

SUMMARY
Chronic infection with hepatitis C virus (HCV) is a common cause of liver cirrhosis and cancer. We performed RNA-sequencing in primary human hepatocytes activated with synthetic dsRNA to mimic HCV infection. Upstream of IFNL3 (IL28B) on chromosome 19q13.13, we discovered a novel, transiently induced region that harbors dinucleotide variant ss469415590 (TT/ΔG), which is in high linkage disequilibrium with rs12979860, a genetic marker strongly associated with HCV clearance. ss469415590-ΔG is a frame-shift variant that creates a novel primate-specific gene, designated interferon lambda 4 (IFNL4), which encodes a protein of moderate similarity with IFNL3. Compared to rs12979860, ss469415590 is more strongly associated with HCV clearance in individuals of African ancestry, whereas it provides comparable information in Europeans and Asians. Transient over-expression of IFNL4 in a hepatoma cell line induced STAT1/STAT2 phosphorylation and expression of interferon-stimulated genes. Our findings provide new insights into the genetic regulation of HCV clearance and its clinical management.
More than 3% of the world population is infected with hepatitis C virus (HCV)1. Up to 80% of acutely-infected individuals fail to clear the virus and develop chronic hepatitis C (CHC)2, with as many as 5% eventually progressing to liver cancer3. Success of CHC treatment with pegylated interferon-α plus ribavirin (pegIFN-α/RBV) depends on HCV genotype and reaches 50–80% in patients of European ancestry, but only 30% in patients of African ancestry. Adding a direct acting antiviral (DAA) agent to this regimen increases the success rate, but is subject to numerous side effects of pegIFN-α/RBV4 and DAA treatment. If this treatment fails, there is an increased risk of selection of resistant HCV strains that may compromise future treatment options5,6.
Recent genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) rs12979860 and rs8099917 on chromosome 19q13.13 near IFNL3 (IL28B) gene as markers associated with both spontaneous HCV clearance7,8 and response to pegIFN-α/RBV treatment7–11. Within this region reside the three interferon-λ genes, IFNL1, IFNL2 and IFNL3 (formerly IL29, IL28A and IL28B, respectively), which encode the type-III IFNs12,13. Type-I IFNs (IFN-α and IFN-β) and type-III IFNs (IFN-λs) induce antiviral activity and suppress HCV replication in vitro14,15 and in vivo16 by activation of the JAK/STAT pathway and up-regulation of interferon-stimulated genes (ISGs)14,17,18. The molecular phenotype of this genetic association remains unclear. The GWAS markers have not been consistently associated with hepatic IFNL3 mRNA expression19–21 and a non-synonymous IFNL3 variant, rs8103142 (Arg70Lys), which is in strong linkage disequilibrium (LD) with rs12979860 in all HapMap populations (r2=0.8–1.0), does not seem to affect the function of the IFNL3 protein22.
A genetic variant creates a novel interferon protein (IFNL4) of similarity with IFNL3
We sequenced mRNA (RNA-seq) from primary human hepatocytes (PHH) treated with PolyI:C, which is a synthetic mimic of double-stranded HCV RNA. The PHH sample was from a liver donor who was heterozygous for rs12979860 (C/T) and uninfected with HCV. The hepatocytes were treated with PolyI:C for 0, 1, 2, 4, 8 or 24 hours, and induction of the IFN-λ genes (IFNL1-3) was confirmed by TaqMan expression analysis prior to RNA-seq. An analysis of RNA-seq data that focused on a 150-Kb region around rs12979860 showed concordance with the TaqMan expression results – there was no expression of IFN-λ genes without PolyI:C treatment, and these genes were induced after 2–24 hours of PolyI:C activation (Fig. 1a).
Figure 1. Identification of a novel transcribed region upstream of IFNL3 gene.

RNA-seq in primary human hepatocytes (PHH) treated with 50 ug/ml PolyI:C for 0, 1, 2, 4, 8 or 24 hours. RNA-seq plot of the 150-Kb region in USCS browser shows expression of IFNL1, IFNL2 and IFNL3, and a novel transcribed region upstream of IFNL3. The number of reads (depth) corresponds to the level of mRNA expression.
a. Detailed view of the RNA-seq results for IFNL3 and the novel transcribed region. A CTCF transcriptional insulator (ENCODE data24) between these regions indicates their independence.
b. Splicing architecture of the ten novel transcripts (NCBI accession numbers are presented in Supplementary Table 2). The GWAS marker, rs12979860, is located within the first intron, while a novel marker, ss469415590-TT/ΔG, is located within the first exon, common for all transcripts. Transcription and translation start sites are marked by black and blue arrows; open reading frames are shaded in blue. * -transcripts which carry premature stop codons and are likely to be eliminated by nonsense-mediated decay. Arrows indicate location of primers (Supplementary Table 1) used to generate PCR-products presented on panel d.
c. PCR results in PHH cDNA using primers from panel c. No distinct PCR product is expected in the TT/TT sample; in the ΔG/ΔG sample these primers capture transcripts producing proteins of p179, 131 and 107 aa, but not of 170 aa; a transcript producing a 93 aa protein fragment is expected to be degraded. IFNL3, IFNL1 and PPIA (endogenous control) were measured in the same samples. Similar amounts of DNaseI-treated high quality RNA was used for all the reactions.
We also observed transient activation of a novel transcribed region upstream of IFNL3, with the highest levels of expression detected at 2 and 4 hours (Fig. 1b). Analysis of paired-end RNA-seq reads identified one major splice-junction site. Using this common sequence as a starting point for 5′ rapid amplification of cDNA ends (5′RACE), we mapped a transcription start site, followed by a unique protein translation start site 277 bp downstream. Within the first exon (at amino acid 22), we detected a novel compound dinucleotide variant, denoted ss469415590 TT/ΔG, comprised of a one-base insertion/deletion (indel) polymorphism (T/Δ, rs67272382) and a one–base substitution variant (T/G, rs74597329). Using PolyI:C-stimulated PHH from 5 additional liver donors and primer presented in Supplementary Table 1, we cloned and annotated ten individual transcripts created by a combination of the ss469415590 alleles and inclusion of several alternative exons (Fig. 1c, d, NCBI accession numbers presented in Supplementary Table 2). The location of these novel transcripts 3 Kb upstream of and in the same orientation as IFNL3, raised the possibility that they are alternative splicing forms of IFNL3 or fusions. However, the presence of a CTCF transcriptional insulator site23,24 between the two transcribed regions (Fig. 1b), the results of the RACE experiments and the failure to generate an RT-PCR product between IFNL3 and the novel transcribed region, confirmed their independence. Despite high overall similarity with a genomic region upstream of IFNL2, the novel transcripts and ss469415590 are specific for the region upstream of IFNL3 (Supplementary Fig. 1).
