

Abstract
Background:
Neurodegenerative diseases are characterized by progressive neuron degeneration in specific functional systems of the central or peripheral nervous system. This study investigated the protective effects of quercetin isolated from onion on neuronal cells and its protective mechanisms against glutamate-induced apoptosis in HT22 cells.
Materials and Methods:
HT22 cells were cultured to study the neuroprotective mechanism of quercetin against glutamate-mediated oxidative stress. The intracellular reactive oxygen species (ROS) level and mitochondrial membrane potential (ΔΨm) were measured. The protein expression of calpain, spectrin, Bcl-2, Bax, Bid, cytochrome c, and mitogen-activated protein kinases (MAPKs) was evaluated by Western blotting.
Results:
Quercetin had a protective effect by reducing both intracellular ROS overproduction and glutamate-mediated Ca2+ influx. These effects were due to the downregulation of several apoptosis-related biochemical markers. Calpain expression was reduced and spectrin cleavage was inhibited by quercetin in glutamate-exposed HT22 cells. Disruption of the mitochondrial membrane potential (ΔΨm), activation of the pro-apoptotic proteins Bid and Bax, and cytochrome c release in response to glutamate-induced oxidative stress were reduced. Quercetin also suppressed phosphorylation of MAPKs.
Conclusion:
This is the first report on the detailed mechanisms of the protective effect of quercetin on HT22 cells. Onion extract and quercetin may be useful for preventing or treating neurodegenerative disorders.
Keywords: Glutamate, neuroprotective effects, onion, oxidative stress, quercetin
INTRODUCTION
Neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's diseases are characterized by the progressive loss of neuronal cells, leading to nervous system dysfunction.[1] Oxidative stress caused by the accumulation of reactive oxygen species (ROS) in neuronal cells plays an important role in the pathogenesis of neurodegenerative disorders.[2] Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system and is normally stored intracellularly. However, a high glutamate concentration causes oxidative stress and contributes to neuronal degeneration.[3] Glutamate cytotoxicity is mediated by receptor-initiated excitotoxicity and non-receptor-mediated oxidative stress. Excitotoxicity refers to the excessive activation of neuronal amino acid receptors,[4] while in the oxidative glutamate toxicity pathway, glutamate reduces cystine uptake via the cystine/glutamate antiporter, resulting in the depletion of intracellular levels of anti-oxidative glutathione (GSH).[5] HT22 cells derived from the mouse hippocampus phenotypically resemble neuronal precursor cells, but they lack functional ionotropic glutamate receptors, which excludes excitotoxicity as a cause of glutamate-triggered cell death. Therefore, HT22 cells are a useful model for studying oxidative glutamate toxicity.[6]
In the mechanism of neuronal cell death, a linear increase in ROS is observed after adding glutamate. This induces Ca2+ influx, and altered Ca2+ homeostasis can cause a stress response. High Ca2+ levels activate proteases such as calpain.[7] After calpain is activated, it cleaves many cytoskeletal proteins and signaling molecules, including spectrin, microtubules-associated protein, tau, and neurofilaments. Spectrin breakdown is a hallmark of calpain activation in neuronal cells. Calpain activation is an early event in the onset of apoptosis; therefore, calpain inhibitors can attenuate this process of cell death.[8]
The massive increase in intracellular Ca2+ mediated by glutamate can result in mitochondrial dysfunction. In neuronal cells, apoptosis is facilitated by the altered expression and mitochondrial localization of apoptotic regulators, involving the downregulation of Bcl-2 and full-length Bid and the upregulation of Bax. This changes the mitochondrial membrane potential (ΔΨm) and leads to mitochondrial permeabilization, which is commonly accompanied by the release of cytochrome c into the cytoplasm. The damaged mitochondria can no longer produce energy, and the released cytochrome c activates apoptotic stimuli.[6]
Oxidative stress induced by glutamate also activates the mitogen-activated protein kinase (MAPK) pathways, which are involved in many cellular processes, including cell growth, differentiation, inflammation, and cell death.[9] The extracellular signal-regulated kinase (ERK) pathway is activated in response to growth factors and tumor promoters, and regulates cell proliferation and differentiation. Although the ERK pathway is implicated mainly in cell survival, recent studies have shown that it also has a detrimental role in oxidative neuronal injury.[10] The c-Jun N-terminal kinase (JNK) and the p38 pathway respond to stress stimuli such as cytokines, ultraviolet irradiation, heat shock, and osmotic shock, and are involved in cell differentiation and apoptosis.[11] Therefore, inhibiting the mechanisms of oxidative stress induced by glutamate may be a promising strategy for the development of drugs to prevent and treat neurodegenerative diseases.
The onion (Allium cepa) is commonly consumed, either fresh or in processed products. It has been used as a traditional medicine in the treatment of several disorders, such as dysentery, scars, helminth infestations, migraine, and vertigo.[12] Recently, it was shown that the consumption of onions has positive effects that reduce the risks for various diseases, including many forms of cancer, vascular and heart diseases, diabetes, and ischemia.[13,14,15,16,17] Pretreatment of Swiss albino mice with the methanolic extract of the outer scales and edible portion of the onion remarkably attenuated ischemia/reperfusion-induced cerebral infarct size and prevented ischemia/reperfusion-induced impairment of short-term memory and motor incoordination. This study concluded that the methanolic extract of the outer scales and edible portion of the A. cepa bulb scavenged ROS and that the anti-oxidative effect of the extract attenuated global cerebral ischemia/reperfusion-induced cerebral injury. In addition, it was thought that the ROS scavenging activity of the onion extract was attributable to components such as flavonoids, anthocyanins, and organosulfur compounds.[17] However, little is known of the biochemical mechanisms of onion extract or onion-derived compounds in glutamate-induced neuronal cell apoptosis.
