

Abstract
Determining the preferred substrate cleavage sequence of proteases is an important step towards understanding their roles in cancer development and progression. Knowledge of this sequence can aid in the design of new experimental tools for study as well as aid in the identification of endogenous protease substrates and signaling pathways. Various investigators have demonstrated a number of techniques in order to uncover these sequences, but most can be very time consuming. We have designed and successfully implemented a complete diverse ACC tetrapeptide positional scanning synthetic combinatorial library that allows for the rapid screening of proteases to determine their preferred residues at positions P1-P4. These sequences can be readily verified through kinetic measurements on single peptide substrates and utilized to further knowledge of the role of proteases in cancer.
Keywords: Peptide Library, Substrate Profiling, Protease Specificity, PS-SCL, ACC
1. Introduction
Understanding the in vivo role and function of proteases has become a promising avenue of research for identifying diagnostics, prognostics, and therapeutic targets in the fight against cancer. Although there have been a number of proteases identified in the various stages of cancer growth and progression, determining the precise role that each one plays has proven to be very challenging. One way to provide insight regarding these aspects is to identify the endogenous substrate(s) of each protease. Biochemical and proteomic methods can be used to identify a preferred cleavage sequence specific to a protease, which can in turn be used to identify the endogenous substrates and signaling pathways that involve the protease of interest. Unfortunately, many of the traditional methods for this can be time consuming. Positional scanning synthetic combinatorial libraries (PS-SCLs) of fluorogenic peptides offer the promise to expedite the initial steps towards identification of these substrates.
Positional scanning synthetic combinatorial libraries contain a mixture of peptides where one or more of the positions are individually fixed at a specific amino acid while the remaining positions are comprised of an equimolar mixture of amino acids. Through the use of combinatorial chemistry, these libraries can be quickly synthesized and analyzed. Dependent upon the number of randomized positions (X) and number of amino acids incorporated in those positions (Y), there will be YX different peptides in each mixture, with each containing the same fixed amino acid(s). The benefit of this preparation is the ability to determine the effect of the fixed amino acids independent of the residues in the remaining positions. Initial studies preparing and utilizing these libraries efficiently screened over 34 million hexapeptides to identify the antigenic determinant of a monoclonal antibody (1). This study demonstrated the utility of these libraries to rapidly screen through large numbers of peptides and determine the optimal sequences for peptide binding.
Because the PS-SCL approach proved to be very useful in scanning through large numbers of peptides, fluorogenic leaving groups were added to screen against proteases. The original libraries were created with an aminomethylcoumarin (AMC) fluorogenic leaving group attached to the C-terminus (the P1 amino acid) of the peptide via an amide bond (2, 3). Cleavage of the amide bond results in increased fluorescence of the free AMC group, providing an ideal reporter for enzyme activity. The initial libraries were successfully used to identify the substrate specificities of caspases 1–9, granzyme B, and interleukin 1β converting enzyme.
The AMC labeled PS-SCLs clearly revealed the potential of PS-SCLs towards understanding protease specificity. However, they were also inherently limited in scope. Synthesis of an AMC peptide substrate requires the AMC leaving group to be attached to the P1 amino acid C-terminus prior to solid phase synthesis. Under normal solid phase peptide synthesis conditions, the C-terminal carboxyl group of this first amino acid would be used for coupling to the resin. However, the presence of AMC on carboxyl terminus necessitates coupling to the resin through the amino acid side chain. This requirement severely limits the number of amino acids that can be incorporated into the P1 position to those with side chains that can be functionalized. Overall, the limitations produced AMC libraries that contained fixed P1 residues throughout while the P2-P4 residues were varied in order to test specificity. By restricting the P1 position to a single amino acid, these initial libraries were ideal for proteases with known P1 specificities, but had limited usefulness towards novel proteases.
The promise of these libraries coupled with this limitation led to the development of libraries utilizing a 7-amino-4-carbamoylmethylcoumarin (ACC) fluorogenic leaving group (4, 5). The bifunctionality of ACC allows for the attachment of an ACC-Fmoc directly to solid phase resin using standard Fmoc chemistry. Furthermonre, the ACC leaving group displays a ~3-fold higher fluorescent yield than AMC, increasing the sensitivity of the assay (4). Once attached to the resin, Fmoc chemistry can be used to incorporate any amino acid at the initial (P1) position, allowing all of the peptide positions to be varied. This development led to the complete diverse ACC-tetrapeptide libraries, which are able to analyze each position of the peptide substrate independently. Most importantly, with the ability to completely vary the P1 position, proteases of unknown specificity could now be assayed.
The complete diverse ACC-tetrapeptide PS-SCL contains 160,000 different sequences that can be assayed in a simple, rapid, and easy to interpret format. In this library, each position of the tetrapeptide is held constant for every amino acid while the remaining three positions are randomized (Figure 1). For each position, this creates 20 sublibraries, with each sublibrary composed of 8000 different peptides. By only holding one position constant, the role of the constant residue can be independently analyzed. Because the remaining positions are randomized, the importance of any particular residue in these positions is masked. The overall result reveals the relative preference of the protease for amino acids at positions P1-P4, as shown in Figure 2 for protease MT-SP1.
Figure 1.
