

Abstract
While breast milk has been known as a cause of neonatal hyperbilirubinemia, the underlying mechanism of breast milk-induced jaundice has not been clarified. Here, the impact of fatty acids on human UDP-glucuronosyltransferase (UGT) 1A1 – the sole enzyme that can metabolize bilirubin – were examined. Oleic acid, linoleic acid, and docosahexaenoic acid (DHA) strongly inhibited UGT1A1 activity. Forty-eight hours after a treatment with a lower concentration of DHA (10 mg/kg), total bilirubin significantly increased in neonatal hUGT1 mice, which are human neonatal jaundice models. In contrast, treatments with higher concentrations of fatty acids (0.1–10 g/kg) resulted in a decrease in serum bilirubin in hUGT1 mice. It was further demonstrated that the treatment with higher concentrations of fatty acids induced UGT1A1, possibly by activation of peroxisome proliferator-activated receptors. Our data indicates that activation of peroxisome proliferator-activated receptors would increase UGT1A1 expression, resulting in reduction of serum bilirubin levels in human infants.
UDP-glucuronosyltransferases (UGTs) are phase II drug-metabolizing enzymes that catalyze glucuronidation of compounds by transferring glucuronic acid from a co-substrate, UDP-glucuronic acid, to substrates1. UGTs are super-family enzymes that are mainly classified into two subfamilies, UGT1 and UGT2, on the basis of evolutionary divergence2. The human UGT1 gene encodes multiple unique exons 1 and common exons 2 to 5, producing nine functional UGT1A isoforms, UGT1A1, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, and UGT1A103. These isoforms exhibit substrate specificity and are expressed tissue specifically4. In the liver, which is known as the most important tissue for detoxification, UGT1A1, UGT1A3, UGT1A4, UGT1A6, and UGT1A9 are expressed4. Accumulating evidence indicates that in addition to the liver, extrahepatic tissues such as the small intestine and kidneys potentially contribute to metabolism of a wide variety of compounds5. While UGTs metabolize various clinically used drugs, they are also involved in metabolism of endogenous compounds, such as bilirubin – a specific substrate of UGT1A16.
Increased or decreased expressions of drug-metabolizing enzymes or the presence of factors that can activate or inhibit drug-metabolizing enzymes can cause accelerated or delayed metabolism of administered drugs or endogenous substrates, which can result in severe adverse effects of the drugs or hormone imbalance. Therefore, it is important to understand the factors that affect the expression and activity of drug-metabolizing enzymes to avoid potential adverse effects. It has been known that breast-fed infants have a 3- to 6-fold greater probability of developing elevated total serum bilirubin levels than formula-fed infants7,8,9. Though controversial findings were subsequently reported, Arias et al. originally found an inhibitory effect of 5β-pregnane-3α,20β-diol – an endogenous compound present in breast milk – on glucuronosyltransferase activity in vitro10. A recent study by Ota et al. revealed that 5β-pregnane-3α,20β-diol inhibited enzyme activity, as well as transcriptional activity of human UGT1A111. In addition to β-pregnane-3α,20β-diol, fatty acids included in breast milk have also been reported as inhibitors of bilirubin glucuronosyltransferase activity. In 1972, Bevan and Holton demonstrated that oleic acid – one of the fatty acids highly present in breast milk – inhibited bilirubin conjugation in rat liver slices12. Subsequently, Hargreaves demonstrated that while saturated fatty acids did not, C18 and C20 unsaturated fatty acids inhibited bilirubin conjugation in rat liver slices13. While several studies revealed that mammalian UGTs, such as monkey UGT2B9 and human UGT2B7, possessed glucuronidation activities toward saturated and unsaturated fatty acids in the 1990s to early 2000s14,15,16,17,18,19,20, the inhibitory effects of fatty acids on human UGT enzymes were not investigated until 2004. A study by Tsoutsikos et al. demonstrated that certain fatty acids strongly inhibited 4-methylumbelliferone (4-MU) glucuronidation in human kidney microsomes, as well as in recombinant human UGT1A9 and UGT2B721. Inhibitory effects of unsaturated fatty acids, oleic acid and linoleic acid, on recombinant human UGT1A9-catalyzed 4-MU and propofol glucuronidation were subsequently demonstrated in 200822. However, inhibitory effects of fatty acids on human UGT1A1-catalyzed glucuronidation have not been investigated.
To understand the potential inhibitory effects of fatty acids on human UGT1A1, in the present study, inhibitory effects of 15 saturated and unsaturated fatty acids on UGT1A1-catalyzed estradiol 3-O-glucuronidation were investigated in recombinant UGT1A1. Furthermore, inhibitory patterns and Ki values of the inhibitors were determined in recombinant UGT1A1, as well as in human liver and small intestine microsomes. Finally, to understand the impact of fatty acids on bilirubin glucuronidation in vivo, docosahexaenoic acid (DHA), oleic acid, and linoleic acid were orally administered to human breast milk-induced jaundice model mice, which are recently developed humanized UGT1 mice23,24.
