhages (Table 6).
Table 6.
Summary of treatment options for polycystic liver disease
| Treatment approach | Treatment type |
| Nonsurgical | Medical |
| Somatostatin analogues | |
| mTOR inhibitors | |
| Interventional radiology: | |
| Arterial embolization | |
| Percutaneous sclerotherapy | |
| Surgical | Fenestration |
| Hepatic resection with fenestration | |
| Liver transplantation |
OCP’s: Oral contraceptive pills; mTOR: Mammalian target of rapamycin.
AVOIDANCE OF EXPOSURE TO ESTROGENS
Observational and experimental studies have shown that PLD may worsen under the influence of estrogen during pregnancy or when patients are prescribed estrogen replacement therapy[1,27,31]. Estrogen can increase both the number of liver cysts and their volume, therefore, hormonal therapy should be stopped in most symptomatic patients when appropriate[27].
NON-SURGICAL TREATMENTS
Medical management may be valuable in symptomatic patients with Gigot’s type II/III.
SOMATOSTATIN ANALOGUES
Somatostatin analogues are inhibitors of cAMP and they reduce the secretion of fluid and the proliferation of many cell types, including cholangiocytes[65-69]. They also suppress the expression of insulin-like growth factor 1 (IGF-1), vascular endothelial growth factor (VEGF), and other cytogenetic growth factors[70]. In addition, somatostatin analogues inhibit the downstream signaling of these receptors[70]. Two randomized controlled trials have recently demonstrated that after 6 to 12 mo, treatment with lanreotide, a long-acting somatostatin analogue, was associated with a significant reduction of liver volume in patients with PLD compared with placebo[28,29]. However, the average hepatic volume reduction was only 3% to 5%. The severity of abdominal symptoms was also not significantly improved[28]. Currently, somatostatin analogues are indicated only for a selected group of patients with symptomatic PLD in whom the risks for surgical intervention are not justified, or in whom the surgical intervention is technically challenging.
MAMMALIAN TARGET OF RAPAMYCIN INHIBITORS
Mammalian target of rapamycin (m-TOR) inhibitors have immunosuppressive and antiproliferative effects[71]. Sirolimus and Everolimus were studied in Phase-II prospective randomised control trials. None of the two drugs showed substantial therapeutic effects both in humans[72-74] and in animal models[75]. Clinical prospective data o the effect of m-TOR inhibitors are currently not available, and this class of medications should not be recommended outside clinical trials.
INTERVENTIONAL RADIOLOGY: ARTERIAL EMBOLIZATION
Trans-catheter arterial embolization has been used since the early 2000s[76]. Hepatic artery branches supplying the hepatic segments replaced by the cysts are targeted by using microcoils or polyvinyl alcohol particles measuring 150-250 μm in diameter[76,77]. For patients with advanced PLD and multilobal disease, trans-catheter arterial embolization can be technically demanding. The largest series of patients treated with this modality included 30 patients who had a significant reduction of the volume of their cysts (6.667 ± 2.978 cm3 down to 4.625 ± 2.299 cm3), whereas the volume of the unaffected hepatic parenchyma increased[76]. After several months, patients reported improvement of their symptoms and no major complications except for occasional post-embolization syndrome[76,77].
PERCUTANEOUS SCLEROTHERAPY
This technique requires radiologically guided percutaneous aspiration of the content of the cysts followed by the injection of a sclerosing agent that inhibit the reaccumulation of fluid by damaging the epithelial lining the cystS[78,79]. Symptomatic patients with one to five large dominant cysts (Gigot’s type I) are suitable for percutaneous sclerotherapy. Most commonly, cysts with a diameter larger than 5 cm are candidates for this treatment[10]. Puncturing of the cyst can be done with a 5 or 7 French catheter[80] and sclerosing agents commonly used include ethanol, ethanolamine oleate, minocycline and tetracycline. Although a single session is often sufficient, some patients require more than one[81]. Aspiration with sclerotherapy has an excellent safety profile, although severe abdominal pain can be caused by peritoneal irritation due to spillage of the sclerosing agent[10]. The majority of patients who undergo percutaneous sclerotherapy has improved symptoms in the immediate period following the procedure[10], but only 20% will have partial, or full regression of their disease[10].
SURGERY
Patient and treatment selection remain a clinical challenge. There is no consensus on selection criteria for surgery, the optimal timing, and technique. Current surgical options include fenestration, partial liver resection and OLT. Fenestration and partial liver resection are options for Gigot’s type I and II patients. For Gigot’s type III disease, fenestration and partial liver resection are often ineffective, and OLT should be considered as it is the only curative treatment. In general, several factors have to be considered before any surgical intervention is recommended: (1) The degree of cystic burden; (2) The distribution of the cysts; and (3) The proximity of the cysts to the main biliary ducts and portal and hepatic vein branches.
SURGICAL PEARLS
In Gigot’s type I or II, symptoms might not be related to the size of the entire liver but to the size of one or two large cysts. These patients can be treated similarly to those with simple cysts. Some hepatic segments such as V and VI are frequently spared and, therefore, surgical resection can be performed if the spared liver parenchyma is thought to be sufficient. Frequently, the right hepatic veins are compressed by cysts causing the formation of collateral circulation between the right and the middle hepatic veins that can be responsible for intraoperative bleeding during the parenchymal transaction.
FENESTRATION
Fenestration is a surgical technique that combines aspiration and surgical unroofing of the cyst. It has the advantage that multiple cysts can be treated in one session[48,82]. Fenestration is effective in symptomatic patients with Gigot’s type I and II disease[83]. Patients with superficial and a limited number of large cysts are the best candidates for this procedure[48]. Fenestration may be achieved by laparotomy or laparoscopy[48]. Patients with the majority of their cysts located in the right posterior segments (VI, VII), or at the dome of the liver (segment VIII) may be better candidates for open fenestration because these cysts are difficult to be visualized and fenestrated by laparoscopic approach[48]. Published series describing open and laparoscopic fenestration are summarized in Table 7. Immediate symptom relief is achieved in 92% of the patients, whereas up to 25% experience recurrence of the cysts or symptoms[10]. Complication rate after fenestration is in the range of 23% while mortality is about 2%[10]. Complications include ascites, pleural effusion, hemorrhage and bile leakage[84]. Factors that predict failure of fenestration are previous abdominal procedures, deep-seated cysts, incomplete unroofing, cysts in segments VII-VIII, and the presence of diffuse PLD[10].
