

Abstract
Objective
Hypoxia is intimately linked to atherosclerosis and has become recognized as a primary impetus of inflammation. We recently demonstrated that the neuroimmune guidance cue, netrin-1 (Ntn1) inhibits macrophage emigration from atherosclerotic plaques thereby fostering chronic inflammation. However, the mechanisms governing netrin-1 expression in atherosclerosis are not well understood. In this study, we investigate the role of hypoxia in regulating expression of netrin-1 and its receptor Unc5b in plaque macrophages, and its functional consequences on these immune cells.
Approach and Results
We show by immunostaining that netrin-1 and Unc5b are expressed in macrophages in hypoxia-rich regions of human and mouse plaques. In vitro, Ntn1 and Unc5b mRNA are upregulated in macrophages treated with oxidized LDL or inducers of oxidative stress (CoCL2, DMOG, 1% O2). These responses are abrograted by inhibiting HIF-1α, indicating a causal role for this transcription factor in regulating Ntn1 and Unc5b expression in macrophages. Indeed, using promoter-luciferase reporter genes, we show that Ntn1- and Unc5b-promoter activities are induced by oxLDL and require HIF-1α. Correspondingly, J774 macrophages overexpressing active HIF-1α show increased netrin-1 and Unc5b expression, and reduced migratory capacity compared to control cells, which was restored by blocking the effects of netrin-1. Finally, we show that netrin-1 protects macrophages from apoptosis under hypoxic conditions in a HIF-1α-dependent manner.
Conclusions
These findings provide a molecular mechanism by which netrin-1 and its receptor Unc5b are expressed in atherosclerotic plaques, and implicate hypoxia and HIF-1α-induced netrin-1/Unc5b in sustaining inflammation by inhibiting the emigration and promoting the survival of lesional macrophages.
Keywords: Macrophage, migration, guidance, HIF-1, Ntn1, apoptosis
Introduction
Atherosclerosis is a disease of chronic inflammation that is distinguished by the persistence of cholesterol-engorged macrophages in arterial plaques leading to disease progression and complications1. These cells are the central mediators of the establishment, progression and ultimately, instability, of atherosclerotic plaques. Unlike in other tissues, macrophages that accumulate in plaques appear to have a diminished capacity to migrate2-5, and go from being chemotactic to chemostatic, thereby contributing to a failure to resolve the inflammatory process in arteries set in motion by the retention of atherogenic lipoproteins. Although the regulatory signals that impair this process remain largely unknown, we recently showed that netrin-1, a laminin-like molecule normally expressed during embryonic development to guide the movement of neurons, is secreted by macrophage foam cells and inhibits the emigration of these cells from atherosclerotic plaques6. In the developing nervous system, netrin-1 can act as either a positive or negative regulator of migration depending on receptor expression by the target cell, and can also serve as a cell survival cue7. We found that netrin-1 differentially regulates cellular constituents of atheroma: netrin-1 blocks the migration of macrophages expressing the Unc5b receptor to chemokines (CCL2, CCL19, CCL21) implicated in the egress of macrophages from plaques, while promoting chemoattraction of smooth muscle cells expressing the Neogenin receptor6. These combined effects of netrin-1 – macrophage retention and smooth muscle cell recruitment – would be predicted to increase plaque size. Notably, targeted deletion of netrin-1 in myeloid cells markedly reduced atherosclerosis burden in Ldlr−/− mice and promoted macrophage emigration from plaques. These findings linked netrin-1 in macrophages to the promotion of chronic inflammation, however the mechanisms regulating expression of this guidance cue in atherosclerotic plaques are poorly understood.
The progression of atherosclerotic lesions is closely associated with micro-environmental changes within lipid- and macrophage-rich areas, particularly shifts in oxygen supply, which can lead to hypoxia8. As plaques grow bigger, the oxygen supply is decreased due to both diffusional limitations and the higher demand for oxygen by highly metabolically active cells within the plaque, leading to regions of hypoxia in both human and mouse atheroma. Furthermore, hypoxia has become recognized as a primary impetus of inflammation, and as inflamed lesions often become hypoxic, this further amplifies the inflammatory milieu of the plaque 9. A recent study reported that netrin-1 is induced by ambient hypoxia in mucosal surfaces of the intestine, where it protects the tissue from transient ischemia by attenuating neutrophil transepithelial migration10. In this setting, epithelial expression of netrin-1 was regulated by the hypoxia-inducible transcription factor (HIF), is a well-known sensor and mediator of hypoxic responses. HIF is a heterodimeric protein consists of 2 subunits: a constitutively expressed HIF-1β subunit, and a HIF-1α or HIF-2α subunit, whose protein levels are regulated by oxygen concentration11. HIF-1α and -2α are rapidly degraded by the ubiquitin/proteasome pathway in the presence of oxygen, but under hypoxic conditions these proteins translocate to the nucleus, dimerize with HIF-1β, and induce transcription of genes that coordinate cellular adaptions to hypoxia.