Of the ten novel transcripts, four were interrupted by premature stop codons and, thus, are likely to be eliminated by nonsense-mediated decay25. The remaining six transcripts were predicted to produce full-length proteins of: 143 amino acids (aa; p143) and 124 aa (p124) from transcripts with the ss469415590-TT allele; 179 aa (p179), 170 aa (p170), 131 aa (p131) and 107 aa (p107) from transcripts with the ss469415590-ΔG allele (Fig. 1c). A global protein BLAST search found homology only for p179, with 29.1% aa identity and 40.8% aa similarity with IFNL3. However, the p179 and IFNL3 cDNA sequences were not similar enough to be aligned using a BLAST bl2seq tool. Based on the protein sequence homology with type-III IFNs (Table 1), we designated p179 as interferon analog protein (IFNL4). IFNL3 and p179 (IFNL4) proteins are most related within the sequences that correspond to the A and F helices of IFNL3, which constitute the core area for interaction of IFNL3 and other type-III IFNs with their primary receptor, IFNLR1 (IL28R1). However, IFNL4 differs in the area corresponding to the D helix of IFNL3, which is the area of interaction of type-III IFNs with the second chain of the IFNL receptor complex, IL10R2 (Fig. 2)26–29.
Table 1.
Comparisons between p179 (IFNL4) and selected members of class-2 cytokine family
| Protein | p179 (IFNL4) | IFNL3 | IFNL1 | IFNL2 | IFN-α1 | IL10 | IL22 | IFN-β1 |
|---|---|---|---|---|---|---|---|---|
| Type | IFN-III | IFN-III | IFN-III | IFN-III | IFN-I | IL10-family | IL10-family | IFN-I |
| mRNA | JN806234 | NM_172139 | NM_172140 | NM_172138 | NM_002175 | NM_000572 | NM_020525 | NM_002176 |
| Protein ID | AFQ38559 | A2BDE1 | Q8IU54 | Q45KQ8 | P05014 | Q6FGW4 | Q9GZX6 | Q5VWC9 |
| Size, aa | 179 | 196 | 200 | 200 | 189 | 178 | 179 | 187 |
| Exons | 5 | 5 | 5 | 6 | 1 | 5 | 5 | 1 |
| Identity with p179, aa, n (%) | -- | 52 (29.1) | 48 (26.8) | 47 (26.3) | 26 (14.5) | 24 (13.4) | 20 (11.1) | 20 (11.1) |
| Similarity with p179, aa, n (%) | -- | 73 (40.8) | 66 (36.8) | 70 (39.1) | 47 (26.2) | 40 (22.3) | 45 (25.1) | 44 (24.5) |
Figure 2. Protein sequence analysis.

ClustalW protein sequence alignment for IFNL4 (p179), p131, p107, IFN-λs (IFNL1, IFNL2 and IFNL3), and IFN-α. Identical amino acids are shaded in black. Exon numbering and the location of ss469415590 and other variants identified by sequencing of 270 HapMap samples, are based on IFNL4 protein sequence. Annotations of protein helices, amino acid numbering and specific amino acids are based on the sequence of mature IFNL3 protein26,27 (without leader peptide). Indicated: cysteins conserved between IFNL4 and all other IFNL proteins (C16, C50, C115, C148 and C174), interaction sites of IFNLs with their first common receptor IFNLR1 (aa 27, 33, 34, 36, 37, 44, 53, 155, 158); and interaction sites with their second receptor, IL10R2 (aa 97, 100).
Association of genetic variants within IFNL4 with HCV clearance
The GWAS markers rs12979860 and rs8099917 are located 367 bp downstream (intron 1) and 4 kb upstream of ss469415590, respectively. Analysis of the HapMap30 (Supplementary Table 3) and the 1000 Genomes project31 data (Supplementary Table 4) showed that the IFNL4-creating ss469415590-ΔG allele is perfectly correlated with the unfavorable rs12979860-T allele in Asians (CHB+JPT sets, r2=1.00) and well correlated in Europeans (CEU set, HapMap, r2=0.92; 1000 Genomes, r2=0.83). In Africans, however, this correlation is only moderate (YRI set, HapMap, r2=0.71; 1000 Genomes, r2=0.65), even though rs12979860 is the best surrogate for ss469415590 of all markers within the 100-Kb region. Correlation between ss469415590 and rs8099917 is high in Asians (r2=0.91), moderate in Europeans (r2=0.44), but very low in Africans (r2=0.008) (Supplementary Table 3).
We assessed the association of ss469415590 and rs12979860 with HCV clearance in 1,436 African-American and 1,480 European-American individuals from four studies. In VirahepC32 and HALT-C33, we evaluated response to pegIFN-α/RBV therapy in patients with CHC (Supplementary Table 5). There were differences in the rates of sustained virological response (SVR) among the subjects from these studies, which reflect well known racial differences in response to treatment and the differing selection criteria for these clinical trials: Virahep C European-American, 52%; Virahep C African-American, 28%; HALT-C European-American, 18%; African-American, 7%. We evaluated spontaneous HCV clearance in injection drug users enrolled in two studies, UHS34 and ALIVE35 (Supplementary Table 6). The decrease in HCV RNA during the first 28 days of treatment is a powerful early predictor of ultimate treatment response that is strongly associated with rs12979860 genotype36,37. In African-American Virahep-C participants, the decline in HCV RNA levels after 28 days of treatment was more strongly associated with ss469415590 genotype than with rs12979860 genotype (p=0.015, difference in mean values; Fig. 3; Supplementary Table 7). In the same study we observed a stronger association for ss469415590 than for rs12979860 with other measures of treatment response (week 24 response, end-of-treatment response and SVR; Table 2), although these differences did not reach statistical significance. The association pattern was similar in African-American patients from the HALT-C study, with a stronger association for ss469415590 than for rs12979860 (week 20 response, end-of-treatment and SVR, Table 2). Spontaneous HCV clearance among African-Americans was evaluated using the area under the receiver operating curve (AUC). In UHS participants, the AUC value was greater for ss469415590 (0.62) than rs12979860 (0.58) (Table 3). In the ALIVE study, which has higher percentage of HCV/HIV co-infected patients, the AUC values were similar for rs12979860 (0.64) and ss469415590 (0.64; Table 3).
Figure 3. Median decrease in HCV RNA (log10 IU/ml) in African-American participants in Virahep-C study during the first 28 days of treatment with pegIFN-α/RBV.

P=0.015 for comparison of mean differences in HCV RNA levels at day 28 for each of the three genotype groups for ss469415590 with the respective group for rs12979860.
Table 2.