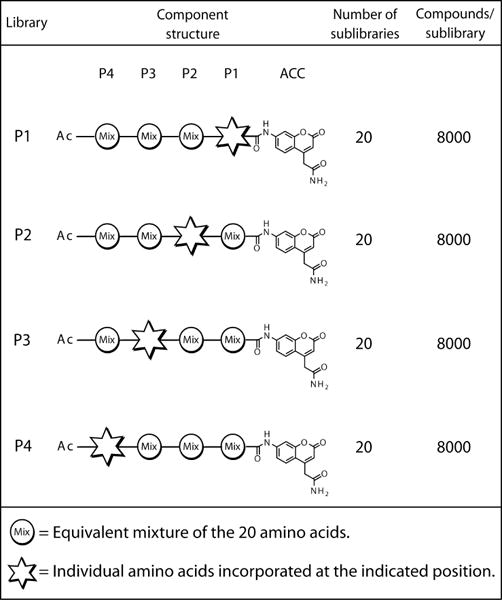
Composition of the complete diverse ACC tetrapeptide PS-SCL libraries. Each library holds one position constant for each of the 20 amino acids while the remaining positions are randomized. This creates 20 sublibraries per library (one per amino acid), with each sublibrary a mixture of 8000 peptides as a result of randomization at the other three positions (203). (Figure adapted from Choe et al. (8))
Figure 2.
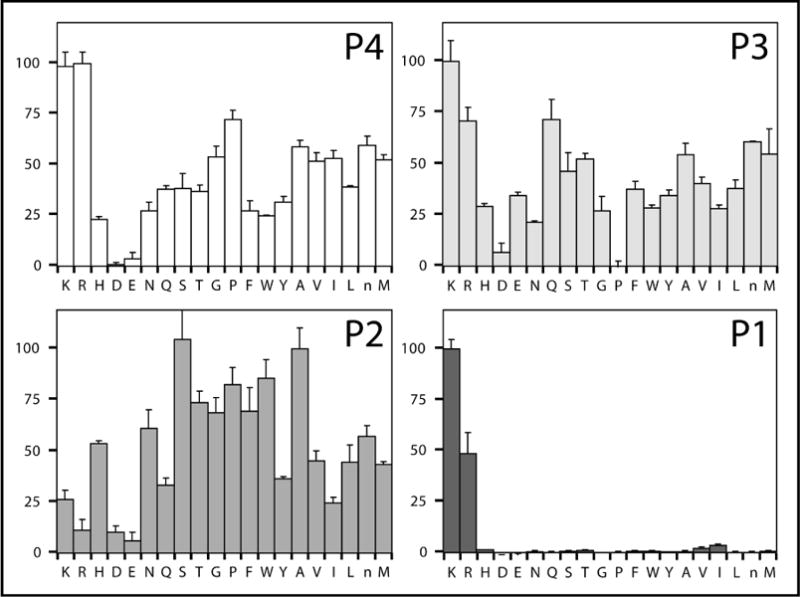
Complete diverse ACC-tetrapeptide PS-SCL results for MT-SP1 for each library P1 through P4. The y-axis is the relative percent activity within each libray. The x-axis indicates the amino acid being held constant.
Although it is extremely useful in determining the preferred residues at P1 through P4 for a given protease, there is an important caveat to interpretation of the data. By randomizing every position other than the position of interest, each sublibrary contains all possible peptides and ensures that if the residue in the constant position supports cleavage, a subset will be cleaved. However, at the same time, this design masks any interdependence between positions. The appearance of incompatible amino acids in positions other than the one being held constant will not be apparent. Returning to the results for MT-SP1 (Figure 2), the preferred substrate appears to be P4 Arg, P3 Lys, P2 Ser, and P1 Lys (RKSK). However, it is possible that each residue, although optimal when the other three positions are completely randomized, may not be compatible with one or more of the other residues when placed in the same sequence. For MT-SP1, this was exactly the case. An interdependence between binding pockets was revealed by phage display libraries that provided discrete peptide substrates to the protease (6). Similar to the library results, the P3 and P4 pockets were shown to prefer basic residues. However, both positions could not be a basic residue in a single peptide. Using the library data alone, this interdependence could not be detected at the P3 and P4 positions.
Alternatively, some proteases do not exhibit cooperativity between binding sites. In a study on granzymes A and B, the library results properly identified the optimal substrates for both, indicating that there was no interdependence between the binding sites (7). As seen in Figure 3, the optimal substrate for granzyme B was identified as P4 Ile, P3 Glu, P2 Pro, and P1 Asp (IEPD). Furthermore, this granzyme B substrate was found to be more sensitive than any previously reported substrate. Because it is difficult to predict if the binding sites will exhibit interdependence, like MT-SP1, or not, like granzymes A and B, the substrate candidate tetrapeptide sequences revealed through library profiling must be individually synthesized and tested to unequivocally show which one is optimal.
Figure 3.