Results
Inhibitory effects of free fatty acids on UGT1A1-catalyzed glucuronidation
Fatty acids present in breast milk have been considered to be the causes of breast milk-induced neonatal hyperbilirubinemia. However, inhibitory effects of fatty acids on human UGT1A1-catalyzed glucuronidation have not been investigated. Oleic acid, linoleic acid, stearic acid, myristic acid, lauric acid, and palmitic acid are the fatty acids highly present in human breast milk25. In the present study, the inhibitory effects of these fatty acids and 9 other fatty acids, which are moderately present in human breast milk, on the enzymatic activity of human UGT1A1 were investigated. Estradiol 3-O glucuronide formation is one of the reactions that are selectively catalyzed by UGT1A126. In our study, the control activity of Estradiol 3-O glucuronidation in UGT1A1 Supersomes was 468 pmol/min/mg at 20 μM estradiol, which was in agreement with the reported catalytic rate. Out of the 15 tested fatty acids, oleic acid, linoleic acid, DHA, EPA, palmitoleic acid, arachidonic acid, and alpha-linolenic acid strongly inhibited UGT1A1 activity with IC50 values of 31.6 μM, 33.1 μM, 11.6 μM, 19.9 μM, 37.1 μM, 22.7 μM, and 26.1 μM, respectively (Fig. 1 and Table 1). While stearic acid and decanoic acid moderately inhibited UGT1A1 activity, their IC50 values were more than 50 μM. UGT1A1 activity was not inhibited at all with 50 μM palmitic acid, behenic acid, arachidic acid, myristic acid, lauric acid, or lignoceric acid.
Figure 1. Inhibitory effects of fatty acids on estradiol 3-O-glucuronidation.

Estradiol 3-O-glucuronidation was determined in the UGT1A1 recombinant system in both the presence and absence of fatty acids. Results are duplicate determinations.
Table 1. IC50 values of fatty acids for estradiol 3-O-glucuronidation.
| Fatty acids | IC50 values |
|---|---|
| Oleic acid | 31.6 μM |
| Linoleic acid | 33.1 μM |
| Palmitoleic acid | 37.1 μM |
| Alpha-linolenic acid | 26.1 μM |
| Arachidonic acid | 22.7 μM |
| DHA | 11.6 μM |
| EPA | 19.9 μM |
IC50 values were directly determined in the UGT1A1 recombinant system from linear regression (Fig. 1). IC50 values of stearic acid, myristic acid, lauric acid, palmitic acid, behenic acid, arachidic acid, lignoceric acid, and decanoic acid were more than 50 μM.
Inhibitory pattern of oleic acid, linoleic acid, and DHA on UGT1A1 activity in the recombinant system
We further determined the inhibitory pattern of oleic acid, linoleic acid, and DHA against estradiol 3-O glucuronidation by UGT1A1. Dixon-plot analyses indicated that oleic acid and linoleic acid non-competitively inhibited UGT1A1 activity (Fig. 2A and B), showing their Ki values of 23.4 μM and 22.1 μM, respectively (Table 2). DHA more potently inhibited UGT1A1 activity than oleic acid and linoleic acid, showing its Ki values of 1.8 μM. The inhibitory pattern of DHA against estradiol 3-O glucuronidation by UGT1A1 was non-competitive, the same as the patterns of oleic acid and linoleic acid (Fig. 2C and Table 2). These data indicate that oleic acid, linoleic acid, and DHA present in breast milk might be the causes of breast milk-induced neonatal hyperbilirubinemia.
Figure 2. Dixon plot analysis of inhibition of estradiol 3-O-glucuronidation in the recombinant UGT1A1.

Inhibitory effects of oleic acid (A), linoleic acid (B), and DHA (C) on estradiol 3-O-glucuronidation were analyzed. Filled circles, open circles, and open triangles are the data obtained with 5, 10, and 20 μM estradiol, respectively.
Table 2. Ki values of fatty acids for estradiol 3-O-glucuronidation.
| Fatty acids | Ki values |
|---|---|
| Recombinant UGT1A1 | |
| Oleic acid | 23.4 μM |
| Linoleic acid | 22.1 μM |
| DHA | 1.8 μM |
| Human liver microsomes | |
| Oleic acid | 29.3 μM |
| Linoleic acid | 24.0 μM |
| DHA | 4.3 μM |
| Human small intestine microsomes | |
| Oleic acid | - |
| Linoleic acid | - |
| DHA | 5.2 μM |
Inhibitory pattern of oleic acid, linoleic acid, and DHA on UGT1A1 activity in human liver and small intestine microsomes
While recombinant systems are convenient tools to determine enzymatic activities of single drug-metabolizing enzymes, differences in the enzymatic properties between recombinant systems and other in vitro tools, such as human liver microsomes or hepatocytes, have been reported27. Investigations have speculated that those differences might be attributed to the differences in composition of endoplasmic reticulum membrane, topology of enzymes, or ratio of active/inactive proteins. Although UGT1A1 is extensively expressed in various tissues, its expression levels are dramatically high in the liver and small intestine28. Indeed, previous studies have revealed that in addition to the liver, the small intestine plays an important role in bilirubin metabolism23,24. Therefore, in the present study, the inhibitory effects of oleic acid, linoleic acid, and DHA on UGT1A1 activity in human liver and small intestine microsomes were examined. In human liver microsomes, Dixon-plot analyses revealed that oleic acid and linoleic acid non-competitively inhibited UGT1A1 activity with Ki values of 29.3 μM and 24.0 μM, respectively (Fig. 3A–B and Table 2). DHA more potently inhibited UGT1A1 activity than oleic acid and linoleic acid, showing its Ki value of 4.3 μM with a non-competitive inhibition pattern (Fig. 3C and Table 2). These data are in agreement with the finding observed in the UGT1A1 recombinant system. In contrast, oleic acid and linoleic acid slightly inhibited estradiol 3-O-glucuronidation in human small intestine microsomes (Fig. 4A). At 50 μM oleic acid and linoleic acid, the residual activities of estradiol 3-O-glucuronidation were more than 80%. However, DHA still potently inhibited intestinal estradiol 3-O-glucuronidation, showing its Ki value of 5.2 μM with a non-competitive inhibition pattern (Fig. 4B and Table 2). This indicates that DHA has a potency to inhibit both hepatic and small intestinal UGT1A1 activities.