Comparison of rs12979860 and ss469415590 genotypes among African-American participants of Virahep-C study and HALT-C Trial for associations with response to treatment with pegIFN-α/RBV
| Variant | Genotype | N | Treatment Time Point
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Week 20/24* | End-of-Treatment | SVR | |||||||||
|
| |||||||||||
| Response Rate (%) | OR | P-value | Response Rate (%) | OR | P-value | Response Rate (%) | OR | P-value | |||
|
| |||||||||||
| Virahep-C (n = 169) | |||||||||||
| rs12979860 | TT | 57 | 40.4 | Ref. | 36.8 | Ref. | 24.6 | Ref. | |||
| CT | 93 | 50.5 | 1.51 | 0.23 | 40.9 | 1.18 | 0.62 | 29.0 | 1.26 | 0.55 | |
| CC | 19 | 63.2 | 2.53 | 0.089 | 52.6 | 1.90 | 0.23 | 36.8 | 1.79 | 0.30 | |
|
| |||||||||||
| ss469415590 | ΔG/ΔG | 71 | 35.2 | Ref. | 32.4 | Ref. | 21.1 | Ref. | |||
| ΔG/TT | 82 | 56.1 | 2.35 | 0.010 | 45.1 | 1.72 | 0.11 | 31.7 | 1.73 | 0.14 | |
| TT/TT | 16 | 68.8 | 4.05 | 0.019 | 56.3 | 2.68 | 0.080 | 43.8 | 2.90 | 0.067 | |
|
| |||||||||||
|
HALT-C Trial (n = 144)
| |||||||||||
| rs12979860 | TT | 61 | 9.8 | Ref. | 8.2 | Ref. | 3.3 | Ref. | |||
| CT | 72 | 16.7 | 1.83 | 0.26 | 12.5 | 1.60 | 0.42 | 6.9 | 2.20 | 0.36 | |
| CC | 11 | 45.5 | 7.64 | 6.2×10−3 | 36.4 | 6.40 | 0.018 | 27.3 | 11.06 | 0.015 | |
|
| |||||||||||
| ss469415590 | ΔG/ΔG | 68 | 8.8 | Ref. | 7.4 | Ref. | 2.9 | Ref. | |||
| ΔG/TT | 68 | 19.1 | 2.44 | 0.090 | 14.7 | 2.17 | 0.18 | 8.8 | 3.19 | 0.16 | |
| TT/TT | 8 | 50.0 | 10.34 | 4.7×10−3 | 37.5 | 7.56 | 0.019 | 25.0 | 11.0 | 0.027 | |
Table 3.
Comparison of rs12979860 and ss469415590 genotypes among African-American participants of the Urban Health Study (UHS) and ALIVE for associations with spontaneous clearance of hepatitis C virus (HCV) infection
| Variant | Genotype | Chronic | %* | Clear | %* | OR | P-value | AUC |
|---|---|---|---|---|---|---|---|---|
| Urban Health Study (n=459) | ||||||||
| rs12979860 | TT | 119 | 34.0 | 26 | 23.9 | Ref. | 0.58 | |
| CT | 183 | 52.3 | 54 | 49.5 | 1.35 | 0.26 | ||
| CC | 48 | 13.7 | 29 | 26.6 | 2.77 | 1.5×10−3 | ||
|
| ||||||||
| ss469415590 | ΔG/ΔG | 137 | 39.1 | 26 | 23.9 | Ref. | 0.62 | |
| ΔG/TT | 168 | 48 | 53 | 48.6 | 1.66 | 0.056 | ||
| TT/TT | 45 | 12.9 | 30 | 27.5 | 3.51 | 7.9×10−5 | ||
|
| ||||||||
|
ALIVE (N=664)
| ||||||||
| rs12979860 | TT | 228 | 38.9 | 15 | 19.2 | Ref. | 0.64 | |
| CT | 275 | 46.9 | 37 | 47.4 | 2.05 | 0.025 | ||
| CC | 83 | 14.2 | 26 | 33.3 | 4.76 | 7.6 ×10−6 | ||
|
| ||||||||
| ss469415590 | ΔG/ΔG | 255 | 43.5 | 19 | 24.4 | Ref. | 0.64 | |
| ΔG/TT | 265 | 45.2 | 36 | 46.2 | 1.82 | 0.043 | ||
| TT/TT | 66 | 11.3 | 23 | 29.5 | 4.68 | 5.5 ×10−6 | ||
Percentage of subjects with indicated genotype; OR - odds ratio; AUC - area under the receiver operating characteristic curve; Urban Health Study34 and ALIVE35 are epidemiological studies of injection drug users who were evaluated for chronic HCV infection on the basis of testing for anti-HCV and HCV RNA.
Virahep-C, HALT-C and UHS also enrolled European-American participants. In these subjects, ss469415590 and rs12979860 showed similar associations for both treatment-induced and spontaneous HCV clearance (Supplementary Tables 7, 8, 9, 10). Taken as a whole, our results show that among African-American individuals, ss469415590 is a better marker than rs12979860 for predicting response to pegIFN-α/RBV treatment of CHC and possibly for spontaneous HCV clearance, while these variants are similarly informative in European-Americans.
By sequencing IFNL4 in 270 HapMap samples we annotated three non-synonymous variants, rs73555604 (Cys17Tyr) in exon 1, rs142981501 (Arg60Pro) and rs117648444 (Pro70Ser) in exon 2, as well as four synonymous variants, rs150891559 (Ala11Ala) and rs4803221 (Ser30Ser) in exon 1, and rs12971396 (Ser149Ser) and rs137902769 (Ser175Ser) in exon 5 (Fig. 2, Supplementary Fig. 2, Supplementary Tables 11, 12). Based on a haplotype analysis of 16 markers from the 8-Kb IFNL3/IFNL4 region, we identified eight markers that captured all haplotypes present in HapMap sets (Supplementary Table 13). These eight markers were also tested in European-American and African-American patients from Virahep-C. In all populations, the unique favorable haplotype included the ss469415590-TT allele, which abrogates the IFNL4 protein. The unfavorable ss469415590-ΔG allele was found on a number of haplotypes, including two haplotypes that were reported as being neutral in Europeans despite carrying the unfavorable rs12979860-T allele38,39; these two haplotypes include minor alleles of either non-synonymous variants rs73555604 or rs11764844. It is possible, therefore, that these variants modify the risk in carriers of the unfavorable ss469415590-ΔG allele and are the source of haplotype heterogeneity previously reported in Europeans38,39, however, data from Virahep C are too sparse to confirm this finding (Supplementary Table 13).
IFNL4 induces expression of ISGs
We evaluated functional properties of the 6 novel protein isoforms created by alleles of ss469415590. For an analysis of 45 signaling pathways, HepG2 hepatoma cells were transiently transfected with expression constructs for all 6 isoforms or treated with recombinant IFN-α, IFNL3 and IFNL4 (the latter two produced in the sfs9 baculoviral expression system). Only transfection with IFNL4 expression construct, as well as treatment with IFN-α or IFNL3, induced activation of an interferon stimulated response element reporter (ISRE-Luc), which contains STAT1/STAT2 binding sites responsive to type-I and type-III IFN signaling, and the IRF1 reporter (Fig. 4a). These results were validated in HepG2 cells transiently (Fig. 4b), as well as stably (Fig. 4c), expressing ISRE-Luc reporter constructs. The effect was comparable when the cells were transfected with IFNL4 expression constructs generating proteins either with a Halo-tag or a FLAG-tag (Supplementary Fig. 3). Similarly, only transient transfection with IFNL4 construct decreased HCV RNA replication in hepatoma cells stably expressing a subgenomic luciferase-expressing hepatitis C virus replicon40 (Fig. 4d), and induced STAT1 and STAT2 phosphorylation (Fig 4 e). Transfection with IFNL4 activated the ISRE reporter in HepG2 and HEK-293T cells, but not in HeLa cells (Supplementary Fig. 4).