In this study, we isolated the active compound quercetin from an ethanol extract of onions, using column chromatography, and investigated its cell protective effects and mechanisms in glutamate-stressed hippocampal neuronal HT22 cells.
MATERIALS AND METHODS
General experimental procedures
All organic solvents, such as ethanol (EtOH), dichloromethane (CH2Cl2), ethyl acetate (EtOAc), methanol (MeOH) and n-butanol (n-BuOH) used for extraction and column chromatography were of analytical grade and purchased from Duksan Chemical (Anseong, Korea). Thin layer chromatography (TLC) was performed on a precoated silica gel plate (Kieselgel 60F254, Merck, NJ, USA) and silica gel column chromatography was carried out using a Kieselgel 60 from Merck. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance Digital 400 NMR spectrometer (400 MHz for 1H) with tetramethylsilane (TMS) as an internal standard. Chemical shift (δ) values are expressed in ppm relative to TMS. All extracts were evaporated below 40°C under reduced pressure. Optical density (OD) was read with a microplate reader (TECAN, Männedorf, Switzerland). Trolox, 17β-estradiol, 2,2-diphenyl-1-picrylhydrazyl and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA). 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) and [5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA)] were from Invitrogen (Eugene, OR, USA) for the determination of the mitochondrial membrane potential (ΔΨm) and ROS levels, respectively.
Plant material, extraction, and isolation of the active compound
Onions (A. cepa) were purchased from a local market in May, 2010, in Daegu, Korea. A voucher specimen has been deposited at the College of Pharmacy, Kyungpook National University (Specimen No. NPC-AC-1).
Onions (5.96 kg) were skinned, chopped, and refluxed with 36 L of 95% EtOH (5 h × 2). The extract was filtered and concentrated to dryness under reduced pressure by a rotary evaporator (EYELA, Tokyo, Japan). The EtOH extract (261.72 g) was suspended in distilled water and partitioned with EtOAc and n-BuOH, consecutively. Each fraction was separately collected and concentrated. The active EtOAc soluble fraction (1.72 g) was chromatographed using medium pressure liquid chromatography (MPLC) system (Biotage Silica, 30 × 145 mm, CH2Cl2:MeOH = 20:1-5:1, 5 ml/min) to yield 4 fractions. Fr. 3 was rechromatographed with Yamazen ODS-S-50A (11 × 300 mm, H2O:MeOH = 1:1, 4 ml/min), to yield 3 fractions, Fr. 3-1 ˜ Fr. 3-3. Fr. 3-2 yielded a yellow powder, compound 1 (50.00 mg). Each fraction was monitored by TLC with acetonitrile: H2O = 2:3 containing 1% acetic acid as a mobile phase, and visualized by 10% sulfuric acid.
Compound 1: 1H NMR (CD3OD, 500 MHz) δ 7.67 (1H, d, J = 2.20 Hz, H-2′), 7.54 (1H, dd, J = 2.20 and 8.56 Hz, H-6′), 6.88 (1H, d, J = 8.56 Hz, H-5′), 6.40 (1H, d, J = 1.96 Hz, H-8), 6.18 (1H, d, J = 1.96 Hz, H-6).
Cell culture
Mouse hippocampal neuronal cells (HT22) were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Welgene, Daegu, Korea) supplemented with 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Welgene) at 37°C in a 5% CO2 humidified atmospheres.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell viability assay
HT22 cells were seeded at a density of 3 × 104 cells/well in 24-well plates (BD Biosciences, San Diego, CA, USA). After incubation for 12 h, each sample was applied to the cells. To induce oxidative stress, cells were treated with 5 mM glutamate for 12 h. After completing the incubation with glutamate, 0.5 mg/ml MTT in phenol red-free medium was applied for 4 h. And then, the medium was removed and the insoluble formazan was dissolved by 1 ml of dimethyl sulfoxide (DMSO). After 1 h of shaking at room temperature, the OD was read at 575 nm using a microplate reader. Cells without samples and glutamate addition served as the control.[18]
Determination of intracellular reactive oxygen species production
Cells were seeded at a density of 2 × 105 cells/well in 6-well plates (BD Biosciences) and incubated for 24 h. They were treated with the samples for 12 h before treatment with 10 mM glutamate. Then cells were trypsinized with 100 μl of 0.25% trypsin-EDTA and centrifuged. Cell pellets were suspended and washed with phenol red-free DMEM. After centrifugation, 10 μM CM-H2DCFDA was added to the cells and incubated for 20 min for 37°C.[19] Cell were rinsed and resuspended in phenol red-free medium before flow cytometric analysis. This analysis was performed on 10,000 viable cells with a fluorescence-activated cell sorting (FACS, BD Biosciences) system. Kaempferol (25 μM) and N-acetyl-cysteine (NAC, 1 mM), well-known antioxidants, were used as positive controls.