Figure 3. Dixon plot analysis of inhibition of estradiol 3-O-glucuronidation in human liver microsomes.

Inhibitory effects of oleic acid (A), linoleic acid (B), and DHA (C) on estradiol 3-O-glucuronidation were analyzed. Filled circles, open circles, and open triangles are the data obtained with 5, 10, and 20 μM estradiol, respectively.
Figure 4. Inhibition of estradiol 3-O-glucuronidation in human small intestine microsomes.

Inhibitory effects of oleic acid (50 μM), linoleic acid (50 μM), and DHA (50 μM) on estradiol 3-O-glucuronidation were shown (A). Dixon plot analysis of inhibition of estradiol 3-O-glucuronidation by DHA was carried out (B). Filled circles, open circles, and open triangles are the data obtained with 5, 10, and 20 μM estradiol, respectively.
DHA inhibits bilirubin glucuronidation in vivo
Our in vitro studies revealed that DHA could be a potent inhibitor of UGT1A1 activity. To understand the impact of the fatty acid in bilirubin glucuronidation in vivo, DHA was orally administered to hUGT1 mice, which are human breast milk-induced jaundice models. HUGT1 mice develop severe hyperbilirubinemia and their serum bilirubin levels reach a peak at 14 days after birth. The model mice were orally treated with DHA (10 mg/kg) at 12 days after birth and their bilirubin levels were measured at 14 days after birth, 48 hours after the treatment. Corn oil is usually used to dissolve hydrophobic compounds; however, it was reported that treatment of hUGT1 mice with corn oil decreased serum bilirubin levels, possibly by an inhibition of enterohepatic circulation of bilirubin. Rapeseed oil, on the other hand, did not affect the serum bilirubin levels of neonatal hUGT1 mice (data not shown). Therefore, in the present study, rapeseed oil was used to dissolve DHA and also to treat mice in the control group. In accordance with the previous report, serum bilirubin levels of neonatal hUGT1 mice in the control group increased by 2.3 ± 3.2 mg/dL from 12 days to 14 days after birth (13.6 mg/dL to 15.9 mg/dL). When DHA was orally given to the mice, the serum bilirubin levels increased by 6.6 mg/dL ± 1.3 from 12 days to 14 days after birth, which was statistically greater than the increased amount in the control group (Fig. 5). In hUGT1 mice, it was demonstrated that intestinal UGT1A1 played an important role in bilirubin metabolism23,24. Thus, the data indicates that DHA inhibited bilirubin glucuronidation in the small intestine, causing increased serum bilirubin levels in the human neonatal jaundice model.
Figure 5. Effects of oral DHA treatments on serum bilirubin levels in hUGT1 mice.

Neonatal hUGT1 mice were orally treated with DHA. Before and forty-eight hours after treatment, their serum bilirubin levels were analyzed. Changes in serum bilirubin levels are shown. Error bars show SD, n > 3. *, P < 0.05; **, P < 0.01 compared to the vehicle treated group.
High concentration of fatty acids induce UGT1A1 in the liver and small intestine
To investigate the concentration-dependent effects of fatty acids on bilirubin clearance in vivo, hUGT1 mice were further treated with high doses of oleic acid, linoleic acid, and DHA. Contrary to our expectation, the fatty acids concentration-dependently decreased serum bilirubin levels (Fig. 5 and 6). Especially, oleic acid and linoleic acid dramatically decreased serum bilirubin levels, from 13.6 mg/dL to 2.4 and 1.9 mg/dL, respectively. Fatty acids have been known to activate peroxisome proliferator-activated receptors (PPARs), which are transcriptional factors of human UGTs29. Indeed, a PPAR agonist, 4-Chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic Acid (WY-14643), has been shown to induce human UGT1A130. UGT1A1 mRNA expression was subsequently determined in fatty acids- and vehicle-treated mice to understand the effects of oleic acid and linoleic acid on the UGT1A1 expression. In the small intestine, oleic acid treatment resulted in 20- to 40-fold increases in UGT1A1 mRNA expression (Fig. 7A). Oleic acid also induced UGT1A1 in the liver, in which the UGT1A1 levels increased 10- to 100-fold (Fig. 7B). While linoleic acid similarly decreased serum bilirubin levels in hUGT1 mice, the induction of UGT1A1 was specifically observed in the small intestine, in which UGT1A1 levels increased 20- to 30-fold (Fig. 7C and D). These data indicate that induction of UGT1A1 in the small intestine might be responsible for the decrease of serum bilirubin in fatty acid-treated hUGT1A1 mice.
Figure 6. Effects of oral linoleic acid and oleic acid treatments on serum bilirubin levels in hUGT1 mice.