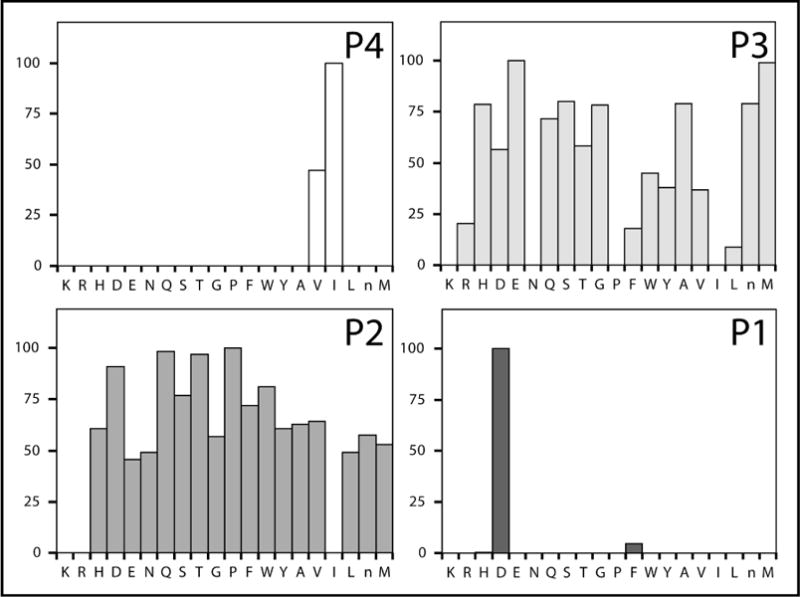
Complete diverse ACC-tetrapeptide PS-SCL results for granzyme B for each library P1 through P4. The y-axis is the relative percent activity within each libray. The x-axis indicates the amino acid being held constant.
Use of the complete diverse ACC tetrapeptide PS-SCL has lead to an increased understanding of the P1 through P4 specificity for a number of proteases implicated in cancer. Furthermore, use of this information has led to the creation of specific inhibitors as well as fulfilling the ultimate goal of assisting in the identification of new in vivo substrates. Examples of these uses can be found in a number of recent papers. In a paper by Choe et al., results from the library were used to design unique tetrapeptide substrates specific for cathepsin K over cathepsins S, L V and B (8). In addition, one of these peptide sequences was incorporated into a mechanism based inhibitor of cathepsin K. Although not as selective as the original peptide substrate, there was no inhibition of cathepsins L and V, while inhibiting B with a kinact about 2-fold slower than for cathepsin K. This indicates that results from the library may be utilized to design selective protease inhibitors.
In a separate paper, Marnett et al. analyzed the substrate specificity of Kaposi’s sarcoma-associated herpesvirus (KSHV) protease with the complete diverse library (9). The results revealed an unexpected preference for aromatic residues at P4. The complete results were successfully used to design both a tetrapeptide ACC substrate that was far superior to anything available as well as a mechanism based diphenyl phosphonate active site inhibitor. These tools helped to uncover a unique dimerization-dependent method of protease activation.
In a third example, the library was used by Bhatt et al. to assist in the identification of novel MT-SP1 substrates (10). The data for MT-SP1 (similar to that shown above in Figure 1) was combined with known endogenous substrate sequences and used to identify novel candidate substrates. Subsequent transcriptional profiling and cell based assays confirmed the identification of MSP-1 and its receptor RON as a MT-SP1 substrate. This in turn revealed an extracellular proteolytic signaling pathway that is important for metastatic breast cancer (11).
These results all demonstrate the usefulness of the complete diverse PS-SCL library in not only determining the substrate specificity of proteases, but also in the development of new tools for protease research. However, many proteases have additional specificity in the prime side pockets, which this library is unable to examine. Of particular importance to cancer research are the metalloproteases which exhibit a large dependence upon prime side binding for activity. For this reason, the development of tools to target the prime side specificity of these proteases is ongoing. One method developed by Barrios et al. has shown promise, but suffers from a relatively high background (12). More work is currently being carried out in order to develop a prime side library that demonstrates the robustness and utility of the complete diverse ACC-tetrapeptide PS-SCL described here.
The methods presented here begin with the synthesis of the P1 through P4 complete diverse ACC-tetrapeptide positional scanning synthetic combinatorial libraries. Each library is synthesized using standard Fmoc chemistry on an ACC linked resin to incorporate the fluorescent leaving group. Each library holds one of the positions of the tetrapeptide constant for all 20 amino acids, while completely randomizing the remaining 3 positions, as shown in the synthetic flow chart in Figure 3. The result is 20 sublibraries (one for each amino acid) per position in the tetrapeptide. The protocol for testing proteases against the newly synthesized libraries is then presented. Finally, the synthesis of individual substrates for confirmation of the library data is described.
2. Materials
2.1. Synthesis of PS-SCL ACC-tetrapeptide library
7-Fmoc-aminocoumarin-4-acetic acid (see Note 1)
Double distilled water (H2O)
Rink amide AM resin
N,N-Dimethylformamide (DMF)
Piperidine
N-Hydroxybenzotriazole (HOBt)
Diisopropylcarbodiimide (DICI)
Tetrahydrofuran (THF)
Phosphorous pentoxide (P2O5)
Methanol
Fmoc amino acids (Fmoc-Ala-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Asp(O-t-Bu)-OH, Fmoc-Glu(O-t-Bu)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Gly-OH, Fmoc-His(Trt)-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Lys(Boc)-OH, Fmoc-Met-OH, Fmoc-Nle-OH, Fmoc-Phe-OH, Fmoc-Pro-OH, Fmoc-Ser(O-t-Bu)-OH, Fmoc-Thr(O-t-Bu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Tyr(O-t-Bu)-OH, Fmoc-Val-OH) (see Note 2)
Collidine
2-(1H-7-Azabenzotriazol-1-yl)–1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium (HATU)
Acetic Acid
Nitrotriazole
Dichloromethane
Trifluoroacetic acid
Empty, fritted Solid Phase Extraction (SPE) column
Vacuum line with trap
Rotary evaporator
Argonaut Quest 210 organic synthesizer (Argonaut Technologies)
FlexChem 48-well reactor block (SciGene, Robbins Scientific)
2.2. Determination of Protease specificity using PS-SCL library
P1, P2, P3 and P4 PS-SCL ACC libraries as synthesized in 3.1.