Measurement of mitochondrial membrane potential (ΔΨm)
After treatment with the samples or 10 mM glutamate, cells were trypsinized and washed. Then, 2 μM JC-1 in phenol red-free DMEM was applied to cells and they were incubated for 20 min at 37°C. Stained cells were washed and resuspended for FACS analysis. In this experiment, 50 μM Trolox and 1 mM NAC served as positive controls. JC-1 aggregates in normal mitochondria emit red fluorescence at 590 nm, whereas JC-1 monomers that are leaked from damaged mitochondria emit green fluorescence at 530 nm.[20] The red and green fluorescence were measured in the green (FL-1) and red (FL-2) channels of the flow cytometer, respectively. For fluorescence microscope imaging, HT22 cells were plated onto poly-l-> Lysine pre-coated coverslips and treated with samples or glutamate. JC-1 stained HT22 cells were mounted on a slide glass with mounting solution (Vactor Laboratories, Burlingame, CA) and images were taken with a fluorescence microscopy, immediately.
Western blotting
After incubation as “Determination of intracellular ROS production”, cells were washed with phenol red-free DMEM and lysed with protein extraction solution (iNtRON Biotechnology, Seongnam, Korea) for 1 h at −20°C. Then, cell lysates were centrifuged at 13,000 rpm for 10 min at 4°C, and the protein content of supernatant was measured by the Bradford method.[21] The protein was mixed with 5 × loading buffer (Biosesang, Seongnam, Korea). The mixture was boiled for 5 min, and proteins were resolved by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membrane was blocked with blocking solution (5% skim milk in TBST [0.01% tween 20 in 1 × Tris buffered saline]) for 1 h. Protein blots were probed with antibodies such as anti-β-Actin, anti-Cytochrome c, anti-ERK, anti-pp38 (Ab Frontier, Seoul, Korea), anti-Calpain, anti-Bid, anti-pJNK (Cell Signaling, Danvers, MA, USA), anti-Spectrin, anti-Bcl-2, anti-pERK, anti-JNK (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and anti-p38 (Bethyl Laboratories, TX, USA). Then, the membranes were exposed to the horseradish peroxidase (HRP)-conjugated goat anti-rabbit or goat anti-mouse secondary antibody (Thermo Scientific, Rockford, IL, USA) followed by a chemiluminescence detection of antibody binding (Thermo Scientific). β-Actin was used as an equal protein loading control. For quantification of Western blot signals, Multi Gauge software (Fuji Film, Tokyo, Japan) was used.
Statistical analysis
Data were expressed as mean±SD and statistical significance was assessed by one-way analysis of variance (ANOVA) with subsequent Turkey's tests. P values of <0.05 were considered to be significant.
RESULTS AND DISCUSSION
An ethanol extract of onions was investigated to identify natural neuroprotective agents. Column chromatography was used to search for bioactive compounds, and compound 1 was isolated. Compound 1 was identified as the flavonoid quercetin based on its chemical structure [Figure 1], which was confirmed by comparing its 1H NMR spectral data with previously reported data.[22]
Figure 1.
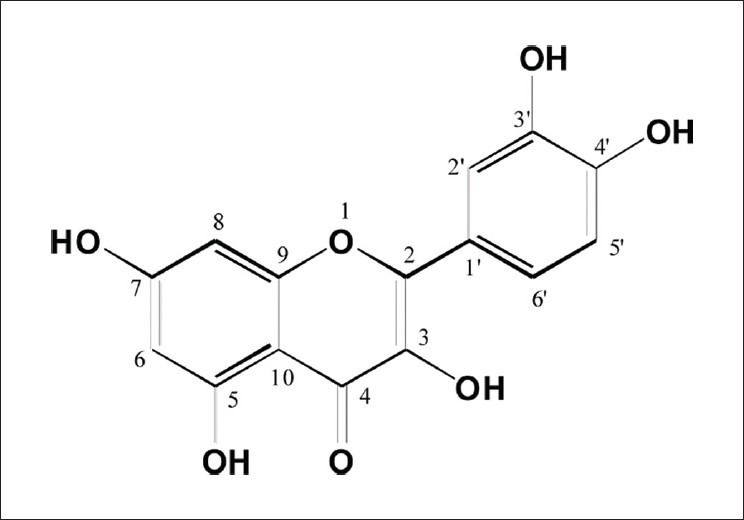
The chemical structure of quercetion
At non-cytotoxic concentrations, quercetin protected HT22 cells against glutamate-induced oxidative stress. The viability of HT22 cells treated with 5 mM glutamate was decreased to 7.23 ± 0.44% compared with untreated cell viability (100.00 ± 3.58%). When the cells were pretreated with quercetin at concentrations of 1-10 μM before treatment with 5 mM glutamate, cell viability increased in a dose-dependent manner, with 10 μM quercetin significantly increasing cell viability to 99.61 ± 1.47% of control cell viability. In addition, quercetin was more effective against glutamate-induced cell death at a concentration lower than that of the positive control, 25 μM 17β-estradiol [67.76 ± 1.74%, Figure 2]. Therefore, we postulated that quercetin protects HT22 cells from glutamate-mediated cell death by inhibiting the overproduction of intracellular ROS.
Figure 2.