Neonatal hUGT1 mice were orally treated with linoleic acid (LA) and oleic acid (OA). Before and forty-eight hours after treatment, their serum bilirubin levels were analyzed. Changes in serum bilirubin levels are shown. Error bars show SD, n > 3. **, P < 0.01 compared to the vehicle treated group.
Figure 7. Effects of oral oleic acid (A) and linoleic acid (B) treatments on UGT1A1 expressions in hUGT1 mice.

Neonatal hUGT1 mice were orally treated with oleic acid and linoleic acid (10 g/kg). Forty-eight hours after the treatment, livers and small intestines were isolated and UGT1A1 expression levels were determined. Fold changes of UGT1A1 levels are shown. Error bars show SD, n = 3. **, P < 0.01 compared to the vehicle treated group.
Discussion
Breast milk has been recognized as a leading cause of neonatal hyperbilirubinemia31. A recent study demonstrated that suppression of the gastrointestinal UGT1A1 expression by breast milk led to the development of neonatal jaundice23. However, inhibition of the UGT1A1-catalyzed bilirubin glucuronidation by the components of breast milk has still been considered the underlying mechanism for breast milk-induced jaundice. To our knowledge, this is the first study examining the inhibitory effects of fatty acids on human UGT1A1 activity, although fatty acids were reported to inhibit mammalian UGT enzymes. In the present study, seven fatty acids out of 15 tested fatty acids inhibited UGT1A1 activity with IC50 values of 11.6 μM to 37.1 μM in the recombinant UGT1A1 system. Oleic acid and linoleic acid also inhibited UGT1A1 activity in the human liver microsomes. More than 30% and, to a lesser amount, 7% of the free fatty acids present in human breast milk are oleic acid and linoleic acid, respectively27. Therefore, those fatty acids could inhibit UGT1A1 activity in breast-fed infants, contributing to elevation of serum bilirubin levels in infants. Interestingly, DHA significantly inhibited the UGT1A1 activity not only in human liver microsomes, but also in small intestine microsomes, with Ki values of 4.3 μM and 5.2 μM, which were much lower than the values of oleic acid and linoleic acid. In the human neonatal hyperbilirubinemia model, intestinal UGT1A1 plays an important role in bilirubin metabolism23. The inhibitory effects of DHA in UGT1A1-catalyzed bilirubin glucuronidation was further confirmed in our in vivo study with hUGT1 mice (Fig. 5). The amount of DHA in human breast milk is less than 1% of the total free fatty acids in breast milk27. Given the strong inhibitory effects of DHA toward intestinal UGT1A1 activity, however, not only oleic acid and linoleic acid, but DHA could also be a factor contributing to the onset of breast milk-induced neonatal jaundice.
To examine whether our observation of the in vivo study was valid or not, the inhibitory effects of DHA on bilirubin clearance in the hUGT1 mice were estimated using equations described in the methods section (Eq 1–4). The Km value of bilirubin glucuronidation by human UGT1A1 was reported to be around 2 μM32. Because the serum fee bilirubin concentration is much lower than the Km value, Equation 4 can be used to estimate the inhibitory effects of DHA on bilirubin clearance. With the parameters of Ki value (5.2 μM) and concentration of DHA used for the oral treatment (0.5 mg/mL), the R value was calculated to be 0.0034 (Equation 4). This maximum inhibitory effect of DHA on bilirubin clearance would be temporal due to the rapid absorption of DHA into the blood stream. However, the increased serum bilirubin levels by DHA in hUGT1 mice was considered to be attributed to the inhibition of bilirubin glucuronidation in the small intestine, because in the hUGT1 mice, hepatic UGT1A1 is not functional due to its very low expression in the liver24. In humans, breast milk of mothers contains on average 3.2% total fat and the DHA concentration of human breast milk was reported to be 0.06% of total fat33. This indicates that the concentration of DHA in human breast milk is around 60 μM, giving an R value of 0.08 (Equation 4). Breast-fed human infants constantly drink breast milk from their mothers; therefore, inhibition of bilirubin glucuronidation in the small intestine by DHA would be the cause of breast milk-induced jaundice. Interindividual variability in the DHA content in breast milk34 might explain the difference of serum bilirubin levels in human infants.
A key question was why higher concentrations of DHA or other fatty acids did not increase, but rather decreased serum bilirubin levels in hUGT1 mice. Our in vivo study demonstrated that UGT1A1 expression was significantly induced with treatment of a high concentration of fatty acids, possibly by activating PPARs. Therefore, it is considered that in conditions in which excessive amounts of fatty acids were treated, the inducible effect of fatty acids on UGT1A1 overcame their inhibitory effects on UGT1A1 activity. In physiological conditions, however, DHA in breast milk would not be involved in induction of UGT1A1, because it was demonstrated that breast-fed hUGT1 mice did not increase the expression level of UGT1A1 in the small intestine compared with that in hUGT1 mice fed with formula in which DHA was not included23. Clinical intervention to treat episodes of severe hyperbilirubinemia calls for extended phototherapy treatment or even blood transfusion. While exchange transfusions using the umbilical vein and phenobarbital treatments are now becoming extinct, phototherapy also has disadvantages; for example, increased insensible water loss, skin rashes, pyrexia, decreased maternal-infant interaction, and lack of visual sensory input are potential disadvantages of this treatment. Unlike the constitutive androstane receptor, which is activated by phenobarbital, PPARs can be activated by various natural compounds such as saturated fatty acids35, which do not function as UGT inhibitors (Table 1). Therefore, supplemental treatment of mothers or direct treatment of newborn infants with natural ligands for PPAR would be an alternative method to increase UGT1A1 expression to accelerate bilirubin conjugation in infants.