The protease of interest, typically less than 25 nmol but possibly more.
25 mL of the optimal assay buffer for protease activity.
Fluorescence microplate reader (e.g. Molecular Devices SpectraMax Gemini)
Round-bottom 96 well plates for fluorescent plate readers (e.g. Thermo Scientific Microfluor 1 Black).
Multi-channel pipette capable of delivering 12 x 100 μL.
2.3. Synthesis of individual tetrapeptide ACC substrates for kinetic verification
Empty, fritted SPE column
DMF
P1 Fmoc amino acids bound to ACC-resin (from 3.1.B)
Individual Fmoc amino acids
Piperidine
HOBt
DICI
Acetic acid
Dichloromethane
TFA
Triisopropylsilane
H2O
DMSO
3. Methods
3.1. Synthesis of the complete diverse PS-SCL ACC-tetrapeptide library
-
ACC-Rink Amide Resin
In an empty round bottom flask, swell 17 mmol (21 g) of Rink Amide AM resin with 200 ml DMF for 30 minutes in order to make the functional groups accessible with gentle overhead stirring.
Vacuum filter to remove the DMF.
Add 200 ml of 20% piperidine in DMF to the resin to remove the Fmoc protecting group and activate the amine. Gently agitate to mix (see Note 3).
After 25 minutes, remove the piperidine by vacuum filtration and wash 3 times with 20 mL of DMF.
Add 2 equivalents (34 mmol, 15 g) of 7-Fmoc-aminocoumarin-4-acetic acid along with 2 equivalents HOBt (34 mmol, 4.6 g) and 150 mL DMF followed by 2 equivalents DICI (34 mmol, 5.3 mL).
Mix with gentle agitation overnight.
Filter the resin mixture and wash 3 times with 200 mL DMF followed by 3 washes with 200 mL tetrahydrofuran and 3 washes with 200 mL methanol.
Dry over P2O5 under vacuum.
Determine the substitution level of the resin by Fmoc analysis following the Millipore procedure (see Note 4)(13).
-
Preparation of Fmoc amino acid substituted ACC-resin
Add 100 mg of the Fmoc-ACC resin to 20 reaction vessels of an Argonaut Quest 210 organic synthesizer and swell with 2 mL of DMF (see Note 5). Calculate the molar quantity according to the substitution level determined above in 3.1.1.9.
Filter resin and add 2 mL of 20% piperidine in DMF to each reaction vessel. Gently agitate for 25 minutes to remove the Fmoc protecting group.
Filter resin and wash 3 times with 2 mL DMF.
To one of the reaction vessels containing the ACC resin add 5 molar equivalents of the Fmoc-Ala-OH along with 0.7 mL DMF, 10 molar equivalents of collidine, and 5 molar equivalents of HATU. Similarly add the remaining 19 Fmoc amino acids to the 19 wells containing ACC-resin (see Note 6). Gently agitate for 20 hours.
Filter the resin and wash 3 times with 2 mL DMF.
Repeat step 4 to ensure coupling occurs at a high efficiency.
Add 0.7 mL DMF, 40 μL acetic acid (0.7 mmol), 110 μL DICI (0.7 mmol), and 80 mg 3-nitro-1,2,4-triazol (0.7 mmol) to each reaction vessel and mix for 24 hours in order to cap any unreacted ACC-resin.
Filter the resin and wash 3 times with 2 mL DMF.
Repeat wash 3 times with 2 mL tetrahydrofuran and 3 times with methanol.
Dry the Fmoc-amino acids bound to ACC-resin over P2O5 under vacuum.
Determine the substitution level by Fmoc analysis as in 3.1.1.9.
-
Synthesis of the P1 sublibraries
According to the calculated Fmoc substitution levels, add 0.1 mmol of each of the 20 single Fmoc amino acids bound to ACC resin (prepared in 3.1.2) to 20 separate wells of a MultiChem 48-well synthesis apparatus. Each of the 20 wells should contain a different Fmoc-amino acid bound to ACC-resin.
Add 4 mL of DMF to each well for 30 minutes to solvate the resin.
Remove the DMF and add 4 mL of 20% piperidine in DMF to each well and gently agitate for 30 minutes to remove the Fmoc protecting group.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
Activate 20 mmol of an isokinetic mixture (see Note 7) of the 20 Fmoc amino acids (enough for all 20 wells at 10 equivalents/well compared to resin) in 80 mL DMF contianing 20 mmol HOBt and 20 mmol DICI.
Add 4 mL of the activated isokinetic Fmoc amino acid mixture to each of the 20 reaction vessels containing an Fmoc amino acid bound to ACC resin. Allow to couple with gentle agitation for 3 hours. This will add a randomized P2 amino acid to each P1 residue.
Drain and wash the resin 3 times with 4 mL DMF.
Repeat steps 3–7 two more times to add a randomized P3 and P4 residue, creating the 20 P1 sublibraries.
Remove the final Fmoc protecting group with 4 mL of 20% piperidine in DMF with gentle agitation for 30 minutes.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
Cap the final peptide with 80 mmol acetic acid, 80 mmol HOBt, and 80 mmol DICI in 4 mL DMF for 4 hours with gentle agitation
Remove the capping solution and wash 3 times with 4 mL DMF followed by 3 washes with 4 mL dichloromethane.