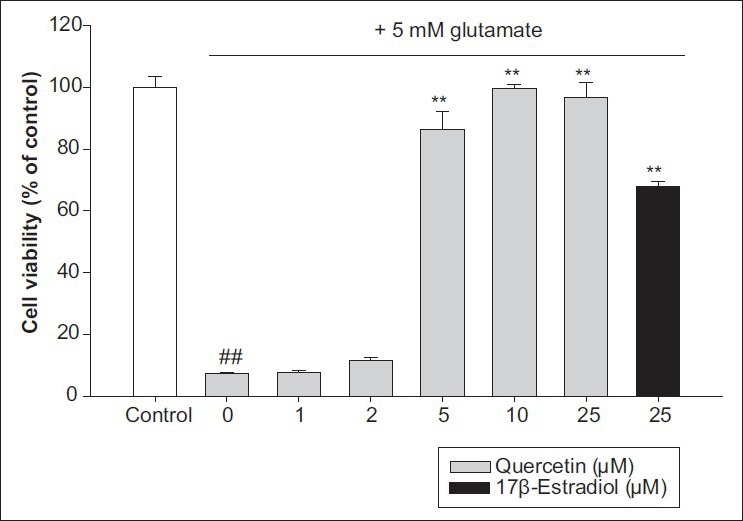
HT22 cell protective effect by quercetin on glutamate-induced cell death. Values represent mean±SD of the relative optical density obtained from three independent experiments. ##P<0.01 compared to control, and **P<0.01 compared to 5 mM glutamate-treated group
At high concentrations, glutamate is a major contributor to neuronal cell apoptosis, and this appears to be mediated by ROS.[2] To address the inhibitory effect of glutamate-mediated ROS generation in HT22 cells, flow cytometry was performed after CM-H2DCFDA staining of the cells. As shown in Figure 3, the green fluorescence intensity due to ROS production was increased significantly, to 2.8-fold the control level of 35.32 ± 5.54%, in HT22 cells exposed to 10 mM glutamate for 12 h (100.00%). By contrast, quercetin pretreatment remarkably suppressed ROS overproduction in a dose-dependent manner. In particular, pretreatment with 10 μM quercetin significantly inhibited ROS formation by about 77.89% compared with ROS formation in glutamate-treated cells without pretreatment. These results indicate that glutamate-induced apoptosis of hippocampal neuronal cells requires intracellular ROS overproduction and that quercetin can rescue glutamate-treated HT22 cells by reducing intracellular ROS.
Figure 3.
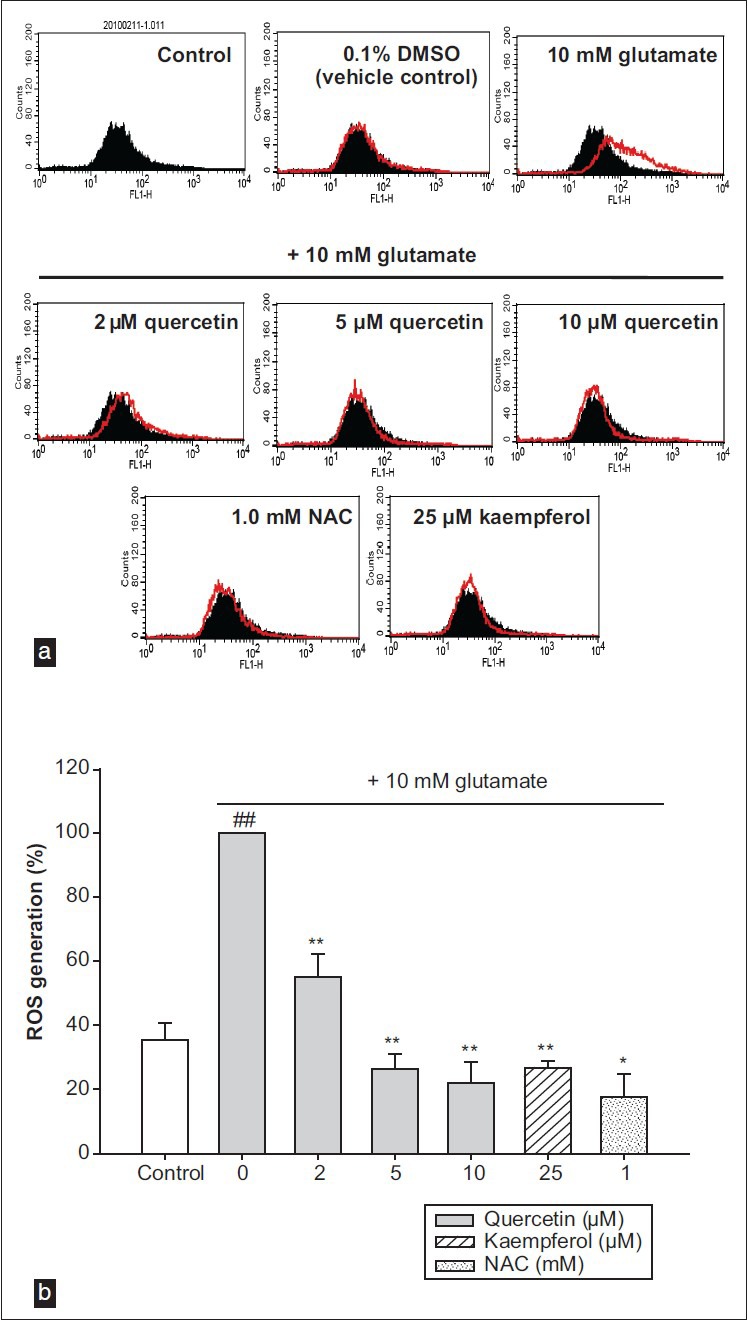
(a) The histograms of flow cytometry analysis. (b) The results of flow cytometry analysis. Values represents the mean ± SD of the three independent experiments, ##P<0.01 compared to control, *P<0.05 and **P<0.01 compared to 10 mM glutamate-treated group
Recent studies have shown that glutamate induces apoptotic cell death in HT22 cells via the caspase-independent apoptotic pathway.[23] Glutamate toxicity appears to involve a rapid Ca2+ influx into the neurons, and high intracellular Ca2+ levels are cytotoxic. In addition, Ca2+ can activate several key enzymes such as nitric oxide synthase and calpain, and can result in mitochondrial dysfunction.