It should be noted that oleic acid and linoleic acid did not inhibit estradiol 3-O-glucuronidation in human small intestine microsomes, although they inhibited the reaction in the UGT1A1 recombinant system and in human liver microsomes. Among functional human UGT enzymes, UGT1A1, UGT1A3, UGT1A8, UGT1A10, UGT2A1, UGT2A2, and UGT2B15 exhibit estradiol 3-O-glucuronide formation activities36. Due to the high expression levels of UGT1A1 among those UGT isoforms in the human liver, 3-O-glucuronidation of estradiol has been used as a probe reaction of UGT1A1 in the human liver25. Because oleic acid and linoleic acid inhibited the 3-O-glucuronidation of estradiol in the UGT1A1 recombinant system, the inhibitory effects of oleic acid and linoleic acid against estradiol 3-O-glucuronidation in liver microsomes were considered to be attributed to the inhibition of UGT1A1. However, the expression pattern of UGT enzymes in other tissues is different from that in the liver. In the small intestine, not only UGT1A1, but also other UGT enzymes such as UGT1A8 and UGT1A10 are highly expressed26. UGT1A8 shows slightly less 3-O-glucuronidation activity toward estradiol compared with UGT1A136. On the other hand, UGT1A10 shows a nearly 10-fold greater Vmax of estradiol 3-O-glucuronidation than UGT1A136. These suggest that estradiol 3-O-glucuronidation might be mainly catalyzed by UGT1A10 in the small intestine, in which oleic acid and linoleic acid weakly inhibit UGT1A10. Bilirubin, in contrast, is a specific substrate of UGT1A16. Therefore, it is still possible that oleic acid and linoleic acid inhibit UGT1A1-catalyzed bilirubin glucuronidation in the small intestine, contributing to breast milk-induced neonatal hyperbilirubinemia. It will be important to determine whether oleic acid and linoleic acid inhibit UGT1A1 in the human small intestine with a UGT1A1 specific substrate, such as bilirubin. In this study, however, we were unable to conduct UGT1A1 enzyme assay with bilirubin due to its lack of stability against light. Although inhibition study with bilirubin as an inhibitor is also promising to understand the contribution ratio of UGT1A1 to estradiol 3-O-glucuronidation in the human small intestine microsomes, it has been demonstrated that bilirubin could inhibit not only UGT1A1, but also other UGT isoforms due to their high amino acid sequence similarities37.
Interestingly, all of the fatty acids that strongly inhibited UGT1A1 activity were unsaturated fatty acids, and fatty acids that did not inhibit UGT1A1 were all saturated fatty acids. This finding is in accordance with previously reported data that most unsaturated fatty acids dramatically inhibited human UGT1A9- and UGT2B7-catalyzed 4-MU glucuronidation in their recombinant systems21. Therefore, it can be speculated that unsaturated fatty acids have the potency to inhibit a wide variety of human UGT enzymes. Unsaturated fatty acids are abundant in food such as vegetable oil and fish oil. Additionally, nowadays certain unsaturated fatty acids, such as DHA and EPA, are widely used as daily supplements, as DHA and EPA have been reported to confer various benefits, which include a protective role against cardiovascular diseases. Human UGT enzymes are involved in metabolism of not only endogenous compounds such as bilirubin and thyroid hormones, but also a wide variety of clinically used drugs38. Therefore, interindividual variability of clearance of drugs in humans could be partially explained by the presence of unsaturated fatty acids in diets or supplements. To prevent food (supplements)-drug interactions, excessive use of unsaturated fatty acid-containing oil or supplements, such as DHA and EPA, might need to be avoided in patients taking drugs.
In conclusion, our results demonstrated for the first time that unsaturated fatty acids, especially oleic acid, linoleic acid, and DHA, inhibited human UGT1A1 activity in vitro and in vivo. While its content in human breast milk is low, DHA strongly inhibited UGT1A1 activity in human liver and small intestine microsomes with Ki values of 4.3 μM and 5.2 μM. Inhibition of UGT1A1 activity by DHA could be an underlying mechanism of breast milk-induced jaundice. Our data also indicate that an excessive amount of fatty acids induced UGT1A1 expression, possibly by activating PPARs, resulting in a reduction of serum bilirubin levels in human newborn infants. Activation of PPARs could be an alternative method of treating neonatal jaundice.
Methods
Chemicals and reagents
UDP-glucuronic acid, estradiol, estradiol 3-O-glucuronide, and decanoic acid were purchased from Sigma–Aldrich (St Louis, MO). The UGT1A1 recombinant system, UGT1A1 Supersomes, human liver microsomes, and human small intestine microsomes were purchased from BD Gentest (Woburn, MA). Oleic acid, linoleic acid, stearic acid, myristic acid, and palmitic acid were obtained from Wako Pure Chemical (Osaka, Japan). Behenic acid and lignoceric acid were purchased from MP Biomedicals (Solon, OH). Lauric acid was purchased from Tokyo Chemical Industry (Tokyo, Japan) and arachidic acid, palmitoleic acid, alpha-linolenic acid, arachidonic acid, eicosapentaenoic acid (EPA), and DHA were purchased from Cayman Chemical Company (Ann Arbor, MI). All other chemicals and solvents were of analytical grade or the highest grade commercially available.