Table 7.
Summary of largest series published on the surgical techniques used for cystic fenestration of symptomatic polycystic liver disease
| Ref. | No. of patients | Technique | Outcome | Complications | Follow-up (mo) |
| van Erpecum et al[35] | 15 | Open fenestration | 0% symptom recurrence | One mortality | Mean of 48 |
| Kabbej et al[37] | 13 | Lap fenestration | 72% symptom recurrence | 54% morbidity | Mean follow-up 26 |
| Gigot et al[38] | 10 | Open fenestration | 11% symptom recurrence | 60% morbidity | 73 mean follow-up |
| van Keimpema et al[82] | 12 | Lap fenestration | Reduction in liver volume by 12.5% | Bile leak, vena cava occlusion and sepsis | - |
| Pirenne et al[92] | 4 | Lap fenestration | 100% symptom relief | 50% cyst recurrence | - |
| Liska et al[95] | 7 | Lap fenestration plus open | - | No mortality | Mean 41 |
| Bai et al[96] | 10 | Lap fenestration | Symptom and cyst recurrence in 20% | 3 patients with minor complications. No mortality | Mean of 57 |
| Palanivelu et al[97] | 4 | Lap fenestration | 100% cyst recurrence | - | - |
| Garcea et al[98] | 6 | Lap/Open fenestration | 16.7% symptom recurrence, 33.3% cyst recurrence | 50% morbidity | 5-36 |
| Neri et al[99] | 3 | Lap fenestration | 100% symptom relief | 50% morbidity | - |
| Kornprat et al[100] | 8 | Lap fenestration | 0% symptom recurrence | - | - |
| Robinson et al[101] | 11 | Lap fenestration | 54.5% symptom recurrence | - | - |
| Fiamingo et al[102] | 6 | Lap fenestration | 30% symptom recurrence | 50% morbidity | 1-64 |
| Tocchi et al[103] | 18 | Lap/open fenestration | - | - | - |
| Koperna et al[104] | 39 | Open fenestration (n = 34); Lap (n = 5) | 21% symptom recurrence | - | 75 mean follow-up |
| Morino et al[105] | 7 | Lap fenestration | 40% symptom recurrence | 44% morbidity rate | - |
| Farges et al[106] | 13 | Open fenestration | 23% symptom recurrence | 69% morbidity | 84 follow-up |
| Ueno et al[118] | 13 | Open fenestration (n = 6); Lap (n = 13) | 71% symptom recurrence | 30% morbidity | 37 mean follow-up |
Lap: Laparoscopic.
HEPATIC RESECTION WITH FENESTRATION
Hepatic resection is usually reserved for highly symptomatic patients who are incapacitated by their disease due to the massive expansion of their livers (Gigot’s type II and III)[38]. In these circumstances fenestration alone is rarely successful because the liver parenchyma is rigid and it does not collapse[10]. Symptom relief is achieved in 86% of cases although cyst recurrence is expected in one third of patients[10]. Overall, most of the patients have an improvement in their quality of life and functional status[36]. The morbidity rate associated with this procedure can be up to 50% and includes ascites, pleural effusions, biliary leakage, and hemorrhage[10]. One of the reasons for these complications is the fact that there is a significant distorsion of the intra-hepatic vasculature and biliary tree which makes these procedures technically very challenging. Mortality rate is around 3%[10]. As subsequent adhesions may complicate future OLT, this surgical treatment is usually preserved for patients with massive hepatomegaly for which OLT is not an option[85,86]. Published series describing hepatic resection with/without fenestration for symptomatic PLD are summarized in Table 8.
Table 8.
Summary of largest series published on the surgical techniques used for cystic fenestration and resection of symptomatic polycystic liver disease
| Ref. | No. | Technique | Outcome | Complications | Follow-up (mo) |
| Que et al[36] | 31 | Fenestration and resection | 3% symptom recurrence | 3% mortality, 58% morbidity | Mean of 28 |
| Schnelldorfer et al[64] | 124 | Fenestration and resection | 93% symptom relief, 72.6% recurrent cyst formation | 72.6% morbidity, 3.2% mortality | Mean of 48 |
| Kornprat et al[100] | 9 | Fenestration and resection | 100% symptom relief, 11% recurrence | 33.35% morbidity | 24-98 |
| Koperna et al[104] | 5 | Fenestration and resection | 0% symptom recurrence | - | - |
| Li et al[107] | 21 | Fenestration and resection | 14.3% cyst recurrence | 76.2% cyst morbidity, 0% mortality | 10-155 |
| Gamblin et al[108] | 51 | Fenestration and resection | 3.9% symptom recurrence | 17.6% morbidity, no mortality | 1-49 |
| Yang et al[109] | 7 | Fenestration and resection | 100% symptom recurrence | 100% morbidity, no mortality | Mean of 20 |
| Vons et al[110] | 12 | Resection | 17% symptom recurrence | 8% mortality, 83% morbidity | Mean of 34 |
| Soravia et al[111] | 10 | Fenestration and resection | 33% symptom recurrence | 10% mortality, 20% morbidity | Mean of 69 |
| Henne-Bruns et al[112] | 8 | Fenestration and resection | 50% symptom recurrence | No mortality, 38% morbidity | Mean of 15 |
| Vauthey et al[113] | 5 | Fenestration and resection | 0% symptom recurrence | 0% mortality, 100% morbidity | Mean of 14 |
| Sanchez et al[114] | 9 | Resection | 100% symptom relief, 100% recurrence | 0% mortality | Mean of 35 |
| Newman et al[115] | 9 | Fenestration and resection | 88.9% symptom relief, 0% recurrence | 11.1% mortality, 55.6% morbidity | 2-44 |
| Iwatsuki et al[116] | 9 | Resection | 44.4% symptom relief, 44.4% recurrence | 0% mortality, 33.3% morbidity | 12-180 |
LIVER TRANSPLANTATION
OLT is the only curative treatment for patients with severe PLD[87]. It is indicated in those patients with disabling symptoms that lead to decreased performance status and quality of life[10]. Patients with PLD usually have normal liver function and the current organ allocation system based on the Model for End-Stage Liver Disease (MELD) is often unable to assist this group of patients. For these patients, MELD exception criteria are needed[88,89]. Because of the shortness of available grafts, the need for life-long immunosuppression and the perioperative risks, OLT is indicated only for symptomatic patients with Gigot’s type II and III disease[12,48,90]. For patients undergoing OLT for PLD, perioperative morbidity is 40%-50%, whereas overall mortality is 10%-17%[10]. In 3% of patients, retransplantation is required[10] and combined renal and liver transplantation are necessary in 42% of patients[91,92]. Expected survival at 1- and 5-year are 93% and 92% for patients undergoing OLT alone while for patients who undergo combined liver and kidney transplant are 86% and 80% respectively[10]. Published series reporting the outcomes of OLT for symptomatic PLD are summarized in Table 9.