In atherosclerosis, HIF-1α is detected in macrophage-rich and hypoxic regions of human and mouse plaques12, 13. HIF-1α has been reported to regulate a number of gene programs relevant to atherosclerosis, including angiogenesis14, glucose metabolism11, apoptosis, nitric oxide metabolism, and the inflammatory response8. Furthermore, HIF-1α in the arterial wall may be stabilized under normoxic conditions by inflammation14, 15, particularly by inflammatory lipids that accumulate in plaques such as oxidized low density lipoprotein (oxLDL)4, 16. We thus postulated that hypoxic stress may promote netrin-1 expression in macrophages in atherosclerotic plaques, thereby increasing macrophage retention and chronic inflammation. We show herein that netrin-1 co-localizes with regions of hypoxia and HIF-1α accumulation in the arterial wall, most prominently in macrophages. In vitro, netrin-1 and its receptor Unc5b are induced in macrophages by hypoxic stressors, including oxLDL, and these responses are dependent on HIF-1α. Overexpression of an active form of HIF-1α reduces macrophage migration and this is reversed by blocking netrin-1, providing a mechanism for macrophage chemostasis during hypoxia. Finally, we show that netrin-1 also promotes the survival of macrophages under hypoxic conditions, which combined with its effects on macrophage retention, would be expected to fuel chronic inflammation in the plaque.
Materials and Methods
Please refer to supplementary materials.
Results
Hypoxia correlates with netrin-1 expression in atherosclerotic plaques
As netrin-1 expression has been shown to be upregulated in models of transient ischemia, we investigated whether netrin-1 is expressed in hypoxic regions of atherosclerotic plaques. We performed immunofluorescence staining of aortic sinus plaques from Ldlr−/− mice fed a western diet (WD) for 12 weeks and injected with a hypoxia probe (pimonidazole hydrochloride). Using double immunostaining on serial sections, we noted that the macrophage marker cluster of differentiation 68 (CD68) co-localized with the hypoxia probe (Fig. 1a). Furthermore, netrin-1 was prominently expressed in these regions of the plaque and showed significant co-localization with the hypoxia probe (Fig. 1b).
Figure 1. Hypoxia correlates with netrin-1 expression in mouse and human atherosclerotic plaques.

(a-b) Immunofluorescence staining of (a) macrophage CD68 (red) or (b) netrin-1 (red) in aortic root plaques from Ldlr−/− mice fed a Western diet for 12 weeks and injected with a hypoxia probe (green) that detects hypoxic regions. Areas of co-localization are shown in yellow in the merged image (arrows). Scale bar, 100 μm. The inset is a 10× zoom of the area of interest. L indicates the lumen. Images are representative of n ≥ 3 mice. Scale bar 500μm.
HIF-1α-mediated induction of Ntn1 in macrophages
To examine the role of hypoxia in the induction of the netrin-1 gene, Ntn1, we incubated BMDMs at a level of oxygen of 1%, which has previously been demonstrated in intraplaque regions13. Macrophages incubated under these conditions showed a 4-fold increase in Ntn1 mRNA, as well as the hypoxiainducible gene Vegf (Fig. 2a). Similar results were seen under normoxic conditions in macrophages treated with cobalt chloride (CoCl2), which mimics hypoxia by stabilizing hypoxia inducible factor 1a (HIF-1α)17 and with dimethyloxalylglycine (DMOG)18, which increases endogenous levels of this transcription factor by inhibiting the prolyl hydroxylases (Fig. 2b). Furthermore, as seen with the chemical inducers of hypoxia, oxLDL, which is believed to promote oxidative stress in the artery wall19, also increased both Ntn1 and Vegf mRNA (Fig. 2c, Suppl. Fig Ia) and increased Netrin-1 protein (Fig. 2d) in macrophages. Notably, the increase in Ntn1 mRNA under these various conditions was blocked by pretreating macrophages with an inhibitor of HIF-1α (Fig. 2c, e, Suppl. Fig. Ib), implicating this transcription factor in the upregulation of Ntn1.
Figure 2. Mediators of hypoxic stress induce Ntn1 in macrophages.

(a-b) qPCR analysis of Ntn1 and Vegf mRNA in BMDMs (a) exposed to 1% oxygen (hypoxia) or (b) stimulated with DMOG (1 mmol/L) and CoCl2 (0.1 mmol/L) for 24 hours. (c) qPCR analysis of Ntn1 and Vegf mRNA expression in peritoneal macrophages treated with oxLDL with or without HIF-1 inhibitor (100 μmol/L). (d) Western blot of netrin-1 in BMDM treated with 50 μg/ml oxLDL or 1% oxygen. (e) qPCR analysis of Ntn1 mRNA in BMDMs under normoxic or hypoxic conditions with or without HIF-1 inhibitor (100 μmol/L). (f) Netrin-1 promoter-luciferase reporter activity in HEK293 cells treated with oxLDL (50μg/ml) in the presence/absence of the HIF-1 (100 μmol/L). (g) qPCR analysis of Ntn1 mRNA in macrophages isolated from normoxic and hypoxic regions of the plaques of Ldlr−/− mice reconstituted with wild type or macrophage-specific Hif1a−/− (Mac-Hif1a−/−) bone marrow (n=4 mice/group). Data in a-f are the mean ± s.d. of triplicate samples in a single experiment and are representative of 3-4 independent experiments. *P<0.05.