Enzyme assays
Estradiol 3-O-glucuronide formation was determined according to the method of Fujiwara et al. (2007) with slight modifications26. Briefly, a typical incubation mixture (200 μl of total volume) contained 50 mM Tris-HCl (pH 7.4), 10 mM MgCl2, 2 mM UDPGA, 25 μg/ml alamethicin, 0.25 mg/ml UGT1A1 Supersomes or micoromes and 10 μM estradiol. For an inhibition study, 1 to 50 μM of inhibitors were included in the reaction mixtures. To estimate the inhibition type of oleic acid, linoleic acid, and DHA for substrate binding, analyses were conducted using a fixed concentration of 2 mM UDPGA and varied concentrations of estradiol (5, 10, and 20 μM). The reaction was initiated by the addition of UDPGA after a 3-min preincubation at 37°C. After incubation at 37°C for 30 min, the reaction was terminated by addition of 200 μl of cold methanol. After removal of the protein by centrifugation at 15,000 rpm for 5 min, a 50 μl portion of the sample was subjected to HPLC. The type of inhibition and the Ki value were determined by a dixon plot analysis.
HPLC conditions
HPLC was performed using an LC-10AD pump (Shimadzu, Kyoto, Japan), a FP-2020 fluorescence detector (JASCO, Tokyo Japan), a SIL-10A autosampler (Shimadzu), a SCL-10A system controller (Shimadzu), and a Mightysil RP-18 GP column (4.6 × 150 mm, 5 μm; Kanto Chemical, Tokyo, Japan). The flow rate was 1.0 mL/min, and detection was accomplished with a fluorescence detector at 280-nm excitation and 310-nm emission. The mobile phase was 10 mM H3PO4-methanol (45:55, v/v). Quantification of estradiol 3-O-glucuronide was performed by comparing the HPLC peak height to that of the authentic standard.
Animals and treatments
Tg(UGT1A1*28)Ugt1−/− (hUGT1) mice were developed previously in a C57BL/6 background23,24. All animals received food and water ad libitum, and mouse handling and experimental procedures were conducted in accordance with our animal care protocol, which was previously approved by Kitasato University. Fatty acids dissolved in rapeseed oil were orally administered to 12-day-old mice. Blood was obtained before and 48 hours after treatment and the serum total bilirubin levels were measured. Rapeseed oil was orally administered to mice in the control group. For tissue collections, mice were anesthetized by diethyl ether inhalation, and the liver was perfused with ice-cold 1.15% KCl. The small intestine and liver were rinsed in cold 1.15% KCl and stored at −80°C.
Bilirubin measurements
Blood was obtained from the submandibular vein and centrifuged at 2000 × g for 5 minutes. Serum samples (20 μL) were measured for total serum bilirubin using a Bilirubinometer (B-105N, Erma, Tokyo, Japan). For each serum sample, the bilirubin value was measured three times and the mean value was used for data analysis. To avoid hemolysis of the serum and photolysis of the bilirubin, the serum samples were analyzed promptly after collection of blood.
Semi-quantitative and quantitative (Q) reverse transcription (RT)-PCR
Total RNA was extracted from tissues with Trizol reagent (Invitrogen). The cDNA was synthesized from total RNA using ReverTra Ace (TOYOBO, Osaka, Japan) according to the manufacturer's protocol. Primers for human UGT1A1 and GAPDH were developed previously28. Q-RT-PCR was performed with Ssofast Evagreen Supermix, and the reactions were run in a CFX96™ Real-Time PCR Detection System (BioRad). Expression of GAPDH mRNA was used as an internal control for the cDNA quantity and quality.
Equations
When an inhibitor exhibits noncompetitive inhibition against enzymes, velocity of the metabolic reaction can be described by the following equation (Eq 1):
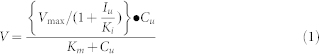 |
where V is the velocity of the metabolic reaction, Cu is the free substrate concentration, and Iu is the free inhibitor concentration. The Vmax is the maximum rate of metabolism and Km is the Michaelis constant, which is defined as the substrate concentration at 1/2 the maximum velocity. Ki is the inhibition constant. When Cu is much lower than the Km value, the equation can be described by the following equation (Eq 2).
 |
In this case, intrinsic clearance (CLint) can be described by the following equation (Eq 3).
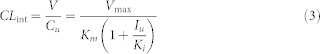 |
Therefore, the ratio of intrinsic clearance (R) in the presence (V+I) and absence (V−I) can be described by the following equation (Eq 4).
 |
This equation was used to estimate the ratio of intrinsic clearance in the presence and absence of an inhibitor.
Statistical analyses
Statistical significances were determined by analysis of variance followed by Dunnett's test. A value of P < 0.05 was considered statistically significant.
Author Contributions
T.I., R.H.T. and R.F. wrote the main manuscript text and A.S. and R.F. conducted experiments and prepared figures 1–7. All authors reviewed the manuscript.
Acknowledgments
This work was supported by a Grant-in-Aid for Research Activity Start-up [24890224].