Table 9.
Summary of largest series published on the outcomes of patients undergoing liver transplantation for symptomatic polycystic liver disease
| Ref. | No. of patients | Previous surgery | Combined liver and kidney transplantation | Morbidity | Mortality | Follow-up (mo) | Re-transplantation |
| Pirenne et al[92] | 16 | 25% | 6% | 38% | 13% | Range 18-120 | 0% |
| Taner et al[117] | 13 | - | 54% | 85% | 31% | - | 0% |
| Ueno et al[118] | 14 | - | 36% | 64% | 21% | - | 0% |
| Ueda et al[119] | 3 | - | 0 | 33% | 0% | Mean of 32 | 0% |
| Gustafsson et al[120] | 7 | 57% | 43% | 57% | 43% | Mean of 4 | 0% |
| Swenson et al[121] | 9 | 44% | 33% | 44% | 11% | Mean of 26 | 11% |
| Lang et al[122] | 17 | 35% | 47% | 47% | 29% | Mean of 12 | 12% |
| Washburn et al[123] | 5 | 90% | 20% | 0% | 20% | Mean of 38 | 0% |
| Starzl et al[124] | 4 | 0% | 25% | 0% | 50% | Mean of 38 | 0% |
HEPATIC RESECTION VS LIVER TRANSPLANTATION
The clinical decision between performing a hepatic resection with or without cyst fenestration[93] and referring the patient for OLT can be extremely difficult (Table 10). Hepatic resection with cyst fenestration implies leaving residual hepatic cysts that will eventually progress[94]. However, hepatic resection is associated with a lower risk of perioperative morbidity and mortality. OLT provides the only option for the cure of these patients but requires lifelong immunosuppression and has higher perioperative risks. Both resection and OLT are technically demanding, and peri-operative care can be complex. The risks and the benefits of each of the possible treatment options have to be carefully evaluated and put in the contest of the clinical presentation and condition of each patient. Referral to a tertiary center with an experienced team of surgeons, hepatologists, and nephrologists is strongly recommended.
Table 10.
Suggested management strategies based on Gigot’s classification
| Gigot’s I | Gigot’s II - III |
| Percutaneous sclerotherapy | Hepatic resection with fenestration if feasible |
| Fenestration | Liver transplantation |
CONCLUSION
For patients with PLD, patients’ selection, timing and choice of treatments can be very challenging even for experienced physicians. For symptomatic patients, treatment strategies should be based on the degree and progression of their symptoms and the severity of other medical conditions. Symptomatic patients with large cysts or limited hepatic involvement might benefit from fenestration or sclerotherapy. Hepatic resection with or without fenestration should be favored in patients with diffuse involvement of the liver but with sufficient spared parenchyma. Finally, in the patient with diffuse disease, OLT is a valid option and should be pursued as primary therapy prior to the development of debilitating disease such as malnutrition and liver dysfunction that can significantly increase the risks of perioperative adverse events.
Footnotes
P- Reviewers Drenth JPH, Hori T, Llado L, Schemmer P S- Editor Wen LL L- Editor A E- Editor Zhang DN
References
- 1.Hoevenaren IA, Wester R, Schrier RW, McFann K, Doctor RB, Drenth JP, Everson GT. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int. 2008;28:264–270. doi: 10.1111/j.1478-3231.2007.01595.x. [DOI] [PubMed] [Google Scholar]
- 2.Bristowe F. Cystic disease of the liver associated with similar disease of the kidneys. Trans Pathol Soc Lond. 1856;7:229–234. [Google Scholar]
- 3.Moschcowitz E. Non-parasitic cysts (congenital) of the liver, with a study of aberrant bile ducts. Am J Med Sci. 1906;131:674–699. [Google Scholar]
- 4.Feldman M. Polycystic disease of the liver. Am J Gastroenterol. 1958;29:83–86. [PubMed] [Google Scholar]
- 5.Tahvanainen P, Tahvanainen E, Reijonen H, Halme L, Kääriäinen H, Höckerstedt K. Polycystic liver disease is genetically heterogeneous: clinical and linkage studies in eight Finnish families. J Hepatol. 2003;38:39–43. doi: 10.1016/s0168-8278(02)00348-3. [DOI] [PubMed] [Google Scholar]
- 6.Torres VE. Treatment of polycystic liver disease: one size does not fit all. Am J Kidney Dis. 2007;49:725–728. doi: 10.1053/j.ajkd.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Arnold HL, Harrison SA. New advances in evaluation and management of patients with polycystic liver disease. Am J Gastroenterol. 2005;100:2569–2582. doi: 10.1111/j.1572-0241.2005.00263.x. [DOI] [PubMed] [Google Scholar]
- 8.Vauthey JN, Maddern GJ, Blumgart LH. Adult polycystic disease of the liver. Br J Surg. 1991;78:524–527. doi: 10.1002/bjs.1800780505. [DOI] [PubMed] [Google Scholar]
- 9.Grünfeld JP, Albouze G, Jungers P, Landais P, Dana A, Droz D, Moynot A, Lafforgue B, Boursztyn E, Franco D. Liver changes and complications in adult polycystic kidney disease. Adv Nephrol Necker Hosp. 1985;14:1–20. [PubMed] [Google Scholar]
- 10.Drenth JP, Chrispijn M, Nagorney DM, Kamath PS, Torres VE. Medical and surgical treatment options for polycystic liver disease. Hepatology. 2010;52:2223–2230. doi: 10.1002/hep.24036. [DOI] [PubMed] [Google Scholar]
- 11.Temmerman F, Missiaen L, Bammens B, Laleman W, Cassiman D, Verslype C, van Pelt J, Nevens F. Systematic review: the pathophysiology and management of polycystic liver disease. Aliment Pharmacol Ther. 2011;34:702–713. doi: 10.1111/j.1365-2036.2011.04783.x. [DOI] [PubMed] [Google Scholar]