[7] As shown in Figure 4, 10 mM glutamate increased the calpain expression compared with the expression in control cells, and quercetin pretreatment effectively reduced the glutamate-induced increase. Because the activation of calpain is triggered by the breakdown of cytoskeletal spectrin, the spectrin levels in the cells were determined. Full-length spectrin was reduced remarkably in glutamate-treated cells, and spectrin cleavage was inhibited by pretreatment with 10 μM quercetin. These data suggest that quercetin acts as a calpain inhibitor and protects cytoskeletal proteins in HT22 cells by suppressing Ca2+ influx.
Figure 4.
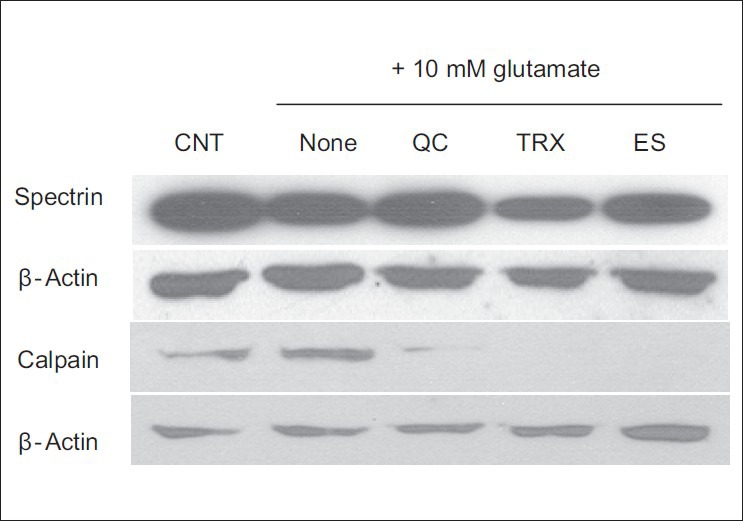
Inhibition of calpain expression and spectrin cleavage by quercetin in glutamate-induced Ca2+ influx
Mitochondrial membrane depolarization, which leads to increased free radical production, is a crucial step in the induction of cellular apoptosis.[24] In most studies with isolated or cultured cells, a disruption of mitochondria membrane potential (ΔΨm) was observed after glutamate-induced Ca2+ influx.[25] Therefore, mitochondria membrane potential (ΔΨm) was measured using JC-1 dye, to estimate the protective effect of quercetin against glutamate-induced mitochondrial damage. Fluorescence microscopy observations showed that treatment with 10 mM of glutamate caused a significant decrease in JC-1 red fluorescence, indicating disruption of mitochondria membrane potential (ΔΨm) by glutamate toxicity. Pretreatment with 10 μM quercetin remarkably restored the red fluorescence [Figure 5a]. In the FACS analysis, glutamate reduced the JC-1 fluorescence intensity (28.96 ± 10.40%) compared with the control intensity (100.00 ± 10.18%), and pretreatment with 10 μM quercetin prevented the reduction (97.49 ± 19.43%). Quercetin had positive effects on mitochondria membrane potential (ΔΨm) at concentrations even lower than those of the positive controls, 1 mM N-acetylcysteine and 50 μM Trolox [Figure 5b]. These results indicate that quercetin effectively protected the mitochondria, which play a critical role in energy production, by inhibiting the glutamate-induced disruption of mitochondria membrane potential (ΔΨm). Consequently, quercetin inhibited the mitochondrial apoptotic pathway.
Figure 5.