Figure 4. Analysis of biological activity of novel proteins.

a. “Pathway finder” analysis using luciferase reporter constructs representing 45 human signaling pathways in HepG2 cells. The cells were transfected with expression constructs or an empty vector (mock), or treated with 10 ng/ml of recombinant purified IFN-α, IFNL3, IFNL4, or with PBS (mock). All results represent mean values of two independent biological transfection/treatment replicates, with standard deviations.
b. Results of transient transfection of the IFNL4, p131 and p107 expression constructs and treatment with recombinant purified IFN-α and IFNL3 proteins in HepG2 cell line transiently co-transfected with ISRE-Luc reporter. The results are normalized to transfection with an empty vector (mock) and represent mean values of 8 biological replicates, with standard errors.
c. Results of transient transfection of the IFNL4, p131 and p107 expression constructs in HepG2 cell line stably expressing the ISRE-Luc reporter. The results are normalized to transfection with an empty vector (mock) and represent mean values of 11 biological replicates, with standard errors.
d. Test for antiviral effects of the IFNL4, p131 and p107 expression constructs transiently transfected into Huh7-Lunet cells stably expressing a subgenomic, luciferase-expressing HCV replicon (HCV-Luc), compared to an empty vector (mock). Results represent mean values of 4 biological replicates, with standard errors.
e. Western blot analysis of Y701-STAT1 and Y689-STAT2 phosphorylation in HepG2 cells transiently transfected with expression constructs for the six protein isoforms. All Halo-tag expression constructs produce proteins detectable with an antibody for the Halo-tag; a rabbit monoclonal anti-IFNL4 antibody recognizes IFNL4 (p179) as well as its nonfunctional isoforms p131 and p107.
The recombinant IFNL4 protein expression was detectable in cells and cell lysates of transfected HepG2 and 293T cells, by confocal imaging and Western blots with antibodies specific for IFNL4 and tag proteins (Supplementary Fig. 5, 6). We also detected weak expression of recombinant IFNL4 in media of transfected HepG2 cells, but not of 293T cells (Supplementary Fig. 7). In PolyI:C- stimulated PHH from liver donors not infected with HCV endogenous protein expression of IFNL4 was detected by confocal imaging in carriers of the unfavorable ss469415590-ΔG allele but not in a homozygous carrier of the favorable ss469415590-TT allele (Fig. 5). In hepatocytes from the donor heterozygous for ss469415590-ΔG, we detected endogenous expression of IFNL4 in cells treated with PolyI:C and after in-vitro infection with the JFH1HCV strain (Supplementary Fig. 6d). In fact, in hepatocytes from one of these donors (ΔG/TT genotype), we observed low IFNL4 expression even without PolyI:C treatment or in-vitro HCV infection (hour 0 time point). Though preliminary, these results suggest that IFNL4 might be expressed in conditions unrelated to HCV infection.
Figure 5. Confocal imaging of IFNL4 in primary human hepatocytes (PHH) from liver donors with different ss469415590 genotypes.
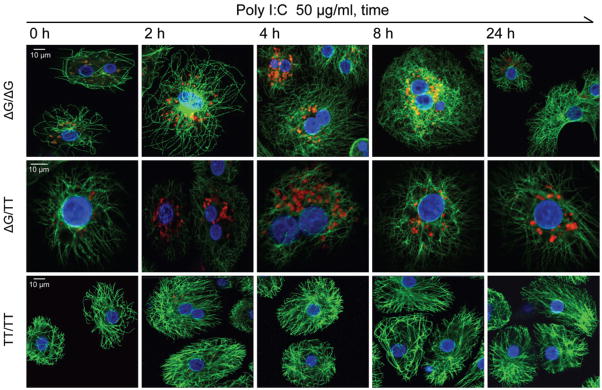
Confocal imaging in PHH treated with 50 ug/ml of PolyI:C for 0, 2, 4, 8 or 24 hours. Red staining – IFNL4, green – cytoskeleton (α-tubulin), blue – nuclei. Confocal imaging of endogenous IFNL4 expression in the same ss469415590-ΔG/TT sample after in-vitro infection with JFH1-HCV strain is presented in Supplementary Fig. 6.
To further explore the functional consequences of IFNL4 expression, we performed RNA-seq in HepG2 cells transiently transfected with IFNL4 or an empty vector (mock) (Supplementary Fig. 8a) and found that the top canonical pathways induced by IFNL4 are related to the activation of interferon signaling (Supplementary Fig. 8b). We validated the RNA-seq results by qRT-PCR analysis and showed that IFNL4 induced expression of many ISGs in a pattern similar to that induced by IFN-α and IFNL3 (Supplementary Fig. 8c). Previously, genes in these pathways have been shown to be expressed at higher levels in pre-treatment liver biopsies from HCV-infected patients who do not respond to pegIFN-α/RBV treatment; these patients tend to carry the unfavorable genotypes of rs12979860 and rs809991719,41–44 which mark the ss469415590-ΔG allele that produces IFNL4. To mimic this clinical phenotype, we transfected HepG2 cells with mock or IFNL4 expression constructs and/or treated cells with 10 ng/ml of recombinant IFN-α or IFNL3. In these samples we validated the RNA-seq data by qRT-PCR analysis and showed that IFNL4 induced expression of selected ISGs (STAT1, ISG15, IFIHI1/MDA5, OAS1, MX1, DHX58/RIG-I) in a pattern similar to that induced by IFN-α and IFNL3 (Supplementary Fig. 8d). Furthermore, treatment by IFN-α or IFNL3 of cells already expressing IFNL4 did not induce additional activation of ISGs (Supplementary Fig. 8d). Some genes known as markers of HCV-induced liver damage, such as chemokine CCL5 (RANTES)45 and the proto-oncogene FOS46–48 were induced by IFNL4 but not by IFNs (Supplementary Fig. 8d), suggesting a functional role for IFNL4 distinct from the roles of IFN-α and IFNL3.
Conclusions
We have identified a novel inducible human protein-coding gene, IFNL4, which is related to, but distinct from known IFNs and other class-2 cytokines. The 179 aa open-reading frame of IFNL4 transcript is created by a common deletion frame-shift allele of ss469415590, which is a dinucleotide variant strongly linked with rs12979860. In individuals of African ancestry, the IFNL4-generating ss469415590-ΔG allele is superior to the rs12979860-T allele for predicting response to pegIFN-α/RBV treatment of CHC. Within IFNL4 we identified three non-synonymous variants, rs73555604 (Cys17Tyr), rs142981501 (Arg60Pro) and rs11764844 (Pro70Ser), present on haplotypes with the ss469415590-ΔG allele. The role of these variants on IFNL4 biological function and their impact on HCV clearance in different populations should be further explored.