Cleave the peptides from the resin in a solution of 2850 μL TFA, 75 μL triisopropylsilane, and 75 μL water (95:2.5:2.5) for 1 hour with gentle agitation.
Collect the cleaved peptides and lyophilize.
Dissolve the peptides in DMSO to a final concentration of 25 mM (see Note 8).
-
Synthesis of the P2 sublibraries
According to the calculated substitution levels, create a 2 mmol mixture containing equal amounts of the 20 Fmoc-amino acids bound to ACC-resin (0.1 mmol each) and combine with 4 mL DMF in an empty SPE column. Mix for 2 hours with gentle agitation.
Remove the DMF and dry the resin mixture.
Split the resin evenly between 20 reaction vessels of a MultiChem 48-well synthesis apparatus (0.1 mmol resin per reaction vessel).
Add 4 mL DMF for 30 minutes to solvate the resin mixtures.
Remove the DMF and add 4 mL of 20% piperidine in DMF to each well and gently agitate for 30 minutes to remove the Fmoc protecting group.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
To insert individual amino acids in the P2 position, separately activate 1 mmol (10 molar equivalents compared to the resin) of each of the 20 Fmoc-amino acids in 4 mL DMF containing 1 mmol HOBt and 1 mmol DICI.
Add one of the activated Fmoc amino acids to one of the 20 reaction vessels containing the mixture of P1 amino acids bound to ACC-resin. Similarly add the 19 remaining activated Fmoc amino acids to the 19 reaction vessels containing resin.
Allow to couple with gentle agitation for 3 hours.
Drain and wash the resin 3 times with 4 mL DMF.
Add 4 mL of 20% piperidine in DMF to each well and gently agitate for 30 minutes to remove the Fmoc protecting group.
Remove the piperidine solution and wash 3 times with 4 mL DMF
Activate 20 mmol of an isokinetic mixture of the 20 Fmoc amino acids (enough for all 20 wells at 10 equivalents/well compared to resin) with 20 mmol HOBt and 20 mmol DICI in 80 mL DMF.
Add 4 mL of the activated isokinetic Fmoc amino acid mixture to each reaction vessel containing resin. Couple with agitation for 3 hours. This will add a randomized P3 residue to each known P2.
Drain and wash the resin 3 times with 4 mL DMF.
Repeat steps 11–15 once more to add a randomized P4 residue to each peptide, creating the P2 sublibraries.
Remove the final Fmoc protecting group with 4 mL of 20% piperidine in DMF with gentle agitation for 30 minutes.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
Cap the final peptide with 80 mmol acetic acid, 80 mmol HOBt, and 80 mmol DICI in 4 mL DMF for 4 hours.
Remove the capping solution and wash 3 times with 4 mL DMF followed by 3 washes with 4 mL dichloromethane.
Cleave the peptides from the resin in a solution of 2850 μL TFA, 75 μL triisopropylsilane, and 75 μL water (95:2.5:2.5) for 1 hour with gentle agitation.
Collect the cleaved peptides and lyophilize.
Dissolve the peptides in DMSO to a final concentration of 25 mM.
-
Synthesis of the P3 sublibraries
As in 3.1.4.1, create a 2 mmol mixture of the 20 Fmoc-amino acids bound to ACC-resin and add it to 4 mL DMF in an empty SPE column. Mix for 2 hours with gentle agitation.
Remove the DMF and dry the resin mixture.
Split the resin evenly between 20 reaction vessels of a MultiChem 48-well synthesis apparatus (0.1 mmol resin per each vessel).
Add 4 mL DMF for 30 minutes to solvate the resin mixtures.
Remove the DMF and add 4 mL of 20% piperidine in DMF to each well and gently agitate for 30 minutes to remove the Fmoc protecting group.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
Activate 20 mmol of an isokinetic mixture of the 20 Fmoc amino acids (enough for all 20 wells at 10 equivalents/well compared to resin) with 20 mmol HOBt and 20 mmol DICI in 80 mL DMF.
Add 4 mL of the activated isokinetic Fmoc amino acid mixture to each reaction vessel containing the mixture of Fmoc amino acids bound to ACC resin. Allow to couple with gentle agitation for 3 hours. This will add a randomized P2 residue.
Drain and wash the resin 3 times with 4 mL DMF.
Add 4 mL of 20% piperidine in DMF to each well and gently agitate for 30 minutes to remove the Fmoc protecting group.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
To insert individual amino acids in the P3 position, separately activate 1 mmol (10 molar equivalents compared to the resin) of each of the 20 Fmoc-amino acids in 4 mL DMF containing 1 mmol HOBt and 1 mmol DICI.
Add one of the activated Fmoc amino acids to one of the 20 reaction vessels containing resin. Similarly add the other 19 activated Fmoc amino acids to the 19 remaining reaction vessels containing resin.
Allow to couple with gentle agitation for 3 hours.
Drain and wash the resin 3 times with 4 mL DMF.
Repeat steps 4–9 once to add a randomized P4 residue to each peptide, creating the P3 sublibraries.
Remove the final Fmoc protecting group with 4 mL of 20% piperidine in DMF with agitation for 30 minutes.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
Cap the final peptide with 80 mmol acetic acid, 80 mmol HOBt, and 80 mmol DICI in 4 mL DMF for 4 hours with gentle agitation.
Remove the capping solution and wash 3 times with 4 mL DMF followed by 3 washes with 4 mL dichloromethane.