- 12.Qian Q. Isolated polycystic liver disease. Adv Chronic Kidney Dis. 2010;17:181–189. doi: 10.1053/j.ackd.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Agata ID, Jonas MM, Perez-Atayde AR, Guay-Woodford LM. Combined cystic disease of the liver and kidney. Semin Liver Dis. 1994;14:215–228. doi: 10.1055/s-2007-1007313. [DOI] [PubMed] [Google Scholar]
- 14.Davila S, Furu L, Gharavi AG, Tian X, Onoe T, Qian Q, Li A, Cai Y, Kamath PS, King BF, et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet. 2004;36:575–577. doi: 10.1038/ng1357. [DOI] [PubMed] [Google Scholar]
- 15.Van Keimpema L, De Koning DB, Van Hoek B, Van Den Berg AP, Van Oijen MG, De Man RA, Nevens F, Drenth JP. Patients with isolated polycystic liver disease referred to liver centres: clinical characterization of 137 cases. Liver Int. 2011;31:92–98. doi: 10.1111/j.1478-3231.2010.02247.x. [DOI] [PubMed] [Google Scholar]
- 16.Desmet VJ. Ludwig symposium on biliary disorders--part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–89. doi: 10.4065/73.1.80. [DOI] [PubMed] [Google Scholar]
- 17.Ramos A, Torres VE, Holley KE, Offord KP, Rakela J, Ludwig J. The liver in autosomal dominant polycystic kidney disease. Implications for pathogenesis. Arch Pathol Lab Med. 1990;114:180–184. [PubMed] [Google Scholar]
- 18.Qian Q, Li A, King BF, Kamath PS, Lager DJ, Huston J, Shub C, Davila S, Somlo S, Torres VE. Clinical profile of autosomal dominant polycystic liver disease. Hepatology. 2003;37:164–171. doi: 10.1053/jhep.2003.50006. [DOI] [PubMed] [Google Scholar]
- 19.Lazaridis KN, Strazzabosco M, Larusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology. 2004;127:1565–1577. doi: 10.1053/j.gastro.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Everson GT, Emmett M, Brown WR, Redmond P, Thickman D. Functional similarities of hepatic cystic and biliary epithelium: studies of fluid constituents and in vivo secretion in response to secretin. Hepatology. 1990;11:557–565. doi: 10.1002/hep.1840110406. [DOI] [PubMed] [Google Scholar]
- 21.Perrone RD, Grubman SA, Rogers LC, Lee DW, Moy E, Murray SL, Torres VE, Jefferson DM. Continuous epithelial cell lines from ADPKD liver cysts exhibit characteristics of intrahepatic biliary epithelium. Am J Physiol. 1995;269:G335–G345. doi: 10.1152/ajpgi.1995.269.3.G335. [DOI] [PubMed] [Google Scholar]
- 22.Nichols MT, Gidey E, Matzakos T, Dahl R, Stiegmann G, Shah RJ, Grantham JJ, Fitz JG, Doctor RB. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40:836–846. doi: 10.1002/hep.20401. [DOI] [PubMed] [Google Scholar]
- 23.Wheatley DN. Primary cilia in normal and pathological tissues. Pathobiology. 1995;63:222–238. doi: 10.1159/000163955. [DOI] [PubMed] [Google Scholar]
- 24.Ong AC, Wheatley DN. Polycystic kidney disease--the ciliary connection. Lancet. 2003;361:774–776. doi: 10.1016/S0140-6736(03)12662-1. [DOI] [PubMed] [Google Scholar]
- 25.Davenport JR, Yoder BK. An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol. 2005;289:F1159–F1169. doi: 10.1152/ajprenal.00118.2005. [DOI] [PubMed] [Google Scholar]
- 26.Harris RA, Gray DW, Britton BJ, Toogood GJ, Morris PJ. Hepatic cystic disease in an adult polycystic kidney disease transplant population. Aust N Z J Surg. 1996;66:166–168. doi: 10.1111/j.1445-2197.1996.tb01148.x. [DOI] [PubMed] [Google Scholar]
- 27.Sherstha R, McKinley C, Russ P, Scherzinger A, Bronner T, Showalter R, Everson GT. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology. 1997;26:1282–1286. doi: 10.1002/hep.510260528. [DOI] [PubMed] [Google Scholar]
- 28.van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, Dekker HM, de Man RA, Drenth JP. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009;137:1661–8.e1-2. doi: 10.1053/j.gastro.2009.07.052. [DOI] [PubMed] [Google Scholar]
- 29.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21:1052–1061. doi: 10.1681/ASN.2009121291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caroli A, Antiga L, Cafaro M, Fasolini G, Remuzzi A, Remuzzi G, Ruggenenti P. Reducing polycystic liver volume in ADPKD: effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol. 2010;5:783–789. doi: 10.2215/CJN.05380709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvaro D, Mancino MG, Onori P, Franchitto A, Alpini G, Francis H, Glaser S, Gaudio E. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. 2006;12:3537–3545. doi: 10.3748/wjg.v12.i22.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Everson GT, Scherzinger A, Berger-Leff N, Reichen J, Lezotte D, Manco-Johnson M, Gabow P. Polycystic liver disease: quantitation of parenchymal and cyst volumes from computed tomography images and clinical correlates of hepatic cysts. Hepatology. 1988;8:1627–1634. doi: 10.1002/hep.1840080626. [DOI] [PubMed] [Google Scholar]
- 33.Dmitrewski J, Olliff S, Buckels JA. Obstructive jaundice associated with polycystic liver disease. HPB Surg. 1996;10:117–120. doi: 10.1155/1996/83547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abascal J, Moya M, Martin F. Infection of hepatic cysts in polycystic disease. World J Surg. 1984;8:424–425. doi: 10.1007/BF01655097. [DOI] [PubMed] [Google Scholar]
- 35.van Erpecum KJ, Janssens AR, Terpstra JL, Tjon A Tham RT. Highly symptomatic adult polycystic disease of the liver. A report of fifteen cases. J Hepatol. 1987;5:109–117. doi: 10.1016/s0168-8278(87)80068-5. [DOI] [PubMed] [Google Scholar]