Analysis of genomic sequences of 45 species available for IFNL4 region in the UCSC genome browser showed that the unfavorable, IFNL4-generating ss469415590-ΔG allele is an ancestral variant present in all the species. The existence of IFNL4 protein could be predicted only in the genomes of macaques (marmoset and rhesus), orangutan, chimpanzee and humans (Supplementary Fig. 9). The beneficial insertion ss469415590-TT allele appears to be a recently derived variant, which became common in all human populations (93% in Asians, 68% in Europeans and 23% in Africans, based on HapMap samples), suggesting positive selection for this allele. Introduction of frame-shifts is considered to be an evolutionary mechanism for the rapid emergence of new proteins49,50, but, in this case, an insertion allele that abrogates IFNL4 seems to have been selected during evolution.
We found that the IFNL4 protein of 179 aa induces STAT1 and STAT2 phosphorylation, activates the ISRE-Luc reporter and ISGs, and generates antiviral response in hepatoma cells. The mechanisms by which IFNL4 induces these responses, but nevertheless impairs HCV clearance, is currently under investigation. IFNL4 and IFNL3 share similarity in the area that is known to interact with the primary receptor of IFNL3 (IFNLR1), but differ in the region of IFNL3 that interacts with the second chain of the IFNL receptor complex, IL10R2. Thus, it is possible that IFNL4 activates JAK-STAT signaling through a unique receptor complex consisting of IFNLR1 and a currently undefined second receptor chain or that IFNL4 functions as a decoy cytokine competing with type-III IFNs for binding of IFNLR1. We also found that the IFNL4-caused pre-activation of interferon signaling prevents further activation by type-I and Type-III IFNs. We used an allele-specific mRNA expression assay (Supplementary Fig. 10) and explored endogenous IFNL4 expression in PHH, where it was induced by PolyI:C, IFN-α (but not with IFNL3) and in vitro infection with HCV (Supplementary Fig. 11). However, no IFNL4 mRNA expression was induced by PolyI:C, IFN-α or IFNL3 in several transformed cell lines that carry the ss469415590-ΔG allele (HepG2, HeLa, HEK293T, A549 and HH29). Experiments aimed at elucidating the triggers of IFNL4 expression in diverse conditions and cell types and its receptor components are ongoing, and may provide greater insight regarding its mechanism of action.
Previous studies found that patients with chronic hepatitis C who carry rs12979860-T, which marks the ss469415590-ΔG allele, have somewhat higher hepatic expression of ISGs before treatment, but poorer ISG response to pegIFN- α/RBV treatment19,41–44. The rs12979860-T variant has also been associated with lower HCV RNA levels in the absence of treatment9,51. Our in vitro experiments in HepG2 cells showed that IFNL4 induced activation of ISGs, which was not further enhanced by exogenous treatment with IFN-α or IFNL3. Together, these data suggest that IFNL4 induces weak expression of ISGs that provides some antiviral effect response which might lower the HCV load, but also reduces the responsiveness to type-I and type-III IFNs that is needed for efficient HCV clearance.
It has been reported that rs12979860 predicts early viral kinetics in HCV-infected patients receiving IFN-α-free treatment52. This genotype has also been associated with response to IFN-α based treatment of chronic hepatitis B virus (HBV) infection in some studies53,54. Furthermore, IFN-α therapy is used for a number of other clinical conditions, including some forms of cancer55. Thus, therapeutic inhibition of IFNL4 might represent a novel biological target for the treatment of HCV and HBV infection and possibly other diseases, and IFNL4 genotype could be used to select patients for this therapy.
ONLINE METHODS
Cells
Fresh primary human hepatocytes (PHH) from HCV un-infected liver donors were purchased from Lonza or Celsis. PHH received in suspension were cultured overnight in collagen-coated plates or chamber slides (BD Biosciences), unattached PHH were removed before treatment. Hepatoma HepG2 cells, cervical carcinoma HeLa and embryonic kidney 293T cells (all from ATCC) were cultured in DMEM with 10% FBS. Hepatoma Huh7-Lunet cells harboring a subgenomic, luciferase-expressing HCV JFH1 replicon were cultured as previously described40. The PHH were treated with 50 μg/ml of PolyI:C (Imgenex), 100 ng/ml of IFN-α (PBL Interferon Source) or IFNL3 (custom); cell lines were treated with 10 ng/ml of IFN-α, IFNL3 or IFNL4 (custom).
Transfections
Transfections were performed using a Lipofectamine™ LTX Reagent and Opti-MEM medium, using standard protocols (Life Technologies).
HCV infection of PHH
PHH (0.35×105/well) were infected with HCV (JFH1 strain)56 for 0, 6 or 24 hours in collagen-coated 24-well plates or chamber slides at a multiplicity of infection (M.O.I.) of 2.
Antiviral assays
Huh7-Lunet cells were transfected with IFNL4, p131 or p107 constructs or an empty GFP vector (mock) in a 48-well plate and luciferase expression was measured 48 hours post-transfection.
Extraction of DNA, RNA and protein
DNA and RNA were prepared using a DNeasy or a RNeasy kit with DNase-I treatment (Qiagen) and evaluated with NanoDrop 8000 (Thermo Scientific) and BioAnalyzer 2100 (Agilent Technologies). Protein was prepared by lysing cells in RIPA buffer with protease inhibitors (Sigma Aldrich). Cell media was concentrated 10x and 100x using the 9K MWCO protein concentrator tubes (Thermo Scientific).
Western blotting
Proteins were resolved on 4–12% tris-glycine SDS-PAGE gels (Life Technologies). Detection was done using the custom anti-IFNL4 mouse and rabbit monoclonal antibodies, rabbit anti-Halo antibody (Promega), and secondary goat anti-rabbit (sc-2030) or goat anti-mouse (sc-2031) antibodies with IgG-HRP (Santa Cruz). The signals were detected with ECL Plus Western blotting detection system (GE Healthcare Life Sciences).
Analysis of STAT1/STAT2 phosporylation
HepG2 cells in 6-well plates were untreated or transfected with expression constructs or empty Halo-tag vector, or were treated for1 hour at 37°C with 50 ng/ml of recombinant IFNL3. Equal amounts (50 μg/lane) of whole-cell lysates prepared 48 hours post-transfection were used for analysis by Western blotting. Detection was performed as previously described57 with rabbit anti-phospho-Tyr701-STAT1 (Cell Signaling Technology) and rabbit anti-phospho-Tyr689-STAT2 (Millipore) antibodies. The blots were stripped and re-probed with rabbit anti-STAT1 and anti-STAT2 antibodies (Santa Cruz Biotechnology) to measure the levels of total STAT1 and STAT2 proteins.