To confirm a role for HIF-1α in the transcriptional regulation of Ntn1 in macrophages, we transfected HEK293 cells with a netrin-1 promoter-luciferase reporter gene, and treated cells with oxLDL or DMOG in the presence or absence a HIF-1α inhibitor. Ntn1 promoter activity was potently induced by oxLDL (Fig. 2f) and DMOG (Suppl. Fig. Ic), and this increase was dependent on HIF-1α activity. To investigate the role of HIF-1α in inducing Ntn1 in vivo, we used laser capture microdissection to isolate macrophages from normoxic and hypoxic regions of the plaques of Ldlr−/− mice transplanted with wild type or macrophage-specific Hif1a−/− (Mac-Hif1a−/−) bone marrow. Notably, there was a 2.5-fold increase in Ntn1 mRNA in macrophages isolated from hypoxic compared to normoxic regions of the plaque. By contrast, no increase in Ntn1 mRNA was measured in macrophages isolated from hypoxic regions of plaques from Ldlr−/−Mac-Hif1a−/− mice (Fig. 2g). Together, these data indicate a central role for HIF-1α in the upregulation of Netrin-1 expression in macrophages under hypoxic conditions relevant to atherosclerosis.
HIF-1α induces the netrin-1 receptor Unc5b in macrophages
To investigate the role of HIF-1α in regulating macrophage expression of netrin-1 receptors and other neuronal guidance molecules, we used a mouse J774 macrophage cell line transduced with a retrovirus expressing HIF-1α engineered to be stable in normoxic conditions13. Gene expression profiling of J774-Con and J774-HIF-1α macrophages using a custom microarray for the analysis of guidance cues of the netrin, slit, semaphorin and ephrin families and their receptors, showed that overexpression of HIF-1α induced expression of Ntn1 and its chemorepulsive receptor Unc5b, but decreased expression of its chemoattractant receptor Dcc (Fig. 3a, b, d). Notably, J774-HIF-1α cells also showed decreased expression of the adenosine A2a and A2b receptors (Adora2a, Adora2b), which have previously been implicated in the neutrophil response to netrin-1 during transient ischemia10. To confirm these changes at the protein level, we performed immunofluorescent staining of J774-HIF-1α and control macrophages. Notably, J774-HIF-1α but not J774-Con cells showed abundant netrin-1 and Unc5b staining (Fig. 3c), indicating that netrin-1 and unc5b are regulated by a common mechanism during hypoxia.
Figure 3. Hypoxia induces select guidance cues and receptors in macrophages.

(a-b) mRNA profiling of J774 macrophages overexpressing a stable form of HIF-1 (J774Hif) versus J774 control (J774Con) cells using a custom qRT-PCR array for the neuronal guidance cue family members (n=3) expressed as (a) a heat map or (b) relative fold change in mRNA expression. (c) Immunofluorescence staining of J774Con and J774Hif cells for HIF-1 netrin-1 and Unc5b. Scale bar = 50 μm. (d) qPCR validation of mRNA expression of Ntn1 and its receptors in J774Hif compared to J774con macrophages. Data are mean ± s.d of triplicate samples in a single experiment and are representative of an experimental n=4. *P<0.05.
To further understand the molecular mechanisms regulating the Unc5b receptor in hypoxic conditions, we incubated macrophages with 1% oxygen, or under normoxic conditions with oxLDL or chemical hypoxia mimics. As seen for Ntn1 and Vegf, macrophages incubated under these conditions showed 3-6-fold increases in Unc5b mRNA (Fig. 4a-c, Suppl. Fig. Id). This increase in Unc5b mRNA was dependent on HIF-1α, as macrophages pre-treated with a HIF-1α inhibitor show no upregulation of Unc5b in response to oxLDL, low oxygen concentration, CoCl2 or DMOG (Fig. 4c, Suppl. Fig. Ie). Furthermore, BMDMs incubated with oxLDL or under hypoxic conditions, showed increases in Unc5b protein (Fig. 4e). To confirm the role of HIF-1α in the transcriptional regulation of Unc5b, we transfected HEK293 cells with an Unc5b promoter-luciferase reporter gene. OxLDL dose-dependently increased luciferase activity in these cells (Fig. 4f), and treatment with a HIF-1α inhibitor abrogated this increase in Unc5b promoter activity (Fig. 4g). A similar HIF-1α-dependent increase in Unc5b promoter activity was also observed in cells treated with DMOG or CoCl2 under normoxic conditions (Fig. 4h), and this was also dependent on NFκB (Fig. 4i). Consistent with these in vitro studies, immunofluorescent staining of mouse atherosclerotic plaques showed co-localization of Unc5b with a hypoxia probe (Fig. 5a). Furthermore, macrophages isolated by laser capture microdissection from hypoxic regions of plaques Ldlr−/− mice show a 5-fold increase in Unc5b mRNA compared to macrophages from normoxic regions (Fig. 5b). By contrast, no increase in Unc5b mRNA was measured in macrophages isolated from hypoxic regions of plaques from Ldlr−/−Mac-Hif1a−/− mice (Fig. 5b). Collectively, these data suggest a key role for HIF-1α in the upregulation of both netrin-1 and its receptor Unc5b in plaque macrophages, which would be predicted to promote an autocrine/paracrine signaling loop.