Cleave the peptides from the resin in a solution of 2850 μL TFA, 75 μL triisopropylsilane, and 75 μL water (95:2.5:2.5) for 1 hour with gentle agitation.
Collect the cleaved peptides and lyophilize.
Dissolve the peptides in DMSO to a final concentration of 25 mM.
-
Synthesis of the P4 sublibraries
As in 3.1.4.1, create a 2 mmol mixture of the 20 Fmoc-amino acids bound to ACC-resin and add it to 4 mL DMF in an empty SPE column. Mix for 2 hours with gentle agitation.
Remove the DMF and dry the resin mixture.
Split the resin evenly between 20 reaction vessels of a MultiChem 48-well synthesis apparatus (0.1 mmol resin per each vessel).
Add 4 mL DMF for 30 minutes to solvate the resin mixtures.
Remove the DMF and add 4 mL of 20% piperidine in DMF to each well and gently agitate for 30 minutes to remove the Fmoc protecting group.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
Activate 20 mmol of an isokinetic mixture of the 20 Fmoc amino acids (enough for all 20 wells at 10 equivalents/well compared to resin) with 20 mmol HOBt and 20 mmol DICI in 80 mL DMF.
Add 4 mL of the activated isokinetic Fmoc amino acid mixture to each reaction vessel containing the mixture of Fmoc amino acids bound to ACC resin. Allow to couple with gentle agitation for 3 hours. This will add a randomized P2 residue.
Drain and wash the resin 3 times with 4 mL DMF.
Repeat steps 5–9 to add a randomized P3 residue.
Add 4 mL of 20% piperidine in DMF to each well and gently agitate for 30 minutes to remove the Fmoc protecting group.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
To insert individual amino acids in the P4 position, separately activate 1 mmol (10 molar equivalents compared to the resin) of each of the 20 Fmoc-amino acids in 4 mL DMF containing 1 mmol HOBt and 1 mmol DICI.
Add one of the activated Fmoc amino acids to one of the 20 reaction vessels containing resin. Similarly add the 19 remaining activated Fmoc amino acids to the 19 reaction vessels containing resin.
Couple with gentle agitation for 3 hours.
Drain and wash the resin 3 times with 4 mL DMF.
Remove the final Fmoc protecting group with 4 mL of 20% piperidine in DMF with agitation for 30 minutes.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
Cap the final peptide with 80 mmol acetic acid, 80 mmol HOBt, and 80 mmol DICI in 4 mL DMF for 4 hours.
Remove the capping solution and wash 3 times with 4 mL DMF followed by 3 washes with 4 mL dichloromethane.
Cleave the peptides from the resin in a solution of 2850 μL TFA, 75 μL triisopropylsilane, and 75 μL water (95:2.5:2.5) for 1 hour with agitation.
Collect the cleaved peptides and lyophilize.
Dissolve the peptides in DMSO at a final concentration of 25 mM.
3.2 Determination of protease specificity using the complete diverse ACC-tetrapeptide PS-SCLs
To ensure that the protease will exhibit high enough activity to observe cleavage throughout the library, the activity of the enzyme should be tested at various concentrations against at least one of the P1 sublibraries that correspond to a known, or expected, P1 preferred residue for the protease. This information can then be used to adjust the protease concentration to give sufficient activity in the assay.
To test the activity, prepare protease samples at various concentrations within the typical working range of 1 to 1000 nM in 100 μL assay buffer optimized for the protease activity.
Spot 1 μL of the selected 25 mM P1 sublibrary in wells of a 96 well plate (see Note 9). (e.g. MT-SP1 (shown in Figure 2) has known trypsin-like activity and would therefore be initially tested against the P1 Arg or Lys sublibrary.)
Add the 100 μL protease samples prepared at various concentrations to the wells containing the sublibrary sample.
Monitor the hydrolysis reaction on a fluorescence plate reader taking readings every 15 seconds for 30 minutes with excitation at 380 nm and emission at 460 nm (see Note 10).
A good working concentration will give an rfu/sec reading of between 1 and 10 for a known substrate P1 residue. This will provide enough activity to measure cleavage of substrates containing less optimal residues at the constant position. Adjust the protease concentration appropriately to have activity within this range.
In order to keep the results consistent, it is best to prepare enough protease to run the entire library, P1 through P4. When performed in triplicate, the assay requires 24 mL of protease in assay buffer (triplicate × 20 residues/library × 100 μL/assay × 4 libraries = 24000 μL).
To ensure enough sample for the entire assay, prepare >24 mL of the protease in assay buffer at the enzyme concentration determined in steps 1–6.
In a 96 well plate, spot 1 μL of each P1 sublibrary substrate in triplicate, giving a total of 60 reactions.
Using a multi-channel pipette, add 100 μL of the protease solution to each of the 60 wells.
Monitor the hydrolysis reaction on the fluorescence plate reader taking readings every 15 seconds for 30 minutes with excitation at 380 nm and emission at 460 nm.
Repeat steps 8–11 for the P2, P3 and P4 libraries.
The results (reaction velocities) for each library can be standardized to the residue within that library giving the maximum activity for general comparison, as shown in Figure 2 (see Note 11).
3.3 Synthesis of individual tetrapeptide ACC substrates for kinetic verification
Because the results from the complete diverse ACC PS-SCL do not take into consideration the effects that neighboring residues contribute to binding pocket specificity, it is important to verify the preferred sequence suggested by the library. To test this, a few of the preferred sequences should be synthesized as ACC substrates following a procedure similar to that outlined for each sublibrary.