RNA-sequencing
Total RNA (1 ug) from PHH or HepG2 cells was used for library preparation with TruSeq PolyA kit (Illumina). Sequencing with Genome Analyzer (GAII) generated 47 million of 107 bp paired-end sequencing reads per sample. The TopHat v1.2.0 settings were changed to allow multiple read alignments (up to 10 regions, UCSC hg19) and 3 nucleotide mismatches per each 25-bp segment. Results were viewed with the UCSC genome browser http://genome.ucsc.edu/ and the Integrative Genomics Viewer (IGV) software: broadinstitute.org/igv/.
Identification and cloning of novel splicing forms
Rapid amplification of cDNA ends (5′ and 3′RACE) and cloning of full-length open reading frames were performed with SMARTer RACE cDNA kit (Clontech,) using a pool of DNAse-I treated RNA samples from PolyI:C-treated PHHs from several liver donors (Supplementary Table 1 for primers). PCR reactions were performed with AmpliTaq Gold 360 Master Mix (Life Technologies) and 360 GC Enhancer (Life Technologies) using the touchdown PCR program: 10 minutes at 95 °C, 20 cycles of touchdown (30 seconds at 95 °C, 45 seconds from 70 to 60°C decreasing by 1 °C after each 2 cycles, 45 seconds at 72 °C); 25 additional cycles (30 seconds at 95 °C, 45 seconds at 60 °C and 45 seconds at 72 °C); and final extension time of 7 minutes at 72 °C. Gel-purified PCR fragments were cloned into a C-terminal pFC14A–Halo tag expression vector (Promega) and sequenced for validation. IFNL3-Halo expression construct was generated using the same approach. p179 was also cloned into a pcDNA3.1-FLAG-tagged expression vector.
Production of recombinant proteins
IFNL4 and IFNL3 bacmids were generated in pFastBac C-terminal His-tag vector (Life Technologies) and expressed in a sf9 baculoviral strain. Using the anti-His-tag antibody (Sigma), expression of IFNL3 was detected in cell media, which was used for protein purification. Expression of p179 was not detectable in cell media and protein was purified from the cell pellet after cultivation of cells for 3–5 days in 2 liters of SF-900 III medium (Life Technologies). Proteins were first purified on HisTrap 5-ml nickel column followed by size-exclusion chromatography preparative column TSK G3000pw of 21.5×600 mm (Tosoh). The purified protein fractions were concentrated and dialyzed into a buffer. High protein purity (>90%) was confirmed by Coomassie staining and Western blot analyses with anti-His antibody (#H1029, Sigma, 1:3000 dilution), anti-IFNL3 and custom mouse and rabbit monoclonal anti-p179 antibodies. The IFNL3 and p179 proteins were custom-produced by Protein One.
Development of anti-IFNL4 antibodies
A mouse monoclonal antibody was custom-developed for a synthetic peptide KALRDRYEEEALSWGQRNCSFRPRRDSPRPS corresponding to amino acids 44–74 of p179 protein, by Precision Antibody. A rabbit monoclonal antibody was custom-developed for a synthetic peptide PGSSRKVPGAQKRRHKPRRADSPRC corresponding to amino acids 128–152 of p179 protein, by Epitomics.
Evaluation of biological activity of novel proteins
Luciferase Cignal 45-Pathway Finder Reporter Arrays were used in HepG2 cells according to instructions (Qiagen, full list of reporters tested is available at http://www.sabiosciences.com/reporter_assay_product/HTML/CCA-901L.html). Cells were transfected with expression constructs for p179, p170, p143, p131, p124 and p107 or treated for 24 h with purified recombinant proteins −10 ng/ml of IFN-α or IFNL3 and/or IFNL4-p179. Validation was performed with an individual interferon-stimulated response element (ISRE) luciferase Cignal reporter. All studies were performed using at least 8 biological replicates. A HepG2 cell line stably expressing the same ISRE-Luc reporter construct was generated by transduction of cells with a Luciferase Cignal Lenti ISRE reporter construct (Qiagen) and selection of positive clones by growth in DMEM + 10% FBS with 1x Antibiotic-Antimycotic (Life Technologies) and 2 ug/mL puromycin. The best HepG2-ISRE-stable clones were identified by testing with purified recombinant IFN-α and IFNL3.
Global analysis of transcriptome and pathway analysis
HepG2 cells were mock-transfected with an empty Halo-tag vector or with IFNL4-Halo expression construct. High-quality RNA (RIN ~10) prepared from transfected cells (48 hours) was used for sequencing with HiSeq 2000 (Illumina), generating ~300M reads per sample. Standard analysis identified 535 transcripts with >2-fold difference in expression and an FDR<0.05. Ingenuity Pathway Analysis (IPA) performed on this set nominated a list of pathways and specific transcripts. mRNA expression of selected transcripts was evaluated in samples transfected with mock, IFNL4, p107, p131 constructs or/and treated with 10 ng/ml of IFN-α or IFNL3, in 4 biological replicates. mRNA expression in all samples was evaluated with pathway-based RT2 Profiler PCR arrays, according to instructions (Qiagen).
Confocal Imaging
PHH cultured on collagen-coated slides were treated with 50μg/ml PolyI:C for 0 h (untreated), 2, 4, 8 and 24 hrs. HepG2 cells were transiently transfected with IFNL4-Halo expression construct. Cells were fixed for 20 min with 4% formaldehyde (Sigma) in PBS, permeabilized with 0.5% TritonX 100 (Sigma) for 5 min, blocked with 4% BSA (Sigma) and then incubated overnight at 4°C with primary antibodies: mouse monoclonal anti-IFNL4 ab (custom), rabbit α-tubulin ab (ab-15246, Abcam), rabbit anti-Halo-tag ab (Promega), mouse α-tubulin ab (ab-7291, Abcam, all at 1:1000 dilution. Secondary antibodies were donkey anti-rabbit or anti-mouse Alexa Fluor 594 and donkey anti-rabbit and anti-mouse Alexa Fluor 488, all at 1:1000 dilutions (Life Technologies). Slides were covered with mounting media (Prolong Gold Antifade Reagent with DAPI). Immmunofluorescent images were obtained with a confocal laser-scanning microscope (LSM 510 META, Carl Zeiss).
Analysis of IFNL4 mRNA expression
All expression assays (Supplementary Table 1 and Supplementary Fig. 10) were purchased from Life Technologies. Expression analysis was performed with gene expression master mix (Life Technologies) on DNAseI-treated RNA samples on ABI SDS 7700 instrument. Expression was measured in Ct values (PCR cycle at detection threshold), which are log 2 values. Expression was normalized to PPIA (endogenous control) and analyzed according to relative quantification method, as ΔCt=Ct PPIA −Ct target. Fold difference between any two samples can be calculated as fold=2^(ΔCt1 − ΔCt2).
Genotyping
Genotyping was performed with custom-designed TaqMan assays (Supplementary Table 1 and Supplementary Fig. 10), using Genotype Master Mix (Qiagen), on ABI SDS7700, with standard conditions. Testing was performed blinded to clinical phenotypes. Sanger sequencing of HapMap samples and subsets of clinical samples showed complete concordance with Taqman genotyping results.