References
- Dutton G. J. Acceptor substrates of UDP glucuronosyltransferase and their assay., in Glucuronidation of Drugs and Other Compounds (Dutton, G. J. ed); pp 69–78, CRC Press, Boca Raton, FL (1980). [Google Scholar]
- Mackenzie P. I., Bock K. W., Burchell B., Guillemette C., Ikushiro S. & Iyanagi T. et al. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics. 15, 677–685 (2005). [DOI] [PubMed] [Google Scholar]
- Ritter J. K., Chen F., Sheen Y. Y., Tran H. M., Kimura S. Yeatman M. T. et al. A novel complex locus UGT1 encodes human bilirubin, phenol, and other UDP-glucuronosyltransferase isozymes with identical carboxyl termini. J Biol Chem. 267, 3257–61 (1992). [PubMed] [Google Scholar]
- Tukey R. H. & Strassburg C. P. Human UDP-glucuronosyltransferases: metabolism, expression and disease. Annu Rev Pharmacol Toxicol. 40, 581–616 (2000). [DOI] [PubMed] [Google Scholar]
- Fisher M. B., Paine M. F., Strelevitz T. J. & Wrighton S. A. The role of hepatic and extrahepatic UDP-glucuronosyltransferases in human drug metabolism. Drug Metab Rev. 33, 273–97 (2001). [DOI] [PubMed] [Google Scholar]
- Bosma P. J., Seppen J., Goldhoorn B., Bakker C., Oude Elferink R. P. & Chowdhury J. R. Bilirubin UDP-glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man. J Biol Chem. 269, 17960–4 (1994). [PubMed] [Google Scholar]
- Newman A. J. & Gross S. Hyperbilirubinemia in breast-fed infants. Pediatrics. 32, 995–1001 (1963). [PubMed] [Google Scholar]
- Arias I. M., Gartner L. M. Seifter S. et al. Prolonged neonatal unconjugated hyperbilirubinemia associated with breast feeding and a steriod, pregnane-3(alpha), 20(beta)-diol, in maternal milk that inhibits glucuronide formation in vitro. J Clin Invest. 43, 2037–2047 (1964). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A. P. Breast milk jaundice in the newborn. A real entity. JAMA. 255, 3270–3274 (1986). [PubMed] [Google Scholar]
- Arias I. M., Gartner L. M., Seifter S. & Furman M. Neonatal unconjugated hyperbilirubinemia associated with breast feeding and a factor in milk that inhibits glucuronide formation in vitro. J Clin Invest. 42, 913 (1963). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota Y., Maruo Y., Matsui K., Mimura Y., Sato H. & Takeuchi Y. Inhibitory effect of 5â-pregnane-3á,20â-diol on transcriptional activity and enzyme activity of human bilirubin UDP-glucuronosyltransferase. Pediatr Res. 70, 453–7 (2011). [DOI] [PubMed] [Google Scholar]
- Bevan B. R. & Holton J. B. Inhibition of bilirubin conjugation in rat liver slices by free fatty acids, with relevance to the problem of breast milk jaundice. Clin Chim Acta. 41, 101–7 (1972). [DOI] [PubMed] [Google Scholar]
- Hargreaves T. Effect of fatty acids on bilirubin conjugation. Arch Dis Child. 48, 446–50 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard M., Fournel-Gigleux S., Siest G., Mackenzie P. & Magdalou J. A recombinant phenobarbital-inducible rat liver UDP-glucuronosyltransferase (UDP-glucuronosyltransferase 2B1) stably expressed in V79 cells catalyses the glucuronidation of morphine, phenols and carboxylic acids. Mol Pharmacol. 45, 42–50 (1994). [PubMed] [Google Scholar]
- Green M. D., Belanger G., Hum D. W., Belanger A. & Tephly T. R. Glucur-onidation of opioids carboxylic acid-containing drugs, and hydroxylated xenobiotics catalysed by expressed monkey UDP-glucuronosyltransfer-ase 2B9 protein. Drug Metab Dispos. 25, 1389–94 (1997). [PubMed] [Google Scholar]
- Green M. D., King C. D., Mojarrabi B., Mackenzie P. I. & Tephly T. R. Glucuronidation of amines and other xenobiotics catalysed by ex-pressed human UDP-glucuronosyltransferase 1A3. Drug Metab Dispos. 26, 507–12 (1998). [PubMed] [Google Scholar]
- Jude A. R., Little J. M., Freeman J. P., Evans J. E., Radominska-Pandya A. & Grant D. F. Linoleic acid diols are novel substrates for human UDP-glucuronosyltransferases. Arch Biochem Biophys. 380, 294–302 (2000). [DOI] [PubMed] [Google Scholar]
- Jude A. R., Little J. M., Czernik P. J., Tephly T. R., Grant D. F. & Radominska-Pandya A. Glucuronidation of linoleic acid diols by human micro-somal and recombinant UDP-glucuronosyltransferases: identification of UGT2B7 as the major isoform involved. Arch Biochem Biophys. 389, 176–86 (2001). [DOI] [PubMed] [Google Scholar]
- Jude A. R., Little J. M., Bull A. W., Podgorski I. & Radominska-Pandya A. 13-Hydroxy- and 13-oxooctadecadienoic acids: novel substrates for human UDP-glucuronosyltransferases. Drug Metab Dispos. 29, 625–55 (2001). [PubMed] [Google Scholar]