According to the calculated Fmoc substitution levels, add 0.1 mmol of the desired P1 Fmoc amino acid bound to ACC resin to an empty fritted SPE column.
Add 4 mL of DMF and gently agitate for 30 minutes to solvate the resin with.
Remove the DMF and add 4 mL of 20% piperidine in DMF and gently agitate for 30 minutes to remove the Fmoc protecting group.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
Activate 1 mmol of the desired P2 Fmoc amino acid (10 equivalents compared to the P1 substituted ACC resin) in 4 mL DMF with 1 mmol HOBt and 1 mmol DICI.
Add the activated Fmoc amino acid to the reaction vessel containing the P1 amino acid bound to ACC resin. Allow to couple with gentle agitation for 3 hours.
Drain and wash the resin 3 times with 4 mL DMF.
Repeat steps 4–8 two more times to add the desired P3 and P4 residues to the peptide.
Remove the final Fmoc protecting group with 4 mL of 20% piperidine in DMF with gentle agitation for 30 minutes.
Remove the piperidine solution and wash 3 times with 4 mL DMF.
Cap the final peptide with 80 mmol acetic acid, 80 mmol HOBt, and 80 mmol DICI in 4 mL DMF for 4 hours.
Remove the capping solution and wash 3 times with 4 mL DMF followed by 3 washes with 4 mL dichloromethane.
Cleave the peptide from the resin in a solution of 2850 μL TFA, 75 μL triisopropylsilane, and 75 μL H2O (95:2.5:2.5) for 1 hour with gentle agitation.
Collect the cleaved peptide and lyophilize.
Dissolve the peptide in DMSO to the desired concentration for kinetic evaluation.
Figure 4.
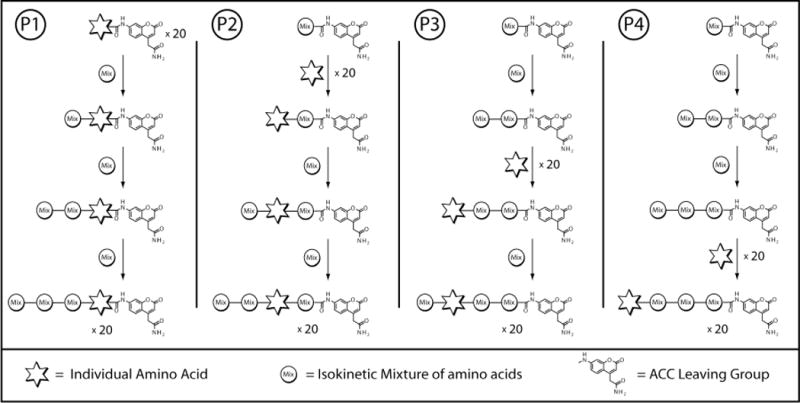
Overview of the synthesis for each library of the Complete Diverse Tetrapeptide-ACC PS-SCL. The steps shown incorporate a specific amino acid in the position labeled with a star and an equal mixture of the 20 amino acids in the other three positions. The final result is one of the 20 sublibraries for each library. The steps are then repeated 19 more times to complete each library for all 20 amino acids.
Acknowledgments
We thank M. D. Lim, M. Zhao, and Y. Choe for helpful discussions and critical reading of the manuscript. Grant support from NIH R01CA128765 to CSC is gratefully acknowledged.
Footnotes
7-Fmoc-aminocoumarin-4-acetic acid can be synthesized according to the procedures outlined by Harris et al. (4), Kanaoka, et al. (14), Besson et al. (15).
Cysteine was excluded due to its propensity to polymerize. Norleucine was included to extend the information revealed by the library. Norleucine has a four carbon side-chain similar to leucine and isoleucine, but unlike those amino acids it is unbranched. This extended form offers additional information about the protease binding pocket.
It is important not to use a stirbar for agitation of the resin in order to avoid damaging the resin. A platform shaker modified to hold the fritted reaction or the FlexChem reactor block upright works well for the gentle agitation required throughout this method. Alternatively an overhead stirrer could be used in some instances.
Addition of the first amino acid to the ACC-resin can also be accomplished in SPE columns following the same procedure. The Argonaut Quest was used to simplify the process.
The 20 amino acids used in the library described here are the 20 common amino acids excluding cysteine and including norleucine.
An isokinetic mixture contains a mixture of amino acids at a ratio based upon the individual reaction kinetics of coupling with a free amine that ensures an equal distribution of each amino acid in the final product. This is based on the findings that individual amino acids react with the free amine of a resin bound amino acid at different rates, but independent of the identity of the resin bound amino acid (4, 16). To create the library described (excluding cysteine and including norleucine) the following ratio was used (Amino Acid, mol %): Fmoc-Ala-OH, 3.3; Fmoc-Arg(Pbf)-OH, 6.4; Fmoc-Asn(Trt)-OH, 5.2; Fmoc-Asp(O-t-Bu)-OH, 3.4; Fmoc-Glu(O-t-Bu)-OH, 3.5; Fmoc-Gln(Trt)-OH, 5.2; Fmoc-Gly-OH, 2.8; Fmoc-His(Trt)-OH, 3.4; Fmoc-Ile-OH, 17.0; Fmoc-Leu-OH, 4.8; Fmoc-Lys(Boc)-OH, 6.1; Fmoc-Met-OH, 2.2; Fmoc-Nle-OH, 3.7; Fmoc-Phe-OH, 2.4; Fmoc-Pro-OH, 4.2; Fmoc-Ser(O-t-Bu)-OH, 2.7; Fmoc-Thr(O-t-Bu)-OH, 4.7; Fmoc-Trp(Boc)-OH, 3.7; Fmoc-Tyr(O-t-Bu)-OH, 4.0; Fmoc-Val-OH, 11.1. These isokinetic ratios were prepared based on those reported by Harris et al. and Ostresh et al. (4, 16).