Figure 4. Mediators of hypoxic stress induce Unc5b in macrophages.

qPCR analysis of Unc5b, Ntn1 or Vegf mRNA in BMDM macrophages treated with (a) DMOG (1 mmol/L) and CoCl2 (0.1 mmol/L), (b) 1% oxygen, or (c) 50 μg/ml oxLDL, and (c-d) with or without HIF-1 inhibitor (100 μmol/L). (e) Western blot of Unc5b in BMDM treated with 50 μg/ml oxLDL or 1% oxygen. (f) Unc5b promoter-luciferase reporter activity in HEK293 cells treated with the indicated concentration of oxLDL. (g-h) Unc5b promoter-luciferase reporter activity in HEK293 cells treated with (g) oxLDL (50μg/ml) or (h) DMOG (1 mmol/L) in the presence/absence of a HIF-1 (100μmol/L). (i) Unc5b promoter-luciferase reporter activity in HEK293 cells treated with DMOG (1 mmol/L) in the presence/absence of an NFkB inhibitor (BAY11-7082). Data are mean ± s.d. of triplicate samples in a single experiment and are representative of 3 independent experiments. *P<0.05.
Figure 5. Unc5b is expressed in hypoxic-rich regions in mouse and human atherosclerotic lesions.
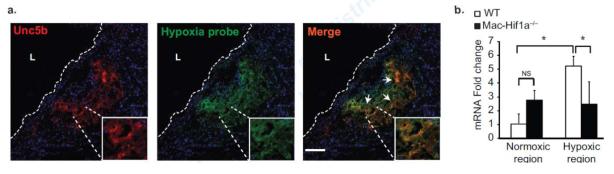
Immunofluorescence staining of (a) Unc5b (red) in aortic root plaques from Ldlr−/− mice fed a Western diet for 12 weeks and injected with a hypoxia probe (green) to detect hypoxic regions. Areas of co-localization are shown in yellow in the merged image (arrows). Scale bar, 100 μm. The inset is a 10× zoom of the area of interest. L indicates the lumen. Images are representative of n ≥ 3 mice. (b) qPCR analysis of Unc5b mRNA in macrophages isolated from normoxic and hypoxic regions of the plaques of Ldlr−/− mice reconstituted with wild type or macrophage-specific Hif1a−/− (Mac-Hif1a−/−) bone marrow (n=4 mice/group). Data are mean ± s.d. *P<0.05.
Netrin-1 and Unc5b are increased by hypoxia in human monocytes and atherosclerotic plaques
To extend these findings in mouse atherosclerotic models to human atherosclerosis, we first treated the human monocytic cell line THP-1 with oxLDL under normoxic conditions or subjected the cells to hypoxic conditions by incubating them in 1% oxygen. As seen in mouse macrophages, these conditions induce 4-5 fold increases in NTN1 (Fig. 6a) and UNC5B (Fig. 6b) mRNA. The upregulation of NTN1 and UNC5B by oxLDL or hypoxia was reduced by treating cells with an inhibitor of HIF-1 (Fig. 6a-b). We next stained human carotid endarterectomy sections for netrin-1, Unc5b and HIF-1α. HIF-1α was detected in the central region of human plaques, in areas maximally distant from the blood supply (Fig. 6c-d) and staining for netrin-1 and Unc5b co-localized with HIF-1α in these regions. These data suggest a common mechanism of HIF-1α-dependent regulation of netrin-1 and Unc5b in macrophages in mouse and human atherosclerosis.
Figure 6. Netrin-1 and Unc5b are induced by hypoxic stress in human monocytes and co-localize with HIF-1α in human atheroma.

qPCR analysis of (a) Ntn1 and (b) Unc5b mRNA in human THP-1 cells treated with 50μg/ml oxLDL under normoxic conditions or exposed to 1% oxygen (hypoxia) for 24 hours, in the presence or absence of a HIF-1α inhibitor (100 μmol/L). Data are mean ± s.d. of triplicate samples in a single experiment and are representative of 3 independent experiments. *P<0.05. (c-d) Immunofluorescence staining of human carotid plaques for (c) netrin-1 (red), (d) Unc5b (red) and HIF-1α (green). DAPI nuclear stain is in blue. Areas of co-localization are shown in yellow in the merged image (arrows). Scale bar of inset images = 500μm. Brightfield image on left indicates the region of staining (boxed area) which is in areas maximally distant from the blood supply. Staining is representative of plaques from 5 subjects.