- Sonka J., Little J., Samokyszyn V. & Radominska-Pandya A. Glucuroni-dation of arachidonic acid, 20-hydroxy-eicosatetraenoic acid and prostaglandin E2 by human hepatic and intestinal UDP-glucuronosyl-transferases and recombinant UGT2B7. Drug Metab Rev. 34(Suppl 1): 194 (2001). [Google Scholar]
- Tsoutsikos P., Miners J. O., Stapleton A., Thomas A., Sallustio B. C. & Knights K. M. Evidence that unsaturated fatty acids are potent inhibitors of renal UDP-glucuronosyltransferases (UGT): kinetic studies using human kidney cortical microsomes and recombinant UGT1A9 and UGT2B7. Biochem Pharmacol. 67, 191–9 (2004). [DOI] [PubMed] [Google Scholar]
- Rowland A., Knights K. M., Mackenzie P. I. & Miners J. O. The “albumin effect” and drug glucuronidation: bovine serum albumin and fatty acid-free human serum albumin enhance the glucuronidation of UDP-glucuronosyltransferase (UGT) 1A9 substrates but not UGT1A1 and UGT1A6 activities. Drug Metab Dispos. 36, 1056–62 (2008). [DOI] [PubMed] [Google Scholar]
- Fujiwara R., Chen S., Karin M. & Tukey R. H. Reduced expression of UGT1A1 in intestines of humanized UGT1 mice via inactivation of NF-kB leads to hyperbilirubinemia. Gastroenterology. 142, 109–18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara R., Nguyen N., Chen S. & Tukey R. H. Developmental hyperbilirubinemia and CNS toxicity in mice humanized with the UDP glucuronosyltransferase 1 (UGT1) locus. Proc Natl Acad Sci U S A. 107, 5024–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson R. A. & Kneebone G. M. Fatty acid composition of human colostrum and mature breast milk. Am J Clin Nutr. 34, 252–7 (1981). [DOI] [PubMed] [Google Scholar]
- Fujiwara R., Nakajima M., Yamanaka H., Nakamura A., Katoh M. Ikushiro S. et al. Effects of coexpression of UGT1A9 on enzymatic activities of human UGT1A isoforms. Drug Metab Dispos. 35, 747–57 (2007). [DOI] [PubMed] [Google Scholar]
- Lin J. H. & Wong B. K. Complexities of glucuronidation affecting in vitro in vivo extrapolation. Curr Drug Metab. 3, 623–46 (2002). [DOI] [PubMed] [Google Scholar]
- Nakamura A., Nakajima M., Yamanaka H., Fujiwara R. & Yokoi T. Expression of UGT1A and UGT2B mRNA in human normal tissues and various cell lines. Drug Metab Dispos. 36, 1461–4 (2008). [DOI] [PubMed] [Google Scholar]
- Duval C., Fruchart J. C. & Staels B. PPAR alpha, fibrates, lipid metabolism and inflammation. Arch Mal Coeur Vaiss. 97, 665–72 (2004). [PubMed] [Google Scholar]
- Senekeo-Effenberger K., Chen S., Brace-Sinnokrak E., Bonzo J. A., Yueh M. F. Argikar U. et al. Expression of the human UGT1 locus in transgenic mice by 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid (WY-14643) and implications on drug metabolism through peroxisome proliferator-activated receptor alpha activation. Drug Metab Dispos. 35, 419–27 (2007). [DOI] [PubMed] [Google Scholar]
- Watchko J. F. Identification of neonates at risk for hazardous hyperbilirubinemia: emerging clinical insights. Pediatr Clin North Am. 56, 671–87 (2009). [DOI] [PubMed] [Google Scholar]
- Fujiwara R., Nakajima M., Yamanaka H., Katoh M. & Yokoi T. Interactions between human UGT1A1, UGT1A4, and UGT1A6 affect their enzymatic activities. Drug Metab Dispos. 35, 1781–7 (2007). [DOI] [PubMed] [Google Scholar]
- Finley D. A., Lönnerdal B., Dewey K. G. & Grivetti L. E. Breast milk composition: fat content and fatty acid composition in vegetarians and non-vegetarians. Am J Clin Nutr. 41, 787–800 (1985). [DOI] [PubMed] [Google Scholar]
- Jensen C. L. & Lapillonne A. Docosahexaenoic acid and lactation. Prostaglandins Leukot Essent Fatty Acids. 81, 175–8 (2009). [DOI] [PubMed] [Google Scholar]
- Berger J. & Moller D. E. The mechanisms of action of PPARs. Annu Rev Med. 53, 409–35 (2002). [DOI] [PubMed] [Google Scholar]
- Itäaho K., Mackenzie P. I., Ikushiro S., Miners J. O. & Finel M. The configuration of the 17-hydroxy group variably influences the glucuronidation of beta-estradiol and epiestradiol by human UDP-glucuronosyltransferases. Drug Metab Dispos. 36, 2307–15 (2008). [DOI] [PubMed] [Google Scholar]
- Watanabe Y., Nakajima M. & Yokoi T. Troglitazone glucuronidation in human liver and intestine microsomes: high catalytic activity of UGT1A8 and UGT1A10. Drug Metab Dispos. 30, 1462–9 (2002). [DOI] [PubMed] [Google Scholar]
- Williams J. A., Hyland R., Jones B. C., Smith D. A., Hurst S. Goosen T. C. et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 32, 1201–8 (2004). [DOI] [PubMed] [Google Scholar]