- 36.Que F, Nagorney DM, Gross JB, Torres VE. Liver resection and cyst fenestration in the treatment of severe polycystic liver disease. Gastroenterology. 1995;108:487–494. doi: 10.1016/0016-5085(95)90078-0. [DOI] [PubMed] [Google Scholar]
- 37.Kabbej M, Sauvanet A, Chauveau D, Farges O, Belghiti J. Laparoscopic fenestration in polycystic liver disease. Br J Surg. 1996;83:1697–1701. doi: 10.1002/bjs.1800831211. [DOI] [PubMed] [Google Scholar]
- 38.Gigot JF, Jadoul P, Que F, Van Beers BE, Etienne J, Horsmans Y, Collard A, Geubel A, Pringot J, Kestens PJ. Adult polycystic liver disease: is fenestration the most adequate operation for long-term management. Ann Surg. 1997;225:286–294. doi: 10.1097/00000658-199703000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waanders E, van Keimpema L, Brouwer JT, van Oijen MG, Aerts R, Sweep FC, Nevens F, Drenth JP. Carbohydrate antigen 19-9 is extremely elevated in polycystic liver disease. Liver Int. 2009;29:1389–1395. doi: 10.1111/j.1478-3231.2009.02055.x. [DOI] [PubMed] [Google Scholar]
- 40.Deschênes M, Michel RP, Alpert E, Barkun JS, Metrakos P, Tchervenkov J. Elevation of CA-125 level is due to abdominal distension in liver transplantation candidates. Transplantation. 2001;72:1519–1522. doi: 10.1097/00007890-200111150-00008. [DOI] [PubMed] [Google Scholar]
- 41.Iwase K, Takenaka H, Oshima S, Yagura A, Nishimura Y, Yoshidome K, Tanaka T. Determination of tumor marker levels in cystic fluid of benign liver cysts. Dig Dis Sci. 1992;37:1648–1654. doi: 10.1007/BF01299853. [DOI] [PubMed] [Google Scholar]
- 42.McCormick SE, Sjogren MH, Goodman ZD. A 22-year-old man with a liver mass and markedly elevated serum alpha fetoprotein. Semin Liver Dis. 1994;14:395–403. doi: 10.1055/s-2007-1007330. [DOI] [PubMed] [Google Scholar]
- 43.Schievink WI, Spetzler RF. Screening for intracranial aneurysms in patients with isolated polycystic liver disease. J Neurosurg. 1998;89:719–721. doi: 10.3171/jns.1998.89.5.0719. [DOI] [PubMed] [Google Scholar]
- 44.Ong AC. Screening for intracranial aneurysms in ADPKD. BMJ. 2009;339:b3763. doi: 10.1136/bmj.b3763. [DOI] [PubMed] [Google Scholar]
- 45.Lumiaho A, Ikäheimo R, Miettinen R, Niemitukia L, Laitinen T, Rantala A, Lampainen E, Laakso M, Hartikainen J. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis. 2001;38:1208–1216. doi: 10.1053/ajkd.2001.29216. [DOI] [PubMed] [Google Scholar]
- 46.Xu HW, Yu SQ, Mei CL, Li MH. Screening for intracranial aneurysm in 355 patients with autosomal-dominant polycystic kidney disease. Stroke. 2011;42:204–206. doi: 10.1161/STROKEAHA.110.578740. [DOI] [PubMed] [Google Scholar]
- 47.Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol. 2009;5:221–228. doi: 10.1038/nrneph.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russell RT, Pinson CW. Surgical management of polycystic liver disease. World J Gastroenterol. 2007;13:5052–5059. doi: 10.3748/wjg.v13.i38.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Torra R, Nicolau C, Badenas C, Navarro S, Pérez L, Estivill X, Darnell A. Ultrasonographic study of pancreatic cysts in autosomal dominant polycystic kidney disease. Clin Nephrol. 1997;47:19–22. [PubMed] [Google Scholar]
- 50.Blyth H, Ockenden BG. Polycystic disease of kidney and liver presenting in childhood. J Med Genet. 1971;8:257–284. doi: 10.1136/jmg.8.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Milutinovic J, Schabel SI, Ainsworth SK. Autosomal dominant polycystic kidney disease with liver and pancreatic involvement in early childhood. Am J Kidney Dis. 1989;13:340–344. doi: 10.1016/s0272-6386(89)80043-5. [DOI] [PubMed] [Google Scholar]
- 52.Levine E, Cook LT, Grantham JJ. Liver cysts in autosomal-dominant polycystic kidney disease: clinical and computed tomographic study. AJR Am J Roentgenol. 1985;145:229–233. doi: 10.2214/ajr.145.2.229. [DOI] [PubMed] [Google Scholar]
- 53.Nicolau C, Torra R, Bianchi L, Vilana R, Gilabert R, Darnell A, Brú C. Abdominal sonographic study of autosomal dominant polycystic kidney disease. J Clin Ultrasound. 2000;28:277–282. doi: 10.1002/1097-0096(200007/08)28:6<277::aid-jcu2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 54.Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM, Baumgarten DA, King BF, Wetzel LH, Kenney PJ, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64–69. doi: 10.2215/CJN.00080605. [DOI] [PubMed] [Google Scholar]
- 55.Vachha B, Sun MR, Siewert B, Eisenberg RL. Cystic lesions of the liver. AJR Am J Roentgenol. 2011;196:W355–W366. doi: 10.2214/AJR.10.5292. [DOI] [PubMed] [Google Scholar]
- 56.Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet. 1994;343:824–827. doi: 10.1016/s0140-6736(94)92026-5. [DOI] [PubMed] [Google Scholar]
- 57.Carrim ZI, Murchison JT. The prevalence of simple renal and hepatic cysts detected by spiral computed tomography. Clin Radiol. 2003;58:626–629. doi: 10.1016/s0009-9260(03)00165-x. [DOI] [PubMed] [Google Scholar]
- 58.Chauveau D, Fakhouri F, Grünfeld JP. Liver involvement in autosomal-dominant polycystic kidney disease: therapeutic dilemma. J Am Soc Nephrol. 2000;11:1767–1775. doi: 10.1681/ASN.V1191767. [DOI] [PubMed] [Google Scholar]