Netrin-1 reduces macrophage migration and promotes survival
As netrin-1 secreted during hypoxia may act in an autocrine/paracrine manner on Unc5b-expressing macrophages, we next examined the functional consequences of this regulation. To understand the role of netrin-1 in regulating macrophage migration under hypoxic conditions, we measured the chemotaxis of control or CoCl2-treated BMDM to MCP-1 (CCL2) in the presence and absence of recombinant Unc5b-FC to block the effects of netrin-1. Macrophages treated with CoCl2 were strongly impaired in their ability to migrate to MCP-1 compared to control macrophages (Fig. 7a). Notably, treatment of macrophages with Unc5b-FC reversed this effect, restoring macrophage migration to MCP-1. Similarly, overexpression of stable HIF-1α, which induces netrin-1 expression, reduced the migratory capacity of J774 macrophages in the absence of exogenous chemokine (Fig. 7b), whereas J774-HIF cell treated with Unc5b-FC exhibited migration levels comparable to control J774 macrophages. Together these data indicate that upregulation of netrin-1 and Unc5b by hypoxic stress promotes macrophage chemostasis.
Figure 7. Hypoxia-induced netrin-1 inhibits the migration and promotes the survival of macrophages.

(a) Real-time migration of BMDMs to media alone, MCP-1 (100 nmol/L) in the presence or absence of CoCl2 (0.1 mmol/L) or Unc5bFC. (b) Real-time migration of J774con or J774Hif macrophages in the presence or absence of Unc5b-Fc to block the effects of netrin-1. (c-d) Apoptosis measured by TUNEL assay of BMDMs cultured in L929 conditional media (CM) or deprived of growth factor (no CM) under normoxic or hypoxic (1% oxygen) conditions. Cells were incubated with (c) a HIF-1α inhibitor (100μM) (d) control IgG or Unc5b-FC. Data are the mean ± s.d of triplicate samples in a single experiment and are representative of an experimental n=3. *P<0.05. (e) BMDM apoptosis as measured by active caspase-3 staining of BMDMs cultured in L929 conditional media (CM) or deprived of growth factor and cultured under normoxic or hypoxic (1% oxygen) conditions in the presence or absence of Unc5b-FC.
In addition to regulating migration, netrin-1 has also been reported to be a survival factor for neurons20, 21. To test whether netrin-1 inhibits macrophage apoptosis under hypoxic conditions, we measured apoptosis of growth-factor deprived BMDM under normoxic and hypoxic conditions. Primary BMDM cultivated in the absence of L929 conditioned media undergo apoptosis under normoxic conditions, with a 5-fold increase in TUNEL staining observed after 3 days (Fig. 7c). However, these cells are largely protected from this apoptosis, as shown by reduced TUNEL staining when cultured under hypoxic conditions, and this protection from apoptosis is reversed in the presence of a HIF-1α inhibitor. To determine whether HIF-1α induction of netrin-1 contributes to macrophage survival under hypoxic conditions, we repeated this experiment in the presence of recombinant Unc5b-FC to block the effects of netrin-1 or a control FC fragment. Under hypoxic conditions, growth factor deprived macrophages treated with control FC remained protected from apoptosis, whereas cells treated with Unc5b-FC showed increased TUNEL and active caspase-3 staining similar to normoxic controls (Fig. 7d-e). These results suggest that hypoxia- and HIF-1α-induced expression of netrin-1 promotes the survival of macrophages, which would be predicted to foster macrophage accumulation in plaques.
Discussion
It is now widely accepted that hypoxia and inflammation are intertwined: hypoxia can induce inflammation and inflamed tissues often become hypoxic, fueling a continuous cycle of inflammation. Hypoxia and chronic inflammation are characteristic features of atherosclerosis, in which macrophages accumulate prominently in arterial plaques. Our data provide a molecular mechanism for macrophage persistence during hypoxia through the upregulation of netrin-1, a neuroimmune guidance cue that inhibits chemokine-directed migration of macrophages in vitro and in vivo, including in atherosclerotic plaques6. We show that netrin-1 and its receptor Unc5b are expressed in macrophages in hypoxic regions of mouse and human atherosclerotic lesions. Furthermore, HIF-1α, a transcription factor that mediates cellular adaptations to hypoxia, regulates the expression of both netrin-1 and Unc5b, leading to macrophage chemostasis and protection from apoptosis under hypoxic conditions. Thus, the regulation of the netrin-1-Unc5b axis under hypoxic conditions would be predicted to promote the accumulation of these highly metabolically active cells, further amplifying oxidative stress and chronic inflammation in atherosclerosis.