Clinical and epidemiological studies
The studies had been approved by the institutional review boards of the participating institutions and all patients gave informed consent for genetic testing. Ancestral designation was self-reported in all studies. Full details of the studies are provided in a Supplementary Note included in Supplementary Materials.
Statistical analysis of genetic association
All statistical comparisons of IFNL4 ss469415590 and rs12979860 were limited to subjects successfully genotyped for both variants. The Kruskal-Wallis test was used to compare median HCV RNA levels between genotypes for each variant (e.g., ss469415590-TT/TT versus ss469415590-ΔG/ΔG). The mean HCV RNA levels in each of the three ss469415590 genotype groups (i.e., ΔG/ΔG, ΔG/TT, TT/TT) were compared with the respective rs12979860 genotype groups (TT, CT, CC). The global null hypothesis assumed no difference in decreases of mean HCV RNA values in ss469415590 compared to corresponding rs12979860 genotype groups. Statistical significance of these three mean differences was based on a Wald statistic (3 df) for which the variance was computed using a bootstrap based on re-sampling subjects with replacement (1000 re-samplings). For dichotomous outcomes (e.g., SVR, spontaneous clearance) the odds ratios and accompanying p-values (Wald chi-square) were calculated using proc logistic (SAS 9.2 TS2M3). The differences in the area under the receiver operating characteristic curve (AUC)58 values were evaluated with p-values based on a chi-square test (1 df) that used a bootstrap variance estimate computed by re-sampling subjects with replacement and then repeating the AUC computations for each bootstrap sample.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (National Cancer Institute, Division of Cancer Epidemiology and Genetics; National Institute for Diabetes, Digestive and Kidney Diseases), as well as the following grants: DA R01 013324 (J.A. and D.L.T.); ALIVE cohort R01-DA-04334 and R01-DA-12568, NIH grants #R01-DA09532, R01-DA12109, R01-DA13245 and R01-DA16159 (B. E.); NCI contracts # N02-CP-91027 and #N01-CO-12400 (B. E.), Substance Abuse and Mental Health Services Administration grant #H79-TI12103 (B. E.), and the NCI Director’s Innovation Award (L.P.-O.).
The Virahep-C and HALT-C studies were conducted by the Virahep-C and HALT-C Investigators and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The data and samples from the Virahep-C and HALT-C studies reported here were supplied by the NIDDK Central Repositories. This manuscript was not prepared in collaboration with the Virahep-C and HALT-C study groups and does not necessarily reflect the opinions or views of the Virahep-C and HALT-C studies, the NIDDK Central Repositories, or the NIDDK.
We thank NCI Sequencing Core Facility for help with RNA-seq. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
COMPETING FINANCIAL INTERESTS
L.P.-O, B. M., R. P. D. and T. R. O’B. are inventors on a patent application filed by the National Cancer Institute on the basis of these findings.
DATA DEPOSITION
Sequences for all novel transcripts JN806234, JN806225, JN806226, JN806232, JN806233, JN806227, JN806228, JN806229, JN806230 and JN806231 are deposited to NCBI database http://www.ncbi.nlm.nih.gov/, ss469415590 and ss539198934 are submitted to dbSNP database http://www.ncbi.nlm.nih.gov/snp/.
Reprints and permissions information is available at www.nature.com/reprints
AUTHOR CONTRIBUTIONS
L. P.-O. and T. R. O’B. conceived and supervised the project, J.A., H. L. B., B. R. E., C. D. H., T. R. M. and D. L. T. performed the clinical and epidemiologic studies that contributed the DNA samples and data; L. P.-O., B. R., R. P. D. designed the experiments, L. P-O., B. M, W. T., H. P., H. D., D. H., P. P.-G., I. K., A. M., N. B., M. T., L. L., F. S., B. R. and R. P. D. performed experiments and analysis, T. R. O’B., R.M.P and S. C. performed statistical analysis of genetic association; L.P.-O. and T.R.O’B. wrote the manuscript. All authors contributed to scientific discussions and approved the final manuscript.
References
- 1.National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C 2002 (June 10–12, 2002) Gastroenterology. 2002;123:2082–99. doi: 10.1053/gast.2002.1232082. [DOI] [PubMed] [Google Scholar]
- 2.NIH Consensus Statement on Management of Hepatitis C: 2002. NIH Consens State Sci Statements. 2002;19:1–46. [PubMed] [Google Scholar]
- 3.El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264–1273. e1. doi: 10.1053/j.gastro.2011.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calvaruso V, Craxi A. 2011 European Association of the Study of the Liver hepatitis C virus clinical practice guidelines. Liver Int. 2012;32 (Suppl 1):2–8. doi: 10.1111/j.1478-3231.2011.02703.x. [DOI] [PubMed] [Google Scholar]
- 5.Sarrazin C, Hezode C, Zeuzem S, Pawlotsky JM. Antiviral strategies in hepatitis C virus infection. J Hepatol. 2012;56 (Suppl 1):S88–100. doi: 10.1016/S0168-8278(12)60010-5. [DOI] [PubMed] [Google Scholar]
- 6.Ciesek S, Manns MP. Hepatitis in 2010: the dawn of a new era in HCV therapy. Nat Rev Gastroenterol Hepatol. 2011;8:69–71. doi: 10.1038/nrgastro.2010.219. [DOI] [PubMed] [Google Scholar]
- 7.Rauch A, et al. Genetic variation in IL28B Is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138:1338–1345. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 8.Thomas DL, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ge D, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 10.Suppiah V, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41:1100–4. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka Y, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41:1105–9. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 12.Kotenko SV, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 13.Sheppard P, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–8. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 14.Marcello T, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131:1887–98. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 15.Robek MD, Boyd BS, Chisari FV. Lambda interferon inhibits hepatitis B and C virus replication. J Virol. 2005;79:3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muir AJ, et al. Phase 1b study of pegylated interferon lambda 1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology. 52:822–32. doi: 10.1002/hep.23743. [DOI] [PubMed] [Google Scholar]
- 17.Kempuraj D, et al. Interleukin-28 and 29 (IL-28 and IL-29): new cytokines with anti-viral activities. Int J Immunopathol Pharmacol. 2004;17:103–6. doi: 10.1177/039463200401700201. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z, et al. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol. 2007;81:7749–58. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Honda M, et al. Hepatic ISG expression is associated with genetic variation in interleukin 28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139:499–509. doi: 10.1053/j.gastro.2010.04.049. [DOI] [PubMed] [Google Scholar]