After solubilizing in DMSO, the libraries should be stored at −80 °C.
Some of the peptide sublibraries are less soluble than others and may require gentle warming to ensure they are completely dissolved. Prior to use, check that all peptides are completely in solution and warm slightly if necessary.
With some proteases the resulting fluorescence readout jumps up and down over time, with an overall upward slope. Many times this can be reduced by addition of 0.01% Brij 35 to the reaction buffer.
Alternatively, the rfu/sec data can be converted to pM/sec and reported. This conversion factor must be experimentally determined for each instrument by measuring the rfu resulting from the complete hydrolysis of a known quantity of ACC-peptide. For the Molecular Devices SpectraMax Gemini, we have measured 1 pM of hydrolyzed ACC is equal to 1380 rfu.
References
- 1.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature. 1991;354:84–6. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 2.Rano TA, Timkey T, Peterson EP, Rotonda J, Nicholson DW, Becker JW, Chapman KT, Thornberry NA. A combinatorial approach for determining protease specificities: application to interleukin-1beta converting enzyme (ICE) Chem Biol. 1997;4:149–55. doi: 10.1016/s1074-5521(97)90258-1. [DOI] [PubMed] [Google Scholar]
- 3.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem. 1997;272:17907–11. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 4.Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS. Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci U S A. 2000;97:7754–9. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maly DJ, Leonetti F, Backes BJ, Dauber DS, Harris JL, Craik CS, Ellman JA. Expedient solid-phase synthesis of fluorogenic protease substrates using the 7-amino-4-carbamoylmethylcoumarin (ACC) fluorophore. J Org Chem. 2002;67:910–5. doi: 10.1021/jo016140o. [DOI] [PubMed] [Google Scholar]
- 6.Takeuchi T, Harris JL, Huang W, Yan KW, Coughlin SR, Craik CS. Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem. 2000;275:26333–42. doi: 10.1074/jbc.M002941200. [DOI] [PubMed] [Google Scholar]
- 7.Mahrus S, Craik CS. Selective chemical functional probes of granzymes A and B reveal granzyme B is a major effector of natural killer cell-mediated lysis of target cells. Chem Biol. 2005;12:567–77. doi: 10.1016/j.chembiol.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Bromme D, Ellman JA, Craik CS. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem. 2006;281:12824–32. doi: 10.1074/jbc.M513331200. [DOI] [PubMed] [Google Scholar]
- 9.Marnett AB, Nomura AM, Shimba N, Ortiz de Montellano PR, Craik CS. Communication between the active sites and dimer interface of a herpesvirus protease revealed by a transition-state inhibitor. Proc Natl Acad Sci U S A. 2004;101:6870–5. doi: 10.1073/pnas.0401613101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhatt AS, Welm A, Farady CJ, Vasquez M, Wilson K, Craik CS. Coordinate expression and functional profiling identify an extracellular proteolytic signaling pathway. Proc Natl Acad Sci U S A. 2007;104:5771–6. doi: 10.1073/pnas.0606514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welm AL, Sneddon JB, Taylor C, Nuyten DS, van de Vijver MJ, Hasegawa BH, Bishop JM. The macrophage-stimulating protein pathway promotes metastasis in a mouse model for breast cancer and predicts poor prognosis in humans. Proc Natl Acad Sci U S A. 2007;104:7570–5. doi: 10.1073/pnas.0702095104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrios AM, Craik CS. Scanning the prime-site substrate specificity of proteolytic enzymes: a novel assay based on ligand-enhanced lanthanide ion fluorescence. Bioorg Med Chem Lett. 2002;12:3619–23. doi: 10.1016/s0960-894x(02)00786-2. [DOI] [PubMed] [Google Scholar]
- 13.Bunin BA. The Combinatorial Index. Academic Press; San Diego: 1998. [Google Scholar]
- 14.Kanaoka Y, Kobayashi A, Sato E, Nakayama H, Ueno T, Muno D, Sekine T. Organic Fluorescent Reagents.10. Multifunctional Cross-Linking Reagents.1. Synthesis And Properties Of Novel Photoactivable, Thiol-Directed Fluorescent Reagents. Chemical & Pharmaceutical Bulletin. 1984;32:3926–3933. doi: 10.1248/cpb.32.3926. [DOI] [PubMed] [Google Scholar]
- 15.Besson T, Joseph B, Moreau P, Viaud MC, Coudert G, Guillaumet G. Synthesis And Fluorescent Properties Of New Heterobifunctional Fluorescent-Probes. Heterocycles. 1992;34:273–291. [Google Scholar]
- 16.Ostresh JM, Winkle JH, Hamashin VT, Houghten RA. Peptide libraries: determination of relative reaction rates of protected amino acids in competitive couplings. Biopolymers. 1994;34:1681–9. doi: 10.1002/bip.360341212. [DOI] [PubMed] [Google Scholar]