- 59.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 60.Migali G, Annet L, Lonneux M, Devuyst O. Renal cyst infection in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2008;23:404–405. doi: 10.1093/ndt/gfm665. [DOI] [PubMed] [Google Scholar]
- 61.Lahiri SA, Halff GA, Speeg KV, Esterl RM. In-111 WBC scan localizes infected hepatic cysts and confirms their complete resection in adult polycystic kidney disease. Clin Nucl Med. 1998;23:33–34. doi: 10.1097/00003072-199801000-00010. [DOI] [PubMed] [Google Scholar]
- 62.Soussan M, Sberro R, Wartski M, Fakhouri F, Pecking AP, Alberini JL. Diagnosis and localization of renal cyst infection by 18F-fluorodeoxyglucose PET/CT in polycystic kidney disease. Ann Nucl Med. 2008;22:529–531. doi: 10.1007/s12149-008-0150-3. [DOI] [PubMed] [Google Scholar]
- 63.Bleeker-Rovers CP, de Sévaux RG, van Hamersvelt HW, Corstens FH, Oyen WJ. Diagnosis of renal and hepatic cyst infections by 18-F-fluorodeoxyglucose positron emission tomography in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2003;41:E18–E21. doi: 10.1016/s0272-6386(03)00368-8. [DOI] [PubMed] [Google Scholar]
- 64.Schnelldorfer T, Torres VE, Zakaria S, Rosen CB, Nagorney DM. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg. 2009;250:112–118. doi: 10.1097/SLA.0b013e3181ad83dc. [DOI] [PubMed] [Google Scholar]
- 65.Alvaro D, Gigliozzi A, Attili AF. Regulation and deregulation of cholangiocyte proliferation. J Hepatol. 2000;33:333–340. doi: 10.1016/s0168-8278(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 66.Møller LN, Stidsen CE, Hartmann B, Holst JJ. Somatostatin receptors. Biochim Biophys Acta. 2003;1616:1–84. doi: 10.1016/s0005-2736(03)00235-9. [DOI] [PubMed] [Google Scholar]
- 67.Heisler S, Srikant CB. Somatostatin-14 and somatostatin-28 pretreatment down-regulate somatostatin-14 receptors and have biphasic effects on forskolin-stimulated cyclic adenosine, 3’,5’-monophosphate synthesis and adrenocorticotropin secretion in mouse anterior pituitary tumor cells. Endocrinology. 1985;117:217–225. doi: 10.1210/endo-117-1-217. [DOI] [PubMed] [Google Scholar]
- 68.Jakobs KH, Gehring U, Gaugler B, Pfeuffer T, Schultz G. Occurrence of an inhibitory guanine nucleotide-binding regulatory component of the adenylate cyclase system in cyc- variants of S49 lymphoma cells. Eur J Biochem. 1983;130:605–611. doi: 10.1111/j.1432-1033.1983.tb07192.x. [DOI] [PubMed] [Google Scholar]
- 69.Tan CK, Podila PV, Taylor JE, Nagorney DM, Wiseman GA, Gores GJ, LaRusso NF. Human cholangiocarcinomas express somatostatin receptors and respond to somatostatin with growth inhibition. Gastroenterology. 1995;108:1908–1916. doi: 10.1016/0016-5085(95)90157-4. [DOI] [PubMed] [Google Scholar]
- 70.Pyronnet S, Bousquet C, Najib S, Azar R, Laklai H, Susini C. Antitumor effects of somatostatin. Mol Cell Endocrinol. 2008;286:230–237. doi: 10.1016/j.mce.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 71.Walz G. Therapeutic approaches in autosomal dominant polycystic kidney disease (ADPKD): is there light at the end of the tunnel. Nephrol Dial Transplant. 2006;21:1752–1757. doi: 10.1093/ndt/gfl246. [DOI] [PubMed] [Google Scholar]
- 72.Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820–829. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 73.Walz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Hörl WH, Obermüller N, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830–840. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- 74.Watnick T, Germino GG. mTOR inhibitors in polycystic kidney disease. N Engl J Med. 2010;363:879–881. doi: 10.1056/NEJMe1006925. [DOI] [PubMed] [Google Scholar]
- 75.Renken C, Fischer DC, Kundt G, Gretz N, Haffner D. Inhibition of mTOR with sirolimus does not attenuate progression of liver and kidney disease in PCK rats. Nephrol Dial Transplant. 2011;26:92–100. doi: 10.1093/ndt/gfq384. [DOI] [PubMed] [Google Scholar]
- 76.Takei R, Ubara Y, Hoshino J, Higa Y, Suwabe T, Sogawa Y, Nomura K, Nakanishi S, Sawa N, Katori H, et al. Percutaneous transcatheter hepatic artery embolization for liver cysts in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2007;49:744–752. doi: 10.1053/j.ajkd.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 77.Park HC, Kim CW, Ro H, Moon JY, Oh KH, Kim Y, Lee JS, Yin YH, Jae HJ, Chung JW, et al. Transcatheter arterial embolization therapy for a massive polycystic liver in autosomal dominant polycystic kidney disease patients. J Korean Med Sci. 2009;24:57–61. doi: 10.3346/jkms.2009.24.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saini S, Mueller PR, Ferrucci JT, Simeone JF, Wittenberg J, Butch RJ. Percutaneous aspiration of hepatic cysts does not provide definitive therapy. AJR Am J Roentgenol. 1983;141:559–560. doi: 10.2214/ajr.141.3.559. [DOI] [PubMed] [Google Scholar]
- 79.Bean WJ, Rodan BA. Hepatic cysts: treatment with alcohol. AJR Am J Roentgenol. 1985;144:237–241. doi: 10.2214/ajr.144.2.237. [DOI] [PubMed] [Google Scholar]