- 20.Marukian S, et al. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology. 2011;54:1913–23. doi: 10.1002/hep.24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dill MT, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140:1021–31. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 22.Urban TJ, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology. 2010;52:1888–96. doi: 10.1002/hep.23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenbloom KR, et al. ENCODE whole-genome data in the UCSC Genome Browser: update 2012. Nucleic Acids Res. 2012;40:D912–7. doi: 10.1093/nar/gkr1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shyu AB, Wilkinson MF, van Hoof A. Messenger RNA regulation: to translate or to degrade. EMBO J. 2008;27:471–81. doi: 10.1038/sj.emboj.7601977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trivella DB, Ferreira-Junior JR, Dumoutier L, Renauld JC, Polikarpov I. Structure and function of interleukin-22 and other members of the interleukin-10 family. Cell Mol Life Sci. 2010;67:2909–35. doi: 10.1007/s00018-010-0380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gad HH, et al. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem. 2009;284:20869–75. doi: 10.1074/jbc.M109.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoon SI, Logsdon NJ, Sheikh F, Donnelly RP, Walter MR. Conformational changes mediate interleukin-10 receptor 2 (IL-10R2) binding to IL-10 and assembly of the signaling complex. J Biol Chem. 2006;281:35088–96. doi: 10.1074/jbc.M606791200. [DOI] [PubMed] [Google Scholar]
- 29.Yoon SI, et al. Structure and mechanism of receptor sharing by the IL-10R2 common chain. Structure. 2010;18:638–48. doi: 10.1016/j.str.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 31.Consortium GP. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conjeevaram HS, et al. Peginterferon and ribavirin treatment in African American and Caucasian American patients with hepatitis C genotype 1. Gastroenterology. 2006;131:470–7. doi: 10.1053/j.gastro.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 33.Di Bisceglie AM, et al. Prolonged therapy of advanced chronic hepatitis C with low-dose peginterferon. N Engl J Med. 2008;359:2429–41. doi: 10.1056/NEJMoa0707615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shebl FM, et al. IL28B rs12979860 genotype and spontaneous clearance of hepatitis C virus in a multi-ethnic cohort of injection drug users: evidence for a supra-additive association. J Infect Dis. 2011;204:1843–7. doi: 10.1093/infdis/jir647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vlahov D, et al. The ALIVE study, a longitudinal study of HIV-1 infection in intravenous drug users: description of methods and characteristics of participants. NIDA Res Monogr. 1991;109:75–100. [PubMed] [Google Scholar]
- 36.Thompson AJ, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology. 2010;139:120–9. e18. doi: 10.1053/j.gastro.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 37.Howell CD, Gorden A, Ryan KA, Thompson AJ, Ibrahim C, Fried M, Afdhal NH, McHutchison JG, Shianna KV, Goldstein DB, Shuldiner AR, Mitchell BD. Single nucleotide polymorphism upstream of interleukin 28B associated with phase 1 and phase 2 of early viral kinetics in patients infected with HCV genotype 1. J Hepatol. 2012;56:557–63. doi: 10.1016/j.jhep.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith KR, et al. Identification of improved IL28B SNPs and haplotypes for prediction of drug response in treatment of hepatitis C using massively parallel sequencing in a cross-sectional European cohort. Genome Med. 2011;3:57. doi: 10.1186/gm273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer J, et al. Combined effects of different interleukin-28B gene variants on the outcome of dual combination therapy in chronic hepatitis C virus type 1 infection. Hepatology. 2012;55:1700–10. doi: 10.1002/hep.25582. [DOI] [PubMed] [Google Scholar]
- 40.Jo J, et al. Analysis of CD8+ T-cell-mediated inhibition of hepatitis C virus replication using a novel immunological model. Gastroenterology. 2009;136:1391–401. doi: 10.1053/j.gastro.2008.12.034. [DOI] [PubMed] [Google Scholar]
- 41.Dill MT, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2010;140:1021–31. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 42.Asahina Y, et al. Association of gene expression involving innate immunity and genetic variation in interleukin 28B with antiviral response. Hepatology. 2012;55:20–9. doi: 10.1002/hep.24623. [DOI] [PubMed] [Google Scholar]
- 43.Onomoto K, et al. Dysregulation of IFN system can lead to poor response to pegylated interferon and ribavirin therapy in chronic hepatitis C. PLoS One. 2011;6:e19799. doi: 10.1371/journal.pone.0019799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarasin-Filipowicz M, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105:7034–9. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katsounas A, Schlaak JF, Lempicki RA. CCL5: a double-edged sword in host defense against the hepatitis C virus. Int Rev Immunol. 2011;30:366–78. doi: 10.3109/08830185.2011.593105. [DOI] [PubMed] [Google Scholar]
- 46.Tappero G, et al. Intrahepatic expression of c-fos, c-myb and c-myc oncogenes: correlation with virus-induced chronic liver disease and response to interferon. Ital J Gastroenterol Hepatol. 1997;29:148–54. [PubMed] [Google Scholar]
- 47.Smeyne RJ, et al. Continuous c-fos expression precedes programmed cell death in vivo. Nature. 1993;363:166–9. doi: 10.1038/363166a0. [DOI] [PubMed] [Google Scholar]
- 48.Kang SM, et al. c-Fos regulates hepatitis C virus propagation. FEBS Lett. 2011;585:3236–44. doi: 10.1016/j.febslet.2011.08.041. [DOI] [PubMed] [Google Scholar]
- 49.Okamura K, Feuk L, Marques-Bonet T, Navarro A, Scherer SW. Frequent appearance of novel protein-coding sequences by frameshift translation. Genomics. 2006;88:690–7. doi: 10.1016/j.ygeno.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 50.White SH. The evolution of proteins from random amino acid sequences: II. Evidence from the statistical distributions of the lengths of modern protein sequences. J Mol Evol. 1994;38:383–94. doi: 10.1007/BF00163155. [DOI] [PubMed] [Google Scholar]
- 51.Uccellini L, et al. HCV RNA levels in a multiethnic cohort of injection drug users: human genetic, viral and demographic associations. Hepatology. 2012;56:86–94. doi: 10.1002/hep.25652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chu TW, et al. Effect of IL28B genotype on early viral kinetics during interferon-free treatment of patients with chronic hepatitis C. Gastroenterology. 2012;142:790–5. doi: 10.1053/j.gastro.2011.12.057. [DOI] [PubMed] [Google Scholar]
- 53.Sonneveld MJ, et al. Polymorphisms near IL28B and serologic response to peginterferon in HBeAg-positive patients with chronic hepatitis B. Gastroenterology. 2012;142:513–520. e1. doi: 10.1053/j.gastro.2011.11.025. [DOI] [PubMed] [Google Scholar]
- 54.Lampertico P, et al. IL28B polymorphisms predict interferon-related HBsAg seroclearance in genotype D HBeAg-negative patients with chronic hepatitis B. Hepatology. 2012 doi: 10.1002/hep.25749. [DOI] [PubMed] [Google Scholar]
- 55.Davar D, Tarhini AA, Kirkwood JM. Adjuvant therapy for melanoma. Cancer J. 2012;18:192–202. doi: 10.1097/PPO.0b013e31824f118b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–6. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dickensheets HL, Venkataraman C, Schindler U, Donnelly RP. Interferons inhibit activation of STAT6 by interleukin 4 in human monocytes by inducing SOCS-1 gene expression. Proc Natl Acad Sci U S A. 1999;96:10800–5. doi: 10.1073/pnas.96.19.10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pepe MS. The statistical evaluation of medical tests for classification and prediction. Oxford University Press; Oxford: 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.