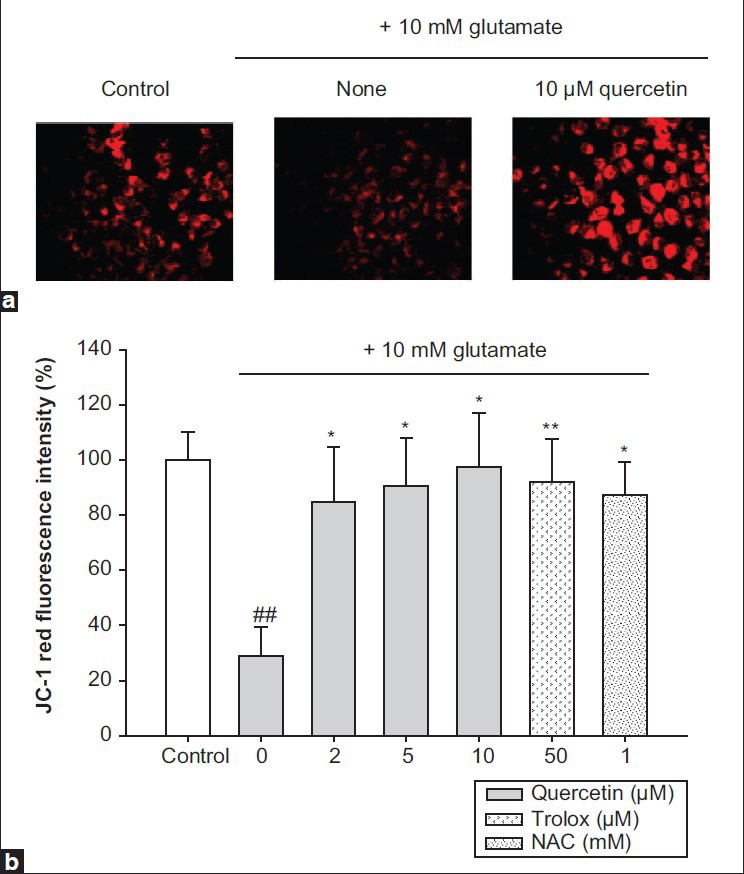
Protection of mitochondria membrane potential (ăÉm) by quercetin against glutamate stress. (a) Microscope images, (b) flow cytometric analysis. Values represents the mean±SD of the three independent experiments, ##P<0.01 compared to control, *P<0.05 and **P<0.01 compared to 10 mM glutamate-treated group
A crucial amplification event in the apoptotic cascades is the nearly complete release of cytochrome c from mitochondria. This is controlled by Bcl-2 family proteins, which include both anti- and pro-apoptotic members. In oxidatively stressed cells, anti-apoptotic Bcl-2 protein is decreased, while pro-apoptotic Bax is increased and full-length Bid is cleaved. These processes promote the release of cytochrome c.[26] To elucidate the molecular mechanism underlying the protective effect of quercetin against the disruption of mitochondrial permeability, the levels of Bcl-2, Bax, Bid, and cytoplasmic cytochrome c were analyzed by Western blotting. In HT22 cells treated with 10 mM glutamate, the Bcl-2 and Bid levels were markedly reduced, and the Bax level was increased slightly. Pretreatment with 10 μM quercetin prevented these changes in Bcl-2, Bid, and Bax levels [Figure 6a]. In response to apoptotic stimuli, cytochrome c was released from mitochondria into the cytosol of glutamate-treated HT22 cells. Pretreatment with 10 μM quercetin noticeably inhibited the cytoplasmic cytochrome c level and inhibited neuronal cell death [Figure 6b]. These data show that the protective effect of quercetin against glutamate-induced mitochondrial damage was attributable to the regulation of Bcl-2 family protein levels and cytochrome c release.
Figure 6.
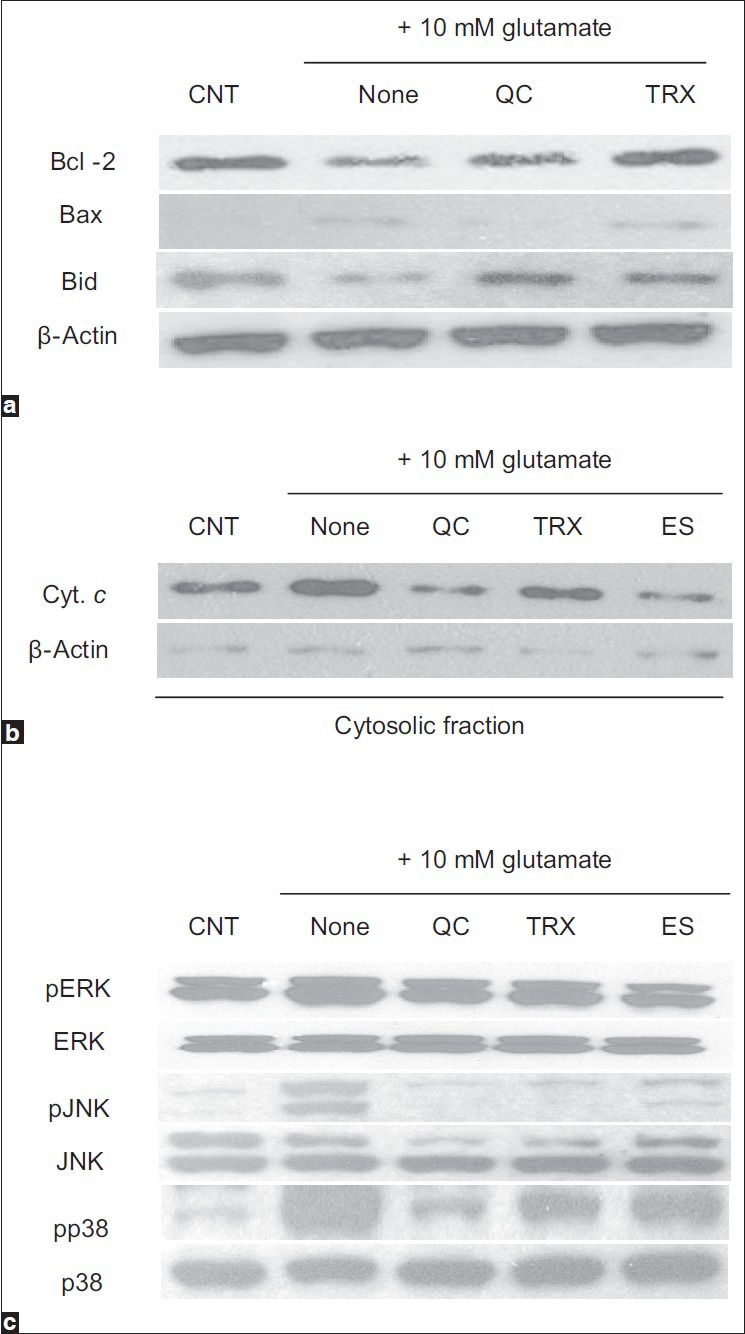
The regulation of the levels of mitochondria-related proteins and the phosphorylation of mitogen-activated protein kinases (MAPKs) by quercetin in HT22. (a) The levels of Bcl-2, Bax, and Bid, (b) the level of cytochrome c in cytosolic fraction, (c) the levels of MAPKs
Western blotting was performed to elucidate the effects of quercetin on MAPKs phosphorylation mediated by glutamate treatment. ERK, JNK, and p38 were phosphorylated in HT22 cells after treatment with 10 mM glutamate, whereas pretreatment with 10 μM quercetin markedly suppressed the activation of these MAPKs [Figure 6c]. Although quercetin may act directly on ERK, JNK, and p38, it is possible that quercetin exerts its protective effect via upstream kinases. As MAPKs have been implicated in promoting apoptosis, the suppression of kinase activation appears to be important in the protection provided by quercetin against glutamate-induced oxidative stress in neuronal cells.
CONCLUSION
Quercetin, a compound found in onions, is a promising biologically active natural compound with anti-inflammatory,[27] anti-cancer,[28] anti-obesity,[29] anti-diabetes,[30] and neuroprotective properties.[31] In particular, quercetin protects against glutamate-mediated HT22 cell death. In one study, quercetin inhibited intracellular ROS accumulation and GSH depletion.[31] However, no study has examined the biochemical mechanism of the neuroprotection by quercetin in HT22 cells. Therefore, our findings provide, for the first time, a mechanistic explanation for the neuroprotective effects of quercetin isolated from onions against the oxidative cytotoxicity of glutamate. Briefly, quercetin reduces oxidative stress in glutamate-treated HT22 cells by inhibiting intracellular ROS overproduction and Ca2+ influx. These major actions of quercetin affect the mitochondrial apoptotic pathway and the MAPKs pathway. Our observations suggest that the inhibition of glutamate-induced neuronal cell apoptosis via decreases in intracellular ROS and Ca2+ levels constitutes a promising strategy for preventing and treating oxidative neuronal injury and degeneration, such as that in Alzheimer's, Parkinson's, and Huntington's diseases. Owing to its antioxidant and cytoprotective effects, quercetin and the ethanol extract of onions may be useful for clinical applications in pathologies involving glutamate-oxidative stress.