- 80.van Keimpema L, de Koning DB, Strijk SP, Drenth JP. Aspiration-sclerotherapy results in effective control of liver volume in patients with liver cysts. Dig Dis Sci. 2008;53:2251–2257. doi: 10.1007/s10620-007-0121-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kairaluoma MI, Leinonen A, Ståhlberg M, Päivänsalo M, Kiviniemi H, Siniluoto T. Percutaneous aspiration and alcohol sclerotherapy for symptomatic hepatic cysts. An alternative to surgical intervention. Ann Surg. 1989;210:208–215. doi: 10.1097/00000658-198908000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.van Keimpema L, Ruurda JP, Ernst MF, van Geffen HJ, Drenth JP. Laparoscopic fenestration of liver cysts in polycystic liver disease results in a median volume reduction of 12.5% J Gastrointest Surg. 2008;12:477–482. doi: 10.1007/s11605-007-0376-8. [DOI] [PubMed] [Google Scholar]
- 83.Garcea G, Rajesh A, Dennison AR. Surgical management of cystic lesions in the liver. ANZ J Surg. 2013;83:E3–E20. doi: 10.1111/j.1445-2197.2012.06096.x. [DOI] [PubMed] [Google Scholar]
- 84.van Keimpema L, Drenth JP. Polycystic liver disease: a critical appraisal of hepatic resection, cyst fenestration, and liver transplantation. Ann Surg. 2011;253:419; author reply 420. doi: 10.1097/SLA.0b013e3182080423. [DOI] [PubMed] [Google Scholar]
- 85.Armitage NC, Blumgart LH. Partial resection and fenestration in the treatment of polycystic liver disease. Br J Surg. 1984;71:242–244. doi: 10.1002/bjs.1800710331. [DOI] [PubMed] [Google Scholar]
- 86.Schindl MJ, Redhead DN, Fearon KC, Garden OJ, Wigmore SJ. The value of residual liver volume as a predictor of hepatic dysfunction and infection after major liver resection. Gut. 2005;54:289–296. doi: 10.1136/gut.2004.046524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Everson GT, Taylor MR, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40:774–782. doi: 10.1002/hep.20431. [DOI] [PubMed] [Google Scholar]
- 88.Freeman RB, Gish RG, Harper A, Davis GL, Vierling J, Lieblein L, Klintmalm G, Blazek J, Hunter R, Punch J. Model for end-stage liver disease (MELD) exception guidelines: results and recommendations from the MELD Exception Study Group and Conference (MESSAGE) for the approval of patients who need liver transplantation with diseases not considered by the standard MELD formula. Liver Transpl. 2006;12:S128–S136. doi: 10.1002/lt.20979. [DOI] [PubMed] [Google Scholar]
- 89.Arrazola L, Moonka D, Gish RG, Everson GT. Model for end-stage liver disease (MELD) exception for polycystic liver disease. Liver Transpl. 2006;12:S110–S111. doi: 10.1002/lt.20974. [DOI] [PubMed] [Google Scholar]
- 90.Kirchner GI, Rifai K, Cantz T, Nashan B, Terkamp C, Becker T, Strassburg C, Barg-Hock H, Wagner S, Lück R, et al. Outcome and quality of life in patients with polycystic liver disease after liver or combined liver-kidney transplantation. Liver Transpl. 2006;12:1268–1277. doi: 10.1002/lt.20780. [DOI] [PubMed] [Google Scholar]
- 91.Rasmussen A, Davies HF, Jamieson NV, Evans DB, Calne RY. Combined transplantation of liver and kidney from the same donor protects the kidney from rejection and improves kidney graft survival. Transplantation. 1995;59:919–921. [PubMed] [Google Scholar]
- 92.Pirenne J, Aerts R, Yoong K, Gunson B, Koshiba T, Fourneau I, Mayer D, Buckels J, Mirza D, Roskams T, et al. Liver transplantation for polycystic liver disease. Liver Transpl. 2001;7:238–245. doi: 10.1053/jlts.2001.22178. [DOI] [PubMed] [Google Scholar]
- 93.Szabó LS, Takács I, Arkosy P, Sápy P, Szentkereszty Z. Laparoscopic treatment of nonparasitic hepatic cysts. Surg Endosc. 2006;20:595–597. doi: 10.1007/s00464-005-0206-6. [DOI] [PubMed] [Google Scholar]
- 94.Martin IJ, McKinley AJ, Currie EJ, Holmes P, Garden OJ. Tailoring the management of nonparasitic liver cysts. Ann Surg. 1998;228:167–172. doi: 10.1097/00000658-199808000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liska V, Treska V, Mírka H, Skalický T, Sutnar A, Ferda J. [Treatment strategy in non-parasitic benign cysts of the liver] Rozhl Chir. 2008;87:512–516. [PubMed] [Google Scholar]
- 96.Bai XL, Liang TB, Yu J, Wang WL, Shen Y, Zhang M, Zheng SS. Long-term results of laparoscopic fenestration for patients with congenital liver cysts. Hepatobiliary Pancreat Dis Int. 2007;6:600–603. [PubMed] [Google Scholar]
- 97.Palanivelu C, Rangarajan M, Senthilkumar R, Madankumar MV. Laparoscopic management of symptomatic multiple hepatic cysts: a combination of deroofing and radical excision. JSLS. 2007;11:466–469. [PMC free article] [PubMed] [Google Scholar]
- 98.Garcea G, Pattenden CJ, Stephenson J, Dennison AR, Berry DP. Nine-year single-center experience with nonparastic liver cysts: diagnosis and management. Dig Dis Sci. 2007;52:185–191. doi: 10.1007/s10620-006-9545-y. [DOI] [PubMed] [Google Scholar]
- 99.Neri V, Ambrosi A, Fersini A, Valentino TP. Laparoscopic treatment of biliary hepatic cysts: short- and medium-term results. HPB (Oxford) 2006;8:306–310. doi: 10.1080/13651820500465766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kornprat P, Cerwenka H, Bacher H, El-Shabrawi A, Tillich M, Langner C, Mischinger HJ. Surgical therapy options in polycystic liver disease. Wien Klin Wochenschr. 2005;117:215–218. doi: 10.1007/s00508-005-0309-z. [DOI] [PubMed] [Google Scholar]
- 101.Robinson TN, Stiegmann GV, Everson GT. Laparoscopic palliation of polycystic liver disease. Surg Endosc. 2005;19:130–132. doi: 10.1007/s00464-004-8813-1. [DOI] [PubMed] [Google Scholar]
- 102.Fiamingo P, Tedeschi U, Veroux M, Cillo U, Brolese A, Da Rold A, Madia C, Zanus G, D’Amico DF. Laparoscopic treatment of simple hepatic cysts and polycystic liver disease. Surg Endosc. 2003;17:623–626. doi: 10.1007/s00464-002-9088-z. [DOI] [PubMed] [Google Scholar]