Recent studies indicate that hypoxia plays a key role in the progression of atherosclerotic plaques to advanced stages by increasing lipid accumulation13, 19, inflammation and angiogenesis. The transcription factor HIF-1α is a central regulator of these cellular programs, which help hypoxic cells adapt to their hostile environment. Previous studies from our group reported that similar to human atheroma, hypoxic regions are present in mouse atherosclerotic plaques and show increased accumulation of HIF-1α13. In vitro, such hypoxic conditions alter macrophage lipid metabolism by increasing macrophage foam cell formation through both induction of sterol synthesis and suppression of cholesterol efflux through a mechanism involving HIF-1α13. Lipid-laden macrophage foam cells are known to have a diminished capacity to migrate2-5 thereby promoting macrophage retention in plaques, and in the current study we identify hypoxia and stabilization of HIF-1α as factors that contribute to this retention by promoting macrophage chemostasis and survival. Hypoxic stress induced by low oxygen concentration, or treatment with oxLDL (a key inflammatory component of plaques that increases oxidative stress) upregulated macrophage expression of netrin-1 and its receptor Unc5b. This neuroimmune guidance cue-receptor interaction was recently shown to block macrophage migration to chemokines implicated in the egress of macrophages from plaques or other inflammatory sites (e.g. CCL19, CCL2)22-24. Furthermore, targeted deletion of netrin-1 in macrophages reduced atherosclerosis burden in Ldlr−/− mice and restored macrophage emigration from plaques6. Notably, we now show that macrophages overexpressing HIF-1α or treated with a hypoxia mimic have impaired migratory capacity, and this hypoxia-induced macrophage chemostasis is reversed by blocking netrin-1. These data suggest that strategies that target hypoxia, thereby reducing netrin-1 and Unc5b, may facilitate macrophage emigration from the plaque and the resolution of inflammation.
In addition to regulating cell migration, as noted above, we now also show that netrin-1 plays an important role in promoting macrophage survival during hypoxic stress. HIF-1α has been reported to promote the survival of neutrophils in hypoxic conditions25, and we show a similar role for HIF-1α in protecting macrophages from apoptosis during hypoxia. Notably, inhibition of netrin-1 in macrophages in vitro under these conditions resulted in a loss of protection from apoptosis. While the effect of netrin-1 on leukocyte survival has not been investigated previously, forced expression of DCC or UNC5 in neuronal and tumor cells in the absence of netrin1 leads to cell apoptosis, which can be rescued by supplying recombinant netrin-126-29. Similarly, netrin-1 has been reported to suppress apoptosis in renal tubular epithelial cells in ischemia/reperfusion injury30. DCC and UNC5 are thought to initiate cell death in the absence of their ligand, as has been described for other dependence receptors including the neurotrophin receptor p75NTR and the androgen receptor31-34.
Whereas the HIF-1α-induced upregulation of netrin-1 and Unc5b in macrophage foam cells in atherosclerotic plaques would be predicted to foster inflammation by both blocking macrophage emigration and sustaining the survival of these trapped cells, other studies report protective roles for HIF-1α and netrin-1 during inflammation. For example, during transient ischemia in the gut, upregulation of netrin-1 on epithelial cells by HIF-1α attenuates neutrophil recruitment and protects the tissue from hypoxia-induced inflammation10. In this model, neutrophil responses to netrin-1 required the adenosine A2B receptor to limit hypoxia-induced inflammation, but not Unc5b, which we found to be the active receptor in macrophages. In fact, using a custom microarray to measure expression of members of the netrin family and its receptors, we find in macrophages that HIF-1α upregulates netrin-1, Unc5a and Unc5b, but highly downregulates other netrin-1 receptors, including the adenosine A2B receptor and DCC. Consistent with this, Unc5b was detected in macrophages in hypoxic regions of the plaque and co-localized with HIF-1α. In vitro, regulation of the Unc5b promoter was also found to depend on NFkB, a master transcriptional regulator of inflammatory genes, which has been shown to upregulate HIF-1α35. In this context, increased secretion of netrin-1 and upregulated Unc5b expression by macrophages would be predicted to inactivate emigration and enhance the survival of these immune cells, thereby promoting chronic inflammation. Although not investigated here, netrin-1 can be expressed by endothelial cells36, and we have previously demonstrated that in this context netrin-1 regulates monocyte adhesion to endothelial cells37. As hypoxia may also affect endothelial cells in atheromata, it may potentially alter the expression of netrin-1 on the endothelium. Thus, as in the nervous system where netrin-1 can have both positive and negative effects on axonal migration, netrin-1 may play multifunctional roles in regulating inflammation depending on the site of its expression (ie. plaque vs endothelium or epithelium) and the receptors it engages.
Together, these studies strongly suggest that netrin-1 and its receptor, Unc5b, initially characterized in the specialized migration of neurons during development, also regulate macrophage trafficking and accumulation during hypoxia in atherosclerosis. These findings also directly implicate HIF-1α regulation of netrin-1 in promoting macrophage accumulation and survival in atherosclerotic plaque and provide further evidence of a role for hypoxia in sustaining inflammation.
Supplementary Material
SIGNIFICANCE.
Our data show that netrin-1 and its receptor Unc5b, initially characterized in the specialized migration of neurons during development, are expressed in macrophages in hypoxic regions of mouse and human atherosclerotic lesions. Furthermore, HIF-1α, a transcription factor that mediates cellular adaptations to hypoxia, regulates the expression of both netrin-1 and Unc5b, leading to macrophage chemostasis and protection from apoptosis under hypoxic conditions. The regulation of the netrin-1-Unc5b axis under hypoxic conditions would be predicted to promote the accumulation of these highly metabolically active cells, further amplifying oxidative stress and chronic inflammation in atherosclerosis. Collectively, these findings implicate HIF-1α regulation of netrin-1 in promoting macrophage accumulation and survival in atherosclerotic plaques and provide further evidence of a role for hypoxia in sustaining inflammation.