ACKNOWLEDGMENTS
The study described here is supported by grants from the BK21 Research Team for Developing Functional Health Food Materials.
Footnotes
Source of Support: This work was supported by the BK21 Research Team for Developing Functional Health Food Materials
Conflict of Interest: None declared.
REFERENCES
- 1.Houghton PJ, Howes MJ. Natural products and derivatives affecting neurotransmission relevant to Alzheimer's and Parkinson's disease. Neurosignals. 2005;14:6–22. doi: 10.1159/000085382. [DOI] [PubMed] [Google Scholar]
- 2.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–95. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 3.Greenwood SM, Connolly CN. Dendritic and mitochondrial changes during glutamate excitotoxicity. Neuropharmacology. 2007;53:891–8. doi: 10.1016/j.neuropharm.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–9. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 5.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–58. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 6.Maher P, Schubert D. Signaling by reactive oxygen species in the nervous system. Cell Mol Life Sci. 2000;57:1287–305. doi: 10.1007/PL00000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duchen MR. Mitochondria and calcium: from cell signaling to cell death. J Physiol. 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Squìer MK, Miller AC, Malkinson AM, Cohen JJ. Calpain activation in apoptosis. J Cell Physiol. 1994;159:229–37. doi: 10.1002/jcp.1041590206. [DOI] [PubMed] [Google Scholar]
- 9.Pearson G, Robinson F, Gibson TB, Xu B, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathway: regulation and physiological functions. Endocr Rev. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 10.Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur J Biochem. 2004;271:2060–6. doi: 10.1111/j.1432-1033.2004.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang L, Karin M. Mammalian MAP kinase signaling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 12.Vol. 1. Geneva: WHO; 1999. World Health Organization. (Chapter: Bulbus Allii Cepae). WHO Monographs on Selected Medicinal Plants; pp. 5–15. [Google Scholar]
- 13.Wu XJ, Stahl T, Hu Y, Kassie F, Mersch-Sundermann V. The production of reactive oxygen species and the mitochondrial membrane potential are modulated during onion oil-induced cell cycle arrest and apoptosis in A549 cells. J Nutr. 2006;136:608–13. doi: 10.1093/jn/136.3.608. [DOI] [PubMed] [Google Scholar]
- 14.Park S, Kim MY, Lee DH, Lee SH, Baik EJ, Moon CH, et al. Methanolic extract of onion (Allium cepa) attenuates ischemia/hypoxia-induced apoptosis in cardiomyocytes via antioxidant effect. Eur J Nutr. 2009;48:235–42. doi: 10.1007/s00394-009-0007-0. [DOI] [PubMed] [Google Scholar]
- 15.Li W, Tang C, Jin H, Du J. Effects of onion extract on endogenous vascular H2S and adrenomedulin in rat atherosclerosis. Curr Pharm Biotechnol. 2011;12:1427–39. doi: 10.2174/138920111798281135. [DOI] [PubMed] [Google Scholar]
- 16.Islam MS, Choi H, Loots du T. Effects of dietary onion (Allium cepa L.) in a high-fat diet streptozotocin-induced diabetes rodent model. Ann Nutr Metab. 2008;53:6–12. doi: 10.1159/000152868. [DOI] [PubMed] [Google Scholar]