- 103.Tocchi A, Mazzoni G, Costa G, Cassini D, Bettelli E, Agostini N, Miccini M. Symptomatic nonparasitic hepatic cysts: options for and results of surgical management. Arch Surg. 2002;137:154–158. doi: 10.1001/archsurg.137.2.154. [DOI] [PubMed] [Google Scholar]
- 104.Koperna T, Vogl S, Satzinger U, Schulz F. Nonparasitic cysts of the liver: results and options of surgical treatment. World J Surg. 1997;21:850–854; discussion 854-855. doi: 10.1007/s002689900316. [DOI] [PubMed] [Google Scholar]
- 105.Morino M, Garrone C, Festa V, Miglietta C. Laparoscopic treatment of non parasitic liver cysts. Ann Chir. 1996;50:419–425; discussion 426-430. [PubMed] [Google Scholar]
- 106.Farges O, Bismuth H. Fenestration in the management of polycystic liver disease. World J Surg. 1995;19:25–30. doi: 10.1007/BF00316975. [DOI] [PubMed] [Google Scholar]
- 107.Li TJ, Zhang HB, Lu JH, Zhao J, Yang N, Yang GS. Treatment of polycystic liver disease with resection-fenestration and a new classification. World J Gastroenterol. 2008;14:5066–5072. doi: 10.3748/wjg.14.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gamblin TC, Holloway SE, Heckman JT, Geller DA. Laparoscopic resection of benign hepatic cysts: a new standard. J Am Coll Surg. 2008;207:731–736. doi: 10.1016/j.jamcollsurg.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 109.Yang GS, Li QG, Lu JH, Yang N, Zhang HB, Zhou XP. Combined hepatic resection with fenestration for highly symptomatic polycystic liver disease: A report on seven patients. World J Gastroenterol. 2004;10:2598–2601. doi: 10.3748/wjg.v10.i17.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vons C, Chauveau D, Martinod E, Smadja C, Capron F, Grunfeld JP, Franco D. [Liver resection in patients with polycystic liver disease] Gastroenterol Clin Biol. 1998;22:50–54. [PubMed] [Google Scholar]
- 111.Soravia C, Mentha G, Giostra E, Morel P, Rohner A. Surgery for adult polycystic liver disease. Surgery. 1995;117:272–275. doi: 10.1016/s0039-6060(05)80201-6. [DOI] [PubMed] [Google Scholar]
- 112.Henne-Bruns D, Klomp HJ, Kremer B. Non-parasitic liver cysts and polycystic liver disease: results of surgical treatment. Hepatogastroenterology. 1993;40:1–5. [PubMed] [Google Scholar]
- 113.Vauthey JN, Maddern GJ, Kolbinger P, Baer HU, Blumgart LH. Clinical experience with adult polycystic liver disease. Br J Surg. 1992;79:562–565. doi: 10.1002/bjs.1800790629. [DOI] [PubMed] [Google Scholar]
- 114.Sanchez H, Gagner M, Rossi RL, Jenkins RL, Lewis WD, Munson JL, Braasch JW. Surgical management of nonparasitic cystic liver disease. Am J Surg. 1991;161:113–18; discussion 113-18;. doi: 10.1016/0002-9610(91)90370-s. [DOI] [PubMed] [Google Scholar]
- 115.Newman KD, Torres VE, Rakela J, Nagorney DM. Treatment of highly symptomatic polycystic liver disease. Preliminary experience with a combined hepatic resection-fenestration procedure. Ann Surg. 1990;212:30–37. doi: 10.1097/00000658-199007000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Iwatsuki S, Starzl TE. Personal experience with 411 hepatic resections. Ann Surg. 1988;208:421–434. doi: 10.1097/00000658-198810000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Taner B, Willingham DL, Hewitt WR, Grewal HP, Nguyen JH, Hughes CB. Polycystic liver disease and liver transplantation: single-institution experience. Transplant Proc. 2009;41:3769–3771. doi: 10.1016/j.transproceed.2009.05.043. [DOI] [PubMed] [Google Scholar]
- 118.Ueno T, Barri YM, Netto GJ, Martin A, Onaca N, Sanchez EQ, Chinnakotla S, Randall HB, Dawson S, Levy MF, et al. Liver and kidney transplantation for polycystic liver and kidney-renal function and outcome. Transplantation. 2006;82:501–507. doi: 10.1097/01.tp.0000231712.75645.7a. [DOI] [PubMed] [Google Scholar]
- 119.Ueda M, Egawa H, Oike F, Taira K, Uryuhara K, Fujimoto Y, Kozaki K, Tanaka K. Living-donor liver transplantation for polycystic liver disease. Transplantation. 2004;77:480–481. doi: 10.1097/01.TP.0000110319.60723.31. [DOI] [PubMed] [Google Scholar]
- 120.Gustafsson BI, Friman S, Mjornstedt L, Olausson M, Backman L. Liver transplantation for polycystic liver disease--indications and outcome. Transplant Proc. 2003;35:813–814. doi: 10.1016/s0041-1345(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 121.Swenson K, Seu P, Kinkhabwala M, Maggard M, Martin P, Goss J, Busuttil R. Liver transplantation for adult polycystic liver disease. Hepatology. 1998;28:412–415. doi: 10.1002/hep.510280218. [DOI] [PubMed] [Google Scholar]
- 122.Lang H, von Woellwarth J, Oldhafer KJ, Behrend M, Schlitt HJ, Nashan B, Pichlmayr R. Liver transplantation in patients with polycystic liver disease. Transplant Proc. 1997;29:2832–2833. doi: 10.1016/s0041-1345(97)00696-9. [DOI] [PubMed] [Google Scholar]
- 123.Washburn WK, Johnson LB, Lewis WD, Jenkins RL. Liver transplantation for adult polycystic liver disease. Liver Transpl Surg. 1996;2:17–22. doi: 10.1002/lt.500020105. [DOI] [PubMed] [Google Scholar]
- 124.Starzl TE, Reyes J, Tzakis A, Mieles L, Todo S, Gordon R. Liver transplantation for polycystic liver disease. Arch Surg. 1990;125:575–577. doi: 10.1001/archsurg.1990.01410170021003. [DOI] [PubMed] [Google Scholar]