ACKNOWLEDGEMENTS
None
SOURCES OF FUNDING: Support for this work came from the National Institutes of Health (RC1HL100815 to K.J.M; R01 HL084312 to EAF), Fondation de la Recherche Medicale (B.R.), American Heart Association (Grant-in-aid 0655840T to K.J.M; 09POST2080250 to J.M.vG), and German Research Foundation (DFG: HE 6092/1-1 to BH).
Abbreviations
- oxLDL
oxidized low density lipoprotein
- Unc5b
uncoordinated-5-B receptor
- DMOG
dimethyloxaloylglycine
Footnotes
DISCLOSURES: none
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ross R. Atherosclerosis is an inflammatory disease. Am Heart J. 1999;138:S419–420. doi: 10.1016/s0002-8703(99)70266-8. [DOI] [PubMed] [Google Scholar]
- 2.Nagao T, Qin C, Grosheva I, Maxfield FR, Pierini LM. Elevated cholesterol levels in the plasma membranes of macrophages inhibit migration by disrupting rhoa regulation. Arterioscler Thromb Vasc Biol. 2007;27:1596–1602. doi: 10.1161/ATVBAHA.107.145086. [DOI] [PubMed] [Google Scholar]
- 3.Park YM, Febbraio M, Silverstein RL. Cd36 modulates migration of mouse and human macrophages in response to oxidized ldl and may contribute to macrophage trapping in the arterial intima. J Clin Invest. 2009;119:136–145. doi: 10.1172/JCI35535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin C, Nagao T, Grosheva I, Maxfield FR, Pierini LM. Elevated plasma membrane cholesterol content alters macrophage signaling and function. Arterioscler Thromb Vasc Biol. 2006;26:372–378. doi: 10.1161/01.ATV.0000197848.67999.e1. [DOI] [PubMed] [Google Scholar]
- 5.Randolph GJ. Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr Opin Lipidol. 2008;19:462–468. doi: 10.1097/MOL.0b013e32830d5f09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Gils JM, Derby MC, Fernandes LR, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13:136–143. doi: 10.1038/ni.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cirulli V, Yebra M. Netrins: Beyond the brain. Nat Rev Mol Cell Biol. 2007;8:296–306. doi: 10.1038/nrm2142. [DOI] [PubMed] [Google Scholar]
- 8.Vink A, Schoneveld AH, Lamers D, Houben AJ, van der Groep P, van Diest PJ, Pasterkamp G. Hif-1 alpha expression is associated with an atheromatous inflammatory plaque phenotype and upregulated in activated macrophages. Atherosclerosis. 2007;195:e69–75. doi: 10.1016/j.atherosclerosis.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 9.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL. Targeting hif-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 12.Sluimer JC, Gasc JM, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn Gelpke MD, Cleutjens JP, van den Akker LH, Corvol P, Wouters BG, Daemen MJ, Bijnens AP. Hypoxia, hypoxiainducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51:1258–1265. doi: 10.1016/j.jacc.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 13.Parathath S, Mick SL, Feig JE, Joaquin V, Grauer L, Habiel DM, Gassmann M, Gardner LB, Fisher EA. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ Res. 2011;109:1141–1152. doi: 10.1161/CIRCRESAHA.111.246363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Celletti FL, Waugh JM, Amabile PG, Brendolan A, Hilfiker PR, Dake MD. Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat Med. 2001;7:425–429. doi: 10.1038/86490. [DOI] [PubMed] [Google Scholar]
- 15.Hellwig-Burgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood. 1999;94:1561–1567. [PubMed] [Google Scholar]
- 16.Bostrom P, Magnusson B, Svensson PA, Wiklund O, Boren J, Carlsson LM, Stahlman M, Olofsson SO, Hulten LM. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol. 2006;26:1871–1876. doi: 10.1161/01.ATV.0000229665.78997.0b. [DOI] [PubMed] [Google Scholar]
- 17.Piret JP, Mottet D, Raes M, Michiels C. Cocl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line hepg2. Ann N Y Acad Sci. 2002;973:443–447. doi: 10.1111/j.1749-6632.2002.tb04680.x. [DOI] [PubMed] [Google Scholar]
- 18.Groenman FA, Rutter M, Wang J, Caniggia I, Tibboel D, Post M. Effect of chemical stabilizers of hypoxia-inducible factors on early lung development. Am J Physiol Lung Cell Mol Physiol. 2007;293:L557–567. doi: 10.1152/ajplung.00486.2006. [DOI] [PubMed] [Google Scholar]
- 19.Shatrov VA, Sumbayev VV, Zhou J, Brune B. Oxidized low-density lipoprotein (oxldl) triggers hypoxia-inducible factor-1alpha (hif-1alpha) accumulation via redox-dependent mechanisms. Blood. 2003;101:4847–4849. doi: 10.1182/blood-2002-09-2711. [DOI] [PubMed] [Google Scholar]