- 17.Shri R, Singh Bora K. Neuroprotective effect of methanolic extracts of Allium cepa on ischemia and reperfusion-induced cerebral injury. Fitoterapia. 2008;79:86–96. doi: 10.1016/j.fitote.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 18.Lee BY, Ban JY, Seong YH. Chronic stimulation of GABAA receptor with muscimol reduces amyloid beta protein (25-35)-induced neurotoxicity in cultured rat cortical cells. Neurosci Res. 2005;52:347–56. doi: 10.1016/j.neures.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Kolárová H, Bajgar R, Tománková K, Krestýn E, Dolezal L, Hálek J. In vitro study of reactive oxygen species production during photodynamic therapy in ultrasound-pretreated cancer cells. Physiol Res. 2007;56:S27–32. doi: 10.33549/physiolres.931298. [DOI] [PubMed] [Google Scholar]
- 20.Grohm J, Plesnila N, Culmsee C. Bid mediates fission, membrane permeabilization and peri-nuclear accumulation of mitochondria as a prerequisite for oxidative neuronal cell death. Brain Behav Immun. 2010;24:831–8. doi: 10.1016/j.bbi.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 21.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 22.Fossen T, Pedersen AT, Andersen ØM. Flavonoids from red onion (Allium cepa) Phytochemistry. 1998;47:281–5. [Google Scholar]
- 23.Zhang Y, Bhavnani BR. Glutamate-induced apoptosis in primary cortical neurons is inhibited by equine estrogens via down-regulation of caspase-3 and prevention of mitochondrial cytochrome c release. BMC Neurosci. 2005;6:13. doi: 10.1186/1471-2202-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isaev NK, Zorov DB, Stelmashook EV, Uzbekov RE, Kozhemyakin MB, Victorov IV. Neurotoxic glutamate treatment of cultured cerebellar granule cells induces Ca2+-dependent collapse of mitochondrial membrane potential and ultrastructural alterations of mitochondria. FEBS Lett. 1996;392:143–7. doi: 10.1016/0014-5793(96)00804-6. [DOI] [PubMed] [Google Scholar]
- 26.Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome C release by proapoptotic Bcl-2 family members. Biochem Biophys Res Commun. 2003;304:437–44. doi: 10.1016/s0006-291x(03)00615-6. [DOI] [PubMed] [Google Scholar]
- 27.Kao TK, Ou YC, Raung SL, Lai CY, Liao SL, Chen CJ. Inhibition of nitric oxide production by quercetin in endotoxin/cytokine-stimulated microglia. Life Sci. 2010;86:315–21. doi: 10.1016/j.lfs.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 28.Senthilkumar K, Elumalai P, Arunkumar R, Banudevi S, Gunadharini ND, Sharmila G, et al. Quercetin regulates insulin like growth factor signaling and induces intrinsic and extrinsic pathway mediated apoptosis in androgen independent prostate cancer cells (PC-3) Mol Cell Biochem. 2010;344:173–84. doi: 10.1007/s11010-010-0540-4. [DOI] [PubMed] [Google Scholar]
- 29.Ahn J, Lee H, Kim S, Park J, Ha T. The anti-obesity effect of quercetin is mediated by the AMPK and MAPK signaling pathways. Biochem Biophys Res Commun. 2008;373:545–9. doi: 10.1016/j.bbrc.2008.06.077. [DOI] [PubMed] [Google Scholar]
- 30.Torres-Piedra M, Ortiz-Andrade R, Villalobos-Molina R, Singh N, Medina-Franco JL, Webster SP, et al. A comparative study of flavonoid analogues on streptozotocin-nicotinamide induced diabetic rats: Quercetin as a potential antidiabetic agent acting via 11beta-hydroxysteroid dehydrogenase type 1 inhibition. Eur J Med Chem. 2010;45:2606–12. doi: 10.1016/j.ejmech.2010.02.049. [DOI] [PubMed] [Google Scholar]
- 31.Ishige K, Schubert D, Sagara Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic Biol Med. 2001;30:433–46. doi: 10.1016/s0891-5849(00)00498-6. [DOI] [PubMed] [Google Scholar]