- 20.Mazelin L, Bernet A, Bonod-Bidaud C, Pays L, Arnaud S, Gespach C, Bredesen DE, Scoazec JY, Mehlen P. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature. 2004;431:80–84. doi: 10.1038/nature02788. [DOI] [PubMed] [Google Scholar]
- 21.Castets M, Coissieux MM, Delloye-Bourgeois C, Bernard L, Delcros JG, Bernet A, Laudet V, Mehlen P. Inhibition of endothelial cell apoptosis by netrin-1 during angiogenesis. Dev Cell. 2009;16:614–620. doi: 10.1016/j.devcel.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 22.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor ccr7 during atherosclerosis regression in apoe-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato N, Ahuja SK, Quinones M, Kostecki V, Reddick RL, Melby PC, Kuziel WA, Ahuja SS. Cc chemokine receptor (ccr)2 is required for langerhans cell migration and localization of t helper cell type 1 (th1)-inducing dendritic cells. Absence of ccr2 shifts the leishmania major-resistant phenotype to a susceptible state dominated by th2 cytokines, b cell outgrowth, and sustained neutrophilic inflammation. J Exp Med. 2000;192:205–218. doi: 10.1084/jem.192.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jimenez F, Quinones MP, Martinez HG, Estrada CA, Clark K, Garavito E, Ibarra J, Melby PC, Ahuja SS. Ccr2 plays a critical role in dendritic cell maturation: Possible role of ccl2 and nf-kappa b. J Immunol. 2010;184:5571–5581. doi: 10.4049/jimmunol.0803494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tadagavadi RK, Wang W, Ramesh G. Netrin-1 regulates th1/th2/th17 cytokine production and inflammation through unc5b receptor and protects kidney against ischemia-reperfusion injury. J Immunol. 2010;185:3750–3758. doi: 10.4049/jimmunol.1000435. [DOI] [PubMed] [Google Scholar]
- 26.Llambi F, Causeret F, Bloch-Gallego E, Mehlen P. Netrin-1 acts as a survival factor via its receptors unc5h and dcc. Embo J. 2001;20:2715–2722. doi: 10.1093/emboj/20.11.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forcet C, Ye X, Granger L, Corset V, Shin H, Bredesen DE, Mehlen P. The dependence receptor dcc (deleted in colorectal cancer) defines an alternative mechanism for caspase activation. Proc Natl Acad Sci U S A. 2001;98:3416–3421. doi: 10.1073/pnas.051378298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams ME, Strickland P, Watanabe K, Hinck L. Unc5h1 induces apoptosis via its juxtamembrane region through an interaction with nrage. J Biol Chem. 2003;278:17483–17490. doi: 10.1074/jbc.M300415200. [DOI] [PubMed] [Google Scholar]
- 29.Chen YQ, Hsieh JT, Yao F, Fang B, Pong RC, Cipriano SC, Krepulat F. Induction of apoptosis and g2/m cell cycle arrest by dcc. Oncogene. 1999;18:2747–2754. doi: 10.1038/sj.onc.1202629. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Reeves WB, Pays L, Mehlen P, Ramesh G. Netrin-1 overexpression protects kidney from ischemia reperfusion injury by suppressing apoptosis. Am J Pathol. 2009;175:1010–1018. doi: 10.2353/ajpath.2009.090224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE. Induction of apoptosis by the low-affinity ngf receptor. Science. 1993;261:345–348. doi: 10.1126/science.8332899. [DOI] [PubMed] [Google Scholar]
- 32.Bredesen DE, Ye X, Tasinato A, Sperandio S, Wang JJ, Assa-Munt N, Rabizadeh S. P75ntr and the concept of cellular dependence: Seeing how the other half die. Cell Death Differ. 1998;5:365–371. doi: 10.1038/sj.cdd.4400378. [DOI] [PubMed] [Google Scholar]
- 33.Ellerby LM, Hackam AS, Propp SS, Ellerby HM, Rabizadeh S, Cashman NR, Trifiro MA, Pinsky L, Wellington CL, Salvesen GS, Hayden MR, Bredesen DE. Kennedy’s disease: Caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J Neurochem. 1999;72:185–195. doi: 10.1046/j.1471-4159.1999.0720185.x. [DOI] [PubMed] [Google Scholar]
- 34.Bordeaux MC, Forcet C, Granger L, Corset V, Bidaud C, Billaud M, Bredesen DE, Edery P, Mehlen P. The ret proto-oncogene induces apoptosis: A novel mechanism for hirschsprung disease. Embo J. 2000;19:4056–4063. doi: 10.1093/emboj/19.15.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by nf-kappab. Biochem J. 2008;412:477–484. doi: 10.1042/BJ20080476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ly NP, Komatsuzaki K, Fraser IP, Tseng AA, Prodhan P, Moore KJ, Kinane TB. Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102:14729–14734. doi: 10.1073/pnas.0506233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Gils JM, Ramkhelawon B, Fernandes L, et al. Endothelial expression of guidance cues in vessel wall homeostasis: Dysregulation under proatherosclerotic conditions. Arterioscler Thromb Vasc Biol. 2013 doi: 10.1161/ATVBAHA.112